

Abstract
Gastrodin (GAS), which is extracted from the Chinese herbal medicine Gastrodia elata Blume, has long been used to improve stroke, epilepsy, dizziness and dementia. However, the effects and underlying mechanisms of GAS on subacute phase cerebral ischemia-reperfusion (I/R) injury remain unknown. The aim of the present study was to investigate the effects and mechanisms of GAS on cerebral I/R injury in rats. The rats were pretreated with GAS by gavage for 7 days followed by I/R surgery, and were then treated with GAS for 7 days after I/R surgery. Neurological deficits were assessed on days 1, 3 and 7 post-cerebral I/R injury. 2,3,5-Triphenyltetrazolium chloride staining was using to measure the infarct volume; morphological alterations were observed by hematoxylin and eosin staining under an optical microscope; apoptosis in the hippocampus and cortex was observed by terminal deoxynucleotidyl transferase dUTP nick end labeling staining; and the level of mRNA and protein expression was tested by reverse transcription-quantitative polymerase chain reation and western blot analysis, respectively. GAS markedly attenuated I/R-induced disability and histological damage, alleviated neuronal apoptosis, and reduced the mRNA and protein expression levels of inflammatory and proapoptotic factors, including interleukin-1β, cyclooxygenase-2, inducible nitric oxide synthase and cleaved caspase-3. These findings suggested that GAS may ameliorate subacute phase cerebral I/R injury by inhibiting inflammation and apoptosis in rats; therefore, GAS may be considered a potential candidate for the treatment of cerebral ischemia.
Keywords: gastrodin, inflammation, apoptosis, neuroprotection, rat
Introduction
Stroke is one of the most common causes of adult disability, and is considered to be the second leading cause of mortality worldwide. The incidence of stroke has increased markedly, particularly in developing countries, over the past four decades (1). Ischemic stroke, which accounts for 70–80% of stroke cases, is a major factor that threatens human life and health (2). Cerebral ischemia, which is induced by a transient or permanent reduction of blood supply, results in disordered homeostasis, including insufficient oxygen and delivery of glucose (3). Cerebral ischemia-reperfusion (I/R) induces serious brain injuries, and is associated with complex underlying mechanisms, including peri-infarct depolarization, oxidative and nitrative stress, excitotoxicity, inflammation and apoptosis, ultimately resulting in cell death (4), mitochondrial dysfunction, release of glutamate and proinflammatory mediators, production of reactive oxygen species (ROS), and/or lipid peroxidation (5). Due to the complex pathological processes of cerebral ischemia, current treatments have not exhibited the expected effects. Therefore, natural products extracted from herbs have received increasing interest with regards to the treatment of cerebral stroke (6).
Gastrodin (GAS; structure presented in Fig. 1) is a natural phenol compound extracted from Gastrodia elata BI (7). GAS has been suggested for the treatment of various ailments, including convulsive illness, headache, vertigo, general paralysis, epilepsy and tetanus (8). GAS is able to pass the blood-brain barrier, and rapidly metabolizes to p-hydroxybenzyl alcohol in the brain, liver and blood (9). It has previously been reported that GAS ameliorates cerebral damage following transient focal cerebral ischemia (10). The potential underlying mechanisms have been identified in in vivo models, by determination of ROS and hippocampal neuronal death assessment, and in vitro models, by assessment of excitotoxicity and neuroprotective effects (10,11). Furthermore, it has been reported that GAS decreases the volume of cerebral infarction and ameliorates cerebral injury in a rat model of I/R, probably by improving the level of amino acids in the striatum (12). In addition, GAS significantly attenuates the levels of neurotoxic proinflammatory mediators and proinflammatory cytokines by inhibiting the nuclear factor-κB signaling pathway and phosphorylation of mitogen-activated protein kinases (MAPKs) in lipopolysaccharide-stimulated microglial cells (13). GAS also ameliorates Parkinson's disease by downregulating connexin 43 (14), and improves anxiety-like behaviors by decreasing the levels of interleukin (IL)-1β and inhibiting p38 MAPK phosphorylation in the hippocampus of a rat model of post-traumatic stress disorder (15). These findings indicated that GAS may exert beneficial effects on diseases of the central nervous system. However, the effects and underlying mechanisms of GAS on subacute phase cerebral I/R injury remain unknown.
Figure 1.
Chemical structure of gastrodin.
The present study investigated the neuroprotective effects of GAS on a rat model of subacute phase focal cerebral I/R injury, and explored the possible underlying mechanisms with regards to neuroinflammation and neuronal apoptotic pathways.
