

Abstract
Human glioma is the most common type of primary brain tumor and one of the most invasive and aggressive tumors, which, even with treatments including surgery, radiotherapy and chemotherapy, often relapses and exhibits resistance to conventional treatment methods. Developing novel strategies to control human glioma is, therefore, an important research focus. The present study investigated the mechanism of apoptosis induction in U251 human glioma cells by capsaicin (Cap) and dihydrocapsaicin (DHC), the major pungent ingredients of red chili pepper, using the Cell Counting Kit-8 assay, transmission electron microscopy analysis, flow cytometry analysis, laser scanning confocal microscope analysis and immunohistochemical staining. Treatment of U251 glioma cells with Cap and DHC resulted in a dose- and time-dependent inhibition of cell viability and induction of apoptosis, whereas few effects were observed on the viability of L929 normal murine fibroblast cells. The apoptosis-inducing effects of Cap and DHC in U251 cells were associated with the generation of reactive oxygen species, increased Ca2+ concentrations, mitochondrial depolarization, release of cytochrome c into the cytosol and activation of caspase-9 and −3. These effects were further confirmed by observations of the anti-tumor effects of Cap and DHC in vivo in a U251 cell murine tumor xenograft model. These results demonstrate that Cap and DHC are effective inhibitors of in vitro and in vivo survival of human glioma cells, and provide the rationale for further clinical investigation of Cap and DHC as treatments for human glioma.
Keywords: glioma, capsaicin, dihydrocapsaicin, mitochondrial pathway, apoptosis
Introduction
The incidence of primary malignant brain tumors has increased by 1.2% each year for the past 30 years, particularly in the elderly (1). Malignant glioma accounts for ~70% of primary brain tumor cases, with an annual incidence of ~5/100,000 and >14,000 new cases each year (2). Despite recent advances in therapy, significant progress in prognosis prediction and improved survival have not been achieved thus far (3), due to its resistance to radio- and chemotherapy, and a high rate of relapse. Therefore, novel strategies are required to treat human glioma.
Dietary agents are currently under consideration as effective and alternative means to control cancer. Previous studies have demonstrated that capsaicin (Cap), tea polyphenols, procyanidins and allicin are protective against various types of human tumor, including prostate cancer, breast cancer, liver cancer and gastric carcinoma (4–7). Cap and dihydrocapsaicin (DHC) are homovanillic acid derivatives with similar structures, and are the principle spicy components of hot chili peppers that are consumed worldwide, particularly in Southeast Asian and Latin American countries (8). Cap has been previously demonstrated to inhibit the proliferation of, or induce apoptosis in, numerous varieties of human cancer, including lung, prostate, pancreatic and breast cancer, in vitro and in vivo (9–12). Cap causes tumor cell cycle arrest in S or G1/G0 phase in NPC-TW 039 human nasopharyngeal carcinoma cells, MCF-7 human breast cells, BT-474 cells, SKBR-3 cells, MDA-MB231 cells and SCC-4 human tongue cells, in model systems in vitro and in vivo (13–15). Furthermore, Cap triggers apoptosis in >40 distinct tumor cell lines, primarily through the mitochondrial pathway or death receptor pathway (16). Cap induced apoptosis in AsPC-1 and BxPC-3 human pancreatic cancer cells through the mitochondrial death pathway, which was initiated by the generation of reactive oxygen species (ROS) and c-Jun N-terminal kinase (JNK) activation (11). In addition, intragastric administration of Cap significantly inhibits the growth of AsPC-1 pancreatic xenograft cells (11), and induces TRPV1-mediated apoptosis in RT4 urothelial cancer cells through the death receptor pathway by activating Fas cell surface death receptor (17). Gil and Kang (18) demonstrated that Cap inhibits the growth of A172 human glioblastoma cells and induces apoptosis by downregulation of B cell lymphoma 2 apoptosis regulator (Bcl-2) and activation of caspase-3. Maity et al (19) reported that Cap induces apoptosis in mouse neuro 2a cells via ubiquitin-proteasome system dysfunction. Notably, normal or noncancerous cells are less sensitive to the anti-proliferative or apoptotic effects of Cap compared with cancerous cells (16). DHC, an analog of Cap, inhibits the proliferation of HCT116, MCF-7 and WI38 cells more potently than Cap, and induces autophagy in HCT 116 cells (20). Furthermore, DHC induces autophagy in A549 cells by downregulation of catalase, which leads to ROS accumulation and attenuation of microtubule-associated protein light chain 3 conversion (21).
However, the molecular mechanisms of Cap and DHC induction of apoptosis in U251 human glioma cells are not sufficiently understood. The present study aimed to investigate the effect of Cap and DHC on U251 human glioma cells and the mechanisms of this effect.
Materials and methods
Chemicals and antibodies
Cap, DHC (purity>99%) and trypsin were purchased from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany). Cell Counting Kit-8 (CCK-8), Fluo-3AM, GENMED mitochondrial permeability transition pore (MPTP) living cell fluorescence detection kit and dimethyl sulfoxide (DMSO) were purchased from Dojindo Molecular Technologies, Inc. (Kumamoto, Japan). U251 cells were obtained from the National Platform of Experimental Cell Resources for Sci-Tech (Beijing, China). L929 cells were obtained from the Institute of Biochemistry and Cell Biology (Shanghai, China). Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit and Cell Cycle Detection kit were purchased from Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China). Caspase-3 activity assay kit, caspase-9 activity assay kit, Rhodamine 123 (Rh123), ROS assay kit and cytochrome C (cyto c) antibody (catalog no. AC909) were purchased from Beyotime Institute of Biotechnology (Haimen, China). UltraSensitive™ surface protein array (mouse/rabbit) immunohistochemistry (IHC) kit and 3,3′-diaminobenzidine (DAB) kit were obtained from Fuzhou Maixin Biotech Co., Ltd. (Fuzhou, China). Inverted fluorescence microscope and confocal laser scanning microscope were obtained from Nikon Corporation (Tokyo, Japan). Flow cytometry equipment was obtained from BD Biosciences (Franklin Lakes, NJ, USA).
