

Abstract
Mucinous tubular and spindle cell carcinoma (MTSCC) is a relatively rare subtype of renal cell carcinoma with distinctive morphologic and cytogenetic features. Here we carry out whole exome and transcriptome sequencing of a multi-institutional cohort of MTSCC (n=22). We demonstrate the presence of either biallelic loss of Hippo pathway tumor suppressor genes (TSGs) and/or evidence of alteration of Hippo pathway genes in 85% of samples. PTPN14 (31%) and NF2 (22%) were the most commonly implicated Hippo pathway genes while other genes such as SAV1 and HIPK2 were also involved in a mutually exclusive fashion. Mutations in the context of recurrent chromosomal losses amounted to bi-allelic alterations in these TSGs. As a read-out of Hippo pathway inactivation, a majority of cases (90%) exhibited increased nuclear YAP1 protein expression. Taken together, nearly all cases of MTSCC exhibit some evidence of Hippo pathway dysregulation.
Keywords: Kidney, mucinous tubular and spindle cell, next-generation sequencing
Introduction
Several large scale sequencing studies have sought to understand the biology of the most common renal cell carcinomas (RCC) including clear cell (KIRC)(1), papillary (KIRP)(2) and chromophobe (KICH)(3) subtypes however, rarer subtypes of RCC have not been characterized comprehensively at the genomic level. Mucinous tubular and spindle cell carcinoma (MTSCC) is a relatively rare, recently characterized, but well established subtype of RCC which contains a biphasic epithelioid and spindle cell component(4). While the majority of patients with these tumors have a favorable prognosis, a small but distinct subset can be associated with an aggressive clinical phenotype and poor outcome.
MTSCC demonstrates a relatively consistent immunophenotype, cytogenetic aberration pattern, and express lineage-specific markers of the kidney such as PAX8(5). These characteristics, combined with the unique morphological phenotype of MTSCC (a variable combination of epithelial spindle cells with focal to abundant stromal mucin), suggest that this tumor may have a convergent mechanism of pathogenesis. To explore this, we assembled a multi-institutional cohort of 22 MTSCCs and carried out whole exome sequencing of tumor matched with adjacent normal as well as capture transcriptome sequencing of the samples(6). This integrative sequencing strategy allowed us to broadly survey single nucleotide variations (SNVs), small insertions-deletions (indels), germline alterations, amplifications and deletions, gene fusions and expression signatures(7, 8).
Results
Mutational Landscape and Recurrent Hippo Pathway Mutations in MTSCC
The mean patient age in our MTSCC cohort was 62 years (range: 44 to 78 years), with a variable tumor size (mean 5.1 cm; range 2.0 cm to 15.0 cm, Supplementary Table S1). All twenty two tumors exhibited classic morphologic features of MTSCC, as described above and illustrated in Figure 1A (confirmed by two genitourinary pathologists, JKM and RM). We obtained genomic DNA from matched tumor and normal formalin fixed paraffin embedded (FFPE) tissue samples and analyzed them by whole exome sequencing (Materials and Methods). Capture transcriptome libraries were generated from total RNA isolated from matched tumor and normal samples (Materials and Methods). We obtained high quality exome sequencing data from all 22 tumor/normal samples as indicated by the high alignment, depth of coverage, and overall low PCR duplication rate (Supplementary Table S2). In addition we also obtained high quality capture transcriptome data implied by the high spliced junctions (indicative of transcriptome library complexity) from 10 matched tumor normal specimens (Supplementary Table S2). Analysis of matched tumor/normal capture genome data revealed a relatively low number of somatic mutations within the coding region in both the discovery (n=10) and validation (n=12) cohorts. MTSCC exhibited an average of 9 non synonymous mutation calls per sample, ranging from 2 to 26. MTSCC contains an average of 0.86 mutations per Mb which is (Fig 1B) consistent with previously reported low mutation rates in kidney cancers of 1.45, 1.1 and 0.4 mutations per Mb in KIRC(1), KIRP(2) and KICH(3) respectively, excluding samples with a hyper-mutator phenotype characteristic of defective DNA mismatch repair. The somatic mutation data was queried for trinucleotide signature patterns reported by Alexandrov et al(9) but no significant pattern was noted primarily due to the low mutation burden in this disease (data not shown).
Figure 1.
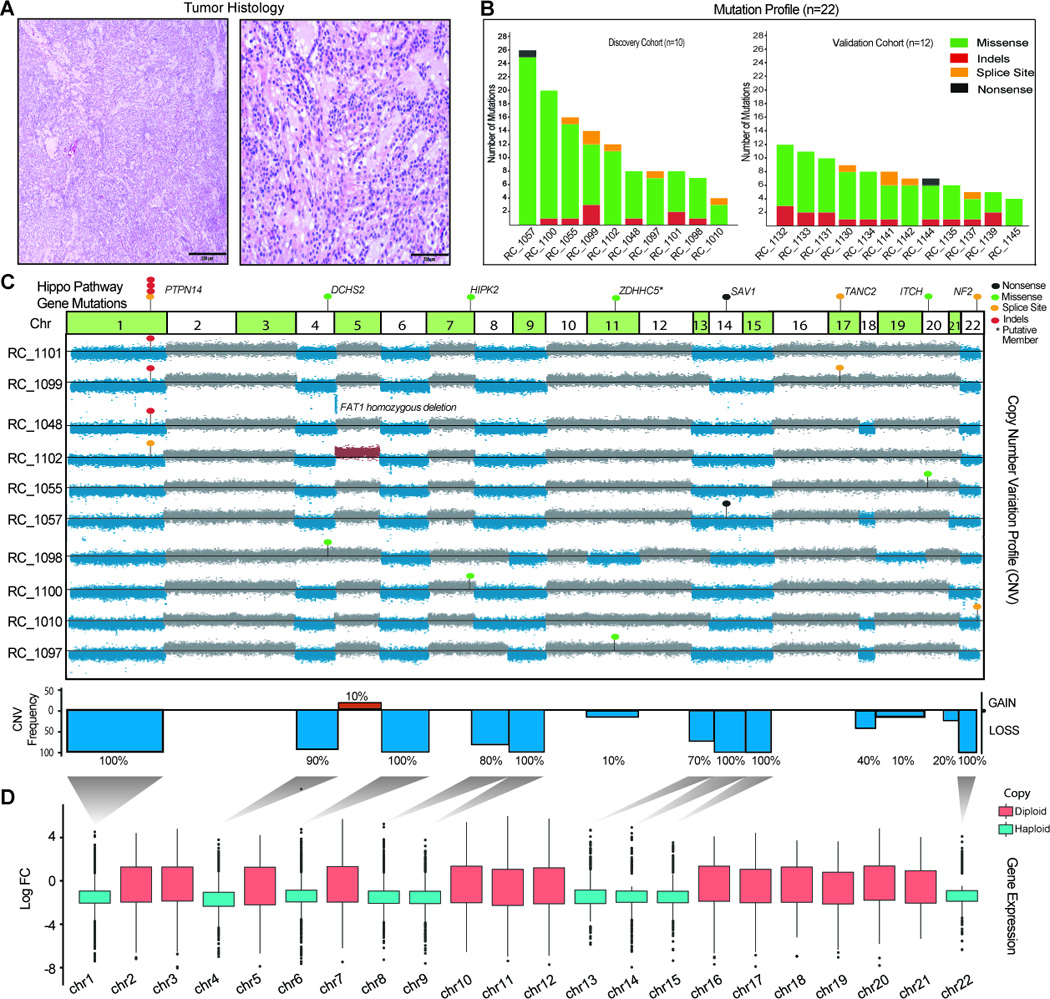
Integrative analysis of genomic alterations in MTSCC A. Representative microscopic images of MTSCC cases that were sequenced. Low magnification, 40× (left panel, scale bar 200 microns) and high magnification, 200× (right panel, scale bar 500 microns). B. Histogram representing total number of non-synonymous somatic mutations identified in each sample in the discovery cohort (n=10) on the left and validation cohort (n=12) on the right. C. Integrated view of the frequencies of chromosomal aneuploidy and Hippo pathway gene mutations identified in MTSCC discovery cohort (n=10). Chromosomes with copy loss (blue) and gain (red) are indicated. Indels (red dots), missense SNVs (green dots), nonsense (black dots) and splice site SNVs (yellow dots) in PTNP14, DCHS2, HIPK2, SAV1, TANC2, ITCH and NF2 genes are indicated. TANC2 and ITCH are recently nominated putative Hippo pathway regulators that require additional experimental validation D. Global reduction of transcript levels due to chromosomal copy loss. Shown is the distribution of fold-changes for all significantly differentially expressed loci per chromosome (chromosomes with copy loss: blue; diploid: red) in the RNAseq data from the discovery cohort (n=4 tumor/normal pairs).
To identify mutations with likely functional significance, somatic calls were ranked according to known activating oncogenic mutations and recurrent inactivating tumor suppressor mutations (Supplementary Table S3). This classification aided in the immediate recognition of highly recurrent Hippo pathway tumor-suppressor gene (TSG) mutations in our list. This included, recurrent somatic alterations in the protein tyrosine phosphatase non-receptor type 14 (PTPN14) in seven cases (7/22, 31%) with a median variant allele frequency (VAF) of 66%, neurofibromin-2 (NF2; merlin) in 5 cases (5/22, 23%) at 58% median VAF besides salvador homolog 1 (SAV1) and Itchy E3 Ubiquitin Protein Ligase (ITCH)(10) in one case each with 77% and 39% VAF respectively (Supplementary Table S3). All PTPN14, NF2, SAV1 mutations observed were loss of function resulting from either frameshift insertions/deletions (indels; 7 cases), splice site mutations (5 cases) or nonsense mutation (1 case). Schramm et al(11) recently reported 2 relapsed neuroblastoma cases with loss-of-function missense mutations in PTPN14 and showed that ectopic expression of the mutant PTPN14 (T42A) protein increased clonogenic growth in the SK-N-SH cell line. Hence PTPN14’s functionality in binding and negatively regulating YAP1 oncogenic function(12) may be compromised in our index cases. NF2 an extensively studied tumor suppressor gene, is an upstream regulator of Hippo pathway and its mutations results in nonmalignant brain tumors in a syndrome termed neurofibromatosis type 2(13). While SAV1 is a core member of the Hippo pathway, most of the mutated genes in this cohort are established associate members(14). SAV1 is known to interact with the MST1 kinase and enhance LATS phosphorylation which in turn negatively regulates YAP(13). Hence losses in NF2, SAV1, and PTPN14 in MTSCC, will likely result in YAP1 activation in the corresponding index tumors.
We also observed additional somatic mutations in independent cases among established members such as Dachsous Cadherin-Related 2 (DCHS2) and Homeodomain Interacting Protein Kinase 2 (HIPK2) and putative Hippo pathway genes involving Zinc Finger DHHC-Type Containing 5 (ZDHHC5), Zinc Finger DHHC-Type Containing 15 (ZDHHC15)(15) (Supplementary Table S3). However, aberrations in HIPK2, DCHS2, ZDHHC5 and ZDHHC15 aberrations may all represent subclonal events as the VAF’s of 16%, 4%, 10%, and 21% respectively are lower compared to the other Hippo pathway calls (Supplementary Table S3). To study this further we obtained exome sequencing data from three independent tumor sites that are spatially well separated, from 3 cases namely, RC_1099, RC_1098 and RC_1097 (Supplementary Fig S1). In RC_1099 all sites sequenced showed high VAF for PTPN14 mutation demonstrating its clonal nature. As expected in both RC_1098 and RC_1097, the VAFs for DCHS2 substantially increased in 2 independent sites and absent in the third, while the VAF for ZDHHC5 showed significant increase in one another site, supporting the subclonal nature of these aberrations (Supplementary Fig S1).
Recurrent Copy Number Alterations and Bi-allelic loss of Hippo Pathway Tumor Suppressor Genes in MTSCC
Owing to widespread chromosomal loss commonly observed in MTSCC we reasoned that a second hit particularly in TSGs may be acquired via copy number variation (CNV). To investigate this, we generated copy number profiles (Materials and Methods) from matched tumor/normal capture genome data and queried for recurrent events in this disease. Our analysis of the 22 cases revealed monosomy of chromosomes 1, 6, 9, 14, 15, and 22 in 100% of cases, and chromosomes 4, 8 and 13 with 90%, 81% and 90% respectively (Fig 1C and Supplementary Fig S2A). Importantly, chromosomes 1, 14 and 22, harbor PTPN14, SAV1 and NF2 genes respectively, and showed one copy loss in all samples which amounts to biallelic loss of these Hippo pathway TSGs in the index tumors. The recurrent chromosomal loss we observed by exome sequencing supports a recent study by Peckova et al(16) that reported similar recurrent chromosomal losses using array CGH and FISH strategies. Importantly none of the samples in this cohort with pure MTSCC histology displayed chromosomes 7 and 17 gains, commonly noted as gains in type I papillary RCC(17). The recent TCGA papillary RCC (KIRP) study reported Hippo pathway mutations (NF2 and SAV1) in 4% of the samples and used CNV to classify tumors into 3 groups such as genome stable, unstable and chromosome 7/17 gains(2). Given the morphologic overlap between MTSCC and a minor subset of PRCC, we used our knowledge of mutations and copy number patterns to identify any possibly misclassified samples in the TCGA KIRP cohort(2). We first investigated the level 3 TCGA copy number data available for 161 KIRP cases by principal component analysis and observed at least 3 outlier samples (TCGA-B3-3926, TCGA-F9-A7QO, TCGA-UN-AAZ9) with the signature MTSCC type chromosomal losses and lacking characteristic genetic alterations associated with type I PRCC(2) (Supplementary Figs S2B and S2C) and subsequent analysis of updated TCGA KIRP cohort (n=288) identified a fourth case (TCGA-G7-A8LC). Interestingly, SAV1 loss of function mutations combined with a haploid status of chromosome 14 observed in the three cases suggested a double hit in this Hippo pathway gene. The fourth sample had a missense mutation in DOCK11, that could be a potential Hippo pathway regulator located on the X chromosome similar to roles proposed for DOCK7, 8 and 9 recently(18, 19) (Supplementary Fig S2C). Our analysis of the TCGA KIRP data showed that Hippo pathway mutations are frequently associated with genome unstable KIRP samples with various cancer subtype classification (mutually exclusive of chr17/7 gain and MET mutations) (Supplementary Fig 2D).
Besides the chromosome level losses observed in our current study, we also observed few focal copy losses such as FAT Tumor Suppressor Homolog 1, FAT1 loss in RC_1099. We also observed rare gains of whole chromosome 5 (2/22 cases) however focal gains were absent. Taken together, integration of somatic mutation and copy number data revealed homozygous loss of the critical Hippo pathway tumor suppressor genes (PTPN14, SAV1, NF2), as a recurrent molecular theme in MTSCC. Importantly, we noticed that most cases in this cohort had strong evidence for mutually exclusive aberration in at least one Hippo pathway gene (except RC_1099, and RC_1139 where we saw additional aberrations among potential Hippo pathway members namely, TANC2, SKIV2L2 (19–21) mutations and FAT1 loss) (Fig 1C).
MTSCC Gene Expression Analysis
To assess whether the pattern of chromosomal loss is reflected at the transcriptome-level, we examined the RNA-seq data obtained in the discovery cohort, from four matched tumor normal pairs generated in this study. Upon identifying loci (genes and intergenic regions) with significant differential expression, we found that on the haploid chromosomes the great majority of loci showed a global decrease in transcript levels relative to the diploid chromosomes (Fig 1D). This link is further accentuated in the Gviz plot which shows a gene wise representation where vast majority of the genes on haploid chromosomes (1, 4, 6, 8, 9, 13, 14, 15 and 22) showed decreased expression as compared to diploid chromosomes (2, 3, 5, 7, 10, 11, 12, 16, 17, 18, 19, 20, 21). (Supplementary Fig S3A). Overall, the majority of differentially expressed genes in MTSCC were down-regulated (Supplementary Fig S3B). To identify transcriptional targets of the Hippo pathway in kidney we performed PTPN14 knockdown followed by RNA-seq in 2 kidney cancer cell lines (CAKI-1 and A-704) and a normal kidney epithelial cell line (HK-2). PTPN14 siRNAs were first functionally validated in a MCF-7 TEAD reporter luciferase stable cell line. Both siRNAs showed comparable knockdown efficiency and significantly increased luciferase reporter activity (Supplementary Fig S4A and Data not shown). In 2 of the kidney cell lines PTPN14 knockdown increased cell proliferation compared to non-target controls (Supplementary Figs S4B and S4C). While we observed excellent correlation between genes dysregulated by either PTPN14 or LATS1 knockdown within each cell line (HK2, CAKI-1 and A704) (Supplementary Fig S4D), the overlap across the 3 cell lines was only 23 genes (Supplementary Figs S4E and Supplementary Table S4). Further, these 23 genes did not show concordant differential expression in MTSCC tumors (Supplementary Fig S4F). Overall, these results illustrate the marked tissue specificity of Hippo pathway targets.
In an alternate approach to assess the functional impact of Hippo pathway inactivation, we performed enrichment analysis and identified several altered transcriptional signatures. We noted enrichment of hepatocyte nuclear factor (HNF) targets where HNF4A/1A targets were significantly down-regulated (Supplementary Fig S5A and Supplementary Table S5). HNF1 and 4 are master regulators of the hepatic transcriptome and considered tumor suppressor genes in liver cancer. Interestingly, a recent study has directly implicated the Hippo pathway in regulating hepatocyte differentiation where enhancer occupancy of HNF4A and FOXA2 was modulated by YAP activation(22). In addition, another previous report showed downregulation of HNF4A, upregulation of HNF1B upon ectopic YAP overexpression in mouse liver organoids(23) (Supplementary Fig S5B). Since hepatocyte nuclear factors are drivers of kidney development and differentiation(24, 25) they may also play a role in the ubiquitous down-regulation of kidney marker expression observed in MTSCC (Supplementary Fig S5C). In the normal human tissue RNA-seq compendia data from “The Genotype Tissue Expression study (GTEX data) (26) significant HNF gene expression was found in liver, colon, pancreas, kidneys and small intestine (Supplementary Fig S6A) and in the kidney, prominent HNF expression was noted in renal proximal tubules(27, 28). We next assessed HNF1A, 1B and 4A transcript expression in MTSCC and the TCGA pan kidney cancer dataset(1–3) (Supplementary Figs S6B and S6C). We found that among major renal cancers, chromophobe RCC (KICH) showed the most significant downregulation of HNF1A and HNF4A similar to MTSCC, which suggests that loss of HNF transcriptional activity is common across renal cancers, as previously reported(29) (Supplementary Figs S6D and S6E).
YAP1 Protein Expression in MTSCC
The deleterious somatic mutations in PTPN14, NF2 and SAV1 genes in the context of their functional protein domains are depicted in Figure 2A. Loss of function of negative regulators of Hippo pathway such as the kinases Large Tumor Suppressor Kinase (LATS), Serine/Threonine Kinase 4 (STK4 also called MST) and factors such as NF2, PTPN14 are known to stabilize the transcription coactivator protein YAP1 and increase its nuclear localization(11, 13, 23). In addition, our cell line experiments demonstrated upregulation of YAP transcriptional activity in an established reporter cell line and increased cell proliferation upon PTPN14 knockdown in various kidney cell lines (Supplementary Fig S4). Nuclear localization of YAP1 will render support to Hippo pathway inactivation caused by loss of the tumor suppressors PTPN14, NF2 and SAV1. Immunohistochemistry (IHC) assessment with a validated anti-YAP1 antibody revealed YAP1 protein nuclear localization in 90% of the MTSCC tumors (Figs 2B, 2C, Supplementary Fig S7A and S7B and Supplementary Table S6). Among the 13 tumors with biallelic loss of hippo pathway tumor suppressors (NF2, SAV1, PTPN14) all samples showed nuclear YAP staining except one where the staining was very weak (Fig 2C). In addition, we also noticed increased YAP1 protein expression in tumors compared to background benign renal parenchyma in 68% of cases (Figs 2B, 2C, Supplementary Fig S7A and S7B and Supplementary Table S6). In comparison, concurrent YAP1 immunohistochemistry performed on a tissue microarray (TMA) consisting of 21 clear cell renal cell carcinomas, 21 papillary renal cell carcinomas, 22 chromophobe renal cell carcinomas, 22 oncocytomas and 10 benign renal tissues demonstrated these cases to be negative for YAP1 overexpression (Supplementary Table S6). Taken together the Hippo pathway alterations in MTSCC predominantly consist of loss of function events in putative tumor suppressor components and missense aberrations in putative oncogenic components likely leading to oncogenic YAP1 activation (Fig 2D). When genetic alteration of the Hippo pathway is considered along with nuclear YAP1 protein expression, nearly all cases evaluated exhibited some evidence of Hippo pathway dysregulation suggesting a convergent mechanism of pathogenesis.
Figure 2.
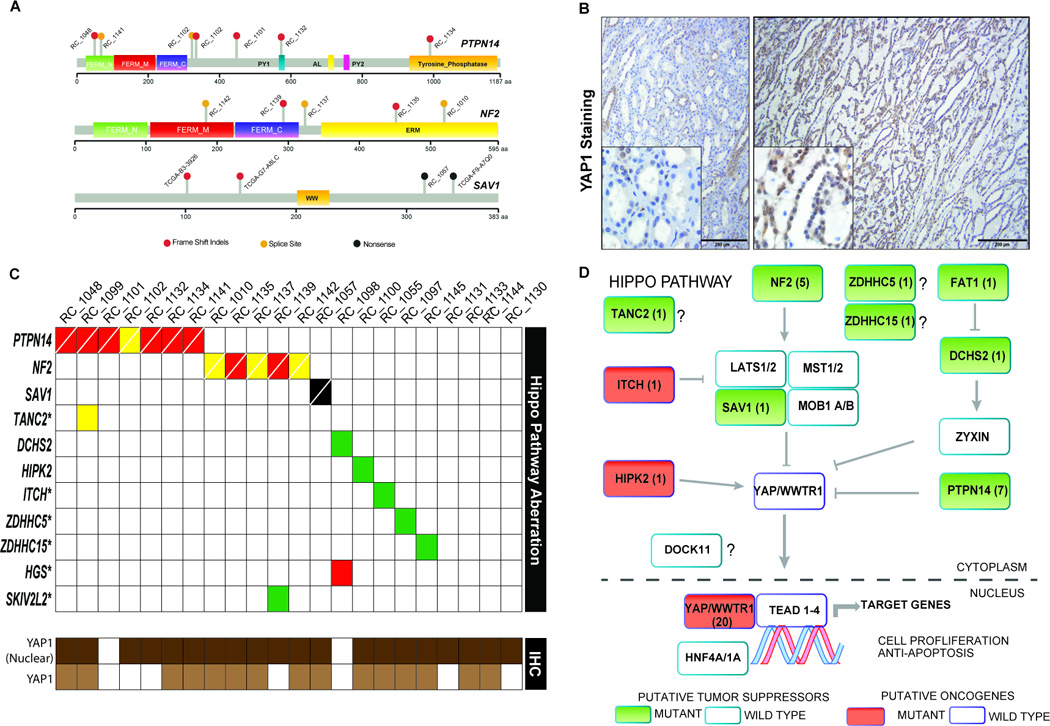
Hippo pathway gene alterations in MTSCC. A. Schematic representation of the Hippo pathway gene aberrations discovered in MTSCC. PFAM domains and scale depicting amino acid numbers are presented for each protein schema. Indels (red dots), missense SNVs (green dots), nonsense (black dots) and splice site SNVs (yellow dots). B. An example of YAP1 nuclear expression in MTSCC case RC_1100 as revealed by immunohistochemistry. Benign background renal parenchyma (left panel, low power view 100× with scale bar 200 microns, and inset 400×) demonstrates patchy and weak predominantly nuclear YAP1 staining (with focal cytoplasmic expression). Tumor areas of MTSCC (right panel, low power view 100× with scale bar 200 microns, and inset 400×) from the same case demonstrate moderate to strong nuclear YAP1 expression (with focal cytoplasmic staining). C. MTSCC Hippo-pathway mutations in the cohort (top panel; red indels, yellow- splice site, black-nonsense, green- missense, cells crossed white line indicates bi-alleic loss) represented along with case wise evaluation of YAP1 protein nuclear localization (bottom panel; dark brown- present; white- absent) and increased YAP1 protein expression in tumor compared to background benign renal parenchyma (bottom panel; light brown- present; white- absent). D. Hippo signaling pathway schematic with alterations identified in this study highlighted. Pathway members with mutations are indicated (filled boxes) and numbers within represent the cases with that particular somatic aberration (except in the nuclear YAP1 box where the numbers represent immunohistochemistry staining data for cases with nuclear localization); putative tumor suppressors are colored green while oncogenes are colored red.
Putative Cellular Origin of MTSCC
To identify nephron sections that could potentially give rise to MTSCC we followed correlation based approaches as proposed by Davis et al. for KICH(3). We leveraged two existing datasets on human (30) and rat (31) dissected nephrons obtained through SAGE and RNA-seq, respectively. To emphasize differences in gene expression patterns that are specific to MTSCC and the various nephron sections all expression profiles were standardized relative to normal kidney tissue (see methods). We also identified marker genes specifically expressed in each rat nephron section). An outline of the analyses is represented in Supplementary Fig S8A. Correlations were calculated either across all genes (Supplementary Fig S8B), all marker genes (n=600, Supplementary Fig S8C and S8D), or specific transcription factors (n=41) (see methods. As expected, strong correlations with proximal tubule, terminal portions of the proximal tubule, and distal nephron sections were noted for TCGA KIRC, KIRP, and KICH, respectively. MTSCC samples showed substantial heterogeneity, but appeared distinct from all other major RCC types. Overall, a trend of positive correlations was observed for some sections of the proximal tubule and loop of Henle, while mostly negative correlations were observed for distal nephron regions for both human and rat datasets (Supplementary Fig S8B, S8D, S8E). To further discriminate between MTSCC and KICH, the two types of RCC with an overlapping pattern of chromosome losses, we restricted the correlation analysis to transcription factors, previously shown to exhibit cell type specificity across nephron sections(31). Interestingly, MTSCC samples positively correlated with the loop of Henle, a region that was previously speculated to be associated with MTSCC(32), while conversely KICH strongly correlated with distal tubule (3) (Supplementary Fig S8F). For example, IRX5 a marker of loop of Henle is highly expressed in MTSCC, and absent in KICH, whereas FOXI1, a marker of the distal nephron, was present in KICH and absent and in MTSCC (Supplementary Fig S8G).
Discussion
Our study now adds MTSCC to a growing list of cancers(13) with recurrent aberrations among Hippo pathway members including somatic mutations, gene fusions and copy number variations. Unlike the basal cell carcinomas involving the skin which harbor a high mutational burden and where Hippo pathway genes LATS1 and PTPN14 loss was observed in 31% of cancers(33), recurrent Hippo pathway aberrations with a low overall mutational burden (Fig 1B), and recurrent chromosomal losses (Fig 1C) seems to define the molecular signature of MTSCC. Current study provides evidence for the first time about the existence of intra tumor mutational heterogeneity in MTSCC. Distinct chromosomal copy number variations is a recurring theme in RCCs such as KICH(3) with losses in chromosomes 1, 2, 6, 10, 13 and 17, KIRP(2) with gains in chromosomes 7 and 17 gain and MTSCC. We believe further subgrouping of renal cancer samples profiled thus far, based on variation in CNV pattern may identify novel disease subtypes, which can then be refined by cross platform integrative data analysis, which along with single cell sequencing of normal kidney cell types and tumors may provide a deeper understanding of the ontology of these cancers.
The mechanisms and causes of these chromosomal losses, whether it is a sequential event and its association with tumor progression, remain as interesting and unanswered question. Chromosomal losses seem to have a major influence on the MTSCC gene expression pattern (Fig 1D) and may likely alter Hippo pathway gene regulation, in a disease and tissue specific manner. Our attempts to delineate Hippo pathway targets in kidney by knockdown experiments showed very low overlap across the 3 different models and genes such as CTGF and CYR61, previously shown to be induced by loss of Hippo pathway in liver were not regulated in kidney. These findings are consistent with the low overlap between YAP transcriptional targets identified in various profiling studies indicating a strong cell type and tissue specific gene regulation by the Hippo pathway(13). Lack of a cell line model for MTSCC hinders our efforts to further characterize the functional effects of Hippo pathway aberrations in this tumor. However our analysis has identified several novel upregulated genes in MTSCC tumors such as VSTM2A (Supplementary Fig S3) both in our cohort and in TCGA index samples identified in Supplementary Fig S2, that could be further evaluated as potential biomarkers.
Finally, the cell of origin for MTSCC remains controversial (16, 32, 34). Our results based on expression correlations with dissected nephron sections suggest loop of Henle as a putative candidate and support previous electron microscopic studies by Srigley J. et al(32). Alternatively, MTSCC may arise form a rare cell type with low representation across the dissected nephron samples. Clearly, further studies are necessary to confirm these observations.
In summary, whole exome and transcriptome sequencing of MTSCC revealed recurrent copy number alterations and somatic mutations of Hippo signaling pathway genes. Bi-allelic loss of Hippo pathway tumor suppressor genes namely PTPN14, NF2 and SAV1 was found in approximately 60% of MTSCCs (Fig 2). Protein expression analysis by IHC further supported our model that implicates nuclear YAP1(90%) and increased YAP protein levels (68%) in tumors, thus rendering further evidence to inactivation of the Hippo pathway in MTSCC. These data suggest that Hippo pathway dysregulation may be a fundamental causal event in the pathogenesis of MTSCC, a finding that may have diagnostic and therapeutic implications for this rare RCC subtype. Small molecule inhibitors of YAP (a key downstream component of the Hippo pathway), such as verteporfin(13), present an attractive opportunity for targeted therapy in the rare MTSCC patients with sarcomatoid differentiation or metastasis. These molecular data highlight the possibility of emerging clinical/therapeutic approaches to rare tumors in this era of precision oncology(35, 36).
Methods
Sample Procurement and Clinical Study
Patient samples were procured from UMHS, Cleveland clinic, Vanderbilt University, Emory School of Medicine, University of Calgary and New York School of Medicine. This study was performed under Institutional Review Board–approved protocols (with waiver of informed consent), and conducted in accordance with the principles of the Declaration of Helsinki. Tumor purity was assessed by study pathologists (R. Mehra, J. McKenney). Tumor and matched adjacent normal kidney sections were utilized to extract both genomic DNA and total RNA using All Prep DNA/RNA FFPE kit. Sample details, including age, gender, and disease stage are summarized in Supplementary Table S1.
Reagents
YAP1 antibody (catalog# 4912s) used for immunohistochemistry and Western blot analysis was purchased from cell signaling technology. YAP1, PTPN14, LATS1 and control siRNAs were purchased from either Thermofisher or Ambion.
Nucleic Acid Isolation
Genomic DNA and total RNA was isolated from five 10 micron sections of matched tumor and normal FFPE specimens using “All Prep FFPE DNA/RNA Isolation” kit (Qiagen, Valencia, CA). The gDNAs and total RNAs were quantitated using Nanodrop spectrophotometer. RNA quality was determined using Bio-Analyzer.
Sequencing Library Preparation and Next Generation Sequencing
Capture genome and capture transcriptome libraries were prepared as recently described with modifications(6, 37). Briefly for capture genome libraries three micrograms of gDNA was subjected to minimum shearing using Covaris. Libraries were prepared with Sciclone G3 NGS workstation (Perkin Elmer) following the Kappa HT library preparation kit protocol (Kappa Biosystems cat#KK8234) using the sheared DNA as input. Libraries were size selected for 200–350bp using dual SPRI bead selection and subjected to PCR amplification with KapaReadyMix regeant in the kit. One microgram of the library was used for capture with Human all exon v4 capture probes following manufacturer’s instructions (Agilent). Post hybridization and wash, the DNA bound to beads was PCR amplified, purified and analyzed by BioAnalyzer before sequencing. For capture transcriptome libraries, 5micrograms of total RNA was used in a reverse transcription reaction and the obtained cDNA was subsequently taken to a second strand DNA synthesis as described previously(6). Following second strand synthesis we used Sciclone G3 NGS workstation (Perkin Elmer) with the Kappa HT library preparation kit to produce the libraries that were then captured with Human all exon v4 capture probes as described above. Both capture genome and transcriptome libraries were sequenced using HiSeq 2500 (Illumina) as described in Cieslik et al(6). All dataset will be deposited in the public repository dbGAP upon acceptance for publication.
Western Blotting
Half a million cells of H1299 cells were plated in 6 well plates the day before transfection. YAP1 or control siRNAs (silencer select) were transfected with RNAiMax (Thermofisher) and 48 hours post transfection cells were lysed to monitor knockdown efficiency by western blotting. For western blotting cell lysates were resolved in 4–12% NuPAGE gels and transferred onto nitrocellulose membranes. Post transfer the membrane was blocked in 5% milk and incubated with either YAP1 (4°C overnight) or anti GAPDH-HRP (1hr room temperature) antibodies. After washing the YAP1 blot was incubated with HRP conjugated secondary antibody and the chemiluminescence signal was obtained with ECL kit (Amersham) was captured on X-Ray films.
Cell Lines, siRNA Transfection, Proliferation, RNA Isolation and Quantitative Reverse Transcription Polymerase Chain Reaction
Caki-1, HK2 and A-704 cells were purchased from American Type Culture Collection (ATCC) in April 2015, and authenticated by STR DNA fingerprinting (on 30th June [Cak1-1 and HK2] and 20th August 2015[A-704]) at the University of Michigan DNA Sequencing Core using Identifiler+kit (Applied Biosystems, Inc) and the resulting amplicons were analyzed in ABI3720XL Genetic Analyzer. Routine mycoplasma testing was also performed using MycoAlert Plus kit (Lonza) every three weeks while in culture, following manufacturers recommendations. The cells were grown in McCoy, Keratinocyte and RPMI media supplemented with 10% FBS, penicillin, and streptomycin respectively. siRNAs were obtained from Ambion and transfected (25nM) using Lipofectamine RNAimax (Invitrogen). For transfection 0.4 million cells were plated in a 6-well plate before transfection. 48 hrs after transfection cells were trypsinized, counted and replated for proliferation assay. Remaining cells were used for RNA isolation using miRNA easy kit (Qiagen). RNA was converted to cDNA using Superscript III (Invitrogen) to perform qRT-PCR.
Luciferase Assay
MCF7 cells stably expressing TEAD regulated luciferase was purchased (BPS Bioscience, San Diego, CA) and maintained in 10% DMEM supplemented with insulin. To study the effect of knockdown of PTPN14 on YAP1/ TEAD regulated luciferase, cells were transfected with 25nM of either non-targeting or PTPN14 siRNAs. After 48 hours of transfection cells were lysed in 1× passive lysis buffer (Promega). Equal amount of protein was used to measure the luciferase activity using luciferase assay reagent (Promega).
Immunohistochemistry
In the current study, IHC were performed on representative whole tumor sections from the MTSCC cases with anti-YAP1 primary antibody (Cell Signaling Technology: / Danvers, MA / 01923 YAP1 Rabbit polyclonal cat# 4912s; 1 in 300 dilution). Formalin fixed, paraffin sections were cut at 5 microns and rehydrated to water. Heat induced epitope retrieval was performed with FLEX TRS Low pH Retrieval buffer (6.1) for 20 minutes (Dako, North America: / Carpentaria, CA / 93013; FLEX TRS Low pH Retrieval Buffer, FLEX HRP EnVision Detection System). After peroxidase blocking, the antibody YAP1 rabbit polyclonal was applied at a dilution of 1:300 at room temperature for 60 minutes. The FLEX HRP EnVision System was used for detection. DAB chromagen was then applied for 10 minutes. Slides were counterstained with Harris Hematoxylin for 5 seconds, dehydrated and coverslipped. IHC was assessed for nuclear and cytoplasmic expression on tumor cells and background benign renal parenchyma by two study pathologists (A.U. and R.M.).
Data Analysis
Exome Analysis
Whole exome tumor/normal paired end sequencing was performed on Illumina HiSeq 2500 instrument and base call files from the instrument were converted to fastq format using Illumina cassava (ref) pipeline. These fastq files were then aligned to human genome build 19 (GRCh37) using novoalign (version 2.08.02, Novocraft Technologies). The sorting and indexing of the bam files were carried out using novosort (1.02.01) and duplicate reads were removed using Picard (version 1.93) furthermore using Picard HsMetrics program we estimated metrics such as on-target and mean coverage for all the libraries. SAMtools (version 0.1.19) was then used to generate the tumor/normal pileup files and were simultaneously used for mutation analysis using VarScan 2 algorithm (version2.3.5)(38, 39). The somatic mutation vcf file generated were then post processed with variant having phred score>q20, with minimum requirement of at least 10× coverage in tumor library to consider a given position for mutation calling and 10 unique variant reads are required in tumor libraries to make a call. The SNVs vcf files were then annotated using annovar (version) and somatic mutations were further filtered out based on their frequency in 1000 genomes (Aug 2015 release), dbsnp (version 142) and 6500 Exome Sequencing Project. SNVs were also annotated with the Catalog of Somatic Mutations in Cancer (COSMIC version 70) and Clinvar for the hotspot/pathogenic mutations(40). Small insertion and deletion (Indels) mutations were identified using Pindel (version 0.2.5). The candidate indels were further filtered by the homopolymer/repeat regions, recurrent sequencing artifacts and high recurrence in 1000 Genomes, followed by manual curation and annotation using annovar(40). The variant allele fraction for Indels presented in Supplementary Table S3 are based on independent estimation of allelic fractions from the BAM files.
Copy number aberrations were quantified and reported for each gene as the segmented, normalized, log2-transformed exon coverage ratio between each tumor sample and its matched normal sample. To account for observed associations between coverage ratios and variation in GC content across the genome, lowess normalization was used to correct per-exon coverage ratios before segmentation analysis. Specifically, mean GC percentage was computed for each targeted region, and a lowess curve was fit to the scatterplot of log2 coverage ratios versus mean GC content across the targeted exome using the lowess function in R (version 2.13.1) with smoothing parameter f = 0.05(41).
RNA-seq Analysis
Following sequencing and base-calling, RNA-seq data was aligned using STAR (2.4.0g1)(42, 43) to the GRCh38.p1 reference genome, using the “basic” version of gencode 22 to construct the splice junction database. To identify chromosome-level differences in gene expression, the genome was divided into genic and inter-genic loci. The total number of reads mapping to each locus was counted using feature Counts(44). To correct for different sequencing depth and effective library size normalization factors we applied the TMM function (default settings) on reads mapped to diploid chromosomes. Expression of protein-coding genes was quantified by counting the reads overlapping exons of annotated protein coding genes in strand-specific mode. Expression level differences between tumor and normal samples was obtained by applying limma(45) with eBayes(46) adjustment on voom transformed count data. Plots of locus-level expression was produced using GViz(47).
To compare MTSCC expression profiles with data sets from dissected nephrons we followed a procedure analogous to Davis et al., 2014, and Chen et al., 2016 (3, 48). Briefly, MTSCC gene expression levels (RPKM) were standardized relative to the mean and standard deviation estimated from normal (benign) renal tissues combined across KIRC, KIRP, and KICH TCGA cohorts. Similarly, expression levels (RPKM) from the rat dissected nephron RNA-seq data (31) were downloaded from GSE56743 and standardized relative to robust estimates of mean and standard deviation across the kidney sections. SAGE data from Cheval et al., 2012 (30) was centered and scaled as described previously by Davis et al., 2014 (3). Concordance between expression profiles is reported as Pearson’s correlation. For human and rat comparisons only 1-to-1 orthologs are used, based on the mappings provided by Ensembl Genes 84. Region specific markers for the rat dataset were nominated as follows: for each region (n=15) the top 40 genes with the most region-specific expression were found. Genes were ranked according to z-scores and sparsity (according to Hoyer)(49) of expression within region. Further, highly correlated markers were removed (PCC > 0.95).
Gene-set Enrichment Analysis (GSEA)
Gene sets in the form of molecular signatures (MSigDb v5.0) and GO-term annotations have been downloaded from MSigDB and NCBI, correspondingly. All enrichment analyses have focused on protein-coding genes. Non-coding genes and genes without a mapping to Ensembl (sep2015.archive.ensembl.org) were removed from the signatures. The randomSet method(50) using shrunken fold-changes from a gene-level score were used to identify significantly up- or down-regulated signatures. RNAseq data from siRNA experiments are deposited in Gene Expression Omnibus (GEO) under the accession number GSE85969.
Supplementary Material
Statement of Significance.
MTSCC is a rare and relatively recently described subtype of renal cell carcinoma. Next generation sequencing of a multi-institutional MTSCC cohort revealed recurrent chromosomal losses and somatic mutations in the Hippo signaling pathway genes leading to potential YAP1 activation. In virtually all cases of MTSCC, there was evidence of Hippo pathway dysregulation suggesting a common mechanistic basis for this disease.
Acknowledgments
Grant Support: A.M.C. is supported by the A. Alfred Taubman Medical Institute, the American Cancer Society, the Howard Hughes Medical Institute and EDRN U01 CA111275. A.A. is supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number 2KL2TR000434. R.M. and A.M.C. are supported by the Prostate Cancer Foundation and UM1 Grant (Number1UM1HG006508).
We thank Tina Fields, the histology staff, Lakshmi Dommeti and Sunita Shankar for technical assistance, Rohit Malik for help with figures, Jyoti Athanikar for manuscript editing and Karen Giles for assistance with manuscript submission.
Footnotes
Disclosures of Potential Conflicts of Interest: None
Author Contributions:
Study concept and design: Mehra, Vats, McKenney, Dhanasekaran and Chinnaiyan.
Acquisition of data: Mehra, Cao, McKenney, Sangoi, Trpkov, Osunkoya, Zhou, Giannico, Shukla, Siddiqui, Dhanasekaran, Pan, Wang and Su.
Analysis and interpretation of data: Mehra, Vats, Cieslik, Udager, Kasaian, Dhanasekaran, McKenney, Chinnaiyan
Drafting of the manuscript: Mehra, Vats, Dhanasekaran, Chinnaiyan.
Critical revision of the manuscript for important intellectual content: Mehra, Vats, Udager, Weizer, Palapattu, Hafez, Wolf Jr., Kumpati, Chinnaiyan
Statistical analysis: Vats, Cieslik, Dhanasekaran.
Obtaining funding: Mehra, Chinnaiyan.
Supervision: Mehra, Chinnaiyan, Dhanasekaran.
REFERENCE
- 1.Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis C, et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. The New England journal of medicine. 2016:135–145. doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fine SW, Argani P, DeMarzo AM, Delahunt B, Sebo TJ, Reuter VE, et al. Expanding the histologic spectrum of mucinous tubular and spindle cell carcinoma of the kidney. The American journal of surgical pathology. 2006;30:1554–1560. doi: 10.1097/01.pas.0000213271.15221.e3. [DOI] [PubMed] [Google Scholar]
- 5.Reuter VE, Argani P, Zhou M, Delahunt B Members of the IIiDUPG. Best practices recommendations in the application of immunohistochemistry in the kidney tumors: report from the International Society of Urologic Pathology consensus conference. The American journal of surgical pathology. 2014;38:e35–e49. doi: 10.1097/PAS.0000000000000258. [DOI] [PubMed] [Google Scholar]
- 6.Cieslik M, Chugh R, Wu YM, Wu M, Brennan C, Lonigro R, et al. The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing. Genome research. 2015;25:1372–1381. doi: 10.1101/gr.189621.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, et al. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. Jama. 2015;314:913–925. doi: 10.1001/jama.2015.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nature genetics. 2013;45:1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salah Z, Melino G, Aqeilan RI. Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer research. 2011;71:2010–2020. doi: 10.1158/0008-5472.CAN-10-3516. [DOI] [PubMed] [Google Scholar]
- 11.Schramm A, Koster J, Assenov Y, Althoff K, Peifer M, Mahlow E, et al. Mutational dynamics between primary and relapse neuroblastomas. Nature genetics. 2015;47:872–877. doi: 10.1038/ng.3349. [DOI] [PubMed] [Google Scholar]
- 12.Liu X, Yang N, Figel SA, Wilson KE, Morrison CD, Gelman IH, et al. PTPN14 interacts with and negatively regulates the oncogenic function of YAP. Oncogene. 2013;32:1266–1273. doi: 10.1038/onc.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nature reviews Cancer. 2013;13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 15.Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development. 2011;138:9–22. doi: 10.1242/dev.045500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peckova K, Martinek P, Sperga M, Montiel DP, Daum O, Rotterova P, et al. Mucinous spindle and tubular renal cell carcinoma: analysis of chromosomal aberration pattern of low-grade, high-grade, and overlapping morphologic variant with papillary renal cell carcinoma. Annals of diagnostic pathology. 2015;19:226–231. doi: 10.1016/j.anndiagpath.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Udager AM, Alva A, Mehra R. Current and proposed molecular diagnostics in a genitourinary service line laboratory at a tertiary clinical institution. Cancer journal. 2014;20:29–42. doi: 10.1097/PPO.0000000000000017. [DOI] [PubMed] [Google Scholar]
- 18.Avruch J, Zhou D, Fitamant J, Bardeesy N, Mou F, Barrufet LR. Protein kinases of the Hippo pathway: regulation and substrates. Seminars in cell & developmental biology. 2012;23:770–784. doi: 10.1016/j.semcdb.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moya IM, Halder G. Discovering the Hippo pathway protein-protein interactome. Cell research. 2014;24:137–138. doi: 10.1038/cr.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Science signaling. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- 21.Wang W, Li X, Huang J, Feng L, Dolinta KG, Chen J. Defining the protein-protein interaction network of the human hippo pathway. Molecular & cellular proteomics : MCP. 2014;13:119–131. doi: 10.1074/mcp.M113.030049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alder O, Cullum R, Lee S, Kan AC, Wei W, Yi Y, et al. Hippo signaling influences HNF4A and FOXA2 enhancer switching during hepatocyte differentiation. Cell reports. 2014;9:261–271. doi: 10.1016/j.celrep.2014.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–1338. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanazawa T, Ichii O, Otsuka S, Namiki Y, Hashimoto Y, Kon Y. Hepatocyte nuclear factor 4 alpha is associated with mesenchymal-epithelial transition in developing kidneys of C57BL/6 mice. The Journal of veterinary medical science / the Japanese Society of Veterinary Science. 2011;73:601–607. doi: 10.1292/jvms.10-0493. [DOI] [PubMed] [Google Scholar]
- 25.Kanazawa T, Konno A, Hashimoto Y, Kon Y. Hepatocyte nuclear factor 4 alpha is related to survival of the condensed mesenchyme in the developing mouse kidney. Developmental dynamics : an official publication of the American Association of Anatomists. 2010;239:1145–1154. doi: 10.1002/dvdy.22276. [DOI] [PubMed] [Google Scholar]
- 26.Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015;348:660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanazawa T, Konno A, Hashimoto Y, Kon Y. Expression of hepatocyte nuclear factor 4alpha in developing mice. Anatomia, histologia, embryologia. 2009;38:34–41. doi: 10.1111/j.1439-0264.2008.00889.x. [DOI] [PubMed] [Google Scholar]
- 28.Suh JM, Yu CT, Tang K, Tanaka T, Kodama T, Tsai MJ, et al. The expression profiles of nuclear receptors in the developing and adult kidney. Molecular endocrinology. 2006;20:3412–3420. doi: 10.1210/me.2006-0312. [DOI] [PubMed] [Google Scholar]
- 29.Saadettin SE, Ryffel T, Drewes GU, T Human renal cell carcinogensis is accompanied by a coordinate loss of the tissue specific transcription factors HNF4A and HNF1A. Cancer Letters. 1996;101:205–210. doi: 10.1016/0304-3835(96)04136-5. [DOI] [PubMed] [Google Scholar]
- 30.Cheval L, Pierrat F, Rajerison R, Piquemal D, Doucet A. Of mice and men: divergence of gene expression patterns in kidney. PloS one. 2012;7:e46876. doi: 10.1371/journal.pone.0046876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JW, Chou CL, Knepper MA. Deep Sequencing in Microdissected Renal Tubules Identifies Nephron Segment-Specific Transcriptomes. Journal of the American Society of Nephrology : JASN. 2015;26:2669–2677. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srigley JK, Reuter L, Amin V, Grignon M, Eble D, Weber J, Moch A, H Phenotypic, molecular and utrastructural studies of a novel low grade renal epithelial neoplasm possibly related to the loop of Henle. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2002;15 [Google Scholar]
- 33.Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nature genetics. 2016;48:398–406. doi: 10.1038/ng.3525. [DOI] [PubMed] [Google Scholar]
- 34.Rakozy C, Schmahl GE, Bogner S, Storkel S. Low-grade tubular-mucinous renal neoplasms: morphologic, immunohistochemical, and genetic features. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2002;15:1162–1171. doi: 10.1097/01.MP.0000031709.40712.46. [DOI] [PubMed] [Google Scholar]
- 35.Harms PW, Vats P, Verhaegen ME, Robinson DR, Wu YM, Dhanasekaran SM, et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer research. 2015;75:3720–3727. doi: 10.1158/0008-5472.CAN-15-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehra R, Vats P, Kalyana-Sundaram S, Udager AM, Roh M, Alva A, et al. Primary urethral clear-cell adenocarcinoma: comprehensive analysis by surgical pathology, cytopathology, and next-generation sequencing. The American journal of pathology. 2014;184:584–591. doi: 10.1016/j.ajpath.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome research. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lonigro RJ, Grasso CS, Robinson DR, Jing X, Wu YM, Cao X, et al. Detection of somatic copy number alterations in cancer using targeted exome capture sequencing. Neoplasia. 2011;13:1019–1025. doi: 10.1593/neo.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dobin A, Gingeras TR. Mapping RNA-seq Reads with STAR. Current protocols in bioinformatics. 2015;51:11 4 1–11 4 9. doi: 10.1002/0471250953.bi1114s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic acids research. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 47.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome biology. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen F, Zhang Y, Senbabaoglu Y, Ciriello G, Yang L, Reznik E, et al. Multilevel Genomics-Based Taxonomy of Renal Cell Carcinoma. Cell reports. 2016;14:2476–2489. doi: 10.1016/j.celrep.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoyer PO. Non-negative Matrix Factorization with Sparseness Constraints. Journal of Machine Learning Research. 2004;5:1457–1469. [Google Scholar]
- 50.Newton MA. Random-set methods identify distinct aspects of the enrichment signal in gene-set analysis. Annals of Applied Statistics. 2007:85–106. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.