Materials and methods
Chemicals
GAS (purity >98% tested using high-performance liquid chromatography) was purchased from Nanjing Zelang Medical Technology Co., Ltd. (Nanjing, China). Nimodipine (Nim) was obtained from Shandong Lukang Pharmaceutical Group Saite Co., Ltd. (Tai'an, China). 2,3,5-Triphenyltetrazolium chloride (TTC) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Animals
Male Sprague-Dawley rats (age, 9 weeks; weight, 250–280 g) were purchased from the Animal Center of the Third Military Medical University [Chongqing, China; Certificate no. SCXK (Jun) 2007-00005]. All animals were caged individually under a 12-h light/dark cycle at 22–24°C with free access to food and water. All procedures complied with the Current Guide for the Care and Use of Laboratory Animals and the study was approved by the Zunyi Medical University Animal Studies Committee (Zunyi, China). All rats were randomly divided into 6 groups: Sham, I/R, I/R+15 mg/kg GAS, I/R+ 30 mg/kg GAS, I/R+60 mg/kg GAS, and I/R+12 mg/kg Nim (n=20/group).
Focal cerebral I/R surgery
The rats were pretreated with GAS for 7 days followed by I/R surgery, and were then treated with GAS for another 7 days following I/R surgery. Nim was used as a positive control. Following pretreatment with GAS or Nim for 7 days, the rats were anesthetized with chloral hydrate (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) via intraperitoneal injection (350 mg/kg). The right common carotid artery, internal carotid artery (ICA), and external carotid artery (ECA) were then exposed through a midline incision in the neck, and a monofilament nylon suture (external diameter, 0.28 mm) with a silicone-coated tip was plugged into the ICA, 16–18 mm from the bifurcation, through the ECA stump, and was gently advanced to cause middle cerebral artery occlusion (MCAO). A total of 2 h after MCAO, the suture was removed to restore blood flow. The sham group underwent the same operation without MCAO.
Evaluation of neurological deficits
Neurological deficits of the experimental animals were graded on a scale of 0–4 on days 1, 3 and 7 post-cerebral I/R injury (16). The criteria were set as follows: 0, no neurological deficits; 1, failure to fully extend right forepaw; 2, forelimb flexion plus decreased resistance to lateral push; 3, falling to the right; 4, unable to walk spontaneously and exhibited depressed levels of consciousness. Rats in model and treated groups with a score between 2 and 3 were selected for the present study.
Measurement of infarct volume
A total of 7 days after reperfusion the rats were sacrificed following appropriate intraperitoneal anesthetization with 350 mg/kg chloral hydrate (Sinopharm Chemical Reagent Co., Ltd.). Brains were removed, immediately frozen in liquid nitrogen, and dissected into 2–3 mm slices. Subsequently, samples were placed in 0.1% TTC solution for 10 min at 37°C in the dark. The red-purple sections were assigned as noninfarcted areas, whereas white sections were assigned as infarcted areas. The percentage of infarction was calculated as follows: Percentage of infarction (%) = (weight of white sections / weight of whole brain) × 100%.
Hematoxylin and eosin (HE) staining
Rats were sacrificed (n=5/group) 7 days after reperfusion, and brains were fixed by transcardial perfusion using 0.9% normal saline and 4% paraformaldehyde. Post-fixation, the tissues were incubated in 4% paraformaldehyde for 24 h at 4°C. Following dehydration in graded xylene and ethanol, the sections were embedded in paraffin. Embedded brain tissue sections were coronally sliced into 4 µm sections, and sections including the hippocampus were analyzed. The sections were then stained with 0.5% hematoxylin for 5 min, followed by 0.5% eosin for 1 min at room temperature. The slices were observed under a light microscope (Leica Microsystems Ltd., Wetzlar, Germany). The hippocampal CA1 region was selected to check for morphological alterations and to assess cerebral injury.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
A TUNEL assay was used to evaluate apoptosis-associated DNA fragmentation using an In Situ Cell Death Detection kit (Roche Molecular Biochemicals, Mannheim, Germany). Following dehydration and rehydration in graded xylene and ethanol, tissue sections were incubated with proteinase K (20 µg/ml) and Tris-HCl (pH 7.4–8.0; 10 mM) for 15 min at room temperature. The sections were rinsed twice with phosphate-buffered saline (PBS), and were incubated with TUNEL reaction mixture for 1 h at 37°C. Subsequently, the sections were rinsed with PBS (3×5 min washes) and incubated with Converter-POD solution for 30 min at 37°C. After further rinsing with PBS (3×5 min washes), the sections were incubated with diaminobenzidine substrate for 10 min. Images of the ischemic penumbra, including the cortex and hippocampal CA1 region, were captured in each section under a light microscope. The density of stained cells was observed under high magnification (×400). Data were presented as cells per square millimeter.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Brain samples were collected from the ischemic hemisphere of rats at day 7 post-reperfusion (n=5), following treatment with various doses of GAS. The gene expression levels of IL-1β, cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) were detected in the ischemic penumbra using RT-qPCR. Briefly, the penumbra of ischemic tissue from the right cortex was homogenized. Total RNA was extracted from the ischemic penumbra using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) followed by chloroform-phenol purification, and was quantified at 280 nm. RNA quality was determined by measuring the ultraviolet absorption ratios at 280/260 nm. The RNA concentration of each sample was adjusted to 50 ng/µl. Purified total RNA was used to prepare cDNA using a RT kit (Takara Biotechnology Co., Ltd., Dalian, China) with MMLV reverse transcriptase and Oligo-(dT) primers. Subsequently, specific regions of target genes were amplified using SYBR Premix Ex Taq (Takara Biotechnology Co., Ltd.) and specific primers (Table I). The reaction volume of PCR was 15 µl containing 7.5 µl SYBR super mix, 0.5 µl of 10 µmol/µl Primers 1/2, 3 µl of 5 ng/µl cDNA and 4 µl of 0.1% DEPC water. The cycling conditions were 1 cycle of 95°C 10 min; 95°C for 10 sec, 60°C for 1 min for 40 cycles; 95°C for 1 min, 55°C for 1 min, and 55°C for 10 sec for 80 cycles. The quantification cycle values (Cq) were used to determine gene expression levels (17). Gene expression levels of glyceraldehyde 3-phosphate dehydrogenase were used as an internal control. Each group was normalized to the sham group, which was set at 100%.
Table I.
Sequence of primers.
| Gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| iNOS | AAGCACATTTGCCAATGGAGAC | TGGAGCCCAGGCCAAATAC |
| COX-2 | AACACGGACTTGCTCACTTTGTTG | AATGGAGGCCTTTGCCACTG |
| IL-1β | GCTGTGGCAGCTACCTATGTCTTG | AGGTCGTCATCATCCCACGAG |
| GAPDH | TAACCAGGCGTCCBATACG | CAGTGCCAGCCTCGTCTCA |
GAPDH, glyceraldehyde 3-phosphate dehydrogenase; COX-2, cyclooxygenase-2; IL-1β, interleukin-1β; iNOS, inducible nitric oxide synthase.
Western blot analysis
A total of 7 days post-reperfusion, the rats were anesthetized using choral hydrate. The brains were then removed and coronally sectioned from −24.3 to +1.7 mm of the bregma, and the right penumbra of ischemic cortex (as the right common carotid artery had been occluded in rats) was separated, weighed and homogenized in cytosolic extraction buffer. Protein was extracted using RIPA lysis buffer (Beyotime Institute of Biotechnology, Nanjing, China) lysis buffer containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 µg/ml aprotinin and 100 µg/ml phenylmethylsulfonyl fluoride. Protein concentration was determined using a Bio-Rad Protein Assay kit (cat. no. 5000002; Bio-Rad Laboratories, Inc., Hercules, CA, USA). Total proteins were diluted in 4X loading buffer and were incubated at 100°C for 5 min. Samples (80 µg) were then loaded onto a loading gel and separated on a Bis-Tris gel (12% gel for IL-1β and cleaved caspase-3; 10% gel for COX-2; and 8% gel for iNOS). Separated proteins were transferred onto a nitrocellulose membrane at 200 mA for 2 h. Nonspecific sites were blocked for 1 h at room temperature in fat-free milk solution [10% in 0.1% Tween-Tris-buffered saline (TTBS)]. Membranes were then incubated overnight at 4°C with the following rabbit polyclonal antibodies: Anti-IL-1β (cat. no. ab9787; 1:1,000; Abcam, Cambridge, UK), anti-COX-2 (cat. no. ab52237 1:1,000; Abcam), anti-iNOS (cat. no. ab3523; 1:1,000; Abcam), anti-cleaved caspase-3 (cat. no. ab2302; 1:1,000; Abcam) and anti-β-actin (cat. no. AF0003; 1:1,000; Beyotime Institute of Biotechnology), in 5% bovine serum albumin TTBS solution. The secondary antibodies (horseradish peroxidase-conjugated goat anti-rabbit IgG (cat. no. A0208) and goat anti-mouse (cat. no. A0216), (Beyotime Institute of Biotechnology) used were diluted to 1:1,000 and incubated for 1 h at room temperature. Subsequently, the membranes were developed using the enhanced chemiluminescence (ECL) reagent Beyo ECL plus (Beyotime Institute of Biotechnology). Images of the blots were captured using a ChemiDoc XRS system (Bio-Rad Laboratories, Inc.). The image was scanned and band intensity was semi-quantified using Quantity One software v4.52 (Bio-Rad Laboratories, Inc.).