Cell culture
U251 human glioma cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2 mM L-glutamine and 1% penicillin-streptomycin solution. L929 murine fibroblast cells were maintained in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS, 10 mM HEPES, 2 mM L-glutamine and 1% penicillin-streptomycin solution. All cultures were maintained at 37°C in a humidified chamber of 95% air and 5% CO2. Cap and DHC were dissolved in 0.5% DMSO solution.
Cell inhibition rate and cell survival
Cell inhibition rate and cell survival were assessed by tetrazolium salt-based colorimetric detection in the CCK-8 assay. Cells were seeded in 96-well plates at an initial density of 5×103 cells/well. Following exposure to 50, 100, 150 and 200 µM Cap or DHC for 12 (U251 only), 24, 48 and 72 h, 10 µl CCK-8 solution was added to each well and the plate was incubated for 1 h. Cell viability was determined with a microplate reader by reading the absorbance at 450 nm (A450). The percentage of viable cells was calculated as follows: Cell viability (%)=[A450(treated)-A450 (blank)]/[A450(control)-A450(blank)]x100%.
Determination of apoptosis
U251 cells (2×105) were plated in 6-well plates and allowed to attach for 24 h prior to exposure to 200 µM Cap and DHC for 12 h at 37°C. Apoptosis induced by Cap or DHC was evaluated using an Annexin V-FITC Apoptosis Detection kit according to the manufacturer's protocol. Briefly, U251 cells were detached, collected and suspended in 500 µl binding buffer, then 5 µl Annexin V-FITC and 5 µl propidium iodide (PI) was added and cells were incubated for 10 min at room temperature in the dark. Apoptotic cells were acquired using a FACS-Calibur flow cytometer (BD Biosciences) and the data was analyzed using CellQuest™ Pro software (BD Biosciences) and ModFit LT™ software (Verity Software House, Inc., Topsham, ME, USE).
Transmission electron microscopy (TEM) analysis
U251 cells (1×105/ml) were exposed to 200 µM Cap or DHC for 12 h at 37°C, detached and collected, postfixed in 1% buffered OsO4 for 2 h at 37°C, dehydrated in graded alcohol and embedded in Epon resin. Sections (60-nm thick) were cut and observed using TEM. A total of 5 slides were visualized, with 10 fields analyzed per slide.
Cell cycle analysis
U251 cells (2×105 cells/well) were plated in 6-well plates and allowed to attach for 24 h prior to exposure to 200 µM Cap or DHC for 12 h at 37°C. Control cells were treated with DMSO only. The cell cycle was analyzed using the Cell Cycle Detection kit according to the manufacturer's protocol. The cells were harvested, washed with phosphate-buffered saline (PBS), resuspended in Reagent A (100 µl), and incubated for 10 min at room temperature. Reagent B (100 µl) was then added, and plates incubated for a further 10 min at room temperature. Cells were stained with 150 µl PI for 15 min at room temperature in the dark, then fluorescence measured with a Becton-Dickinson FACS-Calibur flow cytometer and the data was analyzed using CellQuest Pro software and ModFit LT software.
ROS generation
Changes in intracellular ROS levels were detected by recording the oxidative conversion of cell permeable 2′,7′,-dichlorofluorescin diacetate (DCFH-DA) to fluorescent dichlorofluorescein (DCF) by flow cytometry. Following treatment with 200 µM Cap or DHC for 12 h, 2×105 cells were washed twice with PBS, then incubated with DCFH-DA at 37°C for 20 min. Cells were subsequently washed three times with serum-free DMEM, then collected and resuspended in PBS. The fluorescent signal intensity of DCF was detected using a FACS-Calibur flow cytometer and the data was analyzed using CellQuest Pro software and ModFit LT software.
Measurement of intracellular Ca2+concentration ([Ca2+]i)
Following 12 h treatment with 200 µM Cap or DHC, 2×105 U251 cells were washed 3 times with HEPES-buffered salt solution (HBSS), then incubated with Fluo 3-AM (final concentration, 5 µM) at 37°C for 30 min in the dark. Cells were then washed 3 times with HBSS and incubated in fresh HBSS for a further 30 min to remove the remaining Fluo 3-AM. [Ca2+]i was then detected using a laser scanning confocal microscope, with 6 fields assessed per group.
Measurement of MPTP
MPTP were detected using GENMED MPTP living cell fluorescence detection kits. U251 cells (2×105 cells/well) were plated in 6-well plates and allowed to attach for 24 h, then exposed to 200 µM Cap or DHC for 12 h at 37°C. Following washing with Reagent A, the cells were incubated with 500 µl Reagent B and Reagent C at 37°C for 20 min in the dark. Subsequently, cells were washed twice with Reagent A, and the fluorescence detected with an inverted fluorescence microscope using 488 nm excitation (Ex) and 505 nm emission (Em) filter settings, with 6 fields assessed per group.
Measurement of mitochondrial membrane potential (MMP)
Rh123 was used to estimate MMP. Treated U251 cells (1×105 cells treated with 200 µM Cap or DHC for 12 h at 37°C) were harvested and washed with PBS, then incubated with Rh123 (10 µl/ml) in PBS for 30 min in the dark at 37°C. Following 2 washes with PBS, the fluorescence was measured with the laser scanning confocal microscope using 507 nm Ex and 529 nm Em filter settings.
Caspase-9 and caspase-3 activity measurement
Caspase-9 and caspase-3 activity was detected with caspase-9 and caspase-3 activity kits according to the manufacturer's protocols, based on the ability of the enzymes to hydrolyze acetyl-Asp-Glu-Val-Asp phospho (p)-nitroaniline (caspase-3) or acetyl-Leu-Glu-His-Asp-p-nitroanilide (caspase-9) into the yellow formazan product, p-nitroanilide. U251 cells (2×105 cells/well) were plated in 6-well plates and allowed to attach for 24 h, followed by exposure to 200 µM Cap or DHC for 12 h at 37°C. Treated cells were washed with cold PBS, then lysed with lysis buffer (100 µl per 2×106 cells) for 15 min on ice, and centrifuged for 15 min at 4°C at 16,000 × g. Supernatant (10 µl) was then mixed with 80 µl reaction buffer and 10 µl 2 mM caspase-9 and caspase-3 substrate in 96-well plates, and incubated at 37°C for 2 h. The caspase-9 and caspase-3 relative activity was evaluated with a microplate reader at 405 nm.
