

Abstract
Recent studies have suggested that regulatory T (Treg) cells comprise a heterogeneous population that regulates various aspects of the immune response, and that Treg cells use the factors that are expressed in their target cells to regulate them. We searched for factors that regulate Th1 response in Treg cells using a meta-analysis. In the process, we discovered that transcription factor interferon regulatory factor 8 (IRF8) was selectively expressed in Treg and Th1 cells. IRF8-deficient Treg cells showed defective expression of CXCR3 and aberrant expression of the Il4 and Il17 genes. Upon treatment with alpha galactosyl-C18-ceramide (αGal-C18-Cer), IRF8-deficient mice showed defective Treg cell recruitment in the liver. Eliciting Th1 immune response by anti-CD40 antibody injection in mice induced IRF8 expression in Treg cells. The expression of IRF8 was induced by Foxp3 in Treg cells. IRF8 had no effect on T-bet expression in Treg and vice versa. Thus, our results strongly suggest that IRF8 controls Th1 immune response in Treg cells independent of T-bet.
Keywords: CXCR3, IRF8, regulatory T cell, Th1-like
Introduction
Regulatory T (Treg) cells play a critical role in maintaining immune homeostasis, controlling autoimmune diseases, and inhibiting excessive immune responses against microbes or allergens.1,2 Differentiation and function of Treg are regulated by a master regulator, Foxp3. Mutation of Foxp3 in both human and mouse causes lymphoproliferation and massive multiorgan autoimmune inflammation, demonstrating the importance of Foxp3 in Treg cell function.1,2 Treg cells can be developed in the thymus as a selection process for cells that express a self-reactive T-cell antigen receptor (TCR) (called thymus-derived Treg, or tTreg), can be differentiated from naïve CD4 T cells in the periphery by antigen stimulation (called peripheral Treg or pTreg), or can be induced in vitro by TGF-β and interleukin (IL)-2 (called induced Treg or iTreg).1,2
Recent studies have suggested that Treg cells are composed of heterogeneous populations with different functional properties.1,3,4 T-bet-expressing Treg cells have been shown to accumulate at the Th1 inflammatory sites.5 These cells express CXCR3, induced by anti-CD40 antibody injection, which prompts the Th1 immune response,5,6 and they control the Th1 immune responses induced by Mycobacterium tuberculosis infection.5 Likewise, IRF4 expressed in Treg cells mediates control of Th2 type immune responses.7 Treg-specific deletion of IRF4 causes lymphoproliferative diseases associated with a selective increase of IL-4- and IL-5-producing CD4 T cells.7 Moreover, STAT3 in Treg controls Th17 type immune responses.8 Treg-specific deletion of STAT3 causes spontaneous fatal intestinal inflammation and excessive IL-17 production.8 In addition, Bcl-6-expressing Treg cells control follicular helper T cell (Tfh)-mediated immune function.9,10 These results suggest that Treg cells use different factors to control a variety of immune responses. Studying the specific function of Treg cells is important for understanding Treg cell-mediated immune regulation and for developing Treg cell-mediated immune therapy.
Although we are beginning to understand several transcription factors in the regulation of different Treg cell functions, we are still far from having a complete understanding of the players and mechanisms involved in heterogeneous Treg cell function. Given the previous findings that Treg cells utilize subset-specific factors to control corresponding subset-specific immune response, we hypothesized that Th1-specific factors control Th1-like Treg functions. Therefore, we searched for transcription factors that are highly expressed in both Treg and Th1 cells using meta-analysis. We discovered interferon regulatory factor 8 (IRF8, also called ICSBP (interferon consensus sequence binding protein)) as a candidate transcription factor, and we examined its role in Th1-like Treg cell function. IRF8-deficient Treg cells expressed aberrantly the Il4 and Il17 genes and reduced the expression of CXCR3. Anti-CD40 treatment, which induces a Th1-polarized immune response, elicited IRF8 expression in Treg cells. IRF8 was induced by Foxp3, which binds to IRF8 promoter. However, IRF8 did not induce T-bet, nor was it induced by T-bet. Our results strongly suggest that Foxp3-induced IRF8 mediates Th1-like Treg cell function independent of T-bet.
Materials and methods
Mice
IRF8-deficient mice were described previously.11 IRF8 fl/fl mice were purchased from Jackson laboratory, and C57BL/6 mice from 5∼8 weeks were purchased from Samtako Inc. (Osan, Korea). Experiments with live mice were approved by the Sogang University Institutional Animal Care and Use Committee.
Antibodies
The following antibodies were purchased from BioLegend, Inc. (San Diego, USA): anti-CD3ε (145-2C11, Cat. No. 100331), anti-CD28 (37.51, Cat. No. 102112), anti-IFN-γ (XMG1.2, Cat. No. 505827), anti-IL-4 (11B11, Cat. No. 504115), anti-CD8α (53-6.7, Cat. No. 100735), anti-I-A/I-E (M5/114.15.2, Cat. No. 107610), anti-NK-1.1 (PK136, Cat. No. 108712), anti-CD25 (PC61, Cat. No. 102031), anti-TCRγδ (UC7-13D5, Cat. No. 107510), and anti-CD62L (MEL-14, Cat. No. 104404). Secondary antibodies were purchased from Qiagen N.V. (Hilden, Germany); BioMag goat anti-rat IgG (Cat. No. 310107) and goat anti-mouse IgG (Cat. No. 310007). Anti-IRF8 antibody (sc-6058) was purchased from Santa Cruz (Dallas, USA).
In vitro CD4 T-cell differentiation
Mice were killed and spleens were harvested. Naïve CD4 T cells were isolated from spleens using modified methods from MACS naïve T cell CD4+CD62L+ T Cell Isolation kit II (Miltenyi Biotec, Bergisch Gladbach, Germany, Cat. No. 130-093-227). Briefly, spleens were minced and red blood cells were removed by ACK lysing buffer (Life Technologies, Waltham, USA, Cat. No. A10492-01) treatment. Remaining cells were treated with anti-CD8α, I-A/I-E, NK-1.1, CD25, and TCRγδ antibodies followed by BioMag goat anti-rat IgG and goat anti-mouse IgG antibodies for negative selection. Antibody-bound cells were magnetically separated. Enriched CD4+ T cells were then treated with anti-CD62L-biotin attached antibody followed by anti-biotin microbeads antibody. Cells were passed through an LS column for selection, collected, and resuspended in RPMI1640 (Cat. No. 22400-089) culture media supplemented with 5% FBS, 2-mercaptoethanol (Cat. No. 21985-023), MEM amino acids solution (Cat. No. 11130-051), non-essential MEM amino acids solution (Cat. No. 11140-050), and penicillin–streptomycin solution (Cat. No. 15140-122), all purchased from Life Technologies. Cells were activated in an anti-CD3ε antibody-bound plate with soluble anti-CD28 antibody in common. For differentiation of CD4 T cells, the following cytokines and antibodies were supplemented: 1 ng mL−1 mouse recombinant IL-2 (eBioscience, Santa Clara, USA, Cat. No. 14-8021), 3.5 ng mL−1 mouse recombinant IL-12 p70 (eBioscience, Cat. No. 14-8121), and 2 μg mL−1 anti-IL-4 antibody for Th1; 1 ng mL−1 IL-2, 5 ng mL−1 mouse recombinant IL-4 (eBioscience, Cat. No. 14-8041), and 2 μg mL−1 anti-IFN-γ antibody for Th2; 2 ng mL−1 human recombinant TGF-β1 (eBioscience, Cat. No. 14-8348), 50 ng mL−1 mouse recombinant IL-6 (eBioscience, Cat. No. 14-8061), 10 ng mL−1 mouse recombinant IL-1β (eBioscience, Cat. No. 14-8012), 1 ng mL−1 mouse recombinant TNF-α (eBioscience, Cat. No. 14-8321), anti-IFN-γ and anti-IL-4 antibodies for Th17;1 ng mL−1 IL-2, 5 ng mL−1 TGF-β1, and anti-IFN-γ and anti-IL-4 antibodies for Treg cells. Cells were stimulated for three to four days and used for further experiments.
Microarray analysis
Cells were stimulated as described above and harvested. Then, total RNA was isolated from each subset by Tri-reagent (Molecular Research Center, Inc., Cat. No. TR 118) using protocols provided by the manufacturer. RNA was analyzed in eBiogen Inc. (Seoul, Korea) using an Agilent mouse gene expression microarray (44K).
Retroviral transduction
Packaging cells were transfected with MIEG3 and pCL-eco vectors. The viral supernatant was harvested 48 hours after transfection and filtered through a 0.4 μm filter. Naïve CD4 T cells were activated for 24 hours and transduced by changing stimulation media with polybrene-added viral supernatant. Cells were centrifuged at 2800 rpm for 90 minutes at room temperature, and media were changed back to a stimulation cocktail for further stimulation. Cells were analyzed after 48 hours.
Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated by Tri-reagent (Molecular Research Center, Cincinnati, USA) and reverse-transcribed by Topscript reverse transcriptase (Enzynomics, Daejeon, Korea) using protocols provided by the manufacturer. Quantitative PCR was performed using HiFast Probe Lo-ROX (GenePole, Gwangmyeong, Korea) for a dual-labeled probe, or KAPA SYBR FAST qPCR kits (Kapa Biosystems, Wilmington, USA) for SYBR qPCR primers with protocols provided by the manufacturer.
Intracellular staining
For cytokine staining, cells were restimulated with PMA and ionomycin for 4 hours, and then stained using the Fixation and Permeabilization Solution kit with GolgiStop (BD Biosciences Franklin Lakes, USA). For transcription factor staining, cells were stained with anti-Foxp3 antibody (eBioscience) using the Transcription Factor Staining Buffer Set (eBioscience). Occasionally, the FOXP3 Fix/Perm Buffer Set (BioLegend) was used instead for GFP-expressing cells. For IRF8 intracellular staining, cells were treated with anti-IRF8 antibody followed by anti-goat IgG Alexa Fluor 488 antibody (Invitrogen, Waltham, USA).
Hepatitis induction
The experiment was done as previously described.12 Briefly, 10 μg of alpha galactosyl-C18-ceramide (αGal-C18-Cer) (Toronto Research Chemicals Inc., Toronto, Canada) dissolved in 100 μl phosphate-buffered saline (PBS) was given intraperitoneally to each mouse. Twenty-four hours later, the liver was isolated and minced for Percoll gradient centrifuge, and mononuclear cells were harvested. Isolated cells were stained for CD4, CXCR3, and Foxp3 for flow cytometry.
Administration of anti-CD40 antibody
The experiment was done as previously described.5 Briefly, anti-CD40 antibody (eBioscience) was injected intraperitoneally, 25 μg per mouse on days 0, 2, and 4. Isotype-matching IgG was used as a negative control. On day 6, mice were killed and splenocytes were stained for CD4, Foxp3, and IRF8 for flow cytometry.
Cytokine suppression assay
Purified CD45.1+ naïve CD4+ T cells were cultured under Th1-polarizing conditions (10 μg mL−1 of plate-bound anti-CD3, 2 μg mL−1 anti-CD28, 1 ng mL−1 IL-2, 3.5 ng mL−1 IL-12 and 5 μg mL−1 anti-IL-4 antibodies) for three days. The activated cells were co-cultured with sorted WT or IRF8-deficient iTreg cells at the ratios of 10:0, 9:1, 7:3, and 5:5 under Th1-polarizing conditions for two days. CD45.1+ cells were sorted using a FACSAria III (BD Biosciences) and subjected to total RNA isolation. Total RNA isolation and qRT-PCR were performed as described above.
Delayed-type hypersensitivity (DTH) assay
DTH assay was performed as described previously.13 Briefly, mice were subcutaneously injected with a mixture of 100 μg grade-V ovalbumin (OVA) (Sigma-Aldrich, St. Louis, USA) in PBS and complete Freund's adjuvant (Sigma) at the base of the tails. Four days later, 600 μg of heat-aggregated grade-II OVA (Sigma) in PBS was intradermally injected into the right-hind footpad of mice and PBS in the left as a control. Forty-eight hours after the challenge, swelling of footpad was measured with a Vernier caliper. Increased thickness was calculated by subtracting the thickness of the unimmunized left foot from that of the immunized right foot.
Statistical analysis
Values are shown as mean ± standard deviation. Statistical significance of differences between mean values was determined by Student's t-test. Results were considered significant when the p-value was < 0.05.
Results
IRF8 is selectively expressed in Treg and Th1 cells
In our search for transcription factors that are expressed in a Treg- and Th1-specific manner, we examined a previously published database (meta-analysis) on genome-wide gene expression profiles and histone 3 lysine 4 tri-methylation (H3K4-me3) modification from naïve, Th1, Th2, Th17, and Treg cells.14 We found that gene expression and H3K4-me3 of a transcription factor IRF8 was highly increased in iTreg cells and substantially increased in Th1 cells but not in naïve, Th2, and Th17 cells.14 Th1- and Treg-specific expression was confirmed by quantitative RT-PCR (qRT-PCR) (Figure 1a) and by western blot analysis (Figure 1b). We also found that IRF8 was highly expressed in CD4+CD25+ Treg cells but not in CD4+CD25− cells (Figure 1c).
Figure 1.

Treg- and Th1-specific expression of Irf8. (a) Splenic naïve CD4 T cells from C57BL/6 mice were stimulated with anti-CD3 and anti-CD28 and polarized in vitro under Th1, Th2, Th17, and Treg conditions for four days. RNA was isolated from the cells and subjected to quantitative RT-PCR. (b) Experiments were done as in (a). Cells were harvested at days 1, 2, 3, and 4, and western blotting was performed with an anti-IRF8 or anti-β-actin antibody (c). Splenic CD4 T cells were sorted based on the surface expression of CD4 and CD25. RNA was isolated from the cells and subjected to quantitative RT-PCR. The y-axis shows the relative expression amount of each gene to the internal control Hprt (encoding HPRT).The error bar shows standard deviation (n = 3). Statistical difference was analyzed by Student's t-test. *p < 0.05. Data are representative of three independent experiments with similar results.
Deficiency of IRF8 causes aberrant up-regulation of Il4 and Il17 in Treg
To gain insight into the role of IRF8 in Treg cell differentiation and function, we measured the expression of subset-specific cytokines and transcription factors in in vitro differentiated Treg cells from wild-type or IRF8-deficient mice. Surprisingly, the expression of Il4, Il5, Il13, Il17a, and Il21 genes was aberrantly increased in iTreg cells from IRF8-deficient mice compared to their counterparts of wild-type mice (Figure 2). The expression of Ifng, Foxp3, Tbx21 (encoding T-bet), Gata3, and Rorc (encoding Rorγt) genes was no different between cells of wild-type and IRF8-deficient mice (Figure 2). These results suggest that IRF8 represses Th2 and Th17 gene expression in Treg cells, and that this suppressive effect occurs independently of T-bet, GATA3, and Rorγt.
Figure 2.

Aberrant expression of Il4 and Il17 in IRF8-deficient cells. Splenic naïve CD4 T cells from wild-type or IRF8-deficient mice were differentiated in vitro into Treg for four days. RNA was isolated from the cells and subjected to quantitative RT-PCR. The amount of each gene was normalized to the internal control Hprt (encoding HPRT). The y-axis shows the relative amount between wild-type and IRF8 knockout (KO) (the value of wild type was arbitrarily set 1). The error bar shows standard deviation (n = 3). Statistical difference was analyzed by Student's t-test. *p < 0.05; **p < 0.01; ***p < 0.001. Data are representative of three to five independent experiments with similar results.
IRF8-deficient Treg cells have a defect in CXCR3 expression
To further analyze the gene expression pattern in IRF8-deficient Treg cells, we performed a microarray analysis with RNA isolated from wild-type and IRF8-deficient CD4+CD25+ Treg cells (Figure 3a). Among the differentially expressed genes, we found that a chemokine receptor CXCR3 is selectively down-regulated in IRF8-deficient CD4+CD25+ Treg cells compared to their wild-type counterpart (Figure 3a). Defective expression of CXCR3 in IRF8-deficient CD4+CD25+ Treg cells was confirmed by quantitative RT-PCR (Figure 3b). Next, we examined whether the reduced CXCR3 expression IRF8-deficient Treg cells was a T-cell intrinsic effect using CD4-specific IRF8-deficient (IRF8 fl/fl × CD4-Cre) mice. The frequency of CXCR3+ Treg cells in the spleen from CD4-specific IRF8 KO mice was reduced compared to that from control mice (Figure 3c), suggesting that the effect of IRF8 on CXCR3 expression in Treg cells is a T-cell intrinsic.
Figure 3.

Defective expression of CXCR3 in IRF8-deficient Treg cells. (a) Scatterplot of the microarrary analysis. Cxcr3 gene was down-regulated 4.6-fold in IRF8-deficient CD4+CD25+ cells compared that in wild-type counterpart. (b) Splenic CD4+CD25+ cells were isolated from wild-type or IRF8-deficient mice. RNA was isolated from the cells and subjected to quantitative RT-PCR. The error bar shows standard deviation (n = 3). Statistical difference was analyzed by Student's t-test. **p < 0.01. (c) Splenic cells from control (IRF8 fl/fl) or CD4-specifgic IRF8-deficient (IRF8 fl/fl × CD4-Cre) mice were stained with anti-CD4, anti-Foxp3, and anti-CXCR3 antibodies, and analyzed by fluorescence-activated cell sorting (FACS). Cells were gated on CD4.
To get an insight of the molecular mechanism shows IRF8 controls CXCR3 expression in Treg cells, we examined whether IRF8 binds to the Cxcr3 locus by chromatin immunoprecipitation. IRF8 did not bind to the Cxcr3 locus in Treg cells (data not shown). Therefore, the effect of IRF8 on CXCR3 expression in Treg cells seems to be indirect.
CD4-specific IRF8-deficient mice have a defect in the recruitment of CXCR3+ Treg cells to an inflammation site in an αGal-C18-Cer-induced hepatitis model
CXCR3, which was originally shown to be involved in Th1 cell recruitment to sites of type I inflammation,15,16,17 has also been shown to be important in the recruitment of Treg cells to an inflammation site.5 Next, we examined whether CD4-specific IRF8-deficient mice have a defect in Treg cell recruitment to an inflammation site. For this purpose, we used an αGal-C18-Cer-induced hepatitis model.12 In this model, αGal-C18-Cer-induced natural killer T cells mediate hepatitis and recruitment of Treg cells to the inflammation site.12 We induced hepatitis in wild-type and CD4-specific IRF8-deficient mice by injecting αGal-C18-Cer intraperitoneally. After 24 hours, Treg cells in the liver were isolated and analyzed by flow cytometry. Treg cell recruitment was found to be reduced in CD4-specific IRF8-deficient mice (Figure 4), suggesting that IRF8 is important in the recruitment of CXCR3+ Treg cells to the inflammation site.
Figure 4.

Defective recruitment of CXCR3+ Treg cells to an inflammation site. (a) Wild-type or CD4-specific IRF8-deficient mice were injected with αGal-C18-Cer or vehicle control intraperitoneally. After one day, CD4 T cells were isolated from the liver and the expression of Foxp3 and CXCR3 was analyzed by FACS. Data are representative of three independent experiments with similar results. (b) Ratio of Foxp3+CXCR3+ cells in CD4 T cells from the liver. Three experiments of (a) were combined. The error bar shows standard deviation (n = 3∼8). Statistical difference was analyzed by Student's t-test. *p < 0.05.
IRF8 increases in Treg cells during type I inflammation
A previous study by Koch et al. has shown that T-bet-deficient Treg cells show defective expression of CXCR3, and that T-bet controls Treg function during type I inflammation.5 Reduced expression of CXCR3 in IRF8-deficient Treg cells is reminiscent of T-bet-deficient Treg cells, and suggests that IRF8 may play a role in the regulation of type I inflammation. To test this possibility, we injected wild-type or IRF8-deficient mice with the anti-CD40 antibody, which has been shown to induce a strong Th1 response in mice.5,6 Injection of the anti-CD40 antibody induced an increase in IRF8-expressing Treg cells in the spleen (Figure 5a). We also isolated splenic CD4+CD25− or CD4+CD25+ cells from mice injected with the anti-CD40 antibody or control rat IgG, and measured expression of Irf8, Tbx21, and Cxcr3 by quantitative RT-PCR. Expression of Irf8 was substantially increased in cells from anti-CD40-injected mice compared to those from control IgG-injected mice (Figure 5b). Consistent with the previous report, Tbx21 and Cxcr3 were also increased in cells from anti-CD40-injected mice compared to those from control IgG-injected mice5 (Figure 5b). These results suggest that IRF8 is induced in Treg cells during Th1 immune response.
Figure 5.

IRF8 is induced in Treg upon Th1 immune response. (a) Anti-CD40 antibody was injected into C57BL/6 mice intraperitoneally on days 0, 2, and 4. On day 6, splenic cells were stained with anti-CD4, anti-Foxp3, and anti-CXCR3 antibodies, and analyzed by FACS. Cells were gated on CD4. Data are representative of six independent experiments with similar results. (b) Anti-CD40 antibody was injected as described in (a). On day 6, splenic CD4+CD25+ or CD4+CD25− cells were isolated by FACS. RNA was isolated from the cells and the expression of genes was measured by quantitative RT-PCR. The y-axis shows the relative expression amount of each gene to the internal control Hprt (encoding HPRT). The error bar shows standard deviation (n = 3). Statistical difference was analyzed by Student's t-test. *p < 0.05; ***p < 0.001.
IRF8-expressing Treg cells control cell-mediated immune responses
To examine whether IRF8 plays a role in controlling Th1 response, we first performed a mixed lymphocyte reaction. In vitro-differentiated CD45.1+ Th1 cells were co-cultured with WT- or IRF8-deficient CD45.2+ iTreg cells at the ratios of 10:0, 9:1, 7:3, and 5:5 under Th1-polarizing conditions for two days. CD45.1+ cells were sorted, and mRNA expression of Ifng was examined by qRT-PCR. IRF8-deficient iTreg cells were less efficient than WT iTreg cells in suppressing Ifng expression in Th1 cells (Figure 6), suggesting that IRF8 in Treg cells plays an important role in suppressing Th1 response.
Figure 6.
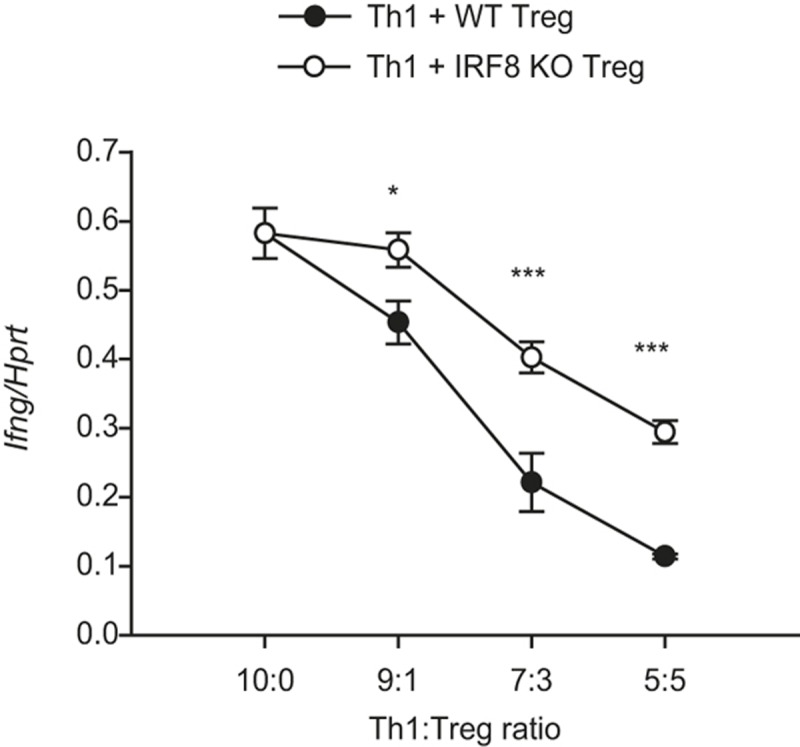
IRF8 is required for Treg cell-mediated suppression of Th1 cells. In vitro-differentiated CD45.1+ Th1 cells were mixed with WT- or IRF8-deficient CD45.2+ iTreg cells at the ratios of 10:0, 9:1, 7:3, and 5:5 and cultured under Th1-polarizing conditions for two days. CD45.1+ cells were sorted, and expression of Ifng was examined by qRT-PCR. The error bar shows standard deviation (n = 3). Statistical difference was analyzed by Student's t-test. *p < 0.05, ***p < 0.001.
We next performed a DTH assay, a well-known in vivo cell-mediated immune response model. Control and CD4-specific IRF8-deficient mice were immunized and challenged on footpad with OVA as an antigen, and then the thickness of swollen footpads was measured after two days. Footpad swelling was exacerbated in CD4-specific IRF8-deficient mice compared to that in control mice (Figure 7a). We also examined Treg cell populations in the draining lymph nodes from the mice. Frequencies of Foxp3+ cells among CD4 T cells and that of CXCR3+ cells among Treg cells were reduced in the draining lymph nodes of the CD4-specific IRF8-deficient mice compared to those of control mice (Figure 7b and 7c). This result suggests that IRF8 plays an important role in controlling cell-mediated immune responses in vivo and in the recruitment of Treg cells into an inflammation site.
Figure 7.

IRF8-expressing Treg cells play a role in controlling cell-mediated immune response. DTH was induced by injecting OVA into control or CD4-specific IRF8-deficient mice as described in the Materials and Methods section. (a) Footpad swelling of the challenged mice was measured, and increased thickness was calculated by subtracting thickness of immunized foot from that of unimmunized foot. ctrl: control mice (IRF8 fl/fl mice), cKO: conditional IRF8 KO mice (IRF8 fl/fl × CD4-Cre mice). (b–c). Mice from (a) were killed, and cells from the right side of inguinal lymph nodes were isolated, stained with anti-CD4, anti-CXCR3, and anti-Foxp3 antibodies, and analyzed by flow cytometry. Cells were gated on CD4. The error bar shows S.E.M. (n = 3). Statistical difference between groups was analyzed by Student's t-test. *p < 0.05; **p < 0.01; ***p < 0.001.
Expression of IRF8 is controlled by Foxp3
Since IRF8 is selectively up-regulated in Treg cells, we examined whether its expression is controlled by Foxp3. We transduced a Foxp3-retroviral vector into naïve CD4 T cells and differentiated them under Th0 conditions in the presence of IL-2. We sorted the transduced cells and measured the expression of Irf8 by quantitative RT-PCR. The expression of Irf8 was found to be 3.5 times higher in Foxp3-transduced cells compared to that in control vector-transduced cells (Figure 8a). We also transfected control or Foxp3 expression vector into an EL4 mouse thymoma cell line, stimulated with anti-CD3 and anti-CD28, and measured the expression of Irf8 by quantitative RT-PCR. Foxp3 induced Irf8 expression in unstimulated cells and a greater extent in stimulated cells (Figure 8b), suggesting that the expression of Irf8 is dependent on both Foxp3 and TCR stimulation.
Figure 8.
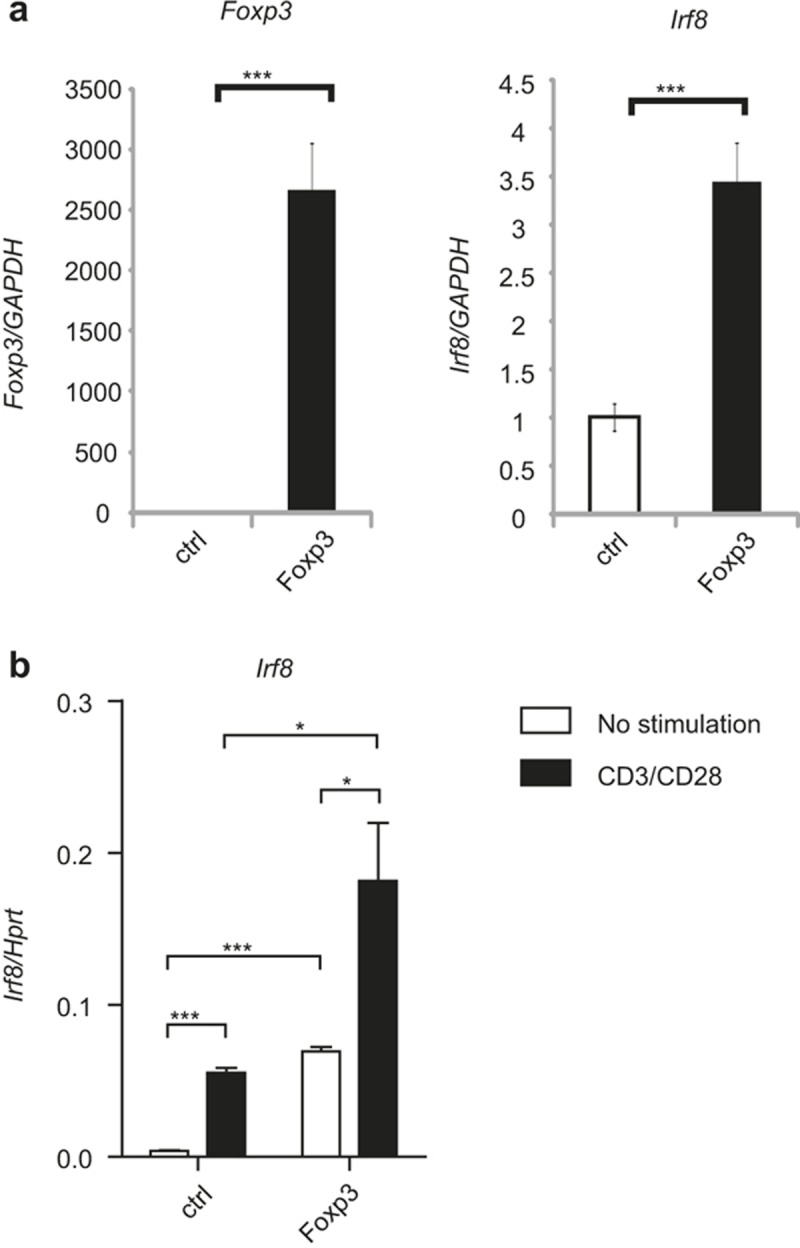
Foxp3 controls IRF8 expression. (a) Naïve CD4 T cells isolated from C57BL/6 mice were transduced with either control (MIEG)- or Foxp3-retroviral vector and stimulated under Th0 conditions. Transduced cells were sorted based on GFP expression, and RNA was isolated from the cells. Expression of Foxp3 and Irf8 was measured by quantitative RT-PCR. The error bar shows standard deviation (n = 3). Statistical difference was analyzed by Student's t-test. ***p < 0.001. (b) EL4 cells were transfected with control (CMV) or Foxp3-expression vector and stimulated with anti-CD3 and anti-CD28. RNA was isolated from the cells and the expression of Irf8 was analyzed by quantitative RT-PCR. The error bar shows standard deviation (n = 3). Statistical difference was analyzed by Student's t-test. *p < 0.05, **p < 0.01. Data are representative of four independent experiments with similar results.
Next, we examined whether IRF8 controls Foxp3 expression. We performed the same retroviral transduction experiment described above with an IRF8-retroviral vector. The ratio of Foxp3+ cells among GFP positive cells in control-retroviral vector (RV) transduced cells was similar to that in IRF8-RV transduced cells (63% vs. 68%), indicating that overexpression of IRF8 has no effect on Foxp3 expression (Supplementary Figure 1). This result suggests that IRF8 is under the control of Foxp3 but not vice versa.
IRF8 and T-bet in Treg cells were independently regulated
Because IRF8-deficient Treg cells show a similar phenotype to T-bet-deficient Treg cells,5 it is plausible that these two transcription factors, IRF8 and T-bet, may influence each other's expression. To test this possibility, we examined whether T-bet induces IRF8, or whether IRF8 induces T-bet in Treg cells, using a retroviral transduction experiment. Transduction of T-bet into naïve CD4 T cells and subsequent Treg cell differentiation was found to have no effect on the expression of the Irf8 gene. Likewise, transduction of IRF8 had no effect on the expression of the Tbx21 gene (Supplementary Figure 2). These results suggest that, even though IRF8 and T-bet exert a similar function in Treg cells, their expression is independently regulated.
Discussion
In the present study, we searched for transcription factors that are selectively expressed in Treg and Th1 cells and discovered IRF8. The IRF8-deficient Treg cells aberrantly expressed the Il4 and Il17 genes, and they exhibited reduced expression of CXCR3, which is a chemokine receptor needed for a Th1 immune response. Anti-CD40 treatment, which induces Th1-polarized immune response, elicited IRF8 expression in Treg cells. IRF8 was induced by Foxp3, which binds to the Irf8 promoter. IRF8 was not found to induce T-bet, nor was it induced by T-bet. These results strongly suggest that Foxp3-induced IRF8 mediates Th1-like Treg cell function independent of T-bet.
The primary finding in the present study is that IRF8 mediates Th1-like Treg function. Our study shows that IRF8-deficient Treg cells almost completely lack CXCR3 expression, and that anti-CD40 injection induces IRF8 expression in Treg cells, supporting the important role of IRF8 in the Th1-like Treg cell property. Previous studies suggested that Treg cells are heterogeneous and regulate many specific immune responses. T-bet-expressing Treg cells have been shown to play an important role in regulating the Th1 immune response;5 IRF4 in Treg mediates regulation of the Th2 immune response,7 STAT3 in Treg mediates the Th17 immune response,8 and Bcl6 expressed in Treg cells regulates the Tfh immune response.9,10 Our present study adds another transcription factor, IRF8, in this category. T-bet and IRF8 mediate similar functions in Treg cells; however, it seems that their expression is independently regulated. The T-bet-independent role of IRF8 has also been reported in the differentiation of naïve CD8 T cells into effector cytotoxic cells.18 Whether IRF8 and T-bet play redundant or non-redundant roles in Treg cells awaits further study. Nonetheless, our study suggests that regulation of subset-specific immune response may be more complex than we currently believe, and that more transcription factors and mechanisms may exist to regulate highly diversified and complex immune responses. This notion is supported by a recent report on complex network of transcription factors cooperating with Foxp3 for optimal Treg cell function.19
IRF8 is a member of the IRF family of transcription factors. It controls many aspects of hematopoietic cell differentiation.20,21,22 IRF8 is required for differentiation of macrophages,11,23 CD8α + DCs,24,25 pDCs,26 B cells,27 eosinophils,28 and effector CD8 T cells.18 It has also been shown to be important in the differentiation of Th1 cells by stimulating macrophages and DCs to produce IL-12.29,30 Our study adds another cell type that is regulated by IRF8, i.e., Treg cells. Thus, it seems that IRF8 regulates differentiation or function of many different cell types in the hematopoietic system, and that its functions are cell type-specific.
Recently, Ouyang et al. have shown that IRF8 inhibits Th17 cell differentiation.31 In our study, IRF8 suppressed IL-17 expression in Treg as well, suggesting that IRF8 plays a similar role in both effector and Treg cells. Ouyang et al. have also shown that the inhibition of Th17 differentiation by IRF8 is not due to reciprocal activation of Treg cells, based on the results that there is no difference in the number and ratio of Treg cells between wild-type and IRF8-deficient mice. There is likewise no difference in the capacity for cure of inflammatory bowel disease by transferring naïve CD4 T cells with wild-type or IRF8-deficient Treg cells into lymphopenic mice.31 We obtained similar results to these authors. Thus, it seems that IRF8 is not responsible for the general function of Treg cells. Our results suggest that IRF8 specifically controls Th1-like Treg cell function but not the general function of Treg cells.
Migration of Treg cells to tissues or inflammatory sites is essential for surveillance of immune responses and inflammation in tissues.3,32 The migration of Treg cells requires interactions of cell adhesion molecules and chemokine–chemokine receptor signaling3,32 A variety of different chemokine-chemokine receptors have been shown to play roles in the homing of Treg cells to different sites with various functional properties.3,32 CXCR3,which binds to Th1-associated chemokines (CXCL9, CXCL10, and CXCL11), originally had been shown to impact in the Th1 immune response,15,16,17 and subsequently it was found to play a role in homing of Th1-like Treg cells to inflammation sites.5 The homing of CXCR3+ Treg cells to tissues has been shown to play a critical role in regulating Th1 immune response induced by infection of M. tuberculosis5 or Toxoplasma gondii.33 In addition, it exerts a protective effect in graft-versus-host-disease (GVHD)34 and has an adversal effect in human ovarian carcinomas.35 Since our study shows that CXCR3 expression is influenced by IRF8 in Treg cells, the controlling of specific IRF8 expression and function or using selected CXCR3+ Treg cell populations in immune therapy may have great potential for regulating the abovementioned immune functions.
In conclusion, we have shown that IRF8 plays an important role in Th1-like Treg cell function. This study provides an insight that controlling Treg cell function by IRF8 has potential therapeutic value in regulating infection, graft rejection, and tumor progression. The detailed mechanism of IRF8 regulation of Treg cell function awaits further study.
Acknowledgments
We thank Drs. Keiko Ozato (National Institutes of Health) and Tomohiko Tamura (Yokohama City University) for providing and transferring IRF8-deficient mice, respectively. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (NRF-2010-0017447 and NRF-2014R1A2A1A11052545).
Footnotes
Supplementary Information of this article can be found on Cellular & Molecular Immunology website: http://www.nature.com/cmi.
Supplementary Information
References
- Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30: 531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008; 133: 775–787. [DOI] [PubMed] [Google Scholar]
- Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol 2011; 11: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BM, Verma ND, Tran GT, Hodgkinson SJ. Distinct regulatory CD4+T cell subsets; differences between naive and antigen specific T regulatory cells. Curr Opin Immunol 2011; 23: 641–647. [DOI] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 2009; 10: 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlin WG, von der Weid T, Cottrez F, Ferrick DA, Coffman RL, Howard MC. The induction of a protective response in Leishmania major-infected BALB/c mice with anti-CD40 mAb. Eur J Immunol 1998; 28: 525–531. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 2009; 458: 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 2009; 326: 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med 2011; 17: 983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med 2011; 17: 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtschke T, Lohler J, Kanno Y, Fehr T, Giese N, Rosenbauer F et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell 1996; 87: 307–317. [DOI] [PubMed] [Google Scholar]
- Santodomingo-Garzon T, Han J, Le T, Yang Y, Swain MG. Natural killer T cells regulate the homing of chemokine CXC receptor 3-positive regulatory T cells to the liver in mice. Hepatology 2009; 49: 1267–1276. [DOI] [PubMed] [Google Scholar]
- Jacysyn JF, Abrahamsohn IA, Macedo MS. Modulation of delayed-type hypersensitivity during the time course of immune response to a protein antigen. Immunology 2001; 102: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 2009; 30: 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med 1998; 187: 2009–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber JM. A macrophage mRNA selectively induced by gamma-interferon encodes a member of the platelet factor 4 family of cytokines. Proc Natl Acad Sci USA 1990; 87: 5238–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster AD, Unkeless JC, Ravetch JV. Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature 1985; 315: 672–676. [DOI] [PubMed] [Google Scholar]
- Miyagawa F, Zhang H, Terunuma A, Ozato K, Tagaya Y, Katz SI. Interferon regulatory factor 8 integrates T-cell receptor and cytokine-signaling pathways and drives effector differentiation of CD8 T cells. Proc Natl Acad Sci USA 2012; 109: 12123–12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS et al. A multiply redundant genetic switch ‘locks in' the transcriptional signature of regulatory T cells. Nat Immunol 2012; 13: 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 2008; 26: 535–584. [DOI] [PubMed] [Google Scholar]
- Savitsky D, Tamura T, Yanai H, Taniguchi T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol Immunother 2010; 59: 489–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistini A. Interferon regulatory factors in hematopoietic cell differentiation and immune regulation. J Interferon Cytokine Res 2009; 29: 765–780. [DOI] [PubMed] [Google Scholar]
- Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, Ozato K. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity 2000; 13: 155–165. [DOI] [PubMed] [Google Scholar]
- Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC, 3rd et al. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med 2002; 196: 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura H, Tamura T, Gongora C, Aliberti J, Reis e Sousa C, Sher A et al. ICSBP/IRF-8 retrovirus transduction rescues dendritic cell development in vitro. Blood 2003; 101: 961–969. [DOI] [PubMed] [Google Scholar]
- Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O'Shea JJ et al. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J Immunol 2005; 174: 2573–2581. [DOI] [PubMed] [Google Scholar]
- Lu R, Medina KL, Lancki DW, Singh H. IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev 2003; 17: 1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanovic M, Terszowski G, Struck D, Liesenfeld O, Carstanjen D. IFN consensus sequence binding protein (Icsbp) is critical for eosinophil development. J Immunol 2008; 181: 5045–5053. [DOI] [PubMed] [Google Scholar]
- Scharton-Kersten T, Contursi C, Masumi A, Sher A, Ozato K. Interferon consensus sequence binding protein-deficient mice display impaired resistance to intracellular infection due to a primary defect in interleukin 12 p40 induction. J Exp Med 1997; 186: 1523–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese NA, Gabriele L, Doherty TM, Klinman DM, Tadesse-Heath L, Contursi C et al. Interferon (IFN) consensus sequence-binding protein, a transcription factor of the IFN regulatory factor family, regulates immune responses in vivo through control of interleukin 12 expression. J Exp Med 1997; 186: 1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Zhang R, Yang J, Li Q, Qin L, Zhu C et al. Transcription factor IRF8 directs a silencing programme for TH17 cell differentiation. Nat Commun 2011; 2: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindebacke H, Stenstad H, Quiding-Jarbrink M, Waldenstrom J, Adlerberth I, Wold AE et al. Dynamic development of homing receptor expression and memory cell differentiation of infant CD4+CD25high regulatory T cells. J Immunol 2009; 183: 4360–4370. [DOI] [PubMed] [Google Scholar]
- Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG et al. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 2012; 37: 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa H, Inoue A, Kohno M, Lei J, Miyazaki T, Yoshie O et al. Therapeutic effect of CXCR3-expressing regulatory T cells on liver, lung and intestinal damages in a murine acute GVHD model. Gene Ther 2008; 15: 171–182. [DOI] [PubMed] [Google Scholar]
- Redjimi N, Raffin C, Raimbaud I, Pignon P, Matsuzaki J, Odunsi K et al. CXCR3+ T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res 2012; 72: 4351–4360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.