Statistical analysis
Data are presented as the mean ± standard error of the mean. Statistical significance was determined using one-way analysis of variance followed by Dunnett's multiple comparisons post-hoc test using SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of GAS on neurological deficits in a rat model of cerebral I/R injury
To investigate the effects of GAS on neurological deficits in a rat model of cerebral I/R injury, a behavioral test was performed. Bederson's score was used as a criterion to evaluate neurological deficits. All sham rats obtained a score of 0, indicating no neurological deficits (Fig. 2). The rats that underwent the right side cerebral I/R surgery exhibited paresis of the left paws. In the model group, the neurological deficit scores were 2.63±0.48, 2.37±0.48 and 2±0.50 on days 1, 3 and 7, respectively. Compared with the model group, the positive control group exhibited improved neurological function, with significantly decreased scores on day 3 (1.63±0.74; P<0.05), and day 7 (1.25±0.46; P<0.01). In the GAS treatment groups, the scores on day 7 at all three doses exhibited significant differences compared with the I/R model group.
Figure 2.
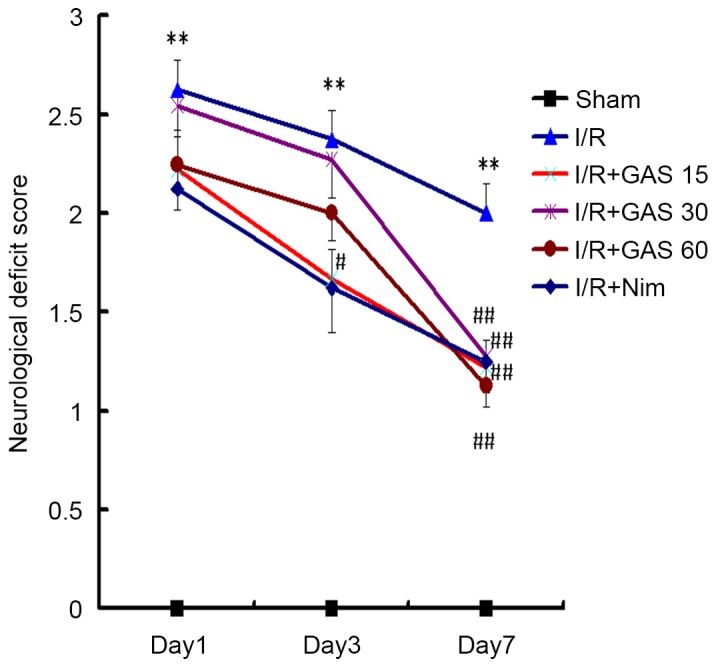
Neurological deficit scores on days 1, 3 and 7 after I/R surgery. The model group exhibited the highest neurological deficit score on each day, whereas the GAS and positive control groups exhibited a significant reduction in neurological deficit scores on day 7. **P<0.01 compared with the sham group; #P<0.05 and ##P<0.01 compared with the I/R model group. GAS, gastrodin; I/R, ischemia-reperfusion; Nim, nimodipine.
Effects of GAS on brain infarct volume in a rat model of subacute phase cerebral I/R injury
In addition to neurological deficit scores, the protective effects of GAS were evaluated with regards to brain infarct volume. No infarcted areas were observed in the sham group (Fig. 3). Conversely, obvious infarcted areas were observed in the model group. The positive control group (Nim) exhibited a significantly smaller infarct volume compared with the model group, and all three doses of GAS exerted a similar effect to the positive control.
Figure 3.

Brain infarct volume in a rat model of cerebral I/R injury. Coronal brain sections (2–3 mm) stained with 0.1% TTC were used to detect infarction. Red staining indicates nonischemic areas and pale sections indicate ischemic areas of the brain. (A) Sham group; (B) middle cerebral artery occlusion group; (C) positive control group; (D) GAS15 (E) GAS30 and (F) GAS60 groups. (G) Total infarct volume following cerebral I/R. **P<0.01 compared with the sham group; ##P<0.01 compared with the model I/R group. GAS, gastrodin; I/R, ischemia-reperfusion; TTC, 2,3,5-triphenyltetrazolium chloride; Nim, nimodipine.
GAS attenuates pathological damage in the hippocampus of the ischemic penumbra
The rat hippocampal CA1 region was observed following HE staining (Fig. 4). Compared with normal neurons in the sham group (Fig. 4A), severe cellular edema, condensed nuclei and nuclear loss were observed in the model group (Fig. 4B). Damaged neurons in the model group exhibited pyknosis and anachromasis of the nucleus, as well as shrunken cell bodies in the hippocampal CA1 region. Nim slightly decreased the damage (Fig. 4C), whereas GAS markedly inhibited cellular edema and nuclear loss in the hippocampal CA1 region (Fig. 4D-F).
Figure 4.

Histological examination of the rat hippocampal CA1 region (magnification, 400x). Representative sections were stained using HE. (A) Normal cellular morphology was present in the sham group. Compared with the (B) model group, a gradual improvement in condensed nuclei (arrows) was detected in the hippocampal CA1 region in the (C) Nim group, (D) GAS 15 mg/kg group, (E) GAS 30 mg/kg group and (F) GAS 60 mg/kg group. Arrows indicate impaired neurons. GAS, gastrodin; HE, hematoxylin and eosin; Nim, nimodipine.
GAS suppresses apoptosis in ischemic hippocampal and cortical penumbra
A TUNEL assay was used to examine fragmented DNA in the hippocampus and cortex in order to determine the potential anti-apoptotic effects of GAS (Figs. 5 and 6). Microscopic inspection of the hippocampal and cortical sections from sham rats revealed morphologically normal neurons with almost no TUNEL staining (Figs. 5A and 6A). A total of 1 week post-reperfusion, the number of TUNEL-positive pyramidal neurons with various degrees of DNA fragmentation was detected in the hippocampal CA1 region and the cerebral cortex of ischemic penumbra (Figs. 5B and 6B). Treatment with Nim and GAS significantly reduced the number of TUNEL-positive neurons (P<0.01; Figs. 5 and 6).
Figure 5.

Effects of GAS on DNA fragmentation in the hippocampus of rats. (A) Almost no TUNEL-reactive cells were detected in the hippocampus of the sham group. (B) Degenerating neurons labeled via the TUNEL technique were detected in the CA1 subfield of the hippocampus of the I/R model group. The number of TUNEL-positive neurons in the (C) Nim, (D) GAS15 (E) GAS30 and (F) GAS60. The number was significantly decreased in the hippocampus of the cerebral I/R injury rats that received Nim or GAS. Arrows indicate degenerating neurons. (A-F, magnification, ×200). (G) Quantitative analysis of the percentage of TUNEL-positive neurons. **P<0.01 compared with the sham group; ##P<0.01 compared with the I/R model group. GAS, gastrodin; I/R, ischemia-reperfusion; Nim, nimodipine; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Figure 6.

Effects of GAS on DNA fragmentation in the cerebral cortex of ischemic penumbra in cerebral I/R injury rats. (A) Almost no TUNEL-reactive cells were detected in the cerebral cortex of the sham group. (B) Degenerating neurons labeled via the TUNEL technique were detected in the cortex of the I/R model group. The number of TUNEL-positive neurons in the (C) Nim, (D) GAS15 (E) GAS30 and (F) GAS60 groups. The number was significantly decreased in the cerebral cortex of middle cerebral artery occlusion rats that received Nim or GAS. Arrows indicate degenerating neurons, the nuclei of which were stained using the TUNEL technique. (G) Quantitative analysis of TUNEL-positive neurons. **P<0.01 compared with the sham group; ##P<0.01 compared with the I/R model group. GAS, gastrodin; I/R, ischemia-reperfusion; Nim, nimodipine; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Effects of GAS on the mRNA expression levels of IL-1β, COX-2 and iNOS in the ischemic penumbra
Due to the important roles of IL-1β, COX-2 and iNOS on cerebral I/R-induced injury, the effects of GAS on the regulation of these genes was investigated. RT-qPCR revealed that all three genes were significantly increased in the model group compared with in the sham group (Fig. 7). However, treatment with Nim or GAS significantly reduced the gene expression levels of IL-1β, COX-2 and iNOS compared with in the model group.
Figure 7.

Effects of GAS on the mRNA expression levels of (A) IL-1β, (B) COX-2 and (C) iNOS in the ischemic penumbra. *P<0.05 and **P<0.01 compared with the sham group; #P<0.05 and ##P<0.01 compared with the I/R model group. GAS, gastrodin; COX-2, cyclooxygenase-2; IL-1β, interleukin-1β; iNOS, inducible nitric oxide synthase; I/R, ischemia-reperfusion; Nim, nimodipine.
Effects of GAS on the protein expression levels of IL-1β, COX-2, iNOS and cleaved caspase-3 in the ischemic penumbra
In addition to the gene expression levels, the protein expression levels of IL-1β, COX-2 and iNOS were determined using western blotting. To confirm the possible role of apoptosis in GAS-induced improvement, the levels of cleaved caspase-3 were also detected. The protein expression levels of IL-1β, COX-2, iNOS and cleaved caspase-3 were significantly higher in the model group compared with in the sham group. As expected, GAS exhibited similar effects to the positive compound Nim; both of which downregulated the expression levels of the aforementioned proteins compared with in the model group (P<0.05; Fig. 8).
Figure 8.

Effects of GAS on the protein expression levels of (A) IL-1β, (B) COX-2, (C) iNOS and (D) cleaved caspase-3 in the ischemic penumbra. **P<0.01 compared with the sham group; #P<0.05 and ##P<0.01 compared with the I/R model group. GAS, gastrodin; COX-2, cyclooxygenase-2; IL-1β, interleukin-1β; iNOS, inducible nitric oxide synthase; I/R, ischemia-reperfusion; Nim, nimodipine.
Discussion
MCAO-induced focal cerebral I/R injury is the most frequently used animal model of stroke (18,19). The present study investigated the effects of GAS on I/R injury 7 days after reperfusion; I/R injury was induced by 2 h MCAO and Nim was used as a positive control. Nim is a dihydropyridine calcium channel blocker, which has been investigated for its therapeutic application in various cerebrovascular trials, including ischemic stroke and cerebral resuscitation (20). Nim has been reported to improve cerebral metabolism in patients with severe head trauma (21), and it has also been shown to exert beneficial effects in the reduction of infarct size in stroke (22). Furthermore, post-ischemic administration of Nim following focal cerebral I/R injury in rats has been reported to alleviate excitotoxicity, neurobehavioral alterations and bioenergetics (23). Similar to Nim, the present study demonstrated that pretreatment with GAS for 7 days followed by I/R surgery, and treatment with GAS for 7 days after I/R surgery significantly attenuated I/R-induced disability and histological damage, alleviated neuronal apoptosis, and reduced the mRNA and protein expression levels of inflammatory and proapoptotic factors, including IL-1β, COX-2, iNOS and cleaved caspase-3.
Although various mechanisms are associated with the pathogenesis of stroke, increasing evidence has demonstrated that inflammatory responses partly account for its pathogenic progression (24). During the subacute phase of ischemic brain injury, 1–7 days after the onset of ischemia, inflammation exacerbates delayed infarct expansion and has a key pathological role in ischemic injury (25). Tissue damage begins with an inflammatory reaction following the interruption of cerebral blood flow, and the administration of GAS exhibited potential neuroprotective effects on MCAO-induced cerebral I/R injury in rats. These effects are likely due to the inhibition of inflammation-associated events. The behavioral test used in the present study was designed to assess impairments consistent with the known functional architecture of the rat brain, and inflammatory mediators have previously been identified through experimental investigations of brain edema (26). In the present study, edema lasted 7 days after MCAO in rats, which was accompanied by an increased infarct size. In addition, behavioral abnormality, infarct size and edema were significantly increased in the I/R group compared with in the sham group. Conversely, in the I/R + GAS groups the development of behavioral abnormality and infarct size was significantly suppressed compared with in the I/R model group.
Inflammatory mediators, including IL-1, iNOS and COX-2, are upregulated following cerebral I/R surgery (27,28). Elevated expression levels of inflammatory mediators may enhance neurological damage via the activation of various downstream pathways (29). A previous study demonstrated that inhibitors of iNOS attenuate infarct size following focal cerebral ischemia (30). In addition, cerebral ischemia has been reported to upregulate the inducible form of COX-2 in the injured brain (31), and attenuating microglial activation-induced expression of IL-1β may provide a neuroprotective effect against transient cerebral ischemic injury (32). GAS, which is a compound isolated from the Chinese herb G. elata BI, was able to significantly ameliorate cerebral I/R surgery-induced infarct size in the present study. In addition, downregulation of the gene and protein expression levels of IL-1β, COX-2 and iNOS were detected in the penumbra of cerebral I/R rats, and may be considered the potential underlying mechanism by which GAS protects the brain from stroke-induced symptoms.
The present study also demonstrated that inhibition of apoptosis was a mechanism by which GAS exerted neuroprotection against cerebral I/R injury. Mitochondrial and death receptor pathways are the two main apoptotic pathways. The caspase family consists of cysteine proteases that induce apoptosis. Among the 12 known caspases in mammals, cleaved caspase-3 is a crucial biomarker of neuronal apoptosis that also acts as an apoptotic executor (33). In the present study, in both the hippocampus and cerebral cortex of the cerebral I/R rats, GAS significantly attenuated neuronal loss and the number of TUNEL-positive cells. Therefore, inhibition of apoptosis is potentially involved in the prevention of neuronal death. In the present study, with regards to activated caspase-3, cerebral ischemia increased its protein expression levels, which is consistent with a previous study that reported elevated caspase-3 gene expression in transient cerebral ischemia (34). Conversely, treatment with GAS significantly decreased the expression levels of cleaved caspase-3. These results suggested that the protective effects of GAS may be associated with the inhibition of apoptotic signaling pathways.
In conclusion, the present study demonstrated that GAS was able to ameliorate subacute phase cerebral I/R injury, and its protective effects may be induced by inhibiting IR-induced upregulation of the inflammatory cytokine IL-1β, by inhibiting the expression of the pro-oxidative enzymes COX-2 and iNOS in the ischemic brain, and by inhibiting neuronal apoptosis via suppression of apoptotic signaling pathways. These findings suggested that GAS exerts a neuroprotective effect in the ischemic brain, due to the inhibition of inflammation and apoptosis. Therefore, GAS may be considered a potential therapeutic agent against inflammation and apoptosis in subacute phase cerebral I/R injury.
Acknowledgements
The present study was supported by the Brainstorm Project on Social Development by Department of Science and Technology of Guizhou Province (grant no. SY [2008] 3040); the Program for Changjiang Scholars and Innovative Research Team in University, China (grant no. IRT1197); the Program for New Century Excellent Talents in University (grant no. NCET-11-0927); and the Key Projects of Guizhou Science and Technology Department [grant no. JZ(2014)2015].
References
- 1.Feigin VL, Lawes CM, Bennett DA, Barker-Collo SL, Parag V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: A systematic review. Lancet Neurol. 2009;8:355–369. doi: 10.1016/S1474-4422(09)70025-0. [DOI] [PubMed] [Google Scholar]
- 2.Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, et al. Heart disease and stroke statistics-2008 update: A report from the American heart association statistics committee and stroke statistics subcommittee. Circulation. 2008;117:e25–e146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- 3.Strosznajder RP, Czubowicz K, Jesko H, Strosznajder JB. Poly (ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol Neurobiol. 2010;41:187–196. doi: 10.1007/s12035-010-8124-6. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez CL, Gharbawie OA, Kolb B. Chronic low-dose administration of nicotine facilitates recovery and synaptic change after focal ischemia in rats. Neuropharmacology. 2006;50:777–787. doi: 10.1016/j.neuropharm.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 5.Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Wu Pf, Zhang Z, Wang F, Chen JG. Natural compounds from traditional medicinal herbs in the treatment of cerebral ischemia/reperfusion injury. Acta Pharmacol Sin. 2010;31:1523–1531. doi: 10.1038/aps.2010.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu H, Dai P, Zhang W, Chen E, Han W, Chen C, Cui Y. Enzymic synthesis of gastrodin through microbial transformation and purification of gastrodin biosynthesis enzyme. Biol Pharm Bull. 2010;33:1680–1684. doi: 10.1248/bpb.33.1680. [DOI] [PubMed] [Google Scholar]
- 8.Kumar H, Kim IS, More SV, Kim BW, Bahk YY, Choi DK. Gastrodin protects apoptotic dopaminergic neurons in a toxin-induced Parkinson's disease model. Evid-Based Compl Alt. 2013;2013:514095. doi: 10.1155/2013/514095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin LC, Chen YF, Lee WC, Wu YT, Tsai TH. Pharmacokinetics of gastrodin and its metabolite p-hydroxybenzyl alcohol in rat blood, brain and bile by microdialysis coupled to LC-MS/MS. J Pharm Biomed Anal. 2008;48:909–917. doi: 10.1016/j.jpba.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Zeng X, Zhang S, Zhang L, Zhang K, Zheng X. A study of the neuroprotective effect of the phenolic glucoside gastrodin during cerebral ischemia in vivo and in vitro. Planta Med. 2006;72:1359–1365. doi: 10.1055/s-2006-951709. [DOI] [PubMed] [Google Scholar]
- 11.Xu X, Lu Y, Bie X. Protective effects of gastrodin on hypoxia-induced toxicity in primary cultures of rat cortical neurons. Planta Med. 2007;73:650–654. doi: 10.1055/s-2007-981523. [DOI] [PubMed] [Google Scholar]
- 12.Bie X, Chen Y, Han J, Dai H, Wan H, Zhao T. Effects of gastrodin on amino acids after cerebral ischemia-reperfusion injury in rat striatum. Asia Pac J Clin Nutr. 2007;16:305–308. [PubMed] [Google Scholar]
- 13.Dai JN, Zong Y, Zhong LM, Li YM, Zhang W, Bian LG, Ai QL, Liu YD, Sun J, Lu D. Gastrodin inhibits expression of inducible NO synthase, cyclooxygenase-2 and proinflammatory cytokines in cultured LPS-stimulated microglia via MAPK pathways. PloS One. 2011;6:e21891. doi: 10.1371/journal.pone.0021891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Wu Z, Liu X, Fu Q. Gastrodin ameliorates Parkinson's disease by downregulating connexin 43. Mol Med Rep. 2013;8:585–590. doi: 10.3892/mmr.2013.1535. [DOI] [PubMed] [Google Scholar]
- 15.Peng Z, Wang H, Zhang R, Chen Y, Xue F, Nie H, Chen Y, Wu D, Wang Y, Wang H, Tan Q. Gastrodin ameliorates anxiety-like behaviors and inhibits IL-1beta level and p38 MAPK phosphorylation of hippocampus in the rat model of posttraumatic stress disorder. Physiol Res. 2013;62:537–545. doi: 10.33549/physiolres.932507. [DOI] [PubMed] [Google Scholar]
- 16.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.STR.17.3.472. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman GE, Merchenthaler I, Zup SL. Neuroprotection by ovarian hormones in animal models of neurological disease. Endocrine. 2006;29:217–231. doi: 10.1385/ENDO:29:2:217. [DOI] [PubMed] [Google Scholar]
- 19.Boyko M, Zlotnik A, Gruenbaum BF, Gruenbaum SE, Ohayon S, Goldsmith T, Kotz R, Leibowitz A, Sheiner E, Shapira Y, Teichberg VI. An experimental model of focal ischemia using an internal carotid artery approach. J Neurosci Methods. 2010;193:246–253. doi: 10.1016/j.jneumeth.2010.08.026. [DOI] [PubMed] [Google Scholar]
- 20.Kakarieka A, Schakel E, Fritze J. Clinical experiences with nimodipine in cerebral ischemia. J Neural Trans Suppl. 1994;43:13–21. [PubMed] [Google Scholar]
- 21.Aslan A, Gurelik M, Cemek M, Goksel HM, Buyukokuroglu ME. Nimodipine can improve cerebral metabolism and outcome in patients with severe head trauma. Pharmacol Res. 2009;59:120–124. doi: 10.1016/j.phrs.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Fogelholm R, Erilä T, Palomäki H, Murros K, Kaste M. Effect of nimodipine on final infarct volume after acute ischemic stroke. Cerebrovasc Dis. 2000;10:189–193. doi: 10.1159/000016055. [DOI] [PubMed] [Google Scholar]
- 23.Babu CS, Ramanathan M. Post-ischemic administration of nimodipine following focal cerebral ischemic-reperfusion injury in rats alleviated excitotoxicity, neurobehavioural alterations and partially the bioenergetics. Int J Dev Neurosci. 2011;29:93–105. doi: 10.1016/j.ijdevneu.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol. 2007;20:334–342. doi: 10.1097/WCO.0b013e32813ba151. [DOI] [PubMed] [Google Scholar]
- 25.Ye YL, Shi WZ, Zhang WP, Wang ML, Zhou Y, Fang SH, Liu LY, Zhang Q, Yu YP, Wei EQ. Cilostazol, a phosphodiesterase 3 inhibitor, protects mice against acute and late ischemic brain injuries. Eur J Pharmacol. 2007;557:23–31. doi: 10.1016/j.ejphar.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007;6:258–268. doi: 10.1016/S1474-4422(07)70055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li F, Gong Q, Wang L, Shi J. Osthole attenuates focal inflammatory reaction following permanent middle cerebral artery occlusion in rats. Biol Pharm Bull. 2012;35:1686–1690. doi: 10.1248/bpb.b12-00133. [DOI] [PubMed] [Google Scholar]
- 29.Maddahi A, Edvinsson L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J Neuroinflammation. 2010;7:14. doi: 10.1186/1742-2094-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang MH, Kaku T, Hada J, Hayashi Y. 7-Nitroindazole reduces nitric oxide concentration in rat hippocampus after transient forebrain ischemia. Eur J Pharmacol. 1999;380:117–121. doi: 10.1016/S0014-2999(99)00555-5. [DOI] [PubMed] [Google Scholar]
- 31.Iadecola C, Forster C, Nogawa S, Clark HB, Ross ME. Cyclooxygenase-2 immunoreactivity in the human brain following cerebral ischemia. Acta Neuropathol. 1999;98:9–14. doi: 10.1007/s004010051045. [DOI] [PubMed] [Google Scholar]
- 32.Lujia Y, Xin L, Shiquan W, Yu C, Shuzhuo Z, Hong Z. Ceftriaxone pretreatment protects rats against cerebral ischemic injury by attenuating microglial activation-induced IL-1β expression. Int J Neurosci. 2014;124:657–665. doi: 10.3109/00207454.2013.856009. [DOI] [PubMed] [Google Scholar]
- 33.Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. P Natl Acad Sci USA. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]