Xenograft tumor model
A total of 40 male BALB/c nude mice (weight, 18–22 g) were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). The use of nude mice and their treatment was approved by the Institutional Animal Care and Use Committee, Tsinghua University (Beijing, China), and all experiments were carried out in strict compliance with their regulations. Nude mouse xenograft models of human glioma were established by injecting 1.0×107 U251 cells subcutaneously in the right hind leg of BALB/c nude mice, with treatment beginning after one week. Mice were divided randomly into 6 groups with 6–7 mice in each group. Cap/DHC was dissolved in 5% ethanol and 5% Tween-80. ‘Control group’ mice received PBS (0.1 ml/20 g) by oral gavage. ‘Vehicle group’ mice received the same volume (0.1 ml/20 g) of vehicle (5% ethanol+5% Tween-80). ‘Cap (5 mg/kg) group’ mice received 5 mg Cap/kg by oral gavage every three days. ‘Cap (10 mg/kg) group’ mice received 10 mg Cap/kg by oral gavage every three days. ‘DHC (5 mg/kg) group’ mice received 5 mg DHC/kg by oral gavage every three days. ‘DHC (10 mg/kg) group’ mice received 10 mg DHC/kg by oral gavage every three days. Tumors were measured by vernier calipers and tumor size was calculated using the following formula: Volume (mm3)=π(axb2)/6, where a is the long diameter and b is the short diameter. Each mouse was weighed every three days. The mice were sacrificed with CO2 3 weeks following treatment. The tumor, stomach, heart, spleen, lung, liver, and kidney were weighed and fixed in 10% formalin solution for histological analysis by hematoxylin and eosin (H&E) staining. Sections (5-µm thick) were stained as previously described (22). Sections were observed under a TH4-200 light microscope (Olympus Corporation, Tokyo, Japan); 10 fields were examined per mouse.
Cyto c expression in tumor xenografts
For IHC analysis of cyto c, sections (5 µm thick) were cut, dewaxed, rehydrated and immersed in a 10 nM sodium citrate buffer (pH 6.0) and boiled for 30 min in a pressure cooker while maintaining the pressure of 181 kpa, then cooled and washed in PBS. Endogenous peroxidase activity was blocked with Immunol Staining Blocking buffer (Beyotime Institute of Biotechnology) for 1 h at room temperature to reduce nonspecific background staining. Subsequently, cyto C primary antibody diluted 1:1,000 in Immunol Staining Primary Antibody Dilution buffer (Beyotime Institute of Biotechnology) was added to the slides, incubated for 1 h at room temperature, then washed in PBS. Biotinylated anti-mouse immunoglobulin diluted 1:1,000 in Immunol Staining Secondary Antibody Dilution buffer (Beyotime Institute of Biotechnology) and streptavidin conjugated to horseradish peroxidase, from the cyto C antibody kit, were subsequently applied for 30 min at room temperature. Finally, DAB was used for color development according to the manufacturer's protocol and sections were counterstained with hematoxylin. Negative control slides in the absence of primary antibody were included for each staining. Sections were observed under a TH4-200 light microscope; 6 fields were examined per mouse.
Statistical analysis
Data are presented as the mean ± standard error for the indicated number of independently performed experiments. Data analyses were performed using SPSS v.20.0 (IBM SPSS, Armonk, NY, USA). Statistical significance was evaluated by one-way analysis of variation, followed by the least significant difference post-hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of Cap and DHC on cell viability and induction of apoptosis in U251 human glioma cells and L929 normal murine fibroblast cells
The effects of Cap and DHC on the viability of U251 cells were examined by CCK-8 assay. Cap and DHC both significantly inhibited the proliferation of U251 cell lines in a dose- and time-dependent manner (Fig. 1A). Treatment of U251 cells for 72 h with 200 µM Cap and 200 µM DHC, resulted in 3.08±1.60 and 3.49±1.50% survival, respectively (P<0.001 and P<0.001, respectively; Fig. 1A), indicating that the majority of cells were killed by exposure to high concentrations of Cap and DHC. Cell viability of vehicle-treated cells (0.5% DMSO) remained >94.17%, indicating that 0.5% DMSO is not toxic to U251 cells (Fig. 1A). However, the viability of L929 normal mouse fibroblasts was not significantly affected by exposure to the equivalent concentrations of Cap and DHC that were highly cytotoxic to cancer cells (Fig. 1B).
Figure 1.

Effect of Cap and DHC on cell viability of U251 cells and L929 cells. (A) Viability of human glioma U251 cells was analyzed by CCK-8 assay following culture in the presence of various concentrations of Cap and DHC (50, 100, 150 and 200 µM) for 12, 24, 48 and 72 h. (B) Viability of L929 normal murine fibroblast cells was analyzed by CCK-8 assay following culture in the presence of various concentration of Cap and DHC (50, 100, 150 and 200 µM) for 24, 48 and 72 h. Data are presented as the mean ± standard error of 3 independent experiments. *P<0.05, **P<0.01 vs. control. CCK-8, Cell Counting Kit-8; DMSO, dimethyl sulfoxide; Cap, capsaicin; DHC, dihydrocapsaicin.
Effects of Cap and DHC on cell cycle and apoptosis of human glioma U251 cells
Following exposure to Cap or DHC (200 µM) for 12 h, the proportion of U251 cells in the G2/M phase significantly decreased from 16.50% in the control group to 5.78% with Cap (P=0.001; Fig. 2A) and 7.43% with DHC (P=0.001; Fig. 2A). However, the proportion of cells in the G0/G1 phase significantly increased from 71.47% in the control group to 75.78% with Cap (P=0.006; Fig. 2A) and 76.32% with DHC (P=0.003; Fig. 2A), while the proportion of cells in S phase also significantly increased from 12.02% in the control group to 18.43% with Cap (P=0.001; Fig. 2A) and 16.25% with DHC (P=0.006; Fig. 2A). The anti-tumor effect of Cap and DHC was, therefore, associated with G0/G1 and S phase cell-cycle arrest.
Figure 2.

Effect of Cap and DHC on the cell cycle and apoptosis in U251 cells following 12 h treatment with 200 µM Cap or DHC. (A) Flow cytometry analysis of cell cycle distributions of control DMSO-, Cap- and DHC-treated cells. (B) Flow cytometry analysis of Cap- and DHC-induced apoptosis in U251 cells stained with Annexin V-FITC/PI. Cells shown in the lower right (Annexin V+/PI−) are undergoing apoptosis. Cells shown in the upper right (Annexin V+/PI+) are undergoing necrosis. (C) Electron microscopy of U251 cells treated with Cap and DHC. The red arrows represent apoptotic bodies. Data presented are representative of three independent experiments. **P<0.01 vs. control. Cap, capsaicin; DHC, dihydrocapsaicin; PI, propidium iodide; FITC, fluorescein isothiocyanate.
Annexin V-FITC/PI double staining assays were also performed to analyze cell apoptosis following 12 h treatment with 200 µM Cap or DHC. The proportion of apoptosed cells significantly increased from 14.16% in the DMSO-treated control group to 50.81% with Cap (P<0.001; Fig. 2B) and 42.79% with DHC (P<0.001; Fig. 2B). Necrosis was also significantly increased from 3.45% in the control group to 7.17% with Cap (P=0.001; Fig. 2B) and 11.15% with DHC (P<0.001; Fig. 2B).
TEM also revealed that treatment with either 200 µM Cap or 200 µM DHC for 12 h resulted in condensed cytoplasm, nuclei displaying peripheral chromatin condensation, which had broken up into rounded bodies, and cells developing into apoptotic bodies, compared with the integrated cellular morphology of the untreated cells (Fig. 2C).
Cap and DHC treatment increases ROS generation and [Ca2+]i in U251 cells
ROS can initiate the loss of MMP, mitochondrial translocation of pro-apoptotic proteins, including Bcl-2 associated protein X apoptosis regulator (Bax) and Bcl-2 associated agonist of cell death (Bad), and release of cyto c (23). Intracellular ROS levels in U251 cells were, therefore, evaluated by DCFH-DA assays and flow cytometry detection. DCFH-DA is intracellularly hydrolyzed to DCFH, which is oxidized by ROS to generate fluorescent DCF. Following 12 h exposure to Cap or DHC, mean DCF fluorescence intensity in U251 cells increased from 3.45 in the control group to 5.84 with Cap and 6.41 with DHC (Fig. 3A).
Figure 3.

Cap and DHC increase ROS and intracellular Ca2+ levels, and activate the mitochondrial pathway in U251 cells following 12 h treatment with 200 µM Cap or DHC. (A) Intracellular ROS levels were assessed through detection of fluorescent DCF by flow cytometry. (B) Intracellular Ca2+ was detected with Fluo 3-AM by laser scanning confocal microscopy. (C) MPTPs were detected using a GENMED MPTP living cell fluorescence detection kit and an inverted fluorescence microscope. (D) Mitochondrial membrane potential was evaluated using Rhodamine 123 and a laser scanning confocal microscope. ROS, reactive oxygen species; MPTP, mitochondrial permeability transition pore; Cap, capsaicin; DHC, dihydrocapsaicin; DCF, dichlorofluorescein.
High levels of Ca2+ can open mitochondrial permeability transition pores, depolarize mitochondrial membrane potential, activate caspase-9 and caspase-3, initiate the mitochondrial apoptosis pathway, to induce cell apoptosis (24). Fluo 3-AM was, therefore, used to observe the intracellular concentration of Ca2+. The mean fluorescence intensity in U251 cells increased from 333.79 in the control group to 1,116.46 (P<0.001) with Cap and 1,240.66 (P<0.001) with DHC, respectively (Fig. 3B), indicating an increase in the concentration of intracellular Ca2+.
Cap and DHC treatment increases MPTP formation and reduces MMP
MPTP formation is one of the principle regulators of the mitochondrion during cell apoptosis. The opening of MPTPs can lead to matrix swelling, rupture of the outer membrane of mitochondrion and a release of intermembrane space proteins into the cytoplasm, ultimately resulting in a loss of MMP. The presence of MPTPs was, therefore, detected using a GENMED MPTP living cell fluorescence detection kit. Following exposure to Cap or DHC for 12 h, the fluorescence intensity in U251 cells was decreased compared with the control group (Fig. 3C), indicating the formation of MPTPs.
Depolarization of the MMP induces release of cyto c and apoptosis-inducing factor (AIF) from the intermembrane space into the cytoplasm, resulting in fragmentation of nucleus and cell apoptosis (25). The mean fluorescence intensity in U251 cells decreased from 980.90 in the control group to 216.13 following 12 h exposure to 200 µM Cap (P<0.001) and 222.48 with 200 µM DHC (P<0.001), respectively (Fig. 3D), indicating the loss of MMP.
Cap and DHC treatment activates caspase-9 and −3
Apoptotic cell death is inevitable following caspase-3 activation, and is a vital factor in mitochondrial pathway-induced apoptosis. Caspase-9 (Fig. 4A and B) and −3 (Fig. 4C and D) activity were dose-dependently increased in U251 cells exposed to Cap or DHC for 12 h, compared with control group, indicating that Cap- and DHC-induced apoptosis is associated with the activation of caspase-9 and −3.
Figure 4.

Effect of Cap and DHC on the caspase-9 and caspase-3 activity of U251 cells following 12 h treatment with 200 µM Cap or DHC. Caspase-9 activity in response to (A) Cap and (B) DHC, and caspase-3 activity in response to (C) Cap and (D) DHC were detected using caspase-9 and caspase-3 activity kits, respectively. Data are presented as the mean ± standard error of 6 experiments. *P<0.05 and **P<0.01 vs. control. Cap, capsaicin; DHC, dihydrocapsaicin; pNA, phosphorylated nitroaniline.
Cap and DHC inhibits the growth of U251 human glioma tumor xenografts
In vitro experiments are valuable for quick, convenient, and large-scale screening of latent anti-tumor agents, however the effects observed in cell-based assays require further investigation in suitable pre-clinical in vivo models prior to clinical trials. Tumor xenografts in nude mice is an accepted animal model for pre-clinical evaluation of potential anti-tumor agents. The effect of Cap and DHC on the development of U251 glioma tumor xenografts was, therefore, evaluated in nude mice. The rate of U251 glioma tumor xenograft development was attenuated in mice administered with Cap or DHC in both the low dose group (5 mg/kg, every 3 days) and high dose group (10 mg/kg, every 3 days; Fig. 5A). The mean tumor volume at 23 days in control mice (2316.48±211.28 mm3) was ~2.8-fold larger than tumors from the Cap (10 mg/kg)-treated group (821.15±215.63 mm3; P<0.001) and ~2.95-fold larger than tumors from the DHC (10 mg/kg)-treated group (785.83±218.31 mm3; P<0.001; Fig. 5A). Similar results were obtained with mice treated with 5 mg/kg Cap (P<0.001) and DHC (P<0.001) treated mice compared with control mice (Fig. 5A). The mean wet weight of tumors from control mice was 2.30±0.44 g, compared with 0.92±0.33 g in Cap (10 mg/kg)-treated mice (P<0.001; Fig. 5B) and 0.71±0.34 g in DHC (10 mg/kg)-treated mice (P<0.001; Fig. 5B). A similarly significant anti-tumor effect was demonstrated in mice treated with 5 mg/kg Cap or DHC compared with control mice (P<0.001 and P<0.001, respectively; Fig. 5B). However, the differences between high-dose and low-dose treatments with the same drug were not statistically significant (Fig. 5B). As previous reported, alcohol may certain antitumor effects (26). In addition, no difference in average body weight was observed between the control group, Cap- and DHC-treated mice throughout the experiment (data not shown). The liver index (liver weight/body weightx100) and spleen index (spleen weight/body weightx100) in Cap- and DHC-treated mice were also not significantly different compared with the control group (data not shown), indicating that Cap and DHC are not toxic to mice. Histological analysis of the tumors by H&E staining revealed liquefaction and necrosis in tumors from Cap- and DHC-treated mice, compared with vigorous growth in control and vehicle-treated mice (Fig. 5C). No ulcers or erosion, were observed in the stomachs of any mice, nor were any apparent side effects observed during the histological analysis of the hearts, spleens, lungs, livers and kidneys of the mice (data not shown). IHC analysis of cyto c expression revealed numerous cyto c-positive cells in the tumors of Cap- and DHC-treated mice, but few in the control or vehicle-treated groups (Fig. 5D), indicating that cyto c was released from mitochondria to the cytoplasmic matrix, inducing U251 cell apoptosis. Cap and DHC have, therefore, been demonstrated to significantly reduce U251 human glioma tumor development with no apparent negative side effects on the organs.
Figure 5.

Cap and DHC inhibit the growth of U251 human glioma tumor xenografts in a BALB/c nude mouse xenograft model. (A) Average tumor volume, measured using vernier calipers. Data are presented as the mean ± standard error (n=6–7 mice per group). (B) Average wet weight of tumors following 3 weeks of treatment. Data are presented as the mean ± standard error (n=6–7 mice per group). *P<0.05, **P<0.01 vs. control. (C) Morphological changes in tumors, visualized by hematoxylin and eosin staining. (D) Immunohistochemical analysis of cytochrome c immunoreactivity in dissected tumors. Cap, capsaicin; DHC, dihydrocapsaicin.
Discussion
Human glioma is the most common type of primary brain tumor, and one of the most invasive and aggressive tumors, which, even with treatments, including surgery, radiotherapy and chemotherapy, often relapses and exhibits resistance to conventional treatment methods. Apoptosis and angiogenesis are two potential targets for novel strategies to treat this disease (27). Resistance to radiotherapy and chemotherapy are common consequences of the dysregulation of apoptosis, therefore, inducing apoptosis is an important feature of various chemotherapy drugs, including temozolomide and cisplatin (28,29). Cap, the most abundant pungent chili pepper component, has been widely examined as an anti-cancer agent, and induces cell apoptosis in numerous cancer cell lines, in vitro and when explanted into rodents (11,30,31). The present study demonstrated that Cap and DHC, the second most abundant capsaicinoids in chili pepper, are anti-cancer agents against U251 human glioma cells and are not toxic to L929 normal murine fibroblast cells. The apoptotic effects of Cap and DHC in U251 cells were associated with the generation of ROS, increased intracellular Ca2+, disruption of MMP and the release of cyto c to the cytosol, which ultimately resulted in the activation of caspase-9 and caspase-3 cascade (Fig. 6).
Figure 6.
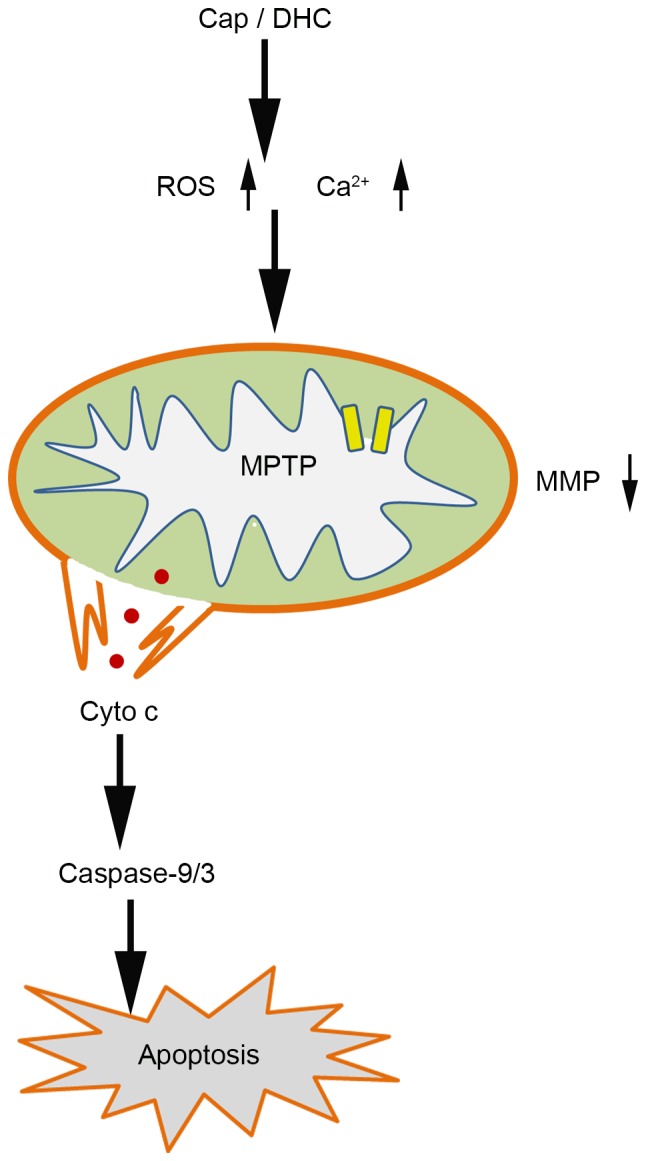
Apoptosis signaling activated by Cap and DHC in U251 cells. Cap- and DHC-induce apoptosis in human glioma cancer cells through a reactive oxygen species and Ca2+-mediated mitochondrial pathway, which is associated with opening the MPTP, dissipating the MMP, releasing cyto c to the cytosol and activating caspase-9 and caspase-3. Cap, capsaicin; DHC, dihydrocapsaicin; ROS, reactive oxygen species; MPTP, mitochondrial permeability transition pore; MMP, mitochondrial membrane potential; cyto C, cytochrome C.
Cell cycle arrest is a prospective therapeutic target in cancer as it is a downstream focal point for oncogenic signaling pathways (32). Previous reports have demonstrated that Cap inhibits cell proliferation by cell-cycle arrest: MCF-7 human breast cancer cells and BT-20 cells were arrested in the S phase following 72 h treatment with 200 µM Cap (12). Thoennissen et al (14) demonstrated that Cap arrests MCF-7, T47D, BT-474, SKBR-3 and MDA-MB231 (ER-positive and -negative) human breast cancer cells in the G0/G1 phase at concentrations of 50–200 µM. Similarly, SCC4 human tongue cancer cells, NPC-TW 039 nasopharyngeal carcinoma cells, RT4 urothelial cancer cells, CE 81 T/VGH esophagus epidermoid carcinoma cells, HL-60 leukemia and NB4 myeloid leukemia cells were also arrested in the G0/G1 phase following treatment with Cap (13,15,17,33–35). In the current study, the anti-proliferation effect of Cap and DHC was associated with G0/G1 and S phase cell cycle arrest following 12 h treatment at a concentration of 200 µM (Fig. 2A).
The most common apoptotic pathway induced by Cap is reported to be the mitochondrial pathway, which is particularly susceptible to ROS and Ca2+. A sustained increase in intracellular Ca2+ increases the production of ROS and eventually triggers cell apoptosis. Excess H2O2 inhibits the activity of Na+/H+ antiporters, which leads to intracellular acidification and creates a microenvironment for apoptosis (36). Furthermore, ROS directly damage the mitochondrial electron transport chain, activate caspases and induce cell apoptosis (29). Cap boosts the generation of ROS in human pancreatic cancer cells by inhibiting mitochondrial complex I and III and destroying mitochondrial functions (37). Initiation of the mitochondrial pathway by excess ROS and sustained an increase in intracellular Ca2+ results in the formation of MPTPs, which leads to the dissipation of the MMP. The opening of MPTPs elevates the permeability of inner mitochondrial membranes, allowing molecules <1.5 kDa to enter the mitochondria, resulting in swelling of the mitochondrial matrix (24,38). The outer mitochondrial membranes subsequently rupture, releasing numerous pro-apoptotic proteins from the intermembrane space into the cytosol (24,38). These pro-apoptotic proteins are divided into two groups: One group includes AIF and endonuclease G, which act as through a caspase-independent pathway to induce cell apoptosis; the other group, containing cyto c and second mitochondrial-derived activator of caspases (Smac/Diablo), activate caspases. Cyto c induces the oligomerization of apoptotic protease activating factor 1 (Apaf-1), which subsequently activates caspase-9. The activated cyto c, Apaf-1 and caspase-9 then form the apoptosome and, ultimately, activate caspase-3, resulting in nuclear fragmentation (39). Once caspase-3 is activated, cell apoptosis is irreversible (29,40). The present study indicated that Cap and DHC induced apoptosis in human glioma cells via a ROS- and Ca2+-mediated mitochondrial pathway, which was associated with MPTP formation, MMP dissipation and the release of cyto c to the cytosol to activate caspase-9 and −3 (Figs. 3 and 4).
The anti-neoplastic activity of Cap and DHC was investigated in vivo by administering Cap or DHC by oral gavage, producing results that were consistent with previous studies. Cap (5 mg/kg) has previously been revealed to reduce the weight and volume of pancreatic xenograft tumors by 30% (11). Thoennissen et al (14) demonstrated that the weight of invasive, epidermal growth factor receptor-positive, p53 mutant orthotopic MDA-MB231 breast cancer tumors in female nude mice were reduced by 70% compared with controls, following oral gavage with Cap (5 mg/kg per day) 3 times per week for 4 weeks, without observable side effects. Another study demonstrated that Cap (50 mg/kg), administered daily for 6 days, reduced the leukemia tumor weight in NOD/SCID mice and increased apoptosis without resulting in organ damage (35). The daily consumption of capsicum spices varies worldwide; 2.5 g/person is consumed per day in India, 5 g/person in Thailand, 15 g/person in Saudi Arabia and 20 g/person in Mexico (41). The Cap content of different capsicums also varies dramatically, with a range from 1–42.5% (42). Furthermore, due to the unclear pharmacokinetics of Cap, it is difficult to determine how much Cap should be administered in an animal model. Accordingly, considering that previous investigations have used 5 mg/kg Cap to estimate the anti-tumor effect in vivo (43), 5 mg/kg and 10 mg/kg were administered as preliminary doses in the present study, with the optimal dose requiring further research.
In conclusion, the present study demonstrated that Cap and DHC induce apoptosis in human glioma cancer cells through ROS and Ca2+-mediated induction of the mitochondrial apoptosis pathway, which caused MPTP formation, dissipation of the MMP, and release of cyto c to the cytosol, resulting in activation of caspase-9 and caspase-3 (Fig. 6). In addition, intragastric administration of Cap and DHC in vivo was demonstrated to suppress the development of U251 human glioma xenograft tumors, with no apparent side effects. Consequently, these findings provide a basis for developing Cap and DHC as potential therapeutic approaches against glioma.
Acknowledgements
The present study was supported by the International Science & Technology Cooperation Program of China (grant no. 2011DFA30620).
References
- 1.Brem SS, Bierman PJ, Brem H, Butowski N, Chamberlain MC, Chiocca EA, DeAngelis LM, Fenstermaker RA, Friedman A, Gilbert MR, et al. Central nervous system cancers. J Natl Compr Canc Netw. 2011;9:352–400. doi: 10.6004/jnccn.2011.0036. [DOI] [PubMed] [Google Scholar]
- 2.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 3.Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 4.Malagarie-Cazenave S, Olea-Herrero N, Vara D, Morell C, Diaz-Laviada I. The vanilloid capsaicin induces IL-6 secretion in prostate PC-3 cancer cells. Cytokine. 2011;54:330–337. doi: 10.1016/j.cyto.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 5.Deb G, Thakur VS, Limaye AM, Gupta S. Epigenetic induction of tissue inhibitor of matrix metalloproteinase-3 by green tea polyphenols in breast cancer cells. Mol Carcinog. 2015;54:485–499. doi: 10.1002/mc.22121. [DOI] [PubMed] [Google Scholar]
- 6.Feng LL, Liu BX, Zhong JY, Sun LB, Yu HS. Effect of grape procyanidins on tumor angiogenesis in liver cancer xenograft models. Asian Pac J Cancer Prev. 2014;15:737–741. doi: 10.7314/APJCP.2014.15.2.737. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Zhu Y, Duan W, Feng C, He X. Allicin induces apoptosis of the MGC-803 human gastric carcinoma cell line through the p38 mitogen-activated protein kinase/caspase-3 signaling pathway. Mol Med Rep. 2015;11:2755–2760. doi: 10.3892/mmr.2014.3109. [DOI] [PubMed] [Google Scholar]
- 8.Luo XJ, Peng J, Li YJ. Recent advances in the study on capsaicinoids and capsinoids. Eur J Pharmacol. 2011;650:1–7. doi: 10.1016/j.ejphar.2010.09.074. [DOI] [PubMed] [Google Scholar]
- 9.Brown KC, Witte TR, Hardman WE, Luo H, Chen YC, Carpenter AB, Lau JK, Dasgupta P. Capsaicin displays anti-proliferative activity against human small cell lung cancer in cell culture and nude mice models via the E2F pathway. PLoS One. 2010;5:e10243. doi: 10.1371/journal.pone.0010243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sánchez AM, Martínez-Botas J, Malagarie-Cazenave S, Olea N, Vara D, Lasunción MA, Díaz-Laviada I. Induction of the endoplasmic reticulum stress protein GADD153/CHOP by capsaicin in prostate PC-3 cells: A microarray study. Biochem Biophys Res Commun. 2008;372:785–791. doi: 10.1016/j.bbrc.2008.05.138. [DOI] [PubMed] [Google Scholar]
- 11.Zhang R, Humphreys I, Sahu RP, Shi Y, Srivastava SK. In vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis. 2008;13:1465–1478. doi: 10.1007/s10495-008-0278-6. [DOI] [PubMed] [Google Scholar]
- 12.Chang HC, Chen ST, Chien SY, Kuo SJ, Tsai HT, Chen DR. Capsaicin may induce breast cancer cell death through apoptosis-inducing factor involving mitochondrial dysfunction. Hum Exp Toxicol. 2011;30:1657–1665. doi: 10.1177/0960327110396530. [DOI] [PubMed] [Google Scholar]
- 13.Ip SW, Lan SH, Lu HF, Huang AC, Yang JS, Lin JP, Huang HY, Lien JC, Ho CC, Chiu CF, et al. Capsaicin mediates apoptosis in human nasopharyngeal carcinoma NPC-TW 039 cells through mitochondrial depolarization and endoplasmic reticulum stress. Hum Exp Toxicol. 2012;31:539–549. doi: 10.1177/0960327111417269. [DOI] [PubMed] [Google Scholar]
- 14.Thoennissen NH, O'Kelly J, Lu D, Iwanski GB, La DT, Abbassi S, Leiter A, Karlan B, Mehta R, Koeffler HP. Capsaicin causes cell-cycle arrest and apoptosis in ER-positive and -negative breast cancer cells by modulating the EGFR/HER-2 pathway. Oncogene. 2010;29:285–296. doi: 10.1038/onc.2009.335. [DOI] [PubMed] [Google Scholar]
- 15.Ip SW, Lan SH, Huang AC, Yang JS, Chen YY, Huang HY, Lin ZP, Hsu YM, Yang MD, Chiu CF, Chung JG. Capsaicin induces apoptosis in SCC-4 human tongue cancer cells through mitochondria-dependent and -independent pathways. Environ Toxicol. 2012;27:332–341. doi: 10.1002/tox.20646. [DOI] [PubMed] [Google Scholar]
- 16.Bley K, Boorman G, Mohammad B, McKenzie D, Babbar S. A comprehensive review of the carcinogenic and anticarcinogenic potential of capsaicin. Toxicol Pathol. 2012;40:847–873. doi: 10.1177/0192623312444471. [DOI] [PubMed] [Google Scholar]
- 17.Amantini C, Ballarini P, Caprodossi S, Nabissi M, Morelli MB, Lucciarini R, Cardarelli MA, Mammana G, Santoni G. Triggering of transient receptor potential vanilloid type 1 (TRPV1) by capsaicin induces Fas/CD95-mediated apoptosis of urothelial cancer cells in an ATM-dependent manner. Carcinogenesis. 2009;30:1320–1329. doi: 10.1093/carcin/bgp138. [DOI] [PubMed] [Google Scholar]
- 18.Gil YG, Kang MK. Capsaicin induces apoptosis and terminal differentiation in human glioma A172 cells. Life Sci. 2008;82:997–1003. doi: 10.1016/j.lfs.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 19.Maity R, Sharma J, Jana NR. Capsaicin induces apoptosis through ubiquitin-proteasome system dysfunction. J Cell Biochem. 2010;109:933–942. doi: 10.1002/jcb.22469. [DOI] [PubMed] [Google Scholar]
- 20.Oh SH, Kim YS, Lim SC, Hou YF, Changl IY, You HJ. Dihydrocapsaicin (DHC), a saturated structural analog of capsaicin, induces autophagy in human cancer cells in a catalase-regulated manner. Autophagy. 2008;4:1009–1019. doi: 10.4161/auto.6886. [DOI] [PubMed] [Google Scholar]
- 21.Choi CH, Jung YK, Oh SH. Selective induction of catalase-mediated autophagy by dihydrocapsaicin in lung cell lines. Free Radic Biol Med. 2010;49:245–257. doi: 10.1016/j.freeradbiomed.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 22.Fallone CA, Loo VG, Lough J, Barkun AN. Hematoxylin and eosin staining of gastric tissue for the detection of helicobacter pylori. Helicobacter. 1997;2:32–35. doi: 10.1111/j.1523-5378.1997.tb00054.x. [DOI] [PubMed] [Google Scholar]
- 23.Cook SA, Sugden PH, Clerk A. Regulation of bcl-2 family proteins during development and in response to oxidative stress in cardiac myocytes: Association with changes in mitochondrial membrane potential. Circ Res. 1999;85:940–949. doi: 10.1161/01.RES.85.10.940. [DOI] [PubMed] [Google Scholar]
- 24.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 25.Kinnallyk W, Peixotop M, Ryu SY, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta. 2011;1813:616–622. doi: 10.1016/j.bbamcr.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology. 2002;122:2049–2063. doi: 10.1053/gast.2002.33613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner L, Marschall V, Karl S, Cristofanon S, Zobel K, Deshayes K, Vucic D, Debatin KM, Fulda S. Smac mimetic sensitizes glioblastoma cells to Temozolomide-induced apoptosis in a RIP1- and NF-κB-dependent manner. Oncogene. 2013;32:988–997. doi: 10.1038/onc.2012.108. [DOI] [PubMed] [Google Scholar]
- 28.Eriksson I, Joosten M, Roberg K, Ollinger K. The histone deacetylase inhibitor trichostatin A reduces lysosomal pH and enhances cisplatin-induced apoptosis. Exp Cell Res. 2013;319:12–20. doi: 10.1016/j.yexcr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Indran IR, Tufo G, Pervaiz S, Brenner C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim Biophys Acta. 2011;1807:735–745. doi: 10.1016/j.bbabio.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 30.Patwardhan CA, Alfa E, Lu S, Chadli A. Progesterone receptor chaperone complex-based high-throughput screening assay: Identification of capsaicin as an inhibitor of the Hsp90 machine. J Biomol Screen. 2015;20:223–229. doi: 10.1177/1087057114549147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewinska A, Jarosz P, Czech J, Rzeszutek I, Bielak-Zmijewska A, Grabowska W, Wnuk M. Capsaicin-induced genotoxic stress does not promote apoptosis in A549 human lung and DU145 prostate cancer cells. Mutat Res Genet Toxicol Environ Mutagen. 2015;779:23–34. doi: 10.1016/j.mrgentox.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 32.Williams GH, Stoeber K. The cell cycle and cancer. J Pathol. 2012;226:352–364. doi: 10.1002/path.3022. [DOI] [PubMed] [Google Scholar]
- 33.Wu CC, Lin JP, Yang JS, Chou ST, Chen SC, Lin YT, Lin HL, Chung JG. Capsaicin induced cell cycle arrest and apoptosis in human esophagus epidermoid carcinoma CE 81T/VGH cells through the elevation of intracellular reactive oxygen species and Ca2+ productions and caspase-3 activation. Mutat Res. 2006;601:71–82. doi: 10.1016/j.mrfmmm.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 34.Tsou MF, Lu HF, Chen SC, Wu LT, Chen YS, Kuo HM, Lin SS, Chung JG. Involvement of Bax, Bcl-2, Ca2+ and caspase-3 in capsaicin-induced apoptosis of human leukemia HL-60 cells. Anticancer Res. 2006;26:1965–1971. [PubMed] [Google Scholar]
- 35.Ito K, Nakazato T, Yamato K, Miyakawa Y, Yamada T, Hozumi N, Segawa K, Ikeda Y, Kizaki M. Induction of apoptosis in leukemic cells by homovanillic acid derivative, capsaicin, through oxidative stress: Implication of phosphorylation of p53 at Ser-15 residue by reactive oxygen species. Cancer Res. 2004;64:1071–1078. doi: 10.1158/0008-5472.CAN-03-1670. [DOI] [PubMed] [Google Scholar]
- 36.Abdel-Salam Omar ME. Capsaicin as a Therapeutic Molecule. Springer; Basel: 2014. pp. 181–203. [PubMed] [Google Scholar]
- 37.Pramanik KC, Boreddy SR, Srivastava SK. Role of mitochondrial electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PLoS One. 2011;6:e20151. doi: 10.1371/journal.pone.0020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kinnally KW, Peixoto PM, Ryu SY, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta. 2011;1813:616–622. doi: 10.1016/j.bbamcr.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeong SY, Seol DW. The role of mitochondria in apoptosis. BMB Rep. 2008;41:11–22. doi: 10.5483/BMBRep.2008.41.1.011. [DOI] [PubMed] [Google Scholar]
- 40.Brunelle JK, Zhang B. Apoptosis assays for quantifying the bioactivity of anticancer drug products. Drug Resist Updat. 2010;13:172–179. doi: 10.1016/j.drup.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 41.O'Neill J, Brock C, Olesen AE, Andresen T, Nilsson M, Dickenson AH. Unravelling the mystery of capsaicin: A tool to understand and treat pain. Pharmacol Rev. 2012;64:939–971. doi: 10.1124/pr.112.006163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al Othman ZA, Ahmed YB, Habila MA, Ghafar AA. Determination of capsaicin and dihydrocapsaicin in capsicum fruit samples using high performance liquid chromatography. Molecules. 2011;16:8919–8929. doi: 10.3390/molecules16108919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.AbdelSalam OME, editor. Capsaicin as a Therapeutic Molecule. In: Capsaicin as a Therapeutic Molecule. Vol. 68. Springer Basel Ag; Picassoplatz 4, Basel, 4052, Switzerland: 2014. [DOI] [Google Scholar]