

Abstract
Finding small molecules that target allosteric sites remains a grand challenge for ligand discovery. In the protein kinase field, only a handful of highly selective allosteric modulators have been found. Thus, more general methods are needed to discover allosteric modulators for additional kinases. Here, we use virtual screening against an ensemble of both crystal structures and comparative models to identify ligands for an allosteric peptide-binding site on the protein kinase PDK1 (the PIF pocket). We optimized these ligands through an analog-by-catalog search that yielded compound 4, which binds to PDK1 with 8 μM affinity. We confirmed the docking poses by determining a crystal structure of PDK1 in complex with 4. Because the PIF pocket appears to be a recurring structural feature of the kinase fold, known generally as the helix αC patch, this approach may enable the discovery of allosteric modulators for other kinases.

INTRODUCTION
Kinase inhibitors are essential research tools and valuable therapeutics. However, most kinase inhibitors are not specific for the intended target because they bind to the highly conserved ATP-binding pocket. The resulting off-target effects are often undesirable for a chemical probe and may cause side effects in patients. One approach to improve the specificity of kinase inhibitors has been to target allosteric sites distinct from the ATP-binding pocket, which are often less conserved among kinases. While this approach is still in its infancy, several striking successes have been reported and exquisitely selective allosteric inhibitors of the kinases AKT, MEK, and ABL are now in clinical trials for the treatment of advanced cancers.1 Despite these successes, developing allosteric inhibitors of protein kinases remains challenging because the kinase of interest either has no known allosteric site or finding ligands for candidate sites is intractable with current technologies.
Here, we explored the druggability of an allosteric site called the helix αC patch. This site is functionally conserved across many evolutionarily distant protein kinases.2-4 The helix αC patch is a hydrophobic pocket formed by the αB/αC helices and the β4/β5 strands in the small N-terminal lobe of the kinase domain (Figure 1). The binding of effector proteins to the helix αC patch activates some kinases and inhibits others.2 We focused on the kinase 3-phosphoinositide-dependent protein kinase 1 (PDK1) as a model system because it is a paradigm for allosteric regulation by the helix αC patch. PDK1 is also an important anticancer target because it phosphorylates and activates more than 20 kinases that regulate cell survival, proliferation, and metabolism, including isoforms of AKT, S6K, RSK, SGK, and PKC.5
Figure 1.

(A) Relative position of the ATP-binding pocket and the helix αC patch on the protein kinase fold. PDK1 with a peptide bound to its helix αC patch, the PIF pocket. (B) Close-up view of the PIF pocket of PDK1. The hydrophobic subpockets targeted by docking are marked with two circles.
PDK1 uses its helix αC patch, named the PDK1-interacting fragment (PIF) pocket, to recruit downstream kinases by engaging a hydrophobic peptide motif in their C-terminal tails.6 Because ATP-competitive inhibitors of PDK1 have so far been incapable of fully suppressing its activity,7-9 small molecules targeting the PIF pocket have been pursued as a secondary strategy aiming to disrupt the recruitment of substrates to PDK1. Several computational and experimental approaches have been used to target this challenging protein–peptide interface, including pharmacophore modeling,10-12 NMR-based fragment screening,3,13 computational design,14 and competitive binding assays.4,15 Here, we investigated the utility of structure-based docking as a site-directed approach to discover new ligands for the PIF pocket of PDK1.
We docked 6300 compounds from the ZINC database against a small conformational ensemble of two crystal structures and four comparative models of PDK1. We prioritized compounds that scored well across at least four of the six target models. Using this consensus ranking approach, we identified two novel ligands, which were subsequently shown to bind to PDK1 with a Kd of ~40 μM. Next, we docked commercially available analogs and discovered compound 4, which binds to PDK1 with an in vitro potency that is comparable to known PIF pocket ligands (Kd of 8 μM).4,12 We solved a 1.4 Å crystal structure of PDK1 bound to 4, validating the predicted binding pose. In summary, we present new scaffolds for the development of allosteric PDK1 inhibitors and demonstrate the utility of virtual screening for targeting a challenging allosteric site that is present in a number of kinases.2
RESULTS
Generation of PDK1 Comparative Models and Chemical Library
The virtual screening workflow is depicted in Figure 2. The conformation of the PIF pocket in PDK1 depends on the bound ligand. Specifically, the αB and αC helices were tightly packed against the kinase when PDK1 was bound to the allosteric activator PS48, but the helices swung away from the kinase by up to 5 Å when PDK1 was bound to 1F8, a covalent PIF pocket ligand.3,11 Moreover, Arg131 repositions to make optimal electrostatic contacts depending on which ligand is bound. Thus, we created a set of six structural models of the PIF pocket to recapitulate this repertoire of ligand-induced conformations. Model 1 (M1) is the crystal structure of PDK1 bound to PS48 (PDB code 3HRF) where the αB and αC helices are in the “closed” conformation. M2 was constructed by grafting the “open” conformation of the αB and αC helices from the structure of PDK1 bound to 1F8 (PDB code 3ORX) onto M1. We then relaxed the αB and αC helices of M2 using MODELLER16 to compute M3, which is an intermediate “open” state between M1 and M2. Finally, we optimized the positioning of the Arg131 side chain using PLOP,17 thereby creating models M1R, M2R, and M3R (Supporting Information Figure 1).17
Figure 2.

Virtual screening workflow.
Next, we generated a chemical library for virtual screening. To avoid rediscovering known ligands, we assembled a list of 112 known PIF pocket ligands from the literature10-15 and our own work.3,4 We then used the DUD-E procedure to identify 6300 commercially available compounds that were topologically distinct from these known ligands but had similar chemical properties, such as molecular weight, calculated log P, and net charge.18
Docking and Experimental Testing
We performed a prospective virtual screen of the 6300 compounds against all 6 structural models using DOCK 3.6.19 To be considered a hit, we required that compounds rank in the top 500 in at least 4 of the 6 docking screens. We further triaged hits by imposing a geometry filter requiring that compounds must (1) make at least two hydrogen bonds to the polar subsite within the PIF pocket defined by residues Arg131, Lys76, or Thr148; (2) occupy the hydrophobic pocket delimited by Ile119 and Leu155; and (3) occupy the hydrophobic pocket with Phe157 at its base (Figure 1B). The geometry filters were derived from crystal structures of PDK1 in complex with PIFtide or various small-molecule ligands. The polar subsite is a critical binding energy hotspot for a negatively charged carboxylate or aspartate. The hydrophobic pockets engage either two phenylalanines or two aryl substituents. Following visual inspection of the hit list, we selected three compounds for experimental testing (Table 1).
Table 1. Top Virtual Screening Hits across the Six PIF Pocket Models.
| Compound structurea | Docking rankb |
Kd
μMc (95% CI) |
LEd | ||||||
|---|---|---|---|---|---|---|---|---|---|
| M1 | M1R | M2 | M2R | M3 | M3R | ||||
| 1 |
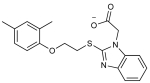
|
6 | 39 | 29 | 66 | 1 | 2 | 39.1 (35.2-43.2) |
0.25 |
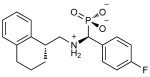
| 2 |
|
109 | 189 | 31 | 297 | 14 | 76 | >200 | - |
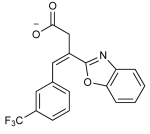
| 3 |
|
2808 | 158 | 125 | 125 | 491 | 294 | 39.4 (36.8-42.2) |
0.26 |
Charged states are depicted assuming a physiological pH of 7.4.
Ranks reported do not consider the molecules discarded by the geometry filter.
Kd was calculated from the IC50 in the FP assay using an equation that accounts for ligand depletion.1
Ligand efficiency (LE) is calculated as binding energy (ΔG, kcal/mol) per non-hydrogen atom.
To determine whether the virtual screening hits bound to PDK1, we used a fluorescence polarization (FP) competitive binding assay that monitors the displacement of a fluorophore-labeled peptide from the PIF pocket of PDK1.4 Compounds 1 and 3 both bound to PDK1 with a Kd of ~40 μM, whereas the binding of 2 was negligible (Figure 3A). To confirm these findings with an orthogonal assay, we also determined the effect of 1 and 3 on the catalytic activity of PDK1 toward a short peptide substrate in vitro. In this assay, substrate recruitment does not depend on the PIF pocket, so allosteric modulation of PDK1 activity by the small-molecule ligand can be observed. We found that compounds 1 and 3 stimulated PDK1 activity up to a maximum of ~1.8-fold with EC50 values of ~40 and ~50 μM, respectively (Figure 3B). Compounds 1 and 3 do not appear to exert their effects through an aggregation-based mechanism: Neither compound formed colloidal aggregates under our assay conditions, as monitored by dynamic light scattering, nor did either compound nonspecifically inhibit an unrelated enzyme–the protease cruzain (Supporting Information Figure 2).20,21
Figure 3.

Dose–response curves for initial hits 1 and 3 and improved analog 4 in the (A) binding and (B) kinase activity assay. A peptide ligand was used as a control (PIFtide, residues 9–23).
Analog-by-Catalog
To improve the potency of 1 and 3, we extracted 518 commercially available analogs from the ZINC database using its analog-by-catalog method with a permissive chemical similarity threshold of 70% (Tanimoto coefficient, Tc),22 using ChemAxon path-based fingerprints. We docked these 518 analogs against the six structural models and found 15 analogs that scored as well as or better than the parent compounds 1 and 3 (Supporting Information Table 1). We tested whether these analogs bound to the PIF pocket using the FP competitive binding assay described above. Of the 15 analogs tested, 8 bound to PDK1 with an affinity 2- to 4-fold worse than their parent compounds, 6 analogs showed very weak or negligible binding, and one (compound 4) bound to PDK1 with a Kd of 8 μM, corresponding to a 5-fold improvement over its parent compound 1 (Figure 3A; Table 2). Compound 4 ranked within the top 26 of 518 analogs across all six models and adopted a slightly different docking pose in each model (Supporting Information Figure 3). Like compounds 1 and 3, compound 4 also enhanced the activity of PDK1 toward a short peptide substrate, with a maximal stimulation of ~1.6-fold and an EC50 value of 2 μM (Figure 3B).
Table 2. Structure–Activity Relationships for the PIF Pocket Ligand Analogs.

|
Crystal Structure
To assess the accuracy of the docking pose of compound 4, we determined a crystal structure of PDK1 bound to ATP and 4. To enable soaking of 4 into PDK1 crystals, we crystallized a double mutant of the kinase domain (Y288A, Q292G) that packs in an arrangement where the PIF pocket is accessible to the solvent.11 The best crystal diffracted to 1.4 Å resolution, and the resulting electron density map showed strong peaks for both the aryl substituents and the carboxylic acid of 4 (Figure 4A). We did not observe clear electron density for the oxyethylsulfanyl linker, indicating it adopts multiple conformations. The only notable difference between the actual binding pose for 4 and the predicted docking pose is the conformation of the flexible linker between the aryl substituents (Figure 4B). The non-hydrogen atom rmsd of compound 4 between the predicted and experimental binding poses ranged from 0.92 to 1.56 Å following least-squares superposition of the six PDK1 models.
Figure 4.

(A) Crystal structure of PDK1 bound to compound 4. Electron density is shown for the ligand (green, Fo – Fc omit map, 3σ) and for key interacting residues (blue, 2Fo – Fc map, 1.25σ). (B) Overlap of the crystallographic binding pose and the docking pose for compound 4, following least-squares superposition of the PDK1 atoms.
DISCUSSION
Using structure-based virtual screening, we have identified novel ligands that bind to the allosteric PIF pocket on PDK1. In contrast to traditional single-structure docking screens, we docked against an ensemble of PDK1 structures and comparative models, allowing for some of the conformational accommodation that we expected from this allosteric site. We hypothesized that ligands that dock well across multiple conformations of a binding site are more likely to be true binders. We also reasoned that docking against multiple conformations of the binding site would identify additional chemotypes compared to docking against a single structure. We were encouraged by previous ensemble docking studies that have increased the hit rate and chemical diversity compared to single target docking.23-25
Here, our ensemble docking and consensus ranking approach showed three advantages over docking against a single crystal structure. First, we correctly identified the phosphonate 3 as a true binder despite its poor rank against the starting model M1 (2808/6300), because 3 scored well across the other 5 models. Moreover, a perturbed model prioritized 6 of the 9 true binders better than the crystal structures during the analog-by-catalog stage. Second, the success rate for identifying true binders exceeded 50% during both the initial screening (66%) and the analog-by-catalog steps (60%). Finally, using the consensus ranking approach to select hits greatly reduced the level of human intervention needed relative to our prior virtual screens against single targets. Docking against multiple models may lessen the need for an experienced scientist to scrutinize hundreds of docking poses, as is now common practice in virtual screening.26,27 The stringency of the consensus ranking rule (i.e., top 500 rank across 4 of 6 models) should be adjusted to match the number of compounds that can be purchased and tested.
We docked a library of only 6300 of the ~4 million commercially available compounds in the ZINC database. We selected this subset of compounds with the goal of retaining the chemical properties that are favorable for binding to the PIF pocket, the most notable being a net negative charge, while avoiding rediscovery of known chemotypes. This goal was accomplished by repurposing the DUD-E method, which was originally designed to identify property-matched “decoy” molecules as negative controls for docking. While the diaryl acid (1) and diaryl phosphonate (3) compounds can be broadly grouped into the same aromatic–charge–aromatic pharmacophore that describes nearly all reported PIF pocket ligands, both compounds represent novel scaffolds that are topologically dissimilar to all known ligands (maximum Tc of 0.25).28 Thus, they would not be discovered by searching for analogs of known ligands in the ZINC database using even a low 50% similarity cutoff. Unexpectedly, the DUD-E method for generating “negative control” molecules can be repurposed for scaffold hopping when the protein target has a set of known ligands.
While 9 of the 15 compounds we selected during the analog-by-catalog step were true binders, only one analog (4) was substantially more potent than the parent compounds 1 and 3. This finding is consistent with common knowledge that computational docking is better at distinguishing true binders from nonbinders than it is at predicting relative potencies. Thus, the success of an analog-by-catalog procedure is bounded by the number of analogs that can be purchased for testing and, indirectly, by the chemical diversity of the commercially available analogs. Nevertheless, we were able to identify analog 4, which was 5-fold more potent than its parent compound 1. The crystal structure of 4 bound to PDK1 revealed that the 2,6-dimethyl substituted phenyl group of 4 packs tightly into its hydrophobic subpocket, suggesting the 2,4-dimethyl substitution pattern of the parent compound 1 was sterically suboptimal. Additionally, the poor electron density observed for the oxyethylsulfanyl linker between the phenyl and benzimidazole rings of 4 suggests this region is highly flexible. Therefore, in future studies the potency of 4 is likely to be further improved by rigidifying this linker with a carbocycle or other conformation-restricting moiety.
In conclusion, structure-based virtual screening against the PIF pocket of PDK1 has identified novel allosteric modulators. The approach is applicable to targeting the helix αC patch of other protein kinases and therefore may enable the discovery of ligands for this broad class of protein–protein and protein–peptide interfaces.
EXPERIMENTAL SECTION
Generation of PDK1 Structural Models
Six structural models of human PDK1 were used in this study. The first model (M1) is the crystal structure of PDK1 bound to an allosteric activator (PDB code 3HRF). Starting from the first structure, the PIF pocket allosteric site was adjusted in three steps to sample a variety of conformations. First, the positions of the αB and αC helices were extracted from another crystal structure with a disulfide-trapped fragment in the PIF pocket (PDB code 3ORX), resulting in the second structure (M2). Second, the αB and αC helices in M2 were refined using conjugate gradient (CG) minimization in MODELLER,16 resulting in different positions of the αB helix and the loop that links the two helices (M3). Finally, the side chain of the Arg131 on the αC helix was optimized in all three models using the “side chain prediction” protocol in PLOP,17 resulting in three more models (M1R, M2R, M3R).
Generation of Virtual Chemical Library
With the goal of identifying novel ligands for the PIF pocket, a list of 112 known ligands was first compiled, consisting of 7 diaryl carboxylates and 105 diaryl sulfonamides. The docking library was then constructed from the ZINC database using the DUD-E procedure18 to identify 6300 “decoy” compounds that had physicochemical properties similar to the known ligands but differed from them topologically.
Virtual Screening
Virtual screening against the six PDK1 models was performed using a semiautomatic docking procedure. The receptor structure was prepared by removing all non-protein atoms from the crystal structures. Receptor-derived spheres were calculated using the program SPHGEN29 (part of the UCSF DOCK suite), while the ligand-derived spheres were generated from the positions of the heavy atoms of the crystallographic ligand in the 3HRF structure. In total, 45 matching spheres were used to orient ligands in the binding site. All docking calculations were performed with DOCK 3.6.19 The docking poses were scored using van der Waals, Poisson–Boltzmann electrostatic, and ligand-desolvation penalty terms.
Chemical Novelty Evaluation and Analog Search
For assessing chemical similarity between two compounds, we relied on the ECFP4 fingerprints28 to calculate Tanimoto coefficients (Tc) using the program Pipeline Pilot (Accelrys). A Tc value of less than 0.4 is commonly accepted as an indication of chemical dissimilarity between two compounds. Commercially available analogs of the initial docking hits were identified using the analog-by-catalog method of the ZINC database22 with a permissive chemical similarity level of 70%, as calculated by JChemBase, using ChemAxon path-based fingerprints (ChemAxon).
Fluorescence Polarization Competitive Binding Assay
Docking hits were experimentally tested for binding to PDK1 using a competitive binding assay that monitored the displacement of a fluorophore-labeled peptide from the PIF pocket.4 The dissociation constant (Kd) for ligands was calculated from their IC50 values using an equation that accounts for ligand depletion.30
Cruzain Assay
Cruzain assays were performed in 100 mM sodium acetate, pH 5.5, containing 5 mM DTT. Triton X-100 was added to 0.01% in reaction mixtures as indicated. Drugs were incubated with 0.8 nM cruzain for 5 min until reactions were initiated by adding fluorogenic substrate Z-Phe-Arg-aminomethylcoumarin (Z-FR-AMC). The final reaction volume was 200 μL, containing 0.4 nM cruzain, 2.5 μM ZF-R-AMC, and 0.5% DMSO. Increase in fluorescence (excitation wavelength of 355 nm, emission wavelength of 460 nm) was recorded for 5 min in a microtiter plate spectrofluorimeter (Molecular Devices, FlexStation). Assays were performed in duplicate in 96-well plates; control samples contained DMSO only.
Dynamic Light Scattering
Concentrated DMSO stocks of drugs were diluted with assay buffer to a final DMSO concentration of 3.2%. Measurements were made using a DynaPro MS/X (Wyatt Technology) with a 55 mW laser at 826.6 nm, laser power of 100%, and detector angle of 90°. Single-point measurements are reported.
Protein Kinase Activity Assay
The effect of PIF pocket ligands on the catalytic activity of PDK1 toward a short peptide substrate (T308tide) was measured using a radioactivity-based kinase assay.3
Crystallization and Structure Determination
Crystals were obtained using a PDK150–359 mutant (Y288G, Q292A) that disrupts a crystal contact that normally prevents ligands from binding to the PIF pocket.11 Conditions for crystallization, compound soaking, harvesting, and data collection were described previously.4 Diffraction data were collected at Advanced Light Source beamline 8.3.1 and were indexed and scaled using HKL-2000.31 Structures were solved by molecular replacement using a PDK1 crystal structure (PDB code 4AW1) as a search model in Phaser.32 Iterative model building and refinement were performed with Coot33 and PHENIX,34 respectively. Structure validation was performed using MOLProbity.35 Final refinement statistics are summarized in Supporting Information Table 2.
Compound Quality Control
Every purchased compound was analyzed by LCMS (Waters 2795 analytical HPLC, and ZQ MS). Every compound yielded a single peak by UV and evaporative light scattering (ELSD) and was within 0.1 Da of the expected mass. The structure of compound 4, 2-(2-((2-(2,6-dimethylphenoxy)ethyl)thio)-1H-benzo[d]imidazol-1-yl)acetic acid, was further confirmed by 1HNMR (400 MHz, DMSO-d6): δ 7.46–7.53 (m, 2H), 7.12–7.17 (m, 2H), 6.97 (d, J = 7.6 Hz, 2H), 6.87 (dd, J = 8.0, 6.8 Hz, 1H), 4.97 (s, 2H), 4.03 (t, J = 6.4 Hz, 2H), 3.68 (t, J = 6.0 Hz, 2H), 2.20 (s, 6H). LCMS (m/z): [M + H]+ calcd, 357.12; found, 357.18.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Wells, Sali, and Shoichet laboratories for helpful suggestions and critical review of the manuscript. We also thank the staff at ALS beamline 8.3.1. This work was supported by the NIH Grants R01 CA136779 (J.A.W.), GM59957 (B.K.S.), and U54 GM093342 (A.S., H.F.). T.J.R. was supported by a NIH predoctoral fellowship (Grant F31 CA180378), a Krevans fellowship, and the UCSF Discovery Fellows Program. We thank ChemAxon for JChemBase, Molinspiration for a license for their molecule properties (mib) and molecule depiction tool, and OpenEye Scientific Software for a free academic license for OEChem.
ABBREVIATIONS USED
- PDK1
3-phosphoinositide-dependent protein kinase 1
- PIF
PDK1-interacting fragment
- MEK
MAPK/ERK kinase
- ABL
Abelson tyrosine kinase
- S6K
p70 ribosomal S6 kinase
- RSK
p90 ribosomal S6 kinase
- SGK
serum/glucocorticoid-regulated kinase
- PKC
protein kinase C
- Kd
equilibrium dissociation constant
- DUD-E
directory of useful decoys—enhanced
- FP
fluorescence polarization
- EC50
effective concentration for half-maximal response
- Tc
Tanimoto coefficient
- PLOP
protein local optimization program
- SPHGEN
sphere-generation program
- ECFP4
extended-connectivity fingerprints 4
- AMC
aminomethylcoumarin
- ELSD
evaporative light scattering detection
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.5b01216.
Molecular formula strings (TXT)
Figures and tables showing structural models, docking ranks and poses, and crystallographic statistics (PDF)
Accession Codes
PDB code for crystal structure of PDK1 in complex with ATP and the PIF-pocket ligand RF4 is 4XX9.
The authors declare no competing financial interest.
REFERENCES
- (1).Fang Z, Grütter C, Rauh D. Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem. Biol. 2013;8:58–70. doi: 10.1021/cb300663j. [DOI] [PubMed] [Google Scholar]
- (2).Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell. 2011;42:9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, Wells JA. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc. Natl. Acad. Sci. U. S. A. 2011;108:6056–6061. doi: 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rettenmaier TJ, Sadowsky JD, Thomsen ND, Chen SC, Doak AK, Arkin MR, Wells JA. A small-molecule mimic of a peptide docking motif inhibits the protein kinase PDK1. Proc. Natl. Acad. Sci. U. S. A. 2014;111:18590–18595. doi: 10.1073/pnas.1415365112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- (6).Biondi RM, Cheung PC, Casamayor A, Deak M, Currie RA, Alessi DR. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 2000;19:979–988. doi: 10.1093/emboj/19.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Medina JR. Selective 3-Phosphoinositide-Dependent Kinase-1 (PDK1) Inhibitors: Dissecting the Function and Pharmacology of PDK1. J. Med. Chem. 2013;56:2726–2737. doi: 10.1021/jm4000227. [DOI] [PubMed] [Google Scholar]
- (8).Najafov A, Sommer EM, Axten JM, Deyoung MP, Alessi DR. Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem. J. 2011;433:357–369. doi: 10.1042/BJ20101732. [DOI] [PubMed] [Google Scholar]
- (9).Najafov A, Shpiro N, Alessi DR. Akt is efficiently activated by PIF-pocket- and PtdIns(3,4,5)P3-dependent mechanisms leading to resistance to PDK1 inhibitors. Biochem. J. 2012;448:285–295. doi: 10.1042/BJ20121287. [DOI] [PubMed] [Google Scholar]
- (10).Engel M, Hindie V, Lopez-Garcia LA, Stroba A, Schaeffer F, Adrian I, Imig J, Idrissova L, Nastainczyk W, Zeuzem S, Alzari PM, Hartmann RW, Piiper A, Biondi RM. Allosteric activation of the protein kinase PDK1 with low molecular weight compounds. EMBO J. 2006;25:5469–5480. doi: 10.1038/sj.emboj.7601416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hindie V, Stroba A, Zhang H, Lopez-Garcia LA, Idrissova L, Zeuzem S, Hirschberg D, Schaeffer F, Jørgensen TJD, Engel M, Alzari PM, Biondi RM. Structure and allosteric effects of low-molecular-weight activators on the protein kinase PDK1. Nat. Chem. Biol. 2009;5:758–764. doi: 10.1038/nchembio.208. [DOI] [PubMed] [Google Scholar]
- (12).Busschots K, Lopez-Garcia LA, Lammi C, Stroba A, Zeuzem S, Piiper A, Alzari PM, Neimanis S, Arencibia JM, Engel M, Schulze JO, Biondi RM. Substrate-selective inhibition of protein kinase PDK1 by small compounds that bind to the PIF-pocket allosteric docking site. Chem. Biol. 2012;19:1152–1163. doi: 10.1016/j.chembiol.2012.07.017. [DOI] [PubMed] [Google Scholar]
- (13).Stockman BJ, Kothe M, Kohls D, Weibley L, Connolly BJ, Sheils AL, Cao Q, Cheng AC, Yang L, Kamath AV, Ding Y-H, Charlton ME. Identification of allosteric PIF-pocket ligands for PDK1 using NMR-based fragment screening and 1H-15N TROSY experiments. Chem. Biol. Drug Des. 2009;73:179–188. doi: 10.1111/j.1747-0285.2008.00768.x. [DOI] [PubMed] [Google Scholar]
- (14).Wei L, Gao X, Warne R, Hao X, Bussiere D, Gu X.-j., Uno T, Liu Y. Design and synthesis of benzoazepin-2-one analogs as allosteric binders targeting the PIF pocket of PDK1. Bioorg. Med. Chem. Lett. 2010;20:3897–3902. doi: 10.1016/j.bmcl.2010.05.019. [DOI] [PubMed] [Google Scholar]
- (15).Bobkova EV, Weber MJ, Xu Z, Zhang Y-L, Jung J, Blume-Jensen P, Northrup A, Kunapuli P, Andersen JN, Kariv I. Discovery of PDK1 kinase inhibitors with a novel mechanism of action by ultrahigh throughput screening. J. Biol. Chem. 2010;285:18838–18846. doi: 10.1074/jbc.M109.089946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- (17).Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- (18).Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012;55:6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mysinger MM, Shoichet BK. Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 2010;50:1561–1573. doi: 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- (20).Jadhav A, Ferreira RS, Klumpp C, Mott BT, Austin CP, Inglese J, Thomas CJ, Maloney DJ, Shoichet BK, Simeonov A. Quantitative analyses of aggregation, autofluorescence, and reactivity artifacts in a screen for inhibitors of a thiol protease. J. Med. Chem. 2010;53:37–51. doi: 10.1021/jm901070c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Coan KED, Shoichet BK. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 2008;130:9606–9612. doi: 10.1021/ja802977h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol. 2011;9:71. doi: 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Rueda M, Bottegoni G, Abagyan R. Recipes for the selection of experimental protein conformations for virtual screening. J. Chem. Inf. Model. 2010;50:186–193. doi: 10.1021/ci9003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Fischer M, Coleman RG, Fraser JS, Shoichet BK. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem. 2014;6:575–583. doi: 10.1038/nchem.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, Heveker N, Shoichet BK, Volkman BF. Structure-based ligand discovery for the protein-protein interface of chemokine receptor CXCR4. Proc. Natl. Acad. Sci. U. S. A. 2012;109:5517–5522. doi: 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kruse AC, Weiss DR, Rossi M, Hu J, Hu K, Eitel K, Gmeiner P, Wess J, Kobilka BK, Shoichet BK. Muscarinicreceptors as model targets and antitargets for structure-based ligand discovery. Mol. Pharmacol. 2013;84:528–540. doi: 10.1124/mol.113.087551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rogers D, Hahn M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- (29).Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- (30).Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 2004;332:261–273. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- (31).Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- (32).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (34).Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.