

Abstract
Gene expression is extensively regulated by the occurrence and distribution of the epigenetic marker 2′‐deoxy 5‐methylcytosine (5mC) in genomic DNA. Because of its effects on tumorigenesis there is an important link to human health. In addition, detection of 5mC can serve as an outstanding biomarker for diagnostics as well as for disease therapy. Our previous studies have already shown that, by processing O 6‐alkylated 2′‐deoxyguanosine triphosphate (dGTP) analogues, DNA polymerases are able to sense the presence of a single 5mC unit in a template. Here we present the synthesis and evaluation of an extended toolbox of 6‐substituted 2‐aminopurine‐2′‐deoxyribonucleoside 5′‐triphosphates modified at position 6 with various functionalities. We found that sensing of 5‐methylation by this class of nucleotides is more general, not being restricted to O 6‐alkyl modification of dGTP but also applying to other functionalities.
Keywords: 5mC, DNA methylation, DNA polymerases, epigenetics, gene expression, modified nucleotides
Introduction
Methylation of DNA in position 5 of cytosine (5mC) occurs predominantly at CpG islands1 that are (largely) made up of CpG dinucleotides in promotor regions in the mammalian genome and is one of the most important epigenetic markers.2 The role of 5mC in the mammalian genome is so distinct that this epigenetic marker is considered to be the “fifth base” of DNA.3 5mC serves as an important repressor of transcription,4 with hypermethylation of CpG islands being associated with gene silencing,4 thereby affecting tumorigenesis.5 Cytosine 5‐methylation is also the key mechanism mediating genomic imprinting6 and silencing of foreign DNA7 and is crucial for cellular development8 and differentiation.9
Even though 5mC is the best‐studied epigenetic modification,9 some aspects are still poorly understood.2 Detection of the occurrence and distribution of 5mC in the genome additionally holds the potential to serve as an important biomarker for diagnosis and disease therapy,10 due to the important link of 5mC to human health.11 Therefore, efficient methods for the detection of 5mC are required.
Different strategies for 5mC detection have already been described and used for genome‐wide 5mC mapping. However, all of these methods—which rely variously on affinity enrichment,12 endonuclease digestion,13 nanopore sequencing,14 specific interactions of proteins with 5mC15 or different chemical behaviour involving redox reactivity16 or selective deamination with sodium bisulfite17—show several drawbacks. The most common method for the detection of 5mC is bisulfite sequencing, which can reveal the sites of epigenetic markers through comparison with the output of conventional sequencing methods.18 Sodium bisulfite reversibly adds to the 5,6 double bond in pyrimidine nucleobases under acidic conditions, but no further reaction to replace the amino groups takes place.19 Adjusting the pH to basic conditions converts non‐5‐methylated cytosine adducts into their uracil counterparts upon elimination.20 In the case of 5mC, this deamination (to thymine) via the corresponding sulfonate adduct is nearly two orders of magnitude slower than that of the unmodified cytosine counterpart.20 Bisulfite sequencing, taking advantage of this rate difference in the deamination of C and of 5mC, was subsequently established.
Although bisulfite sequencing has been used for genome‐wide 5mC detection,6 it has several drawbacks due to its reaction requirements.21 The conditions used during bisulfite treatment are harsh and destroy around 95 % of the genomic DNA,22 so large amounts of sample material are required. The method is time‐consuming and prone to contamination21 because many steps are needed and two sequencing runs are required for comparison. Additionally, deamination of C and 5mC after bisulfite treatment is deficient, leading to an error‐prone output.23
Our goal, therefore, is a method for 5mC detection that can reveal the sites of epigenetic marks without the need to perform modification reactions prior to sequencing, thereby limiting the sources of error.
As reported previously,24 we discovered that 6‐substituted 2‐aminopurine‐2′‐deoxyribonucleoside 5′‐triphosphates are able to sense 5mC in DNA polymerase‐catalysed reactions. After testing several different purine‐based 2′‐deoxynucleotides with regard to their ability to be used in 5mC detection, we found that modification in the 6‐position was most promising. Nucleotides modified in this position were incorporated opposite C by Thermococcus kodakaraensis exo‐ DNA polymerase (KOD exo‐) with notably different efficiencies than they were opposite 5mC, with favoured processing opposite C. In order to investigate whether the observed discrimination is more general and extendable to other modifications, we decided to explore different 6‐substituted 2‐aminopurine‐2′‐deoxyribonucleoside 5′‐triphosphates that are modified at position 6 (Figure 1). We found that the observed effect is more general and not restricted to O 6‐alkyl modification of dGTP.
Figure 1.
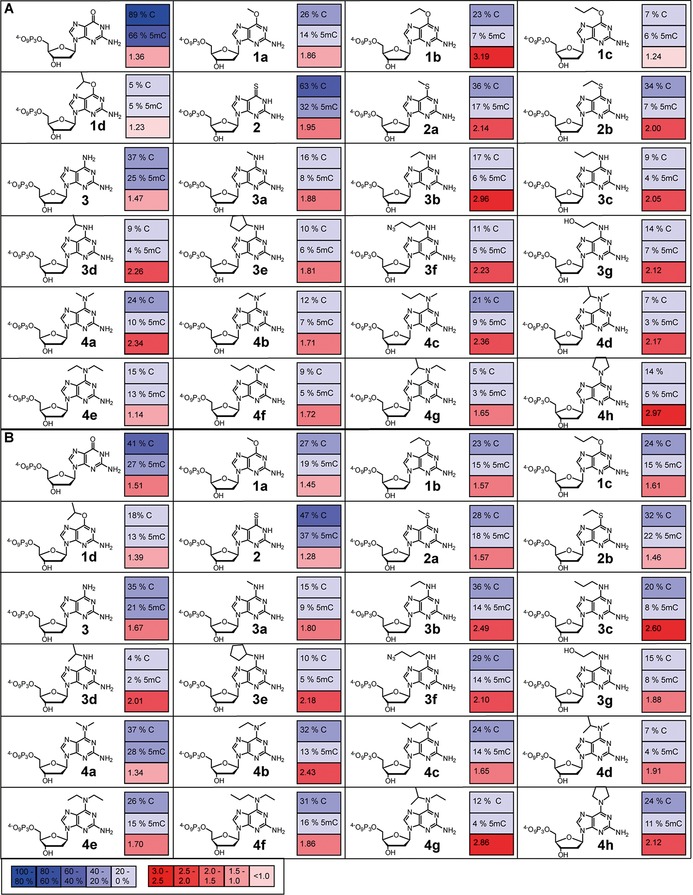
Discrimination observed on utilising 10 nm of A) KOD exo‐ DNA polymerase or B) 9°North exo‐ DNA polymerase with indicated modified nucleotides (50 μm). Reactions were stopped after 5 min; % incorporation values opposite C or 5mC were calculated from the integrated gel band intensities [% incorporation=(100×I extension)/(I primer+I extension)]. Discrimination was determined from the ratio of incorporation opposite C to incorporation opposite 5mC. Experiments were done in triplicate; arithmetic means are given. Values given in (A) for nucleotides 1 a–d have already been published.24
Results and Discussion
In order to investigate whether interruption of the Watson–Crick face of dGTP interferes with DNA polymerase‐catalysed processing opposite C and 5mC, we decided to synthesise thioethers 2 and the secondary and tertiary amines 3 and 4 (Scheme 1).
Scheme 1.
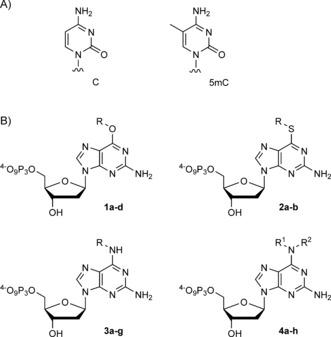
A) Chemical structures of C and 5mC. B) Chemical structures of modified nucleotides. Compounds 1–3: a: R=methyl, b: R=ethyl, c: R= propyl, d: R=isopropyl, e: R=cyclopentyl, f: R=3‐azidopropyl, g: R=2hydroxyethyl K. Compounds 4: a: R1=methyl, R2=methyl, b: R1=methyl, R2=ethyl, c: R1=methyl, R2=propyl, d: R1=methyl, R2=isopropyl, e: R1= ethyl, R2=ethyl, f: R1=ethyl, R2=propyl, g: R1=ethyl, R2=isopropyl, h: R1+R2=pyrrolidine.
The synthesis of the nucleotides 6‐methylthio‐dGTP (2 a) and 6‐ethylthio‐dGTP (2 b) is shown in Scheme 2. Because insertion of the modification by use of a nucleoside or nucleotide precursor failed, we introduced the modifications at the nucleobase level by nucleophilic replacement,25 starting from commercially available 2‐amino‐6‐chloropurine, as already reported for nucleotide 2 a.26 The modified nucleobases 5 a and 5 b were next used for glycosylation27 to yield 6 a and 6 b. Subsequent deprotection of the hydroxy groups under basic conditions26 resulted in nucleosides 7 a and 7 b, which were converted into the corresponding 5′‐O‐triphosphates 2 a and 2 b by standard triphosphorylation methods.28
Scheme 2.
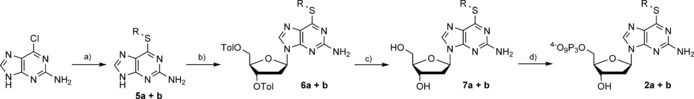
Synthesis of nucleotides 2 a and 2 b. a) 5 a: NaSMe, DMF, RT, 16 h (55 %); 5b: KOtBu, EtSH, DMF, reflux, 16 h (49 %); b) i: NaH (60 % in mineral oil), CH3CN, RT, 30 min; ii: 1‐chloro‐2‐deoxy‐3,5‐di‐O‐toluoyl‐α‐d‐ribofuranose, RT, 20 h (6 a: 64 %, 6 b: 59 %); c) 7 n ammonia in MeOH, 4 °C, 16 h (7 a: 99 %, 7 b: 61 %); d) i: N,N,N′,N′‐tetramethylnaphthalene‐1,8‐diamine, trimethyl phosphate (TMP), POCl3, 0 °C, 30 min; ii: (Bu3NH)2H2P2O7, DMF, nBu3N, RT, 30 min, 3. 0.1 m triethylammonium bicarbonate (TEAB), RT, 30 min (2 a: 28 %, 2 b: 28 %).
To circumvent the need to perform laborious triphosphorylation reactions for every modified nucleotide, we established the synthesis of the 6‐amino‐modified dGTP derivatives 3 a–g and 4 a–h by treating the precursor nucleotide 2‐amino‐6‐chloropurine‐2′‐deoxyriboside triphosphate (10) with aqueous solutions of the appropriate amines; yields ranged from 18–97 % (see Scheme 3). For the synthesis of the chlorinated nucleotide precursor 10, the hydroxy groups of commercially available 2′‐deoxyguanosine required to be masked and afterwards selectively removed prior to 5′‐triphosphorylation of the nucleoside. We employed tert‐butyldimethylsilyl (TBDMS) groups and started the synthesis with 3′,5′‐TBDMS‐protected 2′‐deoxyguanosine. By following known procedures,29 we introduced chlorine in position 6 to obtain nucleoside 8 in a moderate yield of 37 %. The OH groups were subsequently deprotected by treatment with triethylamine trihydrofluoride in THF (93 %). Afterwards, 5′‐triphosphorylation of 8 by standard procedures afforded 9.27
Scheme 3.
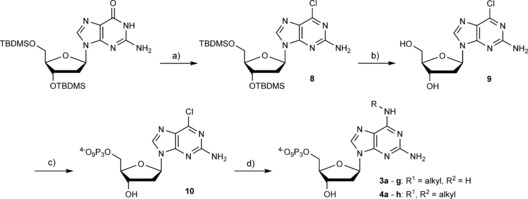
Synthesis of nucleotides 3 a–g and 4 a–g. a) Tetraethylammonium chloride, N,N‐dimethylaniline, POCl3, RT–reflux, 25 min (37 %); b) triethylamine trihydrofluoride, THF, RT, 16 h (93 %); c) i: N,N,N′,N′‐tetramethylnaphthalene‐1,8‐diamine, TMP, POCl3, 0 °C, 30 min; ii: (Bu3NH)2H2P2O7, DMF, nBu3N, RT, 30 min; iii: 0.1 m TEAB, RT, 30 min (13 %); d) aq. amine, RT, 16 h (3 a: 93 %, 3 b: 87 %, 3 c: 63 %, 3 d: 43 %, 3 e: 97 %, 3 f: 86 %, 3 g: 52 %, 4 a: 85 %, 4 b: 73 %, 4 c: 67 %, 4 d: 34 %, 4 e: 65 %, 4 f: 48 %, 4 g: 27 %, 4 h: 18 %).
Next, the modified nucleotides were tested with regard to their action with 5mC. Thus, primer extension by single‐nucleotide incorporation opposite C and 5mC followed by analysis by denaturing PAGE and visualisation by autoradiography were performed with the modified nucleotides in combination with three different DNA polymerases. The KlenTaq DNA polymerase, belonging to the sequence‐family A,30 and the KOD exo‐ DNA polymerase, a member of the sequence‐family B,30 were tested with regard to their ability to incorporate the modified nucleotides with increased differences in incorporation efficiencies opposite C or 5mC. Because previous experiments24 had shown the KOD exo‐ DNA polymerase to be most promising for the desired application, we tested the DNA polymerase 9°North exo‐ as a second sequence‐family B polymerase as well.
As already indicated by previous experiments,24 no significant discrimination between C and 5mC was observed on employing KlenTaq DNA polymerase in combination with any of the modified nucleotides (Figure S1 in the Supporting Information). In addition, the incorporation efficiencies of KlenTaq DNA polymerase for the modified nucleotides decreased considerably with increasing size of the introduced modifications (Figure S1). Consequently, nucleotide 1 a was incorporated with higher efficiencies than nucleotide 1 d. The same holds true for the thio‐ and amino‐modified nucleotides, with nucleotide 4 a being processed with significantly higher efficiencies than nucleotide 4 h bearing the bulky and inflexible pyrrolidine modification.
In contrast, on utilising the sequence‐family B KOD exo‐ and 9°North exo‐ DNA polymerases we found that both DNA polymerases were capable of incorporating all modified nucleotides with only slightly decreased incorporation efficiencies relative to the natural dGMP. Interestingly, we observed a tendency towards more efficient processing of all modified nucleotides opposite C than opposite 5mC by both sequence‐family B DNA polymerases (Figures 1, S2 and S4).
By employing KOD exo‐ DNA polymerase with the 6‐thio‐modified nucleotides 2, 2 a and 2 b we were able to observe enhanced discrimination for the commercially available nucleotide 6‐thio‐dGTP (2). This discrimination increased on introduction of a methylthio (compound 2 a) or ethylthio (compound 2 b) group. Similar effects were observed for the nucleotides 6‐methoxy‐dGTP (1 a) and 6‐ethoxy‐dGTP (1 b).24 For the 6‐amino‐modified nucleotides 3 a–g and 4 a–h we found similar trends. Again, the discrimination increased with introduction of a methylamino (compound 3 a) or ethylamino (compound 3 b) group in comparison with 6‐amino‐dGTP (3). Interestingly, this effect vanishes after introduction of more bulky modifications (3 c–g, Figures 1 A and S2). Consequently, the decrease in discrimination due to increasing size of the modifications, such as in the introduction of a propylamino (compound 3 c) or isopropylamino (compound 3 d) group leads to discrimination ratios of only 2. On increasing the bulkiness of the modification even further through the introduction of a cyclopentyl group (compound 3 e), the discrimination ratio decreased to less than 2. This discrimination ratio could be slightly increased again by employing the 3‐azidopropanamino group (compound 3 f). However, the discrimination ratio still does not surpass the discrimination observed on incorporation of 3 b.
In addition, the difference in incorporation opposite C and 5mC could be further increased by employment of tertiary amines (compounds 4 a–h). Discrimination observed with use of the nucleotide modified with the doubly methylated amino group (compound 4 a) is higher than the discrimination observed for incorporation of 3 a. Consistent with those trends observed above, discrimination increased with increasing size of the introduced modifications up to a certain degree (compound 4 c), to decrease again when the steric hindrance of the employed modifications was enhanced further (compounds 4 d–g). Surprisingly, though, the highest discrimination could be observed with the most rigid, sterically hindered pyrrolidine modification (compound 4 h). In this case, a threefold higher incorporation opposite C could be observed than opposite 5mC (Figures 1, 2 B and S2).
Figure 2.
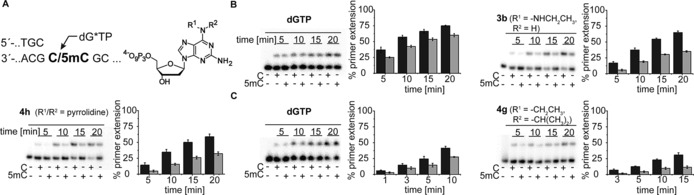
A) Partial primer/template sequences used. B) PAGE analysis of single‐nucleotide incorporation primer extension experiments with dGTP, 6‐(Ethylamino)‐2‐aminopurine‐2′‐deoxyribonucleoside‐5′‐O‐triphosphate (3 b) and 6‐pyrrolidine‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐triphosphate (4 h) opposite a template containing C in comparison with a template containing 5mC, in the presence of KOD exo‐ DNA polymerase. dGTP or dG*TP (100 μm) and KOD exo‐ DNA polymerase (10 nm) were used; reactions were stopped after indicated time points. C) PAGE analysis of single‐nucleotide incorporation primer extension experiments with dGTP and 6‐ethyl‐isopropylamino‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐triphosphate (4 g) opposite a template containing C in comparison with a template containing 5mC in the presence of 9°North exo‐ DNA polymerase. dGTP or dG*TP (100 μm) and 9°North exo‐ (10 nm) were used; reactions were stopped after indicated time points.
As already reported,24 we observed decreased selectivity when employing the dGTP derivatives because most of the modified nucleotides were incorporated opposite T with high efficiency as well (Figure S3).
Similar discrimination between C and 5mC was also observed with employment of 9°North exo‐ DNA polymerase. Consistent with the studies using the KOD exo‐ DNA polymerase, higher incorporation rates opposite C than opposite 5mC could already be seen when using the unmodified dGTP (Figure 1 B). However, in the case of the 9°North exo‐ DNA polymerase, this discrimination could be enhanced only slightly by the introduction of alkoxy modifications at position 6 of the dGTP (compounds 1 a–d). The discrimination increased slightly with growing bulkiness from the methoxy‐modified nucleotide 1 a to the propoxy‐modified nucleotide 1 c, but decreased again for the isopropoxy‐modified nucleotide 1 d. The replacement of the oxygen at position 6 by sulfur in nucleotides 2, 2 a and 2 b led to decreased discrimination for processing of those nucleotides opposite C and 5mC, in comparison with dGTP and the modified nucleotides 1 a–d. However, we were able to enhance the discrimination for the 9°North exo‐ DNA polymerase by utilising the 6‐amino‐modified nucleotides 3, 3 a–g and 4 a–h (Figures 1 B and S4). Enhanced discrimination was already observable with an amino group (compound 3), but introduction of bulkier modifications such as methylamino (compound 3 a), ethylamino (compound 3 b) and propylamino (compound 3 c) groups resulted in higher discrimination, whereas the bulkier modifications of nucleotides 3 d–g led to decreased discrimination (Figures 1 B and S4). Surprisingly, the discrimination could again be further enhanced by employment of the tertiary amine compounds 4 a–h. Increasing the size of the introduced modification from nucleotide 4 a to 4 b leads to a twofold increase in discrimination. Nevertheless, a decline in discrimination ratios can be observed with employment of nucleotides 4 c–f, bearing sterically more demanding modifications. Surprisingly, the highest discrimination, by a factor of 3, can be observed with use of nucleotide 4 g with the sterically most demanding modifications (Figures 1 B, 2 C and S4).
Conclusion
In summary, we have synthesised various 6‐substituted 2‐aminopurine‐2′‐deoxyribonucleoside 5′‐triphosphates and tested them with regard to their ability to discriminate between C and 5mC in the template strand. All modified nucleotides were accepted and processed by all tested DNA polymerases. In accordance with our previous findings, employing the KlenTaq DNA‐polymerase failed to show any promising differences in incorporation efficiencies opposite C or opposite 5mC.24 In contrast, sequence‐family B enzymes KOD exo‐ and 9°North exo‐ DNA polymerases delivered results that are more promising. The different discrimination behaviour of the A‐family and B‐family DNA polymerases during processing of the modified nucleotides is striking. Structural data for both enzyme families bound to a primer/template complex show significantly different interaction patterns of the respective protein with its substrate.31 This difference might be responsible for the observed differences in incorporation behaviour.
We found increased discrimination between C and 5mC, to a certain degree, on enhancing the bulkiness of the introduced modification. Interestingly, discrimination could be further enhanced by the introduction of even bulkier tertiary amine groups. Our results indicate that sequence‐family B DNA polymerases are best suited for the desired purpose. We identified the best discrimination between C and 5mC for KOD exo‐ DNA polymerase with the nucleotide 6‐pyrrolidine‐dGTP (4 h) and for 9°North exo‐ DNA polymerase with nucleotide 6‐(ethyl‐isopropylamino)‐dGTP (4 g). We thereby verified the results of previous studies24 that used the 6‐alkoxy‐modified dGTP derivatives 1 a–d. Interestingly, the degree of discrimination is not exclusively dependent on the bulkiness of the modification. It appears that a subtle interplay between the modification and the DNA polymerase is important for achieving the highest discrimination.
Consistently with our previous studies,24 we observed decreased selectivity of the modified nucleotides between C and T. This surely hinders its application in whole sequencing approaches. However, microarray‐based approaches that address single CpG sites and have been reported for methylation profiling after bisulfite‐mediated conversion might benefit from the findings reported here.10b, 32 The DNA methylation arrays offer low‐cost alternatives for profiling large numbers of samples.10b, 32
This study strongly suggests that sensing of 5‐methylation is a more general phenomenon of this class of nucleotides and not restricted to O 6‐alkyl modification of 2‐aminopurine‐2′‐deoxyribonucleoside 5′‐triphosphates because it applies to other functionalities as well.
We have developed an extended toolbox to examine incorporation efficiencies opposite different nucleotide analogues in RNA and DNA; its biological impact is currently being studied intensively.33 We see great potential in applying this approach to the detection of different modifications in nucleic acids. We are convinced that this approach will help in providing more detailed understanding of the underlying principles resulting in discrimination between C and 5mC by employment of modified nucleotides.
Experimental Section
2‐Amino‐6‐ethylthiopurine (5 b):25 Potassium tert‐butoxide (2.7 g, 24.0 mmol, 10.0 equiv) was dissolved in abs. DMF (20 mL). Ethanethiol (1.7 mL, 24.0 mmol, 10 equiv) was added, and the reaction mixture was stirred at room temperature for 30 min. 2‐Amino‐6‐chloropurine (0.4 g, 2.4 mmol, 1.0 equiv) was then added, and the reaction mixture was heated at reflux in a sealed vessel. After 16 h, the suspension was concentrated in vacuo, and the remaining solid was dissolved in water (10 mL). The aqueous solution was neutralised with acetic acid and extracted with ethyl acetate. The organic layer was dried over MgSO4 and concentrated to dryness, and the crude residue was purified by flash column chromatography with ethyl acetate as solvent to yield the product (0.23 g, 1.17 mmol, 49 %). 1H NMR (400 MHz, [D6]DMSO): δ=12.47 (br s, 1 H), 7.87 (s, 1 H), 6.29 (br s, 2 H), 3.25 (q, J=7.3 Hz, 2 H), 1.32 ppm (t, J=7.3 Hz, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=160.1, 159.3, 152.1, 139.1, 124.3, 22.4, 15.6 ppm; HRMS (ESI): m/z calcd for C7H9N5S: 196.0651 [M+1 H]+; found: 196.0674.
3′,5′‐Di‐O‐toluoyl‐6‐ethylthio‐2′‐deoxyguanosine (6 b):27 2‐Amino‐6‐ethylthiopurine (270 mg, 1.38 mmol, 1 equiv) and sodium hydride (60 % in mineral oil, 58 mg, 1.52 mmol, 1.1 equiv) were suspended in abs. CH3CN (30 mL), and the mixture was stirred for 30 min at room temperature. After 30 min, 1‐chloro‐2‐deoxy‐3,5‐di‐O‐toluoyl‐α‐d‐ribofuranose (591 mg, 1.52 mmol, 1.1 equiv) was added and the suspension was stirred at room temperature for 20 h. The suspension was filtered and the filtrate was concentrated in vacuo. The crude residue was purified by flash column chromatography (n‐hexane/ethyl acetate 3:1) to yield the product (0.43 g, 0.79 mmol, 57 %). 1H NMR (400 MHz, CDCl3): δ=7.97–7.95 (m, 2 H), 7.91–7.89 (m, 2 H), 7.76 (s, 1 H), 7.28 (d, J=8.3 Hz, 2 H), 7.22 (d, J=8.1 Hz, 2 H), 6.38 (dd, J=5.9, 8.4 Hz, 1 H), 5.79 (dt, J=2.1, 6.1 Hz, 1 H), 4.94 (br s, 2 H), 4.81 (dd, J=4.1, 11.4 Hz, 1 H), 4.67–4.60 (m, 2 H), 3.28 (q, J=7.4 Hz, 2 H), 3.16 (ddd, J=6.2, 8.4, 14.4 Hz, 1 H), 2.70 (ddd, J=5.9, 14.2, 2.1 Hz, 1 H), 2.44 (s, 3 H), 2.40 (s, 3 H), 1.40 ppm (t, J=7.4 Hz, 3 H); 13C NMR (100 MHz, CDCl3): δ=166.3, 165.9, 162.2, 158.9, 150.2, 144.4, 144.1, 138.0, 129.8, 129.7, 129.3, 129.3, 126.8, 126.5, 126.3, 84.5, 82.6, 75.3, 64.0, 37.1, 23.0, 21.8, 21.7, 14.8 ppm.
6‐Ethylthio‐2′‐deoxyguanosine (2 b):26 3′,5′‐Di‐O‐toluoyl‐6‐ethanesulfanyl‐2′‐deoxyguanosine (430 mg, 0.79 mmol, 1 equiv) was dissolved in ammonia (50 mL, 7 n) in methanol and stirred at 4 °C for 16 h. The solution was then concentrated to dryness, and the crude residue was purified by flash column chromatography with n‐hexane/ethyl acetate (3:1) to afford the product (0.15 g, 0.48 mmol, 61 %). 1H NMR (400 MHz, [D6]DMSO): δ=8.14 (s, 1 H), 6.48 (br s, 2 H), 6.21 (dd, J=6.0, 7.7 Hz, 1 H), 5.27 (d, J=4.0 Hz, 1 H), 4.96 (t, J=5.5 Hz, 1 H), 4.35 (dq, J=2.9, 6.3 Hz, 1 H), 3.82 (dt, J=2.7, 4.6 Hz, 1 H), 3.57 (dt, J=5.1, 11.7 Hz, 1 H), 3.52–3.48 (m, 1 H), 3.25 (q, J=7.3 Hz, 2 H), 2.59 (ddd, J=5.8, 7.7, 13.3 Hz, 1 H), 2.22 (ddd, J=3.1, 6.0, 13.1 Hz, 1 H), 1.32 ppm (t, J=7.3 Hz, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=160.2, 159.9, 151.0, 139.0, 124.7, 88.1, 83.2, 71.1, 62.1, 40.0, 22.2, 15.5 ppm; HRMS (ESI): m/z calcd for C12H17N5O3S: 312.1125 [M+1 H]+; found: 312.1101.
3′,5′‐Di‐O‐tert‐butyldimethylsilyl‐6‐chloro‐2′‐deoxyguanosine (8):29 Tetraethylammonium chloride (1.42 g, 8.5 mmol, 1.5 equiv) and N,N‐dimethylaniline (4.3 mL, 34.2 mmol, 6.0 equiv) were added to a suspension of 3′,5′‐di‐O‐tert‐butyldimethylsilyl‐2′‐deoxyguanosine (2.82 g, 5.7 mmol, 1.0 equiv) in acetonitrile (40.0 mL). The reaction mixture was cooled to 0 °C, and phosphoryl chloride (3.2 mL, 34.2 mmol, 6.0 equiv) was added dropwise. The reaction mixture was stirred at room temperature for 10 min. The mixture was heated to reflux for 15 min in a preheated oil bath and was afterwards immediately cooled with an ice bath and quickly concentrated to dryness. The remaining phosphoryl chloride was slowly hydrolysed by addition of 30 mL of ice/water with cooling. The solution was stirred for 20 min and extracted with ethyl acetate. The combined organic layers were washed with a saturated solution of sodium hydrogen carbonate, dried over MgSO4 and concentrated in vacuo, and the crude residue was purified by flash column chromatography with methylene chloride with 2 % methanol to afford a white foam in 37 % yield (1.1 g, 2.1 mmol). 1H NMR (400 MHz, [D6]DMSO): δ=8.29 (s, 1 H), 6.94 (br s, 2 H), 6.22 (t, J=6.7 Hz, 1 H), 4.53 (dt, J=3.4, 6.1 Hz, 1 H), 3.85–3.82 (m, 1 H), 3.73 (dd, J=5.7, 11.1 Hz, 1 H), 3.65 (dd, J=4.5, 11.1 Hz, 1 H), 2.77 (ddd, J=5.7, 7.2, 13.1 Hz, 1 H), 2.29 (ddd, J=3.7, 6.2, 13.3 Hz, 1 H), 0.88 (s, 9 H), 0.84 (s, 9 H), 0.10 (s, 6 H), 0.02 (s, 3 H), 0.02 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=160.1, 154.0, 149.0, 140.3, 124.0, 87.4, 83.1, 72.3, 63.0, 38.9, 26.2, 26.1, 18.4, 18.2, −4.3, −4.5, −5.0, −5.1 ppm; HRMS (ESI): m/z calcd for C22H40ClN5O3Si2: 514.2431 [M+1 H]+; found: 514.2399.
6‐Chloro‐2′‐deoxyguanosine (9): 3′,5′‐Di‐O‐tert‐butyldimethylsilyl‐6‐chloro‐2′‐deoxyguanosine (1.0 g, 1.9 mmol, 1 equiv) was dissolved in abs. THF (10 mL) at room temperature. Triethylamine trihydrofluoride (196 μL, 1.2 mmol, 1.7 equiv) was added and the reaction mixture was stirred for 16 h. The solution was then concentrated in vacuo and the crude residue was purified by flash column chromatography with methylene chloride with up to 10 % methanol to afford the product (504.0 mg, 1.7 mmol, 93 %). 1H NMR (400 MHz, [D6]DMSO): δ=8.34 (s, 1 H), 6.94 (br s, 2 H), 6.22 (t, J=6.7 Hz, 1 H), 5.28 (d, J=4.1 Hz, 1 H), 4.92 (t, J=5.5 Hz, 1 H), 4.37 (dq, J=3.5, 6.7 Hz, 1 H), 3.83 (dt, J=2.9, 4.6 Hz, 1 H), 3.58 (dt, J=5.1, 11.7 Hz, 1 H), 3.51 (dt, J=5.0, 11.7 Hz, 1 H), 2.61 (ddd, J=5.8, 7.4, 13.2 Hz, 1 H), 2.26 ppm (ddd, J=3.4, 6.2, 13.2 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=160.2, 154.1, 149.9, 141.5, 124.0, 88.2, 83.5, 71.0, 62.0, 40.0 ppm. HRMS (ESI): m/z calcd: 286.0701 [M+1 H]+; found: 286.0682.
General procedure A:28 Typical reaction scales ranged from 40 to 120 mg of starting nucleoside. The appropriate nucleoside (1.0 equiv) and proton sponge (N,N,N′,N′‐tetramethylnaphthalene‐1,8‐diamine, 1.5 equiv) were dried in vacuo, dissolved in dry trimethyl phosphate (1 mL per 20 mg of starting nucleoside) at room temperature and cooled to 0 °C. Phosphorous oxychloride (1.2 equiv) was added dropwise at 0 °C and the mixture was stirred under nitrogen. After 30 min TLC showed complete conversion of starting material, and a solution (0.5 m) of (Bu3NH)2H2P2O7 in anhydrous DMF (5.0 equiv) and nBu3N (10.0 equiv) were added simultaneously to the mixture. The mixture was allowed to warm to room temperature and stirred for 30 min. Aqueous triethylammonium bicarbonate buffer (pH 7.5, 0.1 m, 10 mL) was then added and the mixture was stirred for a further 30 min. The aqueous layer was washed with ethyl acetate several times to remove trimethyl phosphate and then concentrated to dryness. The residue was dissolved in water and purified by ion‐exchange chromatography [DEAE Sephadex A25, buffer A: TEAB (0.1 m), buffer B: TEAB (1 m), linear gradient: 0 % B to 100 % B] and further purified by reversed‐phase (RP) HPLC [Nucleodur RP 18‐HTec, buffer A: triethylammonium acetate (TEAA, 50 mm), buffer B: acetonitrile, linear gradient: 5 % B to 100 % B]. The triphosphates were concentrated to dryness. To get rid of the TEAA the residues were dissolved in water and freeze‐dried several times.
6‐Thioethyl‐2′‐deoxyguanosine‐5′‐O‐triphosphate (2 b): Yield: 28 % (90.0 μmol). 1H NMR (400 MHz, D2O): δ=8.22 (s, 1 H), 6.31 (t, J=6.9 Hz, 1 H), 4.72–4.63 (m, 1 H), 4.26–4.21 (m, 1 H), 4.18–4.10 (m, 2 H), 3.26–3.17 (m, 2 H), 2.81 (dt, J=6.8, 13.8 Hz, 1 H), 2.55 (ddd, J=3.3, 6.3, 14.2 Hz, 1 H), 1.33 ppm (t, J=7.4 Hz, 1 H); 13C NMR (100 MHz, D2O): δ=158.3, 155.7, 145.7, 135.6, 120.3, 82.0, 79.5, 67.5, 61.9, 34.8, 19.4, 10.1 ppm; 31P NMR (162 MHz, D2O): δ=−10.0 to −11.1 (m, 1 P), −11.1 to −12.0 (m, 1 P), −20.5 to −22.5 ppm (m, 1 P); HRMS (ESI): m/z calcd for C12H20N5O12P3S: 549.9958 [M−H]−; found: 549.9957.
6‐Chloro‐2′‐deoxyguanosine 5′‐O‐triphosphate (10): Yield: 13 % (178.0 μmol). 1H NMR (400 MHz, D2O): δ=8.35 (s, 1 H), 6.32 (t, J=6.8 Hz, 1 H), 4.78–4.75 (m, 1 H), 4.25–4.22 (m, 1 H), 4.18–4.15 (m, 2 H), 2.84 (ddd, J=6.2, 7.5, 13.7 Hz, 1 H), 2.55 ppm (ddd, J=3.6, 6.4, 14.0 Hz, 1 H); 13C NMR (100 MHz, D2O): δ=159.4, 152.8, 150.2, 142.1, 123.5, 85.7, 83.7, 70.8, 65.5, 38.1 ppm; 31P NMR (162 MHz, D2O): δ=−10.86 (d, J=20.1 Hz, 1 P), −11.53 (d, J=20.3 Hz, 1 P), −23.5 ppm (t, J=20.2 Hz, 1 P); HRMS (ESI): m/z calcd for C10H15ClN5O12P3: 523.9535 [M−H]−; found: 523.9525.
General procedure B: An aqueous solution of the appropriate amine (100 equiv) was added to a solution of 6‐chloro‐2′‐deoxyguanosine 5′‐O‐triphosphate (10 mm, 10 μmol) in water and shaken overnight at room temperature. The reaction mixture was concentrated to dryness. The residue was dissolved in water and purified by ion‐exchange chromatography [DEAE Sephadex A25, buffer A: TEAB (0.1 m), buffer B: TEAB (1 m), linear gradient: 0 % B to 100 % B] and further purified by reversed‐phase HPLC [Nucleodur RP 18‐HTec, buffer A: TEAA (50 mm), buffer B: acetonitrile, linear gradient: 5 % B to 100 % B]. The triphosphates were concentrated to dryness and freeze‐dried several times.
6‐(Methylamino)‐2‐aminopurine‐2′‐deoxyribonucleoside‐5′‐O‐triphosphate (3 a): Yield: 93 % (9.3 μmol). 1H NMR (400 MHz, D2O): δ=8.09 (s, 1 H), 6.27 (t, J=6.9 Hz, 1 H), 4.77 (dq, J=3.3, 6.3 Hz, 1 H), 4.26–4.23 (m, 1 H), 4.19–4.12 (m, 2 H), 3.02 (br s, 3 H) 2.76 (ddd, J=6.1, 7.6, 13.8 Hz, 1 H), 2.53 ppm (ddd, J=3.5, 6.2, 13.9 Hz, 1 H); 13C NMR (100 MHz, D2O): δ=159.6, 154.9, 149.1, 136.5, 112.8, 85.4, 82.9, 71.0, 65.5, 38.7, 23.0 ppm; 31P NMR (162 MHz, D2O): δ=−9.72 (d, J=19.3 Hz, 1 P), −11.28 (d, J=19.3 Hz, 1 P), −22.84 ppm (t, J=19.8 Hz, 1 P). HRMS (ESI): m/z calcd for C11H18N6O12P3: 519.0201 [M−H]−; found: 519.0189.
6‐(Ethylamino)‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (3 b): Yield: 87 % (8.7 μmol). 1H NMR (400 MHz, D2O): δ=8.18 (s, 1 H), 6.33 (t, J=6.8 Hz, 1 H), 4.86–4.80 (m, 1 H), 4.33–4.27 (m, 1 H), 4.27–4.22 (m, 2 H), 3.62 (br s, 2 H), 2.75 (dt, J=6.7, 13.8 Hz, 1 H), 2.54 (ddd, J=3.4, 6.2, 14.0 Hz, 1 H), 1.31 ppm (t, J=7.2 Hz, 3 H); 31P NMR (162 MHz, D2O): δ=−10.72 to −10.82 (m, 1 P), −11.19 (d, J=19.6 Hz, 1 P), −22.60 to −22.55 ppm (t, J=19.7 Hz); HRMS (ESI): m/z calcd for C12H21N6O12P3: 533.0358 [M−H]−; found: 533.0433.
6‐(Propylamino)‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (3 c): Yield: 63 % (6.3 μmol); 1H NMR (400 MHz, D2O): δ=8.17 (s, 1 H), 6.34 (t, J=6.9 Hz, 1 H), 4.90–4.82 (m, 1 H), 4.30 (dt, J=3.2, 5.5 Hz, 1 H), 4.26–4.16 (m, 2 H), 3.54 (br s, 2 H), 2.77 (dt, J=6.8, 13.7 Hz, 1 H), 2.54 (ddd, J=3.3, 6.3, 14.0 Hz, 1 H), 1.71 (h, J=7.3 Hz, 2 H), 1.00 ppm (t, J=7.4 Hz, 3 H); 31P NMR (162 MHz, D2O): δ=−10.48 to −10.95 (m, 1 P), −11.2 (d, J=20.2 Hz, 1 P), −22.89 to −23.42 ppm (m, 1 P). HRMS (ESI): m/z calcd for C13H23N6O12P3: 547.0514 [M−H]−; found: 547.0584.
6‐(Isopropylamino)‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (3 d): Yield: 43 % (4.3 μmol). 1H NMR (400 MHz, D2O): δ=8.16 (s, 1 H), 6.40–6.34 (m, 1 H), 4.87–4.80 (m, 1 H), 4.42–4.33 (m, 1 H), 4.31–4.13 (m, 3 H), 2.80 (dt, J=6.7, 13.8 Hz, 1 H), 2.54 (ddd, J=3.1, 6.1, 14.1 Hz, 1 H), 1.32–1.30 ppm (m, 6 H); 31P NMR (162 MHz, D2O): δ=−10.42 to −10.76 (m, 1 P), −11.18 to −11.35 (m, 1 P), −23.00 to −23.42 ppm (m, 1 P); HRMS (ESI) m/z calcd for C13H23N6O12P3: 547.0514 [M−H]−; found: 547.0584.
6‐(Cyclohexylamino)‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (3 e): Yield: 97 % (9.7 μmol). 1H NMR (400 MHz, D2O): δ=8.16 (s, 1 H), 6.38–6.34 (m, 1 H), 4.54–4.31 (m, 1 H), 4.31–4.28 (m, 1 H), 4.27–4.13 (m, 2 H), 2.79 (dt, J=6.4, 13.7 Hz, 1 H), 2.54 (ddd, J=3.0, 6.1, 14.1 Hz, 1 H), 1.82–1.63 ppm (m, 9 H); 31P NMR (162 MHz, D2O): δ=−10.23 to −10.80 (m, 1 P), −11.00 to −11.30 (m, 1 P), −22.80 to −22.67 ppm (m, 1 P); HRMS (ESI): m/z calcd for C15H23N6O12P3: 573.0671 [M−H]−; found: 573.0714.
6‐(3‐Azidopropylamino)‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (3 f): Yield: 86 % (8.6 μmol). 1H NMR (400 MHz, D2O): δ=8.17 (s, 1 H), 6.35 (t, J=7.0 Hz, 1 H), 4.72–4.63 (m, 1 H), 4.30–4.28 (m, 1 H), 4.26–4.18 (m, 2 H), 3.73–3.63 (m, 2 H), 3.50 (t, J=6.6 Hz, 2 H), 2.77 (dt, J=7.0, 14.0 Hz, 1 H), 2.54 (ddd, J=3.2, 6.1, 14.0 Hz, 1 H), 1.99 ppm (d, J=6.7 Hz, 2 H); 31P NMR (162 MHz, D2O): δ=−10.63–10.88 (m, 1 P), −11.15 to −11.31 (m, 1 P), −22.95 to −22.45 ppm (t, J=19.7 Hz, 1 P); HRMS (ESI): m/z calcd for C13H22N9O12P3: 588.0528 [M−H]−; found: 588.0534.
6‐(Hydroxy‐1‐ethanamino)‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (3 g): Yield: 52 % (5.2 μmol). 1H NMR (400 MHz, D2O): δ=8.19 (s, 1 H), 6.36 (t, J=6.9 Hz, 1 H), 4.71–4.66 (m, 1 H), 4.32–4.29 (m, 1 H), 4.26–4.20 (m, 2 H), 3.87–3.85 (m, 2 H), 3.82–3.75 (m, 2 H), 2.78 (dt, J=6.9, 13.8 Hz, 1 H), 2.55 ppm (ddd, J=3.3, 6.2, 14.0 Hz, 1 H); 31P NMR (162 MHz, D2O): δ=−10.86 (m, 1 P), −11.21 (d, J=19.5 Hz, 1 P), −23.00 to −23.33 ppm (m, 1 P); HRMS (ESI): m/z calcd for C12H21N6O13P3: 549.0307 [M−H]−; found: 549.0375.
6‐Dimethylamino‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (4 a): Yield: 85 % (8.5 μmol). 1H NMR (400 MHz, D2O): δ=8.07 (s, 1 H), 6.27 (t, J=6.9 Hz, 1 H), 4.70–4.65 (m, 1 H), 4.27–4.14 (m, 3 H), 3.30 (br s, 6 H), 2.74 (dt, J=6.9, 13.8 Hz, 1 H), 2.53 ppm (ddd, J=3.3, 6.2, 13.9 Hz, 1 H); 13C NMR (100 MHz, D2O): δ=157.3, 153.2, 150.2, 135.9, 112.9, 85.5, 83.2, 71.0, 65.5, 38.8 ppm; 31P NMR (162 MHz, D2O): δ=−10.59 (d, J=19.9 Hz, 1 P), −11.46 (d, J=19.9 Hz, 1 P), −23.25 ppm (t, J=19.8 Hz); HRMS (ESI) m/z calcd for C12H20N6O12P3: 533.0358 [M−H]−; found: 533.0344.
6‐Methylethylamino‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (4 b): Yield: 73 % (7.3 μmol). 1H NMR (400 MHz, D2O): δ=8.03 (s, 1 H), 6.24 (dd, J=6.2, 7.5 Hz, 1 H), 4.73 (dt, J=3.5, 6.4 Hz, 1 H), 4.23–4.19 (m, 1 H), 4.18–4.10 (m, 2 H), 3.75–3.64 (m, 2 H), 3.09–3.05 (m, 3 H), 2.64 (ddd, J=6.1, 7.7, 13.7 Hz, 1 H), 2.42 (ddd, J=3.3, 6.2, 13.9 Hz, 1 H), 1.05 ppm (t, J=7.1 Hz, 3 H); 13C NMR (100 MHz, D2O): δ=158.5, 153.3, 150.8, 135.6, 112.8, 88.5, 82.9, 71.1, 65.5, 45.6, 38.7, 35.9, 12.1 ppm; 31P NMR (162 MHz, D2O): δ=−10.10 (m, 1 P), −11.42 (d, J=19.5 Hz, 1 P), −22.88 ppm (m, 1 P); HRMS (ESI): m/z calcd for C13H23N6O12P3: 547.0514 [M−H]−; found: 547.0499.
6‐Methylpropylamino‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (4 c): Yield: 67 % (6.7 μmol). 1H NMR (400 MHz, D2O): δ=8.15 (s, 1 H), 6.40–6.36 (m, 1 H), 4.74–4.68 (m, 1 H), 4.32–4.17 (m, 3 H), 3.94 (br s, 2 H), 3.36 (br s, 3 H), 2.82–2.78 (m, 1 H), 2.57–2.53 (m, 1 H), 1.73 (d, J=7.5 Hz, 2 H), 0.92 ppm (t, J=7.3 Hz, 3 H); 31P NMR (162 MHz, D2O): δ=−9.47 to −11.93 (m, 2 P), −22.10 to −23.33 (m, Hz). HRMS (ESI): m/z calcd for C14H25N6O12P3: 561.0671 [M−H]−; found: 561.0681.
6‐Methyl‐isopropylamino‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (4 d): Yield: 34 % (3.4 μmol). 1H NMR (400 MHz, CD3OD): δ=8.18 (s, 1 H), 6.49 (t, J=6.8 Hz, 1 H), 4.87 (dt, J=3.4, 6.4 Hz, 1 H), 4.42–4.40 (m, 2 H), 4.39–4.26 (m, 1 H), 3.49 (p, J=1.6 Hz, 1 H), 3.37 (br s, 3 H), 2.93 (dt, J=6.7, 13.5 Hz, 1 H), 2.54 (ddd, J=3.6, 6.3, 13.4 Hz, 1 H), 1.41 ppm (d, J=6.8 Hz, 6 H); 31P NMR (162 MHz, CD3OD): δ=−10.44 (d, J=22.7 Hz, 1 P), −11.26 (d, J=20.4 Hz, 1 P), −23.71 ppm (t, J=21.2 Hz).; HRMS (ESI): m/z calcd for C14H25N6O12P3: 561.0671 [M−H]−; found: 561.0710.
6‐Diethylamino‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (4 e): Yield: 65 % (6.5 μmol). 1H NMR (400 MHz, D2O): δ=8.05 (s, 1 H), 6.27 (dd, J=6.2, 7.6 Hz, 1 H), 4.75 (dq, J=2.8, 3.3 Hz, 1 H), 4.28–4.10 (m, 3 H), 3.79–3.69 (m, 4 H), 2.76–2.69 (m, 1 H), 2.50 (ddd, J=3.4, 6.3, 13.9 Hz, 1 H), 1.16 (t, J=7.0 Hz, 6 H); 13C NMR (100 MHz, D2O): δ=158.7, 153.0, 150.8, 135.5, 112.8, 85.4, 83.0, 70.9, 65.4, 43.2, 38.7, 16.7 ppm; 31P NMR (162 MHz, D2O): δ=−10.02 (m, 1 P), −11.40 (d, J=19.6 Hz, 1 P), −22.89 ppm (t, J=19.7 Hz); HRMS (ESI): m/z calcd: 561.0671 [M−H]−; found: 561.0673.
6‐Ethylpropylamino‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (4 f): Yield: 48 % (4.8 μmol). 1H NMR (400 MHz, CD3OD): δ=7.97 (s, 1 H), 6.29 (t, J=6.8 Hz, 1 H), 4.69 (dt, J=3.3, 6.2 Hz, 1 H), 4.24–4.14 (m, 2 H), 4.11–4.08 (m, 1 H), 3.89 (br s, 2 H), 3.82 (br s, 2 H), 2.73 (dt, J=6.4, 13.6 Hz, 1 H), 2.34 (ddd, J=3.5, 6.2, 13.5 Hz, 1 H), 1.69 (d, J=7.4 Hz, 2 H), 1.21 (t, J=7.0 Hz, 3 H), 0.93 ppm (t, J=7.4 Hz, 3 H); 31P NMR (162 MHz, CD3OD): δ=−10.41 (d, J=21.3 Hz, 1 P), −11.19 (d, J=20.5 Hz, 1 P), −23.64 ppm (t, J=21.5 Hz); HRMS (ESI): m/z calcd for C15H27N6O12P3: 575.0827 [M−H]−; found: 575.0835.
6‐Ethyl‐isopropylamino‐2‐aminopurine‐2′‐deoxyribonucleoside 5′‐O‐triphosphate (4 g): Yield: 27 % (2.7 μmol). 1H NMR (400 MHz, D2O): δ=8.07 (s, 1 H), 6.29 (t, J=6.8 Hz, 1 H), 5.27 (m, 1 H), 4.75 (m, 1 H), 4.24–4.11 (m, 3 H), 3.60 (m, 2 H), 2.71 (dt, J=6.8, 13.7 Hz, 1 H), 2.49 (ddd, J=3.3, 6.4, 14.2 Hz, 1 H), 1.17 (d, J=6.8 Hz, 6 H), 1.13 ppm (t, J=6.3 Hz, 3 H); 31P NMR (162 MHz, D2O): δ=−10.27 (m, 1 P), −11.14 (m, 1 P), −22.93 ppm (m, 1 P); HRMS (ESI): m/z calcd for C15H27N6O12P3: 575.0827 [M−H]−; found: 575.0826.
6‐Pyrrolidine‐2‐aminopurine‐2′‐deoxyribonucleoside‐5′‐O‐triphosphate (4 h): Yield: 18 % (1.8 μmol). 1H NMR (400 MHz, D2O): δ=8.16 (s, 1 H), 6.36 (t, J=6.9 Hz, 1 H), 4.86–4.80 (m, 1 H), 4.31–4.27 (m, 1 H), 4.25–4.17 (m, 2 H), 4.07–3.55 (m, 4 H), 2.80 (dt, J=6.8 Hz, 13.8 Hz, 1 H), 2.54 (ddd, J=3.4, 6.3, 14.0 Hz, 1 H), 2.11–2.01 ppm (m, 4 H); 31P NMR (162 MHz, D2O): δ=−10.56 (d, J=19.7 Hz, 1 P), −11.26 (d, J=19.9 Hz, 1 P), −23.11 ppm (t, J=18.6 Hz); HRMS (ESI): m/z calcd for C14H23N6O12P3: 559.0514 [M−H]−; found: 559.0541.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge the European Research Council (ERC Advanced Grant 339834) and the Konstanz Research School Chemical Biology for financial support.
J. von Watzdorf, A. Marx, ChemBioChem 2016, 17, 1532.
References
- 1.
- 1a. Ramsahoye B. H., Biniszkiewicz D., Lyko F., Clark V., Bird A. P., Jaenisch R., Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Ziller M. J., Muller F., Liao J., Zhang Y., Gu H., Bock C., Boyle P., Epstein C. B., Bernstein B. E., Lengauer T., Gnirke A., Meissner A., PLoS Genet. 2011, 7, e1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xu Y., Wu F., Tan L., Kong L., Xiong L., Deng J., Barbera A. J., Zheng L., Zhang H., Huang S., Min J., Nicholson T., Chen T., Xu G., Shi Y., Zhang K., Shi Y. G., Mol. Cell 2011, 42, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ammerpohl O., Martin-Subero J. I., Richter J., Vater I., Siebert R., Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 847–862. [DOI] [PubMed] [Google Scholar]
- 4. Jones P. A., Takai D., Science 2001, 293, 1068–1070. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Seela F., Winter H., Nucleosides Nucleotides 1995, 14, 129–142; [Google Scholar]
- 5b. Estecio M. R., Gallegos J., Vallot C., Castoro R. J., Chung W., Maegawa S., Oki Y., Kondo Y., Jelinek J., Shen L., Hartung H., Aplan P. D., Czerniak B. A., Liang S., Issa J. P., Genome Res. 2010, 20, 1369–1382; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Jones P. A., Baylin S. B., Nat. Rev. Genet. 2002, 3, 415–428. [DOI] [PubMed] [Google Scholar]
- 6. Suzuki M. M., Bird A., Nat. Rev. Genet. 2008, 9, 465–476. [DOI] [PubMed] [Google Scholar]
- 7. de Koning A. P., Gu W., Castoe T. A., Batzer M. A., Pollock D. D., PLoS Genet. 2011, 7, e1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li E., Bestor T. H., Jaenisch R., Cell 1992, 69, 915–926. [DOI] [PubMed] [Google Scholar]
- 9. Smith Z. D., Meissner A., Nat. Rev. Genet. 2013, 14, 204–220. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Rodríguez-Paredes M., Esteller M., Nat. Med. 2011, 17, 330–339; [DOI] [PubMed] [Google Scholar]
- 10b. Heyn H., Esteller M., Nat. Rev. Genet. 2012, 13, 679–692; [DOI] [PubMed] [Google Scholar]
- 10c. Laird P. W., Nat. Rev. Cancer 2003, 3, 253–266. [DOI] [PubMed] [Google Scholar]
- 11. Law J. A., Jacobsen S. E., Nat. Rev. Genet. 2010, 11, 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cross S. H., Charlton J. A., Nan X., Bird A. P., Nat. Genet. 1994, 6, 236–244. [DOI] [PubMed] [Google Scholar]
- 13. Kaput J., Sneider T. W., Nucleic Acids Res. 1979, 7, 2303–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li W. W., Gong L., Bayley H., Angew. Chem. Int. Ed. 2013, 52, 4350–4355; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4446–4451. [Google Scholar]
- 15. Kubik G., Schmidt M. J., Penner J. E., Summerer D., Angew. Chem. Int. Ed. 2014, 53, 6002–6006; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6113–6117. [Google Scholar]
- 16.
- 16a. Wang T., Hong T., Tang T., Zhai Q., Xing X., Mao W., Zheng X., Xu L., Wu J., Weng X., Wang S., Tian T., Yuan B., Huang B., Zhuang L., Zhou X., J. Am. Chem. Soc. 2013, 135, 1240–1243; [DOI] [PubMed] [Google Scholar]
- 16b. Bareyt S., Carell T., Angew. Chem. Int. Ed. 2008, 47, 181–184; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 187–190. [Google Scholar]
- 17. Frommer M., McDonald L. E., Millar D. S., Collis C. M., Watt F., Grigg G. W., Molloy P. L., Paul C. L., Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Akhtar-Zaidi B., Cowper-Sal-lari R., Corradin O., Saiakhova A., Bartels C. F., Balasubramanian D., Myeroff L., Lutterbaugh J., Jarrar A., Kalady M. F., Willis J., Moore J. H., Tesar P. J., Laframboise T., Markowitz S., Lupien M., Scacheri P. C., Science 2012, 336, 736–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Hayatsu H., Wataya Y., Kazushige K., J. Am. Chem. Soc. 1970, 92, 724–726; [DOI] [PubMed] [Google Scholar]
- 19b. Shapiro R., DiFate V., Welcher M., J. Am. Chem. Soc. 1974, 96, 906–912. [DOI] [PubMed] [Google Scholar]
- 20. Booth M. J., Branco M. R., Ficz G., Oxley D., Krueger F., Reik W., Balasubramanian S., Science 2012, 336, 934–937. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Clark S. J., Statham A., Stirzaker C., Molloy P. L., Frommer M., Nat. Protoc. 2006, 1, 2353–2364; [DOI] [PubMed] [Google Scholar]
- 21b. Pomraning K. R., Smith K. M., Freitag M., Methods 2009, 47, 142–150. [DOI] [PubMed] [Google Scholar]
- 22. Grunau C., Clark S. J., Rosenthal A., Nucleic Acids Res. 2001, 29, E65–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Genereux D. P., Johnson W. C., Burden A. F., Stoger R., Laird C. D., Nucleic Acids Res. 2008, 36, e150; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Harrison J., Stirzaker C., Clark S. J., Anal. Biochem. 1998, 264, 129–132. [DOI] [PubMed] [Google Scholar]
- 24. von Watzdorf J., Leitner K., Marx A., Angew. Chem. Int. Ed. 2016, 55, 3229–3232; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3284–3288. [Google Scholar]
- 25. Huang L. K., Cherng Y. C., Cheng Y. R., Jang J. P., Chao Y. L., Cherng Y. J., Tetrahedron 2007, 63, 5323–5327. [Google Scholar]
- 26. Hanna N. B., Bhattacharya B. K., Robins R. K., Avery T. L., Revankar G. R., J. Med. Chem. 1994, 37, 177–183. [DOI] [PubMed] [Google Scholar]
- 27. Ramasamy K., Robins R. K., Revankar G. R., J. Heterocycl. Chem. 1988, 25, 1043–1046. [Google Scholar]
- 28.
- 28a. Yoshikawa M., Kato T., Takenishi T., Bull. Chem. Soc. Jpn. 1969, 42, 3505; [Google Scholar]
- 28b. Kovács T., Otvos L., Tetrahedron Lett. 1988, 29, 4525–4528. [Google Scholar]
- 29. Woo J. S., Sigurdsson S. T., Hopkins P. B., J. Am. Chem. Soc. 1993, 115, 3407–3415. [Google Scholar]
- 30. Ito J., Braithwaite D. K., Nucleic Acids Res. 1991, 19, 4045–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bergen K., Betz K., Welte W., Diederichs K., Marx A., ChemBioChem 2013, 14, 1058–1062. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Sandoval J., Heyn H., Moran S., Serra-Musach J., Pujana M. A., Bibikova M., Esteller M., Epigenetics 2011, 6, 692–702; [DOI] [PubMed] [Google Scholar]
- 32b. Bibikova M., Barnes B., Tsan C., Ho V., Klotzle B., Le J. M., Delano D., Zhang L., Schroth G. P., Gunderson K. L., Fan J. B., Shen R., Genomics 2011, 98, 288–295; [DOI] [PubMed] [Google Scholar]
- 32c. Dedeurwaerder S., Defrance M., Calonne E., Denis H., Sotiriou C., Fuks F., Epigenomics 2011, 3, 771–784. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Fu Y., He C., Curr. Opin. Chem. Biol. 2012, 16, 516–524; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33b. Roundtree I. A., He C., Curr. Opin. Chem. Biol. 2016, 30, 46–51; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33c. Zhang L., Szulwach K. E., Hon G. C., Song C. X., Park B., Yu M., Lu X., Dai Q., Wang X., Street C. R., Tan H., Min J. H., Ren B., Jin P., He C., Nat. Commun. 2013, 4, 1517; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33d. Wang X., He C., Mol. cell 2014, 56, 5–12; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33e. Zhao B. S., He C., Cell Res. 2015, 25, 153–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary