

ABSTRACT
Hypothalamic AMP-activated protein kinase (AMPK) plays important roles in the regulation of food intake by altering the expression of orexigenic or anorexigenic neuropeptides. However, little is known about the mechanisms of this regulation. Here, we report that hypothalamic AMPK modulates the expression of NPY (neuropeptide Y), an orexigenic neuropeptide, and POMC (pro-opiomelanocortin-α), an anorexigenic neuropeptide, by regulating autophagic activity in vitro and in vivo. In hypothalamic cell lines subjected to low glucose availability such as 2-deoxy-d-glucose (2DG)-induced glucoprivation or glucose deprivation, autophagy was induced via the activation of AMPK, which regulates ULK1 and MTOR complex 1 followed by increased Npy and decreased Pomc expression. Pharmacological or genetic inhibition of autophagy diminished the effect of AMPK on neuropeptide expression in hypothalamic cell lines. Moreover, AMPK knockdown in the arcuate nucleus of the hypothalamus decreased autophagic activity and changed Npy and Pomc expression, leading to a reduction in food intake and body weight. AMPK knockdown abolished the orexigenic effects of intraperitoneal 2DG injection by decreasing autophagy and changing Npy and Pomc expression in mice fed a high-fat diet. We suggest that the induction of autophagy is a possible mechanism of AMPK-mediated regulation of neuropeptide expression and control of feeding in response to low glucose availability.
KEYWORDS: appetite, autophagy, glucoprivation, glucose deprivation, hypothalamic AMPK, neuropeptide
Introduction
Appetite regulation has garnered significant attention due to its importance for maintaining energy balance, which is determined by calorie intake and energy expenditure. Research on appetite control is needed to cure metabolic diseases such as obesity and its complications, which have become a worldwide epidemic.1,2 However, interactions among upstream regulators of appetite-related effectors and regulatory mechanisms underlying their interactions remain to be elucidated.
Feeding behavior is centrally controlled by the hypothalamus, which is located in the medial basal region of the brain. The hypothalamus senses and integrates diverse signals (such as the levels of glucose3-7 and hormones8-11) from blood vessels and through the third ventricle, and thereby regulates food intake. Two major neuronal populations in the arcuate nucleus (ARC) of the hypothalamus play a critical role in controlling food intake.12,13 One of them co-expresses orexigenic NPY (neuropeptide Y) and AGRP (agouti related neuropeptide) and promotes food intake,14,15 whereas the other population co-expresses anorexigenic CARTPT/CART (CART prepropeptide) and POMC (pro-opiomelanocortin-α) and inhibits food intake.16-18
Hypothalamic AMP-activated protein kinase (AMPK) plays a pivotal role in the regulation of energy homeostasis and provides a link between peripheral signals and central control of feeding behavior by sensing nutrient availability.19-21 Hypothalamic AMPK is activated under nutrient deficiencies such as hypoglycemia,22,23 and its activity is strongly affected by peripheral hormones such as LEP (leptin) and GHRL (ghrelin).19,20 Intracerebroventricular administration of the AMPK activator 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) to the hypothalamic paraventricular nucleus significantly increases food intake,19 emphasizing the importance of hypothalamic AMPK in controlling feeding behavior and energy metabolism. The expression of constitutively active AMPK in the medial hypothalamus increases Npy and Agrp mRNA expression levels in fasted mice, whereas the levels of the corresponding neuropeptides are decreased in mice fed ad libitum that express the dominant-negative (DN) PRKAA1/α1 and PRKAA2/α2 subunits of AMPK.20 Food intake and body weight of these mice change significantly in accordance with the alterations in neuropeptide expression. Furthermore, fasted mice with a POMC neuron-specific Prkaa2 knockout have a higher ratio of orexigenic neuropeptides over Pomc mRNA (Npy:Pomc and Agrp:Pomc) than the fasted wild-type control. Although these studies support the view that hypothalamic AMPK exerts its metabolic effects by modulating neuropeptide transcription in the ARC,24 the molecular mechanism by which hypothalamic AMPK regulates neuropeptide expression has not been clearly understood.
Emerging evidence suggests that macroautophagy (hereafter autophagy), a self-degradation process that maintains cellular homeostasis by delivering cytoplasmic components to the lysosome, is closely involved in the regulation of food intake by the hypothalamus.25-30 In mice with a POMC neuron-specific deletion of Atg7 (autophagy-related 7), both food intake and body weight increase,27 and mice lacking Atg12 in hypothalamic POMC neurons show elevated weight gain and adiposity associated with increased food intake.30 Furthermore, hypothalamic POMC neuron-specific loss of autophagy decreases α-MSH (α-melanocyte stimulating hormone) levels and elevates adiposity, which is consistent with increased food consumption.25 In contrast, selective loss of Atg7 in hypothalamic AGRP neurons reduces food consumption during refeeding after 6 or 24 h of fasting, in line with decreased AGRP and increased POMC expression levels.26 Although these studies indicate that hypothalamic autophagy plays a critical role in the regulation of feeding behavior and body metabolism, the physiological conditions that indeed regulate hypothalamic autophagy remain to be elucidated.
ULK1 (unc-51 like kinase 1) is a key initiator of the autophagic process and is inhibited by MTOR (mechanistic target of rapamycin [serine/threonine kinase]), a regulator of cell growth and proliferation.31-34 AMPK phosphorylates RPTOR/raptor (regulatory associated protein of MTOR, complex 1) to inhibit the RPTOR-containing MTOR complex 1 (MTORC1).35 The inhibition of this complex releases ULK1 from MTORC1, leading to autophagy induction.36-38 In addition, AMPK activates autophagy by directly phosphorylating ULK1 under conditions of glucose starvation.31,39-41 Moreover, autophagy induction by AMPK through modulating MTORC1 and ULK1 was also reported in neurons.42
Although these studies suggest that AMPK activity is closely involved in the induction of autophagy, it is not clear whether hypothalamic AMPK-induced autophagy regulates food intake. In this report, we observed that AMPK activation by low glucose availability induced autophagy, leading to changes in Npy and Pomc expression in hypothalamic neuronal cells. Furthermore, in vivo ARC-specific AMPK knockdown suppressed autophagy triggered by glucoprivation induced by intraperitoneal (ip) injection of the glycolysis blocker 2-deoxy-d-glucose (2DG), and thereby significantly decreased food intake and body weight in mice fed a high-fat diet (HFD). To the best of our knowledge, this is the first report demonstrating that hypothalamic AMPK regulates feeding behavior by controlling autophagy-mediated changes in neuropeptide expression in the hypothalamus.
Results
2DG and glucose-free medium activate AMPK and induce autophagy via modulation of ULK1 and MTORC1
Several studies have shown that AMPK induces autophagy under low glucose availability in various cell types.43-46 To examine whether this is true for mouse embryonic hypothalamic cell lines (NPY-expressing mHypoE-N41 and POMC-expressing mHypoE-N43/5), we used 2 conditions of low glucose availability. Glucoprivation was induced by adding 2DG (20 mM) into medium containing 25 mM glucose (the same medium without 2DG was used as control). Glucose deprivation was induced by changing 25 mM glucose medium to glucose-free medium (0 mM glucose). Both 2DG and glucose-free medium increased the level of AMPK phosphorylation at Thr172 (which is an indicator of AMPK activation)47,48 in comparison with the control (Fig. 1A and B). AMPK activation induced by 2DG and glucose-free medium led to phosphorylation of ACAC/ACC (acetyl-coenzyme A carboxylase) at Ser79; ACAC is a well-known AMPK substrate.49,50 The AMPK targets related to autophagy induction, such as ULK1 and RPTOR, were also phosphorylated, in accordance with previous reports.35,37,51 The phosphorylation levels of ULK1 (Ser555) and RPTOR (Ser792) were higher than the control in both cell lines (N41 and N43/5) under low glucose availability. We next examined autophagy induction using NBR1 (neighbor of Brca1 gene 1) and MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3). NBR1 acts as a receptor protein (it brings cargo to the phagophore, the precursor of the autophagosome) and is degraded upon autophagy activation.52,53 LC3 is a well-known marker of autophagic activity; the LC3-I form is converted to LC3-II when autophagy is induced.54-56 2DG and glucose-free medium decreased the levels of NBR1 and increased the levels of LC3-II, indicating the induction of autophagy. Collectively, these results suggest that 2DG or glucose-free medium elevates the phosphorylation of AMPK, ULK1 and RPTOR, leading to induction of autophagy.
Figure 1.

2DG and glucose-free medium induce autophagy through the AMPK signaling pathway. N41 and N43/5 cells were subjected to low glucose conditions (A) by adding 2-deoxy-d-glucose (2DG: 20 mM) into medium containing 25 mM glucose, (B) by replacing 25 mM glucose medium with glucose-free medium (0 mM glucose) for the indicated times. (C) Both cells were treated with 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR: 0.5 mM) for 12 h. (D) Treatment with lysosomal protease inhibitors, E64d (10 μg/ml) and pepstatin A (PepA: 10 μg/ml), for 4 h increased the accumulation of LC3-II compared with nontreated N41 and N43/5 cells under low glucose conditions or AICAR treatment. The levels of p-AMPK (Thr172), total AMPK, p-ACAC (Ser79), total ACAC, p-ULK1 (Ser555), total ULK1, p-RPTOR (Ser792), total RPTOR, NBR1, LC3, and GAPDH were detected by immunoblotting. p-AMPK, p-ACACB (upper band), p-ULK1, and p-RPTOR were normalized to corresponding total proteins. Other protein levels were normalized to GAPDH. Numbers under blots indicate relative quantitative mean values of independent replicates (A and B: n = 2; C and D: n = 3).
To confirm that AMPK activation is sufficient to increase the autophagy flux, hypothalamic neuronal cells were treated with AICAR (0.5 mM), a well-known AMPK activator, for 12 h (Fig. 1C). AICAR increased the levels of phosphorylation of AMPK, ACAC, ULK1, and RPTOR in both N41 and N43/5 cells. AICAR also reduced the levels of NBR1 and elevated the levels of LC3-II. Taken together, these results suggest that AMPK activation by low glucose availability or AICAR induces autophagy in hypothalamic cells.
We confirmed the autophagic flux by assessing a change in LC3-II levels using the lysosomal protease inhibitors E64d and peptstatin A along with the treatment of 2DG, 0 mM glucose and AICAR (Fig. 1D).55 In both cell lines, inhibition of lysosomal activity led to increased LC3-II, indicating that the increase in LC3-II induced by low glucose availability or AICAR reflects an increased autophagy flux.
AMPK inhibition prevents AMPK-induced autophagy
Next, we further verified whether AMPK is required for autophagy induced by low glucose availability. Pharmacological inhibition of AMPK by compound C (16 μM) decreased 2DG- and glucose-free medium-induced elevation of phosphorylation of AMPK, ACAC, ULK1, and RPTOR. In addition, compound C reduced the levels of LC3-II, indicating that autophagy induction by 2DG and glucose-free medium was attenuated (Fig. 2A). We also confirmed the autophagy flux induced by low glucose availability during the treatment with compound C by assessing a change in LC3-II levels using lysosomal protease inhibitors (Fig. 2B). The accumulation of LC3-II caused by E64d and pepstatin A was decreased by compound C in both 2DG and 0 mM glucose conditions.
Figure 2.

Inhibition of hypothalamic AMPK suppresses autophagy induced by low glucose availability. (A) N41 and N43/5 cells were treated with 2DG (20 mM) or exposed to glucose-free medium (0 mM glucose) with or without compound C (CC: 16 μM) for 4 h. The levels of p-AMPK (Thr172), total AMPK, p-ACAC (Ser79), total ACAC, p-ULK1 (Ser555), total ULK1, p-RPTOR (Ser792), total RPTOR, LC3, and GAPDH were determined by immunoblotting. (B) Treatment with lysosomal protease inhibitors, E64d (10 μg/ml) and pepstatin A (PepA: 10 μg/ml), for 4 h in compound C-treated N41 and N43/5 cells confirmed increased autophagy flux induced by low glucose availability. (C) N41 and N43/5 cells transiently expressing mRFP-GFP-LC3 were treated with 2DG or glucose-free medium with or without CC for 4 h. The bottom graphs indicate relative fluorescence intensity (RFI) of RFP and GFP along a white line in the upper enlarged images. The Y-axis shows RFI and the X-axis shows relative distance. Scale bars: 10 μm. (D) Average number of mRFP-GFP-LC3 puncta per cell. At least 30 cells were counted per condition. Red bar and symbols refer to red puncta, yellow bar and symbols to yellow puncta; and white bar and black symbols to total puncta (***, p < 0.001 for 25 mM glucose vs. 2DG or glucose-free medium; ###, p < 0.001 for vehicle vs. CC in 2DG-treated groups; †††, p < 0.001 for vehicle vs. CC in groups exposed to glucose-free medium). (E) N41 and N43/5 cells infected with lentiviruses containing control or both AMPK (against α1 and α2) shRNAs were exposed to low glucose conditions for 4 h, and the levels of p-AMPK (Thr172), total AMPK, p-ACAC (Ser79), total ACAC, p-ULK1 (Ser555), total ULK1, p-RPTOR (Ser792), total RPTOR, LC3, and GAPDH were detected by immunoblotting. p-AMPK, p-ACACB (upper band), p-ULK1, and p-RPTOR were normalized to corresponding total proteins. Protein levels of p-AMPK (in panel D), total AMPK, and LC3-II were normalized to GAPDH. Numbers under blots indicate relative quantitative mean values of independent replicates (A and D: n = 2, B: n = 3).
Observation of mRFP-GFP-LC3 fluorescent puncta further confirmed that autophagy in N41 and N43/5 cells was regulated by AMPK activity (Fig. 2C and D). Due to different stabilities of GFP and RFP at low pH,57 the autophagic flux can be assessed by counting autophagosomes (yellow) and autolysosomes (red). The increased number of total puncta and red puncta per cell in both cell lines upon 2DG addition and glucose-free medium indicated an increased autophagy flux, in accordance with a previous report.58 In contrast, compound C decreased the number of autophagosomes and autolysosomes, indicating that the induction of autophagy by 2DG and glucose-free medium was diminished by AMPK inhibition (Fig. 2D).
Although compound C has been used as a specific AMPK inhibitor, others have reported that compound C could affect autophagy independently of AMPK activity.55,59,60 To further confirm the effect of AMPK inhibition on autophagy, we genetically suppressed AMPK by infecting hypothalamic cells with lentiviruses expressing short hairpin RNAs (shRNAs) targeting PRKAA1 and PRKAA2. We knocked down the expression of both PRKAA1 and PRKAA2 because we observed a compensatory response between the 2 isoforms (data not shown) in line with other reports.61,62 To control for off-target effects, 2 different shRNAs against each isoform were used and effective knockdown was confirmed by reduced levels of total AMPK and its phosphorylation. AMPK knockdown dramatically reduced phosphorylation of ACAC, ULK1, and RPTOR (Fig. 2E). Induction of LC3-II by 2DG or glucose-free medium was diminished in AMPK knockdown cells (Fig. 2E). AMPK knockdown also significantly decreased phosphorylation of ACAC, ULK1 and RPTOR, and the levels of LC3-II in the 25 mM glucose control. Of note, quantification of immunoreactivity for LC3-II in Fig. 2E showed that the reduction of LC3-II by shAmpk was greater in the N43/5 cells than in N41 cells. We assume that this differential effect is due to the different efficiency of AMPK knockdown and its effects on activation of downstream targets, ULK1 and RPTOR, between N41 and N43/5 cells. As shown in the immunoreactivity quantification of Fig. 2E, knocked down N43 cells showed relatively lower levels of phosphorylated (p)-ULK1 and p-RPTOR compared to N41 cells under low glucose conditions, which may contribute to more reduced levels of LC3-II. Taken together, pharmacological or genetic inhibition of AMPK diminished autophagy, suggesting that autophagy induction by low glucose availability as well as basal autophagy is AMPK-dependent.
AMPK upregulates Npy and downregulates Pomc expression
As N41 and N43/5 cells express NPY and POMC, respectively, we next ascertained whether AMPK affects neuropeptide expression in these cell lines. AICAR treatment significantly increased Npy mRNA expression in N41 cells and decreased Pomc mRNA expression in N43/5 cells (Fig. 3A).
Figure 3.
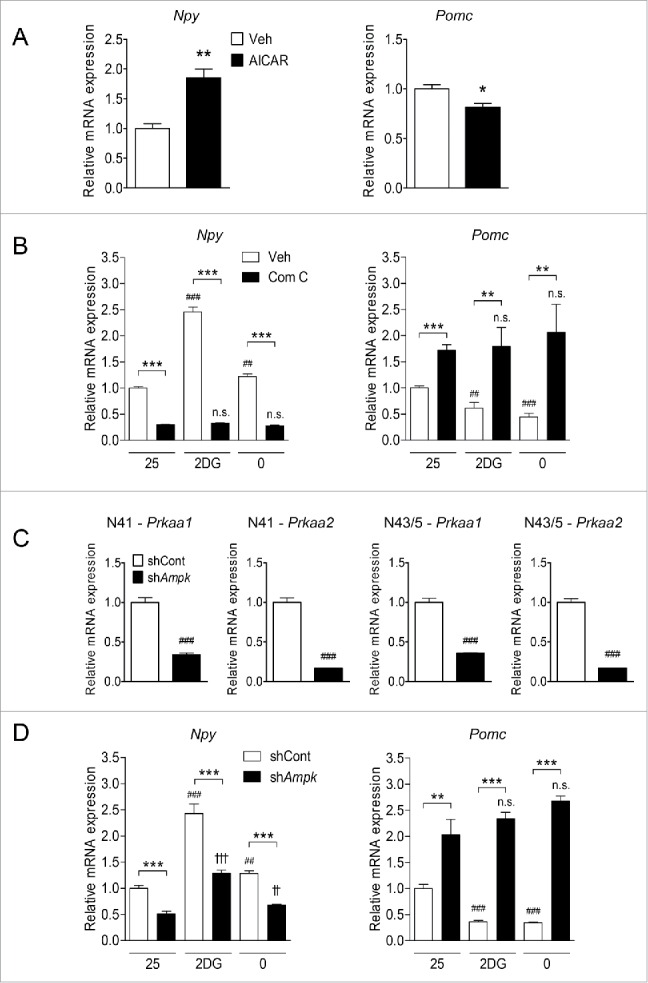
Hypothalamic AMPK regulates Npy and Pomc expression in response to low glucose availability. (A) Npy and Pomc mRNA expression levels in hypothalamic neuronal cells were measured by quantitative real-time PCR. N41 and N43/5 cells were treated with AICAR (0.5 mM) for 12 h (*, p< 0.05; **, p < 0.01 for vehicle [Veh] vs. AICAR; N41: n = 6, N43/5: n = 9). (B) Levels of Npy and Pomc mRNA in N41 and N43/5 cells treated with 2DG (20 mM) or glucose-free medium (0 mM glucose) with or without compound C (Com C; 16 μM) for 4 h (**, p < 0.01; ***, p < 0.001 for vehicle [Veh] vs. compound C; ##, p < 0.01; