

Abstract
In the past decade, new approaches have been explored that are aimed at restoring functional β cell mass as a treatment strategy for diabetes. The two most intensely pursued strategies are β cell replacement through conversion of other cell types and β cell regeneration by enhancement of β cell replication. The approach closest to clinical implementation is the replacement of β cells with human pluripotent stem cell–derived (hPSC-derived) cells, which are currently under investigation in a clinical trial to assess their safety in humans. In addition, there has been success in reprogramming developmentally related cell types into β cells. Reprogramming approaches could find therapeutic applications by inducing β cell conversion in vivo or by reprogramming cells ex vivo followed by implantation. Finally, recent studies have revealed novel pharmacologic targets for stimulating β cell replication. Manipulating these targets or the pathways they regulate could be a strategy for promoting the expansion of residual β cells in diabetic patients. Here, we provide an overview of progress made toward β cell replacement and regeneration and discuss promises and challenges for clinical implementation of these strategies.
Introduction
Diabetes mellitus is a chronic disease affecting an estimated 422 million people worldwide in 2014 (1). Characterized by elevated blood sugar levels, diabetes occurs in two major forms, type 1 (T1D) and type 2 diabetes (T2D). T1D results from autoimmune destruction of the insulin-producing β cells in the pancreas, while T2D is characterized by insulin resistance and inadequate insulin secretion by the β cells. Recent studies suggest that β cell dysfunction occurs early in T2D and precedes the reduction in β cell mass observed later during disease progression (2). Because both types of diabetes eventually lead to β cell loss, research has focused on developing β cell replacement strategies to compensate for insulin deficiency. Islet transplantation has proven to be a successful therapy (3), but its clinical application is limited because of the shortage of donor cadaveric islets and the requirement for lifelong immune suppression. In the past decade, there have been intense efforts to identify alternative sources of β cells. β cell replacement strategies based on the in vitro differentiation of human pluripotent stem cells (hPSCs) toward insulin-producing cells have led to an ongoing human clinical trial (Figure 1). In addition, there have been exciting advances in in vivo regeneration approaches aimed at replenishing β cell mass either by converting related cell types into β cells, or by promoting the expansion of residual β cells in diabetic patients (Figure 2). In this Review, we focus on the recent progress toward clinically relevant therapeutic approaches for regenerating β cells.
Figure 1. β cell replacement from human pluripotent stem cell sources.

Currently pursued approaches include implantation of in vitro–generated pancreatic progenitor cells or β-like cells. In vitro–produced pancreatic progenitor cells differentiate into β cells within 16 weeks after implantation. Cell delivery in an encapsulation device prevents immune cells from contacting implanted cells derived from human embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs), while allowing free exchange of nutrients and hormones, including oxygen, glucose, and insulin. Precursors to β cells are depicted in yellow and insulin-producing cells in green.
Figure 2. Reprogramming approaches for generating replacement β cells.
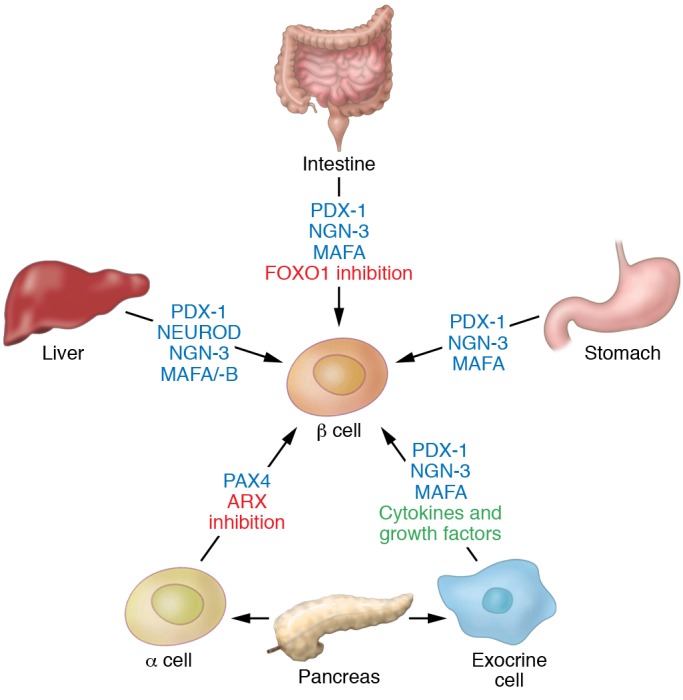
Cells and organs of similar developmental origin to that of pancreatic β cells, such as liver, stomach, intestine, or other pancreatic cell types, can be converted into β cells by reprogramming with transcription factors or in some instances by exposure to cytokines and growth factors.
β cell replacement by implantation of hPSC-derived cells
In the past decade, protocols have been developed that allow for the generation of pancreatic cells from hPSCs (4–7). These multistep protocols, which are based on developmental paradigms, use sequential stimulation or inhibition of key signaling pathways through small molecules and growth factors to differentiate hPSCs toward β cells. Early protocols support the in vitro differentiation of hPSCs up to the pancreatic progenitor cell stage (4, 6). Sixteen weeks after implantation of these progenitors into mice, they spontaneously differentiate into islet-like structures that contain β and non-β islet cell types (4, 5). When endogenous mouse β cells are ablated after in vivo differentiation of the hPSC-derived progenitor cell grafts, the mice are protected from developing diabetes. These findings in mice have provided the basis for the ongoing human phase I/II trial for patients with T1D (ViaCyte Inc. clinical trials identifier: NCT02239354). There is, however, a risk of immature cells having tumorigenic potential, and teratoma-like lesions have been observed around grafts after pancreatic progenitor cell engraftment into mice (4). To mitigate this risk, as well as to protect the implanted cells from alloimmune and autoimmune attack, in the current clinical trial hPSC-derived progenitors are placed in an encapsulation device (Figure 1). The employed macroencapsulation device holds large numbers of cells and is made of a semipermeable membrane that allows diffusion of oxygen, nutrients, and hormones while also creating a barrier against immune cells. In mice, encapsulated pancreatic progenitor cells are able to mature into functional β cells capable of reversing hyperglycemia (8–10). Microencapsulation technology, which uses hydrogel polymers to create a semipermeable sphere around cell aggregates, has also been explored for implanting cells without immune suppression. While earlier microencapsulation materials evoked a foreign body response that led to device failure (11, 12), recent improvements in the hydrogel composition mitigate this response (13). hPSC-derived pancreatic cells encapsulated in these new microencapsulation devices survive and function for at least 6 months after implantation into immunocompetent mice (14). Both types of encapsulation methods could provide immune protection to hPSC-derived cells in patients without the need for immunosuppression. Whether encapsulated pancreatic progenitor cells will differentiate into functional β cells in humans, as seen in mice, is still unclear and will emerge from ViaCyte’s ongoing trial.
Recent in vitro differentiation protocols can now also produce β-like cells from hPSCs, including from T1D patient–derived human induced pluripotent stem cells (hiPSCs) (7, 15, 16). Compared with earlier differentiation protocols, a critical feature of the recent improvements was to delay the induction of endocrine cell differentiation until the pancreatic progenitor cell program is fully established. This was accomplished by first suppressing expression of the proendocrine transcription factor neurogenin 3 (NGN-3) by addition of vitamin C, and then transiently inducing NGN-3 at subsequent stages with thyroid hormone and specific inhibitors of the TGF-β signaling pathway. These alterations to the protocol promoted the formation of insulin-positive cells that express a full spectrum of β cell–specific transcription factors (15). However, even with these improvements, in vitro–produced β-like cells still exhibit an insufficient insulin secretory response to a glucose challenge (7, 15). Upon implantation, these β-like cells require a shorter in vivo maturation period than pancreatic progenitor cells until acquiring the capacity to reverse hyperglycemia in mice (7, 15). It is still unclear whether engrafting β-like cells will prove advantageous to the engraftment of progenitor cells. Although β-like cells could reverse hyperglycemia more immediately, their high oxygen demand might also render β cells particularly prone to death early after implantation. While this may be mitigated by encapsulation in devices with an integrated oxygen reservoir (17), transplantation of progenitor cells offers distinct advantages. For one, the lower oxygen demand of progenitor cells could ensure improved graft survival in the first 8 weeks following implantation, during which the encapsulation device becomes vascularized. Moreover, the other endocrine cell types that develop alongside β cells in progenitor grafts could aid in optimizing blood glucose control in patients with diabetes. Paracrine signaling that occurs within islets is important for adequate regulation of hormone secretion. For example, insulin inhibits the release of glucagon from α cells, glucagon stimulates insulin release, and somatostatin from islet δ cells inhibits the release of both insulin and glucagon (18). These paracrine signaling mechanisms between endocrine cell types act as a feedback mechanism and help maintain glucose homeostasis by preventing excessive fluctuations in pancreatic hormone levels.
β cell replacement by transdifferentiation
Transdifferentiation, the direct conversion of one differentiated cell type into another, could provide an alternative to hPSCs as a source of new β cells. A large body of work suggests that developmentally related cells, including other pancreatic cell types and cells of the liver and gastrointestinal tract, have the greatest potential to convert into β cells (refs. 19–41 and Figure 2).
Transdifferentiation of pancreatic cells.
There is significant plasticity among the different endocrine cell types of the pancreas. In particular, α and δ cells have been shown to spontaneously convert into β cells after near-total β cell ablation in mice (26, 27, 38). Whether spontaneous transdifferentiation of non-β islet cells also occurs in humans in response to extreme β cell loss is still unknown. In patients with T1D, β cell regeneration through transdifferentiation or other mechanisms could be masked by autoimmune destruction of newly generated β cells.
Reprogramming of α cells to β cells can also occur through manipulation of a single transcription factor, either by inactivation of ARX, which is important for specifying α cell fate, or by ectopic expression of PAX4, an essential regulator of β cell development (refs. 19, 28, 29, 40, and Figure 2). Because a single gene is sufficient to induce α-to-β-cell transdifferentiation, it may be possible to identify small molecules that mimic this effect. Recent studies have identified compounds that can promote α-to-β-cell reprogramming, at least in cultured cells. Using a high-content screen for small-molecule inducers of insulin expression, Schreiber, Wagner, and colleagues identified two kinase inhibitors as compounds that upregulate insulin expression in cultured α cells (31, 32). These inhibitors, which target ribosomal S6 kinase (RSK) and cyclin-dependent kinase-2 (CDK2), were shown to upregulate multiple β cell markers, including PAX4, in a dose-dependent manner. Although these compounds have only been tested in vitro, the studies suggest that pharmacologic agents could promote pancreatic cell plasticity without the need for genetic modification. Screens to identify chemical inducers of PAX4 or inhibitors of ARX may reveal compounds that promote α-to-β-cell transdifferentiation and reverse diabetes.
While reprogramming of α cells has proven to restore functional β cell mass and cure diabetes in animal models (19, 28, 29, 40), it also results in a severe depletion of α cells and a corresponding decrease in the production of glucagon (28, 42). Depletion of α cells in mice does not have overt effects on health or lifespan (42), and even a small number of α cells (2% of total population) is sufficient to maintain glucagon signaling (43). Thus, converting α cells into β cells could be a viable approach to treat diabetes; however, it is currently not known what effect α cell depletion will have in humans. It may be that glucagon deficiency due to α cell loss puts patients at risk for hypoglycemia. While the α cell response is important in recovery from insulin-induced hypoglycemia in humans, epinephrine can compensate for insufficient glucagon secretion (44). Therefore, an intact epinephrine response to hypoglycemia is an important prerequisite when considering α-to-β-cell reprogramming in patients.
Owing to their abundance, pancreatic exocrine cells, which comprise acinar and ductal cells, have been explored as an alternative population for reprogramming. In contrast to α cells, acinar cells harbor extensive repressive histone modifications at the promoters of endocrine genes, which might constitute a barrier for endocrine reprogramming (24). Indeed, the in vivo conversion of acinar cells requires the expression of not only one but three transcription factors (PDX-1, NGN-3, and MAFA) that are important for β cell development (ref. 41 and Figure 2). More relevant for possible clinical translation, there is also evidence that exocrine cells can be converted into β-like cells without genetic manipulation through in vivo cytokine/growth factor treatment (21, 35, 45). When β cells are destroyed by injection of a β cell toxin, systemic administration of EGF and CNTF induces conversion of acinar cells into β cells and restores β cell mass and normoglycemia (21). It has been observed that the success of exocrine cell reprogramming may depend on the glycemic environment. A recent study showed that hyperglycemia can inhibit in vivo reprogramming of acinar cells by PDX-1, NGN-3, and MAFA (46). A better understanding of how glucose levels influence cell plasticity and the response to reprogramming factors will be critical to assess the strategy’s potential for future clinical application. One important consideration is that manipulations promoting transdifferentiation could also have undesired effects. For example, loss of the tumor suppressor menin 1 in α cells triggers their transdifferentiation into insulin-expressing cells, but also the development of insulinomas from reprogrammed cells (47). Therefore, before these therapies are implemented, extensive studies are necessary to fully evaluate the state of maturity and stability of reprogrammed cells.
Transdifferentiation of liver and gastrointestinal cells.
Reprogramming-based strategies to generate β cells from somatic cells are not limited to cells of the pancreas, but extend to other gut tube-derived organs, such as the liver, intestine, and stomach. In the past decade, most studies have focused on the liver as a potential source of β cells. Adenoviral delivery of pancreatic transcription factors, including PDX-1 (22, 30) and/or NEUROD (33), NGN-3 (37, 39), and more recently MAFA/MAFB (34), has been shown to induce emergence of insulin-producing cells in the liver (Figure 2). While these insulin-producing cells were to some extent able to correct hyperglycemia in diabetic mice, it is less clear whether these cells are glucose-responsive and thus function as true β cells.
Studies have demonstrated that simultaneous transgenic expression of PDX-1, NGN-3, and MAFA can promote rapid conversion of gastrointestinal endocrine cells in the antral stomach and intestine into insulin-expressing cells in vivo (refs. 20, 25, and Figure 2). These reprogrammed cells exhibit ultrastructural features of β cells, are glucose-responsive, and are able to ameliorate hyperglycemia in diabetic mice. Similarly, ablation of the transcription factor FOXO1 in endocrine progenitors of the intestine results in the generation of insulin-expressing cells that are able to reverse hyperglycemia in mice (36). This finding has been reproduced in human gut organoids (23), suggesting that FOXO1 inhibition in gut organoids could offer a source of insulin-producing cells to treat human diabetes. As human gastrointestinal tissue is readily accessible by noninvasive techniques, ex vivo reprogramming of gastrointestinal cells could be a source of autologous β cells for implantation in patients with diabetes.
Recent studies in an animal model of T1D indicate that insulin-producing reprogrammed liver cells are susceptible to autoimmune attack (37). Therefore, to prevent autoimmune destruction of β cells produced by reprogramming, immunosuppressive therapy will likely be necessary to achieve lasting normoglycemia in T1D patients. Alternatively, ex vivo reprogrammed cells could be implanted in an encapsulation device. Despite remaining hurdles, the progress made in converting other cell types into β cells is remarkable and suggests avenues for clinical translation.
β cell replacement by promotion of β cell expansion
Another, and perhaps the therapeutically most tangible, method for inducing β cell regeneration in vivo is to target pathways that regulate β cell proliferation (Figure 3). The predominant mechanism for adapting β cell mass to increased metabolic demand, as observed during pregnancy or obesity, is through modulation of β cell replication (48, 49). Thus there has been considerable interest in understanding the mechanisms that regulate replication of β cells with the goal of discovering new therapeutic targets to promote their regeneration. This approach could benefit patients with T2D as well as T1D, as residual β cells are frequently found even decades after the onset of T1D (50, 51).
Figure 3. Select signaling pathways known to regulate β cell proliferation.

Growth factors (PDGF), nutrients (glucose), and hormones (GLP-1 and insulin) have been shown to induce β cell proliferation by activating both canonical and noncanonical mitogenic pathways. Activation of their respective receptors induces signaling via the ERK (also called MAPK) signaling pathway either directly (in the case of PDGF) or via crosstalk mechanisms. Glucose, GLP-1, and insulin have been shown to activate mitogenic signaling via the PI3K/AKT/mTOR pathway. Glucose signals through the calcineurin/NFAT and ERK pathways, while signaling via GLP-1 induces cAMP synthesis to induce proliferation. Small molecules/compounds have been identified that target proteins in these signaling pathways, inhibiting either DYRK1a or GSK3β, both of which phosphorylate NFAT, sequestering it in the cytoplasm. Black and gray arrows indicate canonical and noncanonical signaling, respectively. The depicted pathways regulating β cell proliferation are mainly based on evidence in rodents and do not represent a comprehensive overview of all published work. Compounds shown to induce human β cell replication are indicated in red. AP, aminopyrazine; 5-IT, 5-iodotubercidin; Ca2+, calcium; CN, calcineurin.
Aging and β cell replication.
Replication of β cells is the predominant mechanism that ensures the rapid expansion of β cell mass early in life; however, the regenerative capacity of β cells rapidly declines with advancing age (52–56). This age-dependent decline in β cell proliferation is regulated by p16Ink4a, a cyclin-dependent kinase inhibitor encoded by the Cdkn2a gene (57). Multiple age-dependently regulated factors, including p38MAPK and PcG/trithorax group proteins, have been shown to epigenetically modify the Cdkn2a locus and repress p16Ink4a expression (58–62). In young β cells, PDGF receptor signaling increases the abundance of the PcG/trithorax group protein EZH2, thereby repressing p16Ink4a expression. However, in aged β cells, decline of PDGF receptor expression leads to p16Ink4a derepression and β cell cycle arrest (58).
To more globally define age-associated changes in the β cell epigenome, Kaestner and colleagues carried out a genome-wide analysis of β cells from young and old mice and found that the proliferative decline of β cells correlated with increased de novo promoter methylation and decreased expression of cell cycle regulators (63). This suggests that manipulation of epigenetic regulators could reverse β cell senescence and promote regeneration. Interestingly, this group also observed upregulation of genes involved in β cell function and improved insulin secretory function with age (63). While these results contradict early studies showing a decline of β cell function with age (64–67), they align with more recent work demonstrating sustained or improved β cell secretory function in older animals (68, 69). Because β cell proliferation and β cell function are tightly linked (70), additional studies are needed to determine whether β cell proliferation can be safely increased without compromising function.
β cell replication in pregnancy.
During pregnancy, β cell mass expands in order to adapt the organism to increasing insulin demand (71–74). Multiple factors, including lactogens, serotonin, and components of the EGFR signaling pathway, have been shown to increase β cell replication in pregnant rodents (71, 75–78). There is, however, controversy as to whether adaptive β cell proliferation during pregnancy occurs to the same extent in humans (72). Moreover, conflicting reports have been published regarding the conservation of molecular pathways regulating β cell mass expansion between pregnant rodents and humans. While one study found β cell proliferation to be induced by prolactin and placental lactogen in two human islet samples (75), more recently, Stewart and colleagues were unable to induce human β cell proliferation with prolactin in six independent human islet samples (79). This could be explained, at least in part, by the lack of prolactin receptor expression on human β cells (79). While prolactin receptor agonists may not be effective for stimulating human β cell proliferation, downstream signaling pathways may be conserved and could provide insight into therapeutic targets. In support of this idea, Vasavada and colleagues found that treatment of human islets with recombinant osteoprotegerin, a lactogen target, induced human β cell proliferation (80). Importantly, the FDA-approved osteoporosis drug denosumab mimicked the activity of osteoprotegerin and enhanced human β cell replication in vitro and after engraftment of human islets into mice. The pro-proliferative effect of denosumab suggests that there is potential for repurposing this drug for the treatment of diabetes.
With the goal of discovering novel targets for enhancing β cell proliferation, Ahnfelt-Rønne and colleagues took a proteomic approach to identify proteins that change in abundance during pregnancy in mice (81). The analysis not only confirmed regulation of targets previously shown to be controlled at the mRNA level (77, 82–84), but also identified proteins not previously associated with pregnancy-induced β cell expansion. Two examples are stathmin 1 and nuclear chloride ion channel 1, which have known roles in the regulation of cell proliferation and are being evaluated as drug targets in cancer (85–88). While follow-up studies will be necessary, this study highlights the importance of global approaches to identify novel molecular targets for enhancing β cell proliferation.
β cell replication in hyperglycemia and insulin resistance.
Apart from pregnancy, β cell proliferation is also regulated by diet and changes in metabolic state. A recent study suggests that nutritional cues have immediate effects on the capacity of β cells to mount a regenerative response. When mice were prematurely weaned from fat-rich milk to carbohydrate-rich chow, the potential of β cells for compensatory proliferation increased (89). Although it remains to be studied whether a similar mechanism operates during adulthood, this finding suggests that diet composition could have effects on β cell mass. Metabolic regulation of β cell proliferation is also evident during a state of insulin resistance, which is known to trigger compensatory β cell proliferation. This has been demonstrated in multiple rodent models of diabetes, including ob/ob mice (90), db/db mice (91), and Zucker fatty rats (92), and in high-fat-diet feeding (93). Increased β cell mass is also observed in hyperinsulinemic humans with obesity and insulin resistance (94–96); however, whether proliferation is increased is less clear.
Glucose and insulin have been identified as inducers of β cell replication (Figure 3). Multiple studies have demonstrated increased proliferation of rodent and human β cells following glucose infusion (97–103). Glucose metabolism is required for β cell proliferation, as lack of glucokinase, a key enzyme in glycolysis that converts glucose to glucose-6-phosphate, decreases β cell proliferation whereas treatment with a small-molecule glucokinase activator stimulates β cell proliferation (104). However, the pro-proliferative effect of glucose is only observed in the short term, while sustained exposure of β cells to high glucose can cause glucotoxicity, resulting in DNA damage and apoptosis, as also seen in β cells from T2D patients (105). Therefore, there is a need to better understand where the mitogenic and DNA damage pathways diverge before the glucose-induced mitogenic pathway can be considered for therapeutic intervention.
While glucose can increase β cell replication, β cell hyperplasia occurs in ob/ob and db/db mice prior to the onset of hyperglycemia and is also observed in mouse models of insulin resistance in the absence of hyperglycemia (91, 106–108). These observations suggest that factors other than glucose contribute to β cell mass expansion in the face of insulin resistance. Insulin levels are highly elevated in the insulin-resistant state, and insulin signaling has been shown to account for compensatory β cell growth during insulin resistance. Ablation of the insulin receptor in an insulin-resistant mouse model impaired β cell proliferation and rendered mice prematurely diabetic (109). In contrast, deletion of the IGF receptor had little effect on β cell growth, suggesting that compensatory β cell mass expansion predominantly depends on insulin rather than IGF signaling.
Circulating factors and β cell replication.
In recent years, significant effort has been put forth into identifying systemic regulators of β cell proliferation in the context of aging, pregnancy, and metabolic challenge. Circulating factors that are regulated during metabolic adaptation are particularly attractive therapeutic targets, as manipulating their activity might mitigate the risk for inducing tumors in other tissues. Studies have shown that circulating factors from young mice improve regeneration of aged islets. This has been demonstrated in the context of parabiosis experiments, in which a young and an old mouse are surgically joined to develop a shared circulatory system, or after revascularization of an islet graft from aged mice in a young host (68, 110). Likewise, β cell replication increases when islets from metabolically normal mice are grafted under the kidney capsule of insulin-resistant mice (111). While these experiments clearly illustrate the importance of circulating factors in β cell regeneration, the specific factor or factors that account for the effects have remained elusive. Multiple circulating factors, including glucagon-like peptide-1 (GLP-1), secreted by the intestinal L cells (112–114); thyroid hormone (115, 116); the osteoblast-derived hormone osteocalcin (117–120); liver-derived angiopoietin-like protein 8 (ANGPTL8, also known as betatrophin) (121); and recently the liver-secreted protease inhibitor SERPINB1 (122), have been identified as potentially pro-proliferative for β cells, at least in rodents (Figure 3). It is less clear whether these circulating factors can also stimulate human β cell growth. Controversial reports exist regarding effects of GLP-1 analogs on human β cell growth, with one study finding no effect (123), while another reports stimulation (124). Illustrating the difficulty of controlled studies using primary human cells, subsequent work showed that the age of the islet donor might be a factor contributing to responsiveness of β cells to GLP-1 analogs (125). The original report describing ANGPTL8 as a β cell growth factor received much attention (121). However, further studies on ANGPTL8 have called its role in β cell mass expansion into question. Genetic loss- and gain-of-function experiments with Angptl8 revealed no effect on β cell mass in insulin-resistant mice (126, 127), showing that ANGPTL8 is not the long-sought-after liver-derived factor that stimulates β cell growth. Consistent with findings by Gromada and colleagues (127), ANGPTL8 also failed to exert a pro-proliferative effect on transplanted human β cells (128). More promising are recent findings on osteocalcin (120) and SERPINB1 (122), which indicate that these hormones could be effective in stimulating human β cell proliferation. Proliferation of β cells was increased after treatment of human islets with decarboxylated osteocalcin or small molecules mimicking SERPINB1 activity both ex vivo and after transplantation into mice (122). Preliminary analysis of mice treated with small-molecule mimics of SERPINB1 suggests that the effects on proliferation of extrapancreatic tissues are limited, which raises hope that it might be possible to identify growth-stimulating agents that are selective for β cells.
Intracellular signaling pathways regulating β cell replication.
Many groups have used high-throughput screening methods to discover novel molecules and pathways that could stimulate β cell mass expansion (129–138). Some of these screens have led to the discovery of novel compounds with therapeutic potential. For example, a high-throughput chemical screen recently identified aminopyrazine compounds, harmine, INDY, and 5-iodotubercidin as pro-proliferative in rodent β cell lines, and these compounds were subsequently shown to also augment human β cell proliferation (129, 134, 137, 139). Interestingly, all 4 molecules inhibit the kinase DYRK1A, which blocks nuclear localization of NFAT, a transcription factor that activates expression of cell cycle genes in β cells (refs. 140, 141, and Figure 3). Aminopyrazine compounds have a larger effect on β cell proliferation than harmine, which is explained by the additional inhibition of glycogen synthase kinase-3β (GSK3β) by aminopyrazine compounds (137). Like DYRK1A, GSK3β prevents nuclear localization of NFAT (142) and inhibits β cell proliferation (143–146). Interestingly, osteoprotegerin and SERPINB1 have both been shown to inhibit GSK3β activity (refs. 80, 122, and Figure 3), suggesting that their effect on β cell proliferation may, at least in part, be mediated through GSK3β inhibition. A significant hurdle for advancing β cell therapeutics for these pathways is the unclear specificity of many of the small molecules as well as their effects on multiple tissues.
Additional potentially druggable intracellular regulators of β cell proliferation have been identified through candidate approaches. The literature on intracellular signaling in β cell proliferation has recently been comprehensively reviewed (147–150), and we refer to these reviews for a comprehensive description of all pathways shown to regulate β cell proliferation. From the numerous studies, the MAPK and PI3K/AKT pathways have emerged as critical regulators of β cell proliferation also in humans (Figure 3). The MAPK pathway via ERK1/2 phosphorylation is the key mitogenic pathway that separates metabolic regulation of β cell function from the regulation of β cell proliferation, as ERK1/2 phosphorylation is not required for glucose-stimulated insulin secretion (151). The MAPK pathway mediates the β cell mitogenic effect of multiple growth factors, hormones, and nutrients, including PDGF, GLP-1, prolactin, insulin, and glucose (58, 152–159). The second major pathway responsible for transducing β cell proliferative signals is the PI3K/AKT/mTOR pathway, which is activated by insulin, GLP-1, and glucose (153, 159–162). AKT activation is an important component that links growth signals to its downstream target mTOR, which coordinates a cell growth response directly through its effect on cell cycle regulators (143, 163). Numerous studies have demonstrated a role of this pathway in promoting β cell proliferation in vitro and increasing β cell mass in vivo (143, 160, 163–167). Further illustrating its pro-proliferative role, AKT/mTOR signaling is active in pancreatic endocrine tumors (168). Notably, PI3K signaling can induce β cell proliferation not only by activating AKT, but also through AKT-independent PKCζ, which mediates the proliferative effect of glucose on human β cells (169–172).
It is important to consider that significant crosstalk exists between the signaling pathways. For example, high glucose and GLP-1 levels activate both mTOR and MAPK signaling (153, 158, 160). A recent study nicely illustrates how the balance between different signaling arms determines the β cell response to insulin. Knockdown of PI3K resulted in rerouting of the insulin signal from PI3K-mediated metabolic signaling to ERK-mediated mitogenic signaling, which induced a switch of β cells from highly glucose-responsive to proliferative (173). Extensive feedback inhibition and amplification constitute a further layer of complexity, exemplified by mTOR-mediated negative feedback on insulin signaling via IRS2 (174).
All of these intracellular signals converge to regulate the core G1/S cell cycle machinery (147–150). Successful targeting of β cell proliferation will hinge on the downregulation of cell cycle inhibitors and upregulation of cell cycle activators. The example of aminopyrazine compounds, which target DYRK1A and GSK3β, illustrates that targeting more than one pathway will likely have a more robust effect on β cell proliferation than targeting one pathway alone. Given the extremely low proliferation rate of human β cells (54), hitting multiple targets might be necessary to produce clinically relevant effects. Furthermore, as regenerative and oncogenic pathways share similar effector proteins, a major challenge will be to enhance β cell proliferation without inducing aberrant growth of β cells or other tissues.
Challenges for therapeutic implementation
Both in vivo reprogramming approaches and the induction of β cell proliferation will likely require cell type–specific delivery systems for application in humans. Local delivery of reprogramming or regeneration factors could be achieved by ultrasound destruction of microbubbles carrying plasmid DNA administered into the pancreatic microcirculation (175). Tethering the molecule to the ligand of a β cell–specific receptor could be an alternative approach for delivering molecules directly to the β cell. The efficacy of this approach has recently been demonstrated for GLP-1–estrogen conjugates in β cells (176).
Perhaps the most challenging issue is protecting the newly generated β cells from autoimmune destruction in T1D. Similarly, the autoimmune and alloimmune responses impose a hurdle for hPSC-based replacement therapies. hiPSC-derived β cells from T1D patients can serve as an autologous source for cell replacement therapy that would obviate the need for systemic immune suppression (16); however, these cells are not protected from autoimmune destruction in T1D patients, and likely would still need to be implanted within an encapsulation device. Recent studies suggest that gene editing strategies can be used to generate hPSCs that are invisible to the immune system and could escape at least allogenic rejection (177). Similar strategies could perhaps also allow the cells to evade autoimmune destruction. Overall, significant progress has been made in the past decade, and the coming decade will show which strategy will hold most promise for translation into clinical therapies.
Acknowledgments
We apologize to our colleagues whose references were omitted owing to space constraints. Work in the Sander laboratory is supported by grants from the NIH, the Juvenile Diabetes Research Foundation, the Helmsley Charitable Trust, and the California Institute of Regenerative Medicine. JB was supported by the NIH/National Institute of General Medical Sciences UCSD Genetics Training Program T32 GM008666 and the Glenn/American Federation for Aging Research Scholarship for Research in the Biology of Aging.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(10):3651–3660. doi:10.1172/JCI87439.
References
- 1. World Health Organization. Global report on diabetes. http://www.who.int/diabetes/global-report/en Published April 7, 2016. Accessed August 2, 2016.
- 2.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150(6):1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shapiro AM, et al. International trial of the Edmonton protocol for islet transplantation. N Engl J Med. 2006;355(13):1318–1330. doi: 10.1056/NEJMoa061267. [DOI] [PubMed] [Google Scholar]
- 4.Kroon E, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26(4):443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- 5.Rezania A, et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes. 2012;61(8):2016–2029. doi: 10.2337/db11-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Amour KA, et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24(11):1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- 7.Pagliuca FW, et al. Generation of functional human pancreatic β cells in vitro. Cell. 2014;159(2):428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruin JE, et al. Maturation and function of human embryonic stem cell-derived pancreatic progenitors in macroencapsulation devices following transplant into mice. Diabetologia. 2013;56(9):1987–1998. doi: 10.1007/s00125-013-2955-4. [DOI] [PubMed] [Google Scholar]
- 9.Kirk K, Hao E, Lahmy R, Itkin-Ansari P. Human embryonic stem cell derived islet progenitors mature inside an encapsulation device without evidence of increased biomass or cell escape. Stem Cell Res. 2014;12(3):807–814. doi: 10.1016/j.scr.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Xie R, et al. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell. 2013;12(2):224–237. doi: 10.1016/j.stem.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Groot M, Schuurs TA, van Schilfgaarde R. Causes of limited survival of microencapsulated pancreatic islet grafts. J Surg Res. 2004;121(1):141–150. doi: 10.1016/j.jss.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 12.Tuch BE, et al. Safety and viability of microencapsulated human islets transplanted into diabetic humans. Diabetes Care. 2009;32(10):1887–1889. doi: 10.2337/dc09-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vegas AJ, et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat Biotechnol. 2016;34(3):345–352. doi: 10.1038/nbt.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vegas AJ, et al. Long-term glycemic control using polymer-encapsulated human stem cell-derived β cells in immune-competent mice. Nat Med. 2016;22(3):306–311. doi: 10.1038/nm.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rezania A, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32(11):1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- 16.Millman JR, Xie C, Van Dervort A, Gürtler M, Pagliuca FW, Melton DA. Generation of stem cell-derived β-cells from patients with type 1 diabetes. Nat Commun. 2016;7: doi: 10.1038/ncomms11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ludwig B, et al. Transplantation of human islets without immunosuppression. Proc Natl Acad Sci U S A. 2013;110(47):19054–19058. doi: 10.1073/pnas.1317561110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caicedo A. Paracrine and autocrine interactions in the human islet: more than meets the eye. Semin Cell Dev Biol. 2013;24(1):11–21. doi: 10.1016/j.semcdb.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Hasani K, et al. Adult duct-lining cells can reprogram into β-like cells able to counter repeated cycles of toxin-induced diabetes. Dev Cell. 2013;26(1):86–100. doi: 10.1016/j.devcel.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 20.Ariyachet C, et al. Reprogrammed stomach tissue as a renewable source of functional β cells for blood glucose regulation. Cell Stem Cell. 2016;18(3):410–421. doi: 10.1016/j.stem.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baeyens L, et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol. 2014;32(1):76–83. doi: 10.1038/nbt.2747. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Ber I, et al. Functional, persistent, and extended liver to pancreas transdifferentiation. J Biol Chem. 2003;278(34):31950–31957. doi: 10.1074/jbc.M303127200. [DOI] [PubMed] [Google Scholar]
- 23.Bouchi R, et al. FOXO1 inhibition yields functional insulin-producing cells in human gut organoid cultures. Nat Commun. 2014;5: doi: 10.1038/ncomms5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bramswig NC, et al. Epigenomic plasticity enables human pancreatic α to β cell reprogramming. J Clin Invest. 2013;123(3):1275–1284. doi: 10.1172/JCI66514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen YJ, et al. De novo formation of insulin-producing “neo-β cell islets” from intestinal crypts. Cell Rep. 2014;6(6):1046–1058. doi: 10.1016/j.celrep.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chera S, et al. Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers. Nature. 2014;514(7523):503–507. doi: 10.1038/nature13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung CH, Hao E, Piran R, Keinan E, Levine F. Pancreatic β-cell neogenesis by direct conversion from mature α-cells. Stem Cells. 2010;28(9):1630–1638. doi: 10.1002/stem.482. [DOI] [PubMed] [Google Scholar]
- 28.Collombat P, et al. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell. 2009;138(3):449–462. doi: 10.1016/j.cell.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Courtney M, et al. The inactivation of Arx in pancreatic α-cells triggers their neogenesis and conversion into functional β-like cells. PLoS Genet. 2013;9(10): doi: 10.1371/journal.pgen.1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferber S, et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6(5):568–572. doi: 10.1038/75050. [DOI] [PubMed] [Google Scholar]
- 31.Fomina-Yadlin D, et al. GW8510 increases insulin expression in pancreatic alpha cells through activation of p53 transcriptional activity. PLoS ONE. 2012;7(1): doi: 10.1371/journal.pone.0028808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fomina-Yadlin D, et al. Small-molecule inducers of insulin expression in pancreatic alpha-cells. Proc Natl Acad Sci USA. 2010;107(34):15099–15104. doi: 10.1073/pnas.1010018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kojima H, et al. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9(5):596–603. doi: 10.1038/nm867. [DOI] [PubMed] [Google Scholar]
- 34.Nagasaki H, et al. Generation of insulin-producing cells from the mouse liver using β cell-related gene transfer including Mafa and Mafb. PLoS ONE. 2014;9(11): doi: 10.1371/journal.pone.0113022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rooman I, Bouwens L. Combined gastrin and epidermal growth factor treatment induces islet regeneration and restores normoglycaemia in C57Bl6/J mice treated with alloxan. Diabetologia. 2004;47(2):259–265. doi: 10.1007/s00125-003-1287-1. [DOI] [PubMed] [Google Scholar]
- 36.Talchai C, Xuan S, Kitamura T, DePinho RA, Accili D. Generation of functional insulin-producing cells in the gut by Foxo1 ablation. Nat Genet. 2012;44(4):406–412. doi: 10.1038/ng.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang DQ, et al. Genetically reprogrammed, liver-derived insulin-producing cells are glucose-responsive, but susceptible to autoimmune destruction in settings of murine model of type 1 diabetes. Am J Transl Res. 2013;5(2):184–199. [PMC free article] [PubMed] [Google Scholar]
- 38.Thorel F, et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464(7292):1149–1154. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yechoor V, et al. Neurogenin3 is sufficient for transdetermination of hepatic progenitor cells into neo-islets in vivo but not transdifferentiation of hepatocytes. Dev Cell. 2009;16(3):358–373. doi: 10.1016/j.devcel.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Fava GE, Wang H, Mauvais-Jarvis F, Fonseca VA, Wu H. PAX4 gene transfer induces α-to-β cell phenotypic conversion and confers therapeutic benefits for diabetes treatment. Mol Ther. 2016;24(2):251–260. doi: 10.1038/mt.2015.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455(7213):627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hancock AS, Du A, Liu J, Miller M, May CL. Glucagon deficiency reduces hepatic glucose production and improves glucose tolerance in adult mice. Mol Endocrinol. 2010;24(8):1605–1614. doi: 10.1210/me.2010-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorel F, et al. Normal glucagon signaling and β-cell function after near-total α-cell ablation in adult mice. Diabetes. 2011;60(11):2872–2882. doi: 10.2337/db11-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cryer PE, Gerich JE. Relevance of glucose counterregulatory systems to patients with diabetes: critical roles of glucagon and epinephrine. Diabetes Care. 1983;6(1):95–99. doi: 10.2337/diacare.6.1.95. [DOI] [PubMed] [Google Scholar]
- 45.Valdez IA, Dirice E, Gupta MK, Shirakawa J, Teo AK, Kulkarni RN. Proinflammatory cytokines induce endocrine differentiation in pancreatic ductal cells via STAT3-dependent NGN3 activation. Cell Rep. 2016;15(3):460–470. doi: 10.1016/j.celrep.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cavelti-Weder C, et al. Hyperglycaemia attenuates in vivo reprogramming of pancreatic exocrine cells to beta cells in mice. Diabetologia. 2016;59(3):522–532. doi: 10.1007/s00125-015-3838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu J, et al. Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology. 2010;138(5):1954–1965. doi: 10.1053/j.gastro.2010.01.046. [DOI] [PubMed] [Google Scholar]
- 48.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429(6987):41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 49.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest. 2007;117(9):2553–2561. doi: 10.1172/JCI32959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keenan HA, et al. Residual insulin production and pancreatic β-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes. 2010;59(11):2846–2853. doi: 10.2337/db10-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weir GC, Bonner-Weir S. Islet β cell mass in diabetes and how it relates to function, birth, and death. Ann N Y Acad Sci. 2013;1281:92–105. doi: 10.1111/nyas.12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perl S, et al. Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab. 2010;95(10):E234–E239. doi: 10.1210/jc.2010-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gregg BE, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab. 2012;97(9):3197–3206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meier JJ, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57(6):1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Köhler CU, Olewinski M, Tannapfel A, Schmidt WE, Fritsch H, Meier JJ. Cell cycle control of β-cell replication in the prenatal and postnatal human pancreas. Am J Physiol Endocrinol Metab. 2011;300(1):E221–E230. doi: 10.1152/ajpendo.00496.2010. [DOI] [PubMed] [Google Scholar]
- 56.Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54(9):2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 57.Krishnamurthy J, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443(7110):453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 58.Chen H, et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature. 2011;478(7369):349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen H, et al. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009;23(8):975–985. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009;23(8):906–911. doi: 10.1101/gad.1742609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wong ES, et al. p38MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Dev Cell. 2009;17(1):142–149. doi: 10.1016/j.devcel.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 62.Zhou JX, et al. Combined modulation of polycomb and trithorax genes rejuvenates β cell replication. J Clin Invest. 2013;123(11):4849–4858. doi: 10.1172/JCI69468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Avrahami D, et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab. 2015;22(4):619–632. doi: 10.1016/j.cmet.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Järvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J Clin Endocrinol Metab. 1999;84(3):863–868. doi: 10.1210/jcem.84.3.5542. [DOI] [PubMed] [Google Scholar]
- 65.Chang AM, Halter JB. Aging and insulin secretion. Am J Physiol Endocrinol Metab. 2003;284(1):E7–E12. doi: 10.1152/ajpendo.00366.2002. [DOI] [PubMed] [Google Scholar]
- 66.Basu R, et al. Mechanisms of the age-associated deterioration in glucose tolerance: contribution of alterations in insulin secretion, action, and clearance. Diabetes. 2003;52(7):1738–1748. doi: 10.2337/diabetes.52.7.1738. [DOI] [PubMed] [Google Scholar]
- 67.Bergamini E, Bombara M, Fierabracci V, Masiello P, Novelli M. Effects of different regimens of dietary restriction on the age-related decline in insulin secretory response of isolated rat pancreatic islets. Ann N Y Acad Sci. 1991;621:327–336. doi: 10.1111/j.1749-6632.1991.tb16989.x. [DOI] [PubMed] [Google Scholar]
- 68.Almaça J, et al. Young capillary vessels rejuvenate aged pancreatic islets. Proc Natl Acad Sci USA. 2014;111(49):17612–17617. doi: 10.1073/pnas.1414053111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Helman A, et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med. 2016;22(4):412–420. doi: 10.1038/nm.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klochendler A, et al. The genetic program of pancreatic β-cell replication in vivo. Diabetes. 2016;65(7):2081–2093. doi: 10.2337/db16-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology. 1992;130(3):1459–1466. doi: 10.1210/endo.130.3.1537300. [DOI] [PubMed] [Google Scholar]
- 72.Butler AE, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia. 2010;53(10):2167–2176. doi: 10.1007/s00125-010-1809-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas in human pregnancy. Br J Obstet Gynaecol. 1978;85(11):818–820. doi: 10.1111/j.1471-0528.1978.tb15835.x. [DOI] [PubMed] [Google Scholar]
- 74.Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29(6):301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- 75.Brelje TC, et al. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology. 1993;132(2):879–887. doi: 10.1210/endo.132.2.8425500. [DOI] [PubMed] [Google Scholar]
- 76.Vasavada RC, et al. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. J Biol Chem. 2000;275(20):15399–15406. doi: 10.1074/jbc.275.20.15399. [DOI] [PubMed] [Google Scholar]
- 77.Kim H, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med. 2010;16(7):804–808. doi: 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hakonen E, Ustinov J, Palgi J, Miettinen PJ, Otonkoski T. EGFR signaling promotes β-cell proliferation and survivin expression during pregnancy. PLoS ONE. 2014;9(4): doi: 10.1371/journal.pone.0093651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen H, et al. Augmented Stat5 signaling bypasses multiple impediments to lactogen-mediated proliferation in human β-cells. Diabetes. 2015;64(11):3784–3797. doi: 10.2337/db15-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kondegowda NG, et al. Osteoprotegerin and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of nf-κb ligand pathway. Cell Metab. 2015;22(1):77–85. doi: 10.1016/j.cmet.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Horn S, et al. Research resource: a dual proteomic approach identifies regulated islet proteins during β-cell mass expansion in vivo. Mol Endocrinol. 2016;30(1):133–143. doi: 10.1210/me.2015-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rieck S, et al. The transcriptional response of the islet to pregnancy in mice. Mol Endocrinol. 2009;23(10):1702–1712. doi: 10.1210/me.2009-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schraenen A, et al. mRNA expression analysis of cell cycle genes in islets of pregnant mice. Diabetologia. 2010;53(12):2579–2588. doi: 10.1007/s00125-010-1912-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Layden BT, et al. Regulation of pancreatic islet gene expression in mouse islets by pregnancy. J Endocrinol. 2010;207(3):265–279. doi: 10.1677/JOE-10-0298. [DOI] [PubMed] [Google Scholar]
- 85.Gritti M, et al. Metformin repositioning as antitumoral agent: selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget. 2014;5(22):11252–11268. doi: 10.18632/oncotarget.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rubin CI, Atweh GF. The role of stathmin in the regulation of the cell cycle. J Cell Biochem. 2004;93(2):242–250. doi: 10.1002/jcb.20187. [DOI] [PubMed] [Google Scholar]
- 87.Sun R, et al. Overexpression of stathmin is resistant to paclitaxel treatment in patients with non-small cell lung cancer. Tumour Biol. 2015;36(9):7195–7204. doi: 10.1007/s13277-015-3361-y. [DOI] [PubMed] [Google Scholar]
- 88.Schimmack S, et al. Stathmin in pancreatic neuroendocrine neoplasms: a marker of proliferation and PI3K signaling. Tumour Biol. 2015;36(1):399–408. doi: 10.1007/s13277-014-2629-y. [DOI] [PubMed] [Google Scholar]
- 89.Stolovich-Rain M, et al. Weaning triggers a maturation step of pancreatic β cells. Dev Cell. 2015;32(5):535–545. doi: 10.1016/j.devcel.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 90.Tomita T, Doull V, Pollock HG, Krizsan D. Pancreatic islets of obese hyperglycemic mice (ob/ob) Pancreas. 1992;7(3):367–375. doi: 10.1097/00006676-199205000-00015. [DOI] [PubMed] [Google Scholar]
- 91.Gapp DA, Leiter EH, Coleman DL, Schwizer RW. Temporal changes in pancreatic islet composition in C57BL/6J-db/db (diabetes) mice. Diabetologia. 1983;25(5):439–443. doi: 10.1007/BF00282525. [DOI] [PubMed] [Google Scholar]
- 92.Tokuyama Y, et al. Evolution of beta-cell dysfunction in the male Zucker diabetic fatty rat. Diabetes. 1995;44(12):1447–1457. doi: 10.2337/diab.44.12.1447. [DOI] [PubMed] [Google Scholar]
- 93.Terauchi Y, et al. Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest. 2007;117(1):246–257. doi: 10.1172/JCI17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mezza T, et al. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes. 2014;63(3):994–1007. doi: 10.2337/db13-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-Cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36(1):111–117. doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):32–42. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 97.Alonso LC, et al. Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes. 2007;56(7):1792–1801. doi: 10.2337/db06-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic beta-cells in adult rats after short-term glucose infusion. Diabetes. 1989;38(1):49–53. doi: 10.2337/diab.38.1.49. [DOI] [PubMed] [Google Scholar]
- 99.Chick WL. β Cell replication in rat pancreatic monolayer cultures. Effects of glucose, tolbutamide, glucocorticoid, growth hormone and glucagon. Diabetes. 1973;22(9):687–693. doi: 10.2337/diab.22.9.687. [DOI] [PubMed] [Google Scholar]
- 100.Chick WL, Like AA. Effects of diet on pancreatic beta cell replication in mice with hereditary diabetes. Am J Physiol. 1971;221(1):202–208. doi: 10.1152/ajplegacy.1971.221.1.202. [DOI] [PubMed] [Google Scholar]
- 101.Metukuri MR, et al. ChREBP mediates glucose-stimulated pancreatic β-cell proliferation. Diabetes. 2012;61(8):2004–2015. doi: 10.2337/db11-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sharma RB, et al. Insulin demand regulates β cell number via the unfolded protein response. J Clin Invest. 2015;125(10):3831–3846. doi: 10.1172/JCI79264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Levitt HE, et al. Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia. 2011;54(3):572–582. doi: 10.1007/s00125-010-1919-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Porat S, et al. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 2011;13(4):440–449. doi: 10.1016/j.cmet.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 105.Tornovsky-Babeay S, et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in β cells. Cell Metab. 2014;19(1):109–121. doi: 10.1016/j.cmet.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 106.Brüning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell. 1997;88(4):561–572. doi: 10.1016/S0092-8674(00)81896-6. [DOI] [PubMed] [Google Scholar]
- 107.Dubuc PU. The development of obesity, hyperinsulinemia, and hyperglycemia in ob/ob mice. Metab Clin Exp. 1976;25(12):1567–1574. doi: 10.1016/0026-0495(76)90109-8. [DOI] [PubMed] [Google Scholar]
- 108.Edvell A, Lindström P. Initiation of increased pancreatic islet growth in young normoglycemic mice (Umeå +/?) Endocrinology. 1999;140(2):778–783. doi: 10.1210/endo.140.2.6514. [DOI] [PubMed] [Google Scholar]
- 109.Okada T, et al. Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci USA. 2007;104(21):8977–8982. doi: 10.1073/pnas.0608703104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Salpeter SJ, Khalaileh A, Weinberg-Corem N, Ziv O, Glaser B, Dor Y. Systemic regulation of the age-related decline of pancreatic β-cell replication. Diabetes. 2013;62(8):2843–2848. doi: 10.2337/db13-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Flier SN, Kulkarni RN, Kahn CR. Evidence for a circulating islet cell growth factor in insulin-resistant states. Proc Natl Acad Sci USA. 2001;98(13):7475–7480. doi: 10.1073/pnas.131192998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48(12):2270–2276. doi: 10.2337/diabetes.48.12.2270. [DOI] [PubMed] [Google Scholar]
- 113.Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest. 1987;79(2):616–619. doi: 10.1172/JCI112855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Perfetti R, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology. 2000;141(12):4600–4605. doi: 10.1210/endo.141.12.7806. [DOI] [PubMed] [Google Scholar]
- 115.Furuya F, Shimura H, Yamashita S, Endo T, Kobayashi T. Liganded thyroid hormone receptor-alpha enhances proliferation of pancreatic beta-cells. J Biol Chem. 2010;285(32):24477–24486. doi: 10.1074/jbc.M109.100222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Aguayo-Mazzucato C, et al. Thyroid hormone promotes postnatal rat pancreatic β-cell development and glucose-responsive insulin secretion through MAFA. Diabetes. 2013;62(5):1569–1580. doi: 10.2337/db12-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ferron M, Hinoi E, Karsenty G, Ducy P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci USA. 2008;105(13):5266–5270. doi: 10.1073/pnas.0711119105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee NK, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130(3):456–469. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wei J, Hanna T, Suda N, Karsenty G, Ducy P. Osteocalcin promotes β-cell proliferation during development and adulthood through Gprc6a. Diabetes. 2014;63(3):1021–1031. doi: 10.2337/db13-0887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sabek OM, Nishimoto SK, Fraga D, Tejpal N, Ricordi C, Gaber AO. Osteocalcin effect on human β-cells mass and function. Endocrinology. 2015;156(9):3137–3146. doi: 10.1210/EN.2015-1143. [DOI] [PubMed] [Google Scholar]
- 121.Yi P, Park JS, Melton DA. Betatrophin: a hormone that controls pancreatic β cell proliferation. Cell. 2013;153(4):747–758. doi: 10.1016/j.cell.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 122.El Ouaamari A, et al. SerpinB1 promotes pancreatic β cell proliferation. Cell Metab. 2016;23(1):194–205. doi: 10.1016/j.cmet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Parnaud G, et al. Proliferation of sorted human and rat beta cells. Diabetologia. 2008;51(1):91–100. doi: 10.1007/s00125-007-0855-1. [DOI] [PubMed] [Google Scholar]
- 124.Rutti S, Sauter NS, Bouzakri K, Prazak R, Halban PA, Donath MY. In vitro proliferation of adult human β-cells. PLoS One. 2012;7(4): doi: 10.1371/journal.pone.0035801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tian L, et al. Comparison of exendin-4 on beta-cell replication in mouse and human islet grafts. Transpl Int. 2011;24(8):856–864. doi: 10.1111/j.1432-2277.2011.01275.x. [DOI] [PubMed] [Google Scholar]
- 126.Wang Y, et al. Mice lacking ANGPTL8 (Betatrophin) manifest disrupted triglyceride metabolism without impaired glucose homeostasis. Proc Natl Acad Sci USA. 2013;110(40):16109–16114. doi: 10.1073/pnas.1315292110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gusarova V, et al. ANGPTL8/betatrophin does not control pancreatic beta cell expansion. Cell. 2014;159(3):691–696. doi: 10.1016/j.cell.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jiao Y, Le Lay J, Yu M, Naji A, Kaestner KH. Elevated mouse hepatic betatrophin expression does not increase human β-cell replication in the transplant setting. Diabetes. 2014;63(4):1283–1288. doi: 10.2337/db13-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang G, et al. First quantitative high-throughput screen in zebrafish identifies novel pathways for increasing pancreatic β-cell mass. Elife. 2015;4: doi: 10.7554/eLife.08261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Andersson O, et al. Adenosine signaling promotes regeneration of pancreatic β cells in vivo. Cell Metab. 2012;15(6):885–894. doi: 10.1016/j.cmet.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tsuji N, et al. Whole organism high content screening identifies stimulators of pancreatic beta-cell proliferation. PLoS ONE. 2014;9(8): doi: 10.1371/journal.pone.0104112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang W, et al. Identification of small-molecule inducers of pancreatic beta-cell expansion. Proc Natl Acad Sci USA. 2009;106(5):1427–1432. doi: 10.1073/pnas.0811848106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shen W, et al. Small-molecule inducer of β cell proliferation identified by high-throughput screening. J Am Chem Soc. 2013;135(5):1669–1672. doi: 10.1021/ja309304m. [DOI] [PubMed] [Google Scholar]
- 134.Annes JP, et al. Adenosine kinase inhibition selectively promotes rodent and porcine islet β-cell replication. Proc Natl Acad Sci USA. 2012;109(10):3915–3920. doi: 10.1073/pnas.1201149109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Walpita D, et al. A human islet cell culture system for high-throughput screening. J Biomol Screen. 2012;17(4):509–518. doi: 10.1177/1087057111430253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Walpita D, Wagner BK. Evaluation of compounds in primary human islet cell culture. Curr Protoc Chem Biol. 2014;6(3):157–168. doi: 10.1002/9780470559277.ch140088. [DOI] [PubMed] [Google Scholar]
- 137.Shen W, et al. Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat Commun. 2015;6: doi: 10.1038/ncomms9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Robitaille K, et al. High-throughput functional genomics identifies regulators of primary human beta cell proliferation. J Biol Chem. 2016;291(9):4614–4625. doi: 10.1074/jbc.M115.683912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dirice E, et al. Inhibition of DYRK1A stimulates human β-cell proliferation. Diabetes. 2016;65(6):1660–1671. doi: 10.2337/db15-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heit JJ, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. 2006;443(7109):345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- 141.Goodyer WR, Gu X, Liu Y, Bottino R, Crabtree GR, Kim SK. Neonatal β cell development in mice and humans is regulated by calcineurin/NFAT. Dev Cell. 2012;23(1):21–34. doi: 10.1016/j.devcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275(5308):1930–1934. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 143.Liu H, et al. Glycogen synthase kinase-3 and mammalian target of rapamycin pathways contribute to DNA synthesis, cell cycle progression, and proliferation in human islets. Diabetes. 2009;58(3):663–672. doi: 10.2337/db07-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu Y, et al. Conditional ablation of Gsk-3β in islet beta cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetologia. 2010;53(12):2600–2610. doi: 10.1007/s00125-010-1882-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu Z, Tanabe K, Bernal-Mizrachi E, Permutt MA. Mice with beta cell overexpression of glycogen synthase kinase-3beta have reduced beta cell mass and proliferation. Diabetologia. 2008;51(4):623–631. doi: 10.1007/s00125-007-0914-7. [DOI] [PubMed] [Google Scholar]
- 146.Mussmann R, et al. Inhibition of GSK3 promotes replication and survival of pancreatic beta cells. J Biol Chem. 2007;282(16):12030–12037. doi: 10.1074/jbc.M609637200. [DOI] [PubMed] [Google Scholar]
- 147.Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, García-Ocaña A, Stewart AF. Diabetes mellitus—advances and challenges in human β-cell proliferation. Nat Rev Endocrinol. 2015;11(4):201–212. doi: 10.1038/nrendo.2015.9. [DOI] [PubMed] [Google Scholar]
- 148.Stewart AF, et al. Human β-cell proliferation and intracellular signaling: part 3. Diabetes. 2015;64(6):1872–1885. doi: 10.2337/db14-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocaña A. Human β-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes. 2014;63(3):819–831. doi: 10.2337/db13-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF. Human β-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes. 2012;61(9):2205–2213. doi: 10.2337/db12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Khoo S, Cobb MH. Activation of mitogen-activating protein kinase by glucose is not required for insulin secretion. Proc Natl Acad Sci USA. 1997;94(11):5599–5604. doi: 10.1073/pnas.94.11.5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cousin SP, Hügl SR, Myers MG, White MF, Reifel-Miller A, Rhodes CJ. Stimulation of pancreatic beta-cell proliferation by growth hormone is glucose-dependent: signal transduction via janus kinase 2 (JAK2)/signal transducer and activator of transcription 5 (STAT5) with no crosstalk to insulin receptor substrate-mediated mitogenic signalling. Biochem J. 1999;344(Pt 3):649–658. [PMC free article] [PubMed] [Google Scholar]
- 153.Briaud I, Lingohr MK, Dickson LM, Wrede CE, Rhodes CJ. Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic beta-cells. Diabetes. 2003;52(4):974–983. doi: 10.2337/diabetes.52.4.974. [DOI] [PubMed] [Google Scholar]
- 154.Arnette D, et al. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic beta cells. J Biol Chem. 2003;278(35):32517–32525. doi: 10.1074/jbc.M301174200. [DOI] [PubMed] [Google Scholar]
- 155.Amaral ME, et al. Participation of prolactin receptors and phosphatidylinositol 3-kinase and MAP kinase pathways in the increase in pancreatic islet mass and sensitivity to glucose during pregnancy. J Endocrinol. 2004;183(3):469–476. doi: 10.1677/joe.1.05547. [DOI] [PubMed] [Google Scholar]
- 156.Bordin S, et al. Prolactin-modulated gene expression profiles in pancreatic islets from adult female rats. Mol Cell Endocrinol. 2004;220(1–2):41–50. doi: 10.1016/j.mce.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 157.Lingohr MK, Dickson LM, McCuaig JF, Hugl SR, Twardzik DR, Rhodes CJ. Activation of IRS-2-mediated signal transduction by IGF-1, but not TGF-α or EGF, augments pancreatic β-cell proliferation. Diabetes. 2002;51(4):966–976. doi: 10.2337/diabetes.51.4.966. [DOI] [PubMed] [Google Scholar]
- 158.Friedrichsen BN, et al. Stimulation of pancreatic beta-cell replication by incretins involves transcriptional induction of cyclin D1 via multiple signalling pathways. J Endocrinol. 2006;188(3):481–492. doi: 10.1677/joe.1.06160. [DOI] [PubMed] [Google Scholar]
- 159.Beith JL, Alejandro EU, Johnson JD. Insulin stimulates primary beta-cell proliferation via Raf-1 kinase. Endocrinology. 2008;149(5):2251–2260. doi: 10.1210/en.2007-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kwon G, Marshall CA, Pappan KL, Remedi MS, McDaniel ML. Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes. 2004;53 Suppl 3:S225–S232. doi: 10.2337/diabetes.53.suppl_3.s225. [DOI] [PubMed] [Google Scholar]
- 161.Rhodes CJ, White MF, Leahy JL, Kahn SE. Direct autocrine action of insulin on β-cells: does it make physiological sense? Diabetes. 2013;62(7):2157–2163. doi: 10.2337/db13-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McDaniel ML, Marshall CA, Pappan KL, Kwon G. Metabolic and autocrine regulation of the mammalian target of rapamycin by pancreatic beta-cells. Diabetes. 2002;51(10):2877–2885. doi: 10.2337/diabetes.51.10.2877. [DOI] [PubMed] [Google Scholar]
- 163.Balcazar N, et al. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J Biol Chem. 2009;284(12):7832–7842. doi: 10.1074/jbc.M807458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zahr E, et al. Rapamycin impairs in vivo proliferation of islet beta-cells. Transplantation. 2007;84(12):1576–1583. doi: 10.1097/01.tp.0000296035.48728.28. [DOI] [PubMed] [Google Scholar]
- 165.Niclauss N, Bosco D, Morel P, Giovannoni L, Berney T, Parnaud G. Rapamycin impairs proliferation of transplanted islet β cells. Transplantation. 2011;91(7):714–722. doi: 10.1097/TP.0b013e31820c10c8. [DOI] [PubMed] [Google Scholar]
- 166.Fraenkel M, et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008;57(4):945–957. doi: 10.2337/db07-0922. [DOI] [PubMed] [Google Scholar]
- 167.Alliouachene S, et al. Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. J Clin Invest. 2008;118(11):3629–3638. doi: 10.1172/JCI35237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Missiaglia E, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28(2):245–255. doi: 10.1200/JCO.2008.21.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Buteau J, Foisy S, Rhodes CJ, Carpenter L, Biden TJ, Prentki M. Protein kinase Czeta activation mediates glucagon-like peptide-1-induced pancreatic beta-cell proliferation. Diabetes. 2001;50(10):2237–2243. doi: 10.2337/diabetes.50.10.2237. [DOI] [PubMed] [Google Scholar]
- 170.Vasavada RC, et al. Protein kinase C-zeta activation markedly enhances beta-cell proliferation: an essential role in growth factor mediated beta-cell mitogenesis. Diabetes. 2007;56(11):2732–2743. doi: 10.2337/db07-0461. [DOI] [PubMed] [Google Scholar]
- 171.Velazquez-Garcia S, et al. Activation of protein kinase C-ζ in pancreatic β-cells in vivo improves glucose tolerance and induces β-cell expansion via mTOR activation. Diabetes. 2011;60(10):2546–2559. doi: 10.2337/db10-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lakshmipathi J, et al. PKCζ is essential for pancreatic β-cell replication during insulin resistance by regulating mTOR and cyclin-D2. Diabetes. 2016;65(5):1283–1296. doi: 10.2337/db15-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Leibiger B, et al. PI3K-C2α knockdown results in rerouting of insulin signaling and pancreatic beta cell proliferation. Cell Rep. 2015;13(1):15–22. doi: 10.1016/j.celrep.2015.08.058. [DOI] [PubMed] [Google Scholar]
- 174.Elghazi L, et al. Decreased IRS signaling impairs beta-cell cycle progression and survival in transgenic mice overexpressing S6K in beta-cells. Diabetes. 2010;59(10):2390–2399. doi: 10.2337/db09-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chen S, et al. Regeneration of pancreatic islets in vivo by ultrasound-targeted gene therapy. Gene Ther. 2010;17(11):1411–1420. doi: 10.1038/gt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Finan B, et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat Med. 2012;18(12):1847–1856. doi: 10.1038/nm.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rong Z, et al. An effective approach to prevent immune rejection of human ESC-derived allografts. Cell Stem Cell. 2014;14(1):121–130. doi: 10.1016/j.stem.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]