

Abstract
Macrocyclization can be used to constrain peptides in their bioactive conformations, thereby supporting target affinity and bioactivity. In particular, for the targeting of challenging protein–protein interactions, macrocyclic peptides have proven to be very useful. Available approaches focus on the stabilization of α‐helices, which limits their general applicability. Here we report for the first time on the use of ring‐closing alkyne metathesis for the stabilization of an irregular peptide secondary structure. A small library of alkyne‐crosslinked peptides provided a number of derivatives with improved target affinity relative to the linear parent peptide. In addition, we report the crystal structure of the highest‐affinity derivative in a complex with its protein target 14‐3‐3ζ. It can be expected that the alkyne‐based macrocyclization of irregular binding epitopes should give rise to new scaffolds suitable for targeting of currently intractable proteins.
Keywords: macrocyclization, peptide secondary structures, peptidomimetics, protein–protein interactions, ring-closing alkyne metathesis
Peptide‐based drugs have recently experienced a renaissance, with several approvals and many candidates in clinical trials.1 Protein–protein interactions (PPIs), which are mainly mediated by shallow and extensive interaction surfaces, are hardly addressable by small‐molecule entities, and so peptides and peptidomimetics are stepping up to fill the void.2, 3 For the targeting of PPIs, peptides feature many advantages over classic small‐molecule compounds because they can cover larger surfaces, provide straightforward access to structural diversity, and show low toxicity in humans.
However, unmodified short l‐peptides often suffer from low target affinity and bioactivity, hampering their therapeutic potential. These drawbacks are mainly caused by the high flexibility of short peptides in their unbound state.4 In this context, peptide macrocyclization has emerged as an important strategy for enforcing conformational constraint.3 Preorganization of the peptide in solution prior to binding reduces the entropic penalty upon target engagement, thereby increasing the stability of the resulting complex.5 Macrocyclization of natural peptide sequences yields class A peptidomimetics, which are defined as modified peptides that harbor only minor backbone and side chain alterations.3
Class A peptidomimetics have been widely used to mimic secondary structure elements for the inhibition of PPIs through the application of different cyclization strategies.3, 6 Helix stabilization has often been pursued by use of the hydrocarbon peptide stapling approach7, 8 and hydrogen bond surrogates (HBSs),9 two methods that use ring‐closing olefin metathesis (RCM) for the formation of a hydrophobic crosslink. Recently, we reported a new synthetic method utilizing ring‐closing alkyne metathesis10 (RCAM, Scheme 1) for the formation of constrained helices.11 Importantly, the linear and rigid alkyne moiety within the macrocycle allows the installation of a hydrophobic crosslink, known to be beneficial for bioavailability,7 and gives access to new molecular geometries. Especially in cases in which the crosslink mediates the interaction with the target protein, an extended set of synthetically accessible architectures can be crucial for the identification of high‐affinity binders. Here we report the use of RCAM for the stabilization of an irregular peptide secondary structure, to yield macrocyclic peptides with improved target affinities relative to their linear parent peptide. In addition, a crystal structure of the highest‐affinity alkyne derivative in a complex with its protein target 14‐3‐3ζ is presented.
Scheme 1.
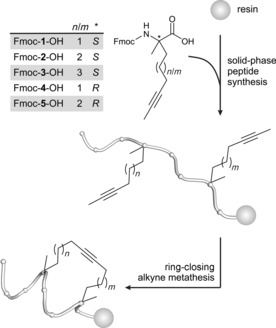
Synthesis of alkyne‐macrocyclized peptides. α‐Methyl, α‐alkynyl amino acids Fmoc‐1–5‐OH are introduced into the peptide sequence by means of SPPS. Macrocyclization is performed on solid phase through molybdenum‐mediated alkyne metathesis.
As a basis for the design of alkyne‐crosslinked peptides we used the so‐called ESp peptide (14‐3‐3 binding motif of exoenzyme S).12 Exoenzyme S is a virulence factor of the bacterium Pseudomonas aeruginosa, and it inactivates various small GTPases through ADP‐ribosylation upon interaction with its cofactor 14‐3‐3 to cause severe cellular damage.13 ESp resembles an irregularly structured binding motif that recognizes 14‐3‐3 mainly through hydrophobic contacts.12, 14 Inhibition of the 14‐3‐3–exoenzyme S interaction would be expected to reduce the pathogenicity of P. aeruginosa.12
Previously, we had developed two crosslinked peptides—βSS12 and βRS8—that stabilize the bioactive irregular peptide structure of ESp and bind 14‐3‐3 with sub‐micromolar affinities.14 In each case a fully saturated hydrocarbon crosslink was applied for conformational restriction. The recently reported alkyne‐based macrocyclization of peptides through on‐resin RCAM enables the introduction of a rigid alkyne into the crosslink to provide additional constraints (Scheme 1).11 To investigate the consequences of crosslink rigidification on 14‐3‐3 binding and the applicability of RCAM for the stabilization of irregular secondary structures, we designed a series of alkyne‐macrocyclized peptides based on the two peptides βSS12 and βRS8 (Scheme 2 A). The two peptides each contain two unnatural amino acids, at positions i and i+3, for crosslinking [Scheme 2 A, amino acids 422 (X) and 425 (Y) based on the exoenzyme S sequence]. Peptide βRS8 features an eight‐carbon linker (absolute configurations: X: R, Y: S) whereas βSS12 has a 12‐carbon linker (absolute configurations: X: S, Y: S). A total of eight alkyne‐macrocyclized ESp derivatives based on these two architectures were synthesized. They can be divided into two families (Scheme 2 B): one is based on βRS8 and contains an R‐ and an S‐configured building block (X and Y, respectively) with carbon linkers varying between eight and ten atoms (Scheme 2 B, peptides A–D). The other family, based on βSS12, incorporates two S‐configured building blocks (X and Y) and crosslinks with between ten and twelve carbon atoms (Scheme 2 B, peptides E–H). Linker lengths and configurations were inspired by previously reported results.14, 15
Scheme 2.
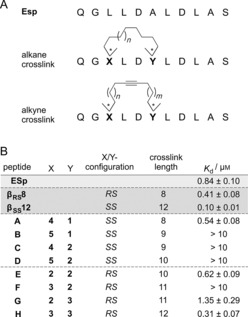
A) Sequence of the unmodified 14‐3‐3‐binding motif of exoenzyme S (ESp, aas 420–430) and a representation of its alkane‐ and alkyne‐crosslinked ESp analogues. B) Binding affinities and sequences of ESp, βRS8, βSS12, and alkyne‐macrocyclized derivatives A–H. Peptides were synthesized by Fmoc‐based SPPS, cyclized, and N‐terminally functionalized with a FITC‐PEG conjugate. Dissociation constants (K d) of peptide⋅14‐3‐3ζ complexes were determined by FP (triplicate, error=1σ).
In the design process of the alkyne‐crosslinked derivatives, we aimed to position the alkyne moiety in the center of the hydrocarbon crosslink. Peptides with even numbers of carbon atoms have the triple bond located in the middle of the crosslink (A, D, E, and H, Scheme 2 B), whereas peptides with odd numbers of linker atoms exist as two regioisomers (B/C and F/G, Scheme 2 B). All peptides were synthesized by Fmoc‐based solid‐phase peptide synthesis (SPPS) with incorporation of unnatural α‐methyl, α‐alkynyl amino acids 1–5 (Scheme 1, Figure S1 in the Supporting Information) at positions X and Y (Scheme 2 B). RCAM was performed according to established protocols with use of the latest versions of molybdenum‐based alkyne metathesis complexes16 (Figure S1).11 Finally, peptides were N‐terminally functionalized with a fluorescein‐polyethylene glycol (FITC‐PEG) conjugate (Table S1), cleaved from solid support, and purified.
Binding affinities towards 14‐3‐3ζ were determined in a fluorescence polarization (FP) assay (Scheme 2 B, Figures S2–S4). Dissociation constants (K d values) of alkyne‐macrocyclized peptides A–H with 14‐3‐3ζ show a broad range depending on linker length and absolute configuration of building blocks at positions X and Y. Of the βRS8‐derived peptides (A–D, R/S‐configured), peptide A appears to be the best 14‐3‐3 binder (K d[A]=0.54 μm), showing a 1.6‐fold improved affinity in relation to the unmodified ESp sequence (K d[ESp]=0.84 μm, Scheme 2 B). In this respect it is interesting to note that the crosslink in peptide A has the same number of carbon atoms (eight) as in alkane‐crosslinked peptide βRS8. Increases in linker length (peptides B–D) result in a tremendous loss in target affinity (K d>10 μm). The βSS12‐derived peptides (E–H, S/S‐configured) show generally higher affinity for 14‐3‐3ζ than their R/S‐configured analogues. Peptides E and H reveal K d values in the sub‐micromolar range (Scheme 2 B). Interestingly, whereas one of the regioisomers (peptide G) binds 14‐3‐3ζ with a K d value in the low micromolar range (K d[G]=1.35 μm), the other one (peptide F) does not show any binding to 14‐3‐3ζ in the FP assay (K d[F]>10 μm). Of all the alkyne‐cyclized peptides, H (Figure 1 A) is the highest‐affinity binder (K d[H]=0.31 μm), exhibiting a dissociation constant in the same range as the alkane‐crosslinked peptide βSS12 (K d[βSS12]=0.10 μm) and showing higher affinity for 14‐3‐3 than βRS8 and unmodified peptide ESp. Peptide H bears the S‐configured building block 3 at positions X and Y (Figure 1 A), forming a 12‐carbon crosslink in analogy to the unsaturated parent peptide βSS12. The binding affinity of peptide H was verified in an orthogonal binding assay based on microscale thermophoresis (MST), providing affinities in good agreement with the FP results (K d=0.44 μm, Figures S5 and S6 and Table S2).
Figure 1.
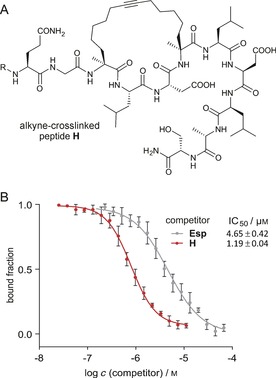
A) Chemical structure of alkyne‐crosslinked 14‐3‐3ζ binding peptide H (R=FITC‐PEG or acetyl). B) FP‐based displacement assay of N‐terminally acetylated peptides [ESp (gray) and H (red)] competing with FITC‐PEG‐labeled ESp (10 nm) from 14‐3‐3ζ (full length, 2 μm). Measurements were performed in triplicate (error=1σ).
To verify the binding site of peptide H on 14‐3‐3ζ, FP competition experiments were performed. Treating a complex of 14‐3‐3ζ and fluorescein‐labeled ESp with increasing concentrations of unlabeled peptide H resulted in full displacement of ESp [half maximal inhibitory concentration (IC50)=1.19 μm, Figure 1 B], thus confirming an overlapping binding site of ESp and H. Using unlabeled ESp as competitor, we observed less efficient displacement (IC50=4.65 μm), which is in agreement with its lower affinity for 14‐3‐3ζ.
To elucidate the details of molecular recognition between peptide H and 14‐3‐3ζ, we attempted to acquire a crystal structure of peptide H in complex with 14‐3‐3ζ. For this reason, H was co‐crystalized with 14‐3‐3ζΔC (aas 1–230), providing crystals that diffract in space group P212121. The resulting crystal structure (PDB ID: 5J31) provides the H–14‐3‐3ζΔC complex at 2.4 Å (Figure 2, Table S3). Each asymmetric unit contains two complexes with both 14‐3‐3 monomers showing a U‐shaped structure formed by nine α‐helices. This leads to the characteristic W‐shape of dimeric 14‐3‐3 (Figure 2 A).12 The hydrophobic groove of each 14‐3‐3ζ monomer is occupied by one molecule of peptide H (Figure 2 A). The corresponding 2 F o−F c density map of H allows the identification of the entire peptide (including the alkyne crosslink) except for N‐terminal amino acid Q420, which is not resolved (Figures S7–S9). In analogy to the unmodified binder ESp and the alkane‐crosslinked βSS12, peptide H adopts an irregular secondary structure binding to the same interface of 14‐3‐3ζ (Figure 2 B, C).14
Figure 2.
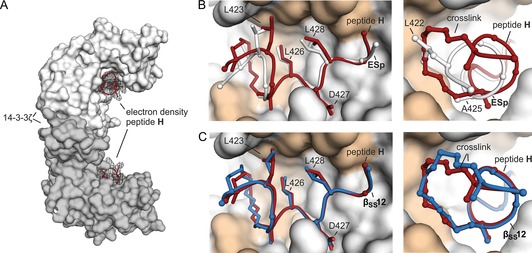
A) 14‐3‐3ζ dimer with 2 F o−F C electron density of peptides H (red, PDB ID: 5J31). B, C) Left: Side view of superimposed structures of peptide H (red) and ESp (white) or βSS12 (blue) in complex with 14‐3‐3ζ (gray/orange). Interacting side chains of peptides are shown in stick presentation (crosslink, L423, L426, D427, Y, L428). Hydrophobic pocket of 14‐3‐3ζ is highlighted orange (N42, S45, V46, F117, P165, I166, L172, N173, D213, L216, I217, L220). Right: Front view of superimposed peptides in complex with 14‐3‐3ζ showing exclusively the amino acid side chains at positions X and Y (ESp: L422, A425) used to incorporate crosslinks.
The C‐terminal part of peptide H (amino acids 426–429) maintains the overall configuration of ESp; this is evident in an overlay of their backbones and side chains (Figure 2 B). This preserves crucial C‐terminal hydrophobic interactions of L426 and L428 as well as the electrostatic attraction of D427 with Y128 and N174 in 14‐3‐3. Overall, peptide H primarily interacts with the hydrophobic groove of 14‐3‐3ζ (N42, S45, V46, F117, P165, I166, L172, N173, D213, L216, I217 and L220) through its alkyne crosslink (422/X and 425/Y) and amino acid side chains (L423, L426, L428, Figure 2 B, C). However, a backbone drift of amino acids 421–425 is evident on comparison with ESp, resulting in a slightly altered conformation of the N terminus. This shift is presumably caused by the sterically demanding 12‐carbon alkyne macrocycle replacing and thereby mimicking amino acids L422 (X) and A425 (Y) of linear ESp. In fact, the macrocycle exhibits extended hydrophobic contacts with 14‐3‐3ζ (Figures S7 and S8) relative to its natural analogues (L442 and A425). The overlay of peptide H with βSS12 when complexed to 14‐3‐3ζ shows excellent backbone and side chain superimposition both of the C‐terminal and of the N‐terminal amino acids with only minor variations of the crosslink conformation (Figure 2 C). This might explain their similar affinities towards 14‐3‐3. Overall, peptide H maintains the conserved C‐terminal conformation of ESp, while increasing the N‐terminal interaction surface with 14‐3‐3ζ through additional contacts arising from the introduced hydrocarbon alkyne macrocycle. Most likely, the alkyne crosslink supports binding by stabilization of the irregular peptide structure as well as formation of additional hydrophobic contacts, relative to ESp, with the 14‐3‐3ζ binding groove.
Here we report the first crystal structure of an alkyne‐crosslinked peptide—peptide H—in complex with its target protein 14‐3‐3ζ. Peptide H proved to be the highest‐affinity binder of a panel of eight alkyne‐crosslinked peptides derived from the 14‐3‐3 binding motif ESp. The binding affinity of peptide H (K d=0.31 μm) was determined by FP and validated by MST measurements. Further investigation of binding properties by competition assays and co‐crystallization confirmed the expected binding site on 14‐3‐3ζ. The crystal structure indicates that the alkyne macrocycle in peptide H constrains the irregular bioactive conformation, and engages in additional hydrophobic contacts while maintaining most of the interactions between ESp and 14‐3‐3ζ. In the presented setup, alkyne macrocyclization allows the stabilization of an irregular peptide structure, thereby expanding the scope of this powerful synthetic method. The introduction of a rigid and linear alkyne moiety into a peptide sequence enables the formation of previously inaccessible architectures with potential for the advancement of other class A peptidomimetics as therapeutic agents and tools in chemical biology research.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
P.M.C. is grateful to the Studienstiftung des Deutschen Volkes for a Fellowship. This work was supported by the German Research Foundation (DFG, Emmy Noether program GR3592/2‐1 and SFB1093), the European Research Council (ERC starting grant, no. 678623), AstraZeneca, Bayer CropScience, Bayer HealthCare, Boehringer Ingelheim, Merck KGaA, and the Max Planck Society. We appreciate the help of the beamline staff at Swiss Light Source PXII‐X10SA.
P. M. Cromm, K. Wallraven, A. Glas, D. Bier, A. Fürstner, C. Ottmann, T. N. Grossmann, ChemBioChem 2016, 17, 1915.
References
- 1. Kaspar A. A., Reichert J. M., Drug Discovery Today 2013, 18, 807–817. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Craik D. J., Fairlie D. P., Liras S., Price D., Chem. Biol. Drug Des. 2013, 81, 136–147; [DOI] [PubMed] [Google Scholar]
- 2b. Nevola L., Giralt E., Chem. Commun. 2015, 51, 3302–3315; [DOI] [PubMed] [Google Scholar]
- 2c. Spiegel J., Cromm P. M., Zimmermann G., Grossmann T. N., Waldmann H., Nat. Chem. Biol. 2014, 10, 613–622; [DOI] [PubMed] [Google Scholar]
- 2d. Cromm P. M., Spiegel J., Grossmann T. N., Waldmann H., Angew. Chem. Int. Ed. 2015, 54, 13516–13537; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13718–13741. [Google Scholar]
- 3. Pelay-Gimeno M., Glas A., Koch O., Grossmann T. N., Angew. Chem. Int. Ed. 2015, 54, 8896–8927; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9022–9054. [Google Scholar]
- 4. Bock J. E., Gavenonis J., Kritzer J. A., ACS Chem. Biol. 2013, 8, 488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Henchey L. K., Jochim A. L., Arora P. S., Curr. Opin. Chem. Biol. 2008, 12, 692–697; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Houk K. N., Leach A. G., Kim S. P., Zhang X., Angew. Chem. Int. Ed. 2003, 42, 4872–4897; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 5020–5046. [Google Scholar]
- 6.
- 6a. Wang Y., Chou D. H.-C., Angew. Chem. Int. Ed. 2015, 54, 10931–10934; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11081–11084; [Google Scholar]
- 6b. Assem N., Ferreira D. J., Wolan D. W., Dawson P. E., Angew. Chem. Int. Ed. 2015, 54, 8665–8668; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8789–8792; [Google Scholar]
- 6c. Mendive-Tapia L., Preciado S., García J., Ramón R., Kielland N., Albericio F., Lavilla R., Nat. Commun. 2015, 6, 7160; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Lau Y. H., Wu Y., Rossmann M., Tan B. X., de Andrade P., Tan Y. S., Verma C., McKenzie G. J., Venkitaraman A. R., Hyvönen M. et al., Angew. Chem. Int. Ed. 2015, 54, 15410–15413; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15630–15633; [Google Scholar]
- 6e. Lau Y. H., de Andrade P., McKenzie G. J., Venkitaraman A. R., Spring D. R., ChemBioChem 2014, 15, 2680–2683. [DOI] [PubMed] [Google Scholar]
- 7. Cromm P. M., Spiegel J., Grossmann T. N., ACS Chem. Biol. 2015, 10, 1362–1375. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Kim Y.-W., Grossmann T. N., Verdine G. L., Nat. Protoc. 2011, 6, 761–771; [DOI] [PubMed] [Google Scholar]
- 8b. Walensky L. D., Bird G. H., J. Med. Chem. 2014, 57, 6275–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patgiri A., Jochim A. L., Arora P. S., Acc. Chem. Res. 2008, 41, 1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fürstner A., Angew. Chem. Int. Ed. 2013, 52, 2794–2819; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2860–2887. [Google Scholar]
- 11. Cromm P. M., Schaubach S., Spiegel J., Fürstner A., Grossmann T. N., Waldmann H., Nat. Commun. 2016, 7, 11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ottmann C., Yasmin L., Weyand M., Veesenmeyer J. L., Diaz M. H., Palmer R. H., Francis M. S., Hauser A. R., Wittinghofer A., Hallberg B., EMBO J. 2007, 26, 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Masters S. C., Pederson K. J., Zhang L., Barbieri J. T., Fu H., Biochemistry 1999, 38, 5216–5221; [DOI] [PubMed] [Google Scholar]
- 13b. Henriksson M. L., Sundin C., Jansson A. L., Forsberg Å., Palmer R. H., Hallberg B., Biochem. J. 2002, 367, 617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glas A., Bier D., Hahne G., Rademacher C., Ottmann C., Grossmann T. N., Angew. Chem. Int. Ed. 2014, 53, 2489–2493; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2522–2526. [Google Scholar]
- 15. Glas A., Grossmann T., Synlett 2015, 26, 1–5. [Google Scholar]
- 16.
- 16a. Heppekausen J., Stade R., Goddard R., Fürstner A., J. Am. Chem. Soc. 2010, 132, 11045–11057; [DOI] [PubMed] [Google Scholar]
- 16b. Heppekausen J., Stade R., Kondoh A., Seidel G., Goddard R., Fürstner A., Chem. Eur. J. 2012, 18, 10281–10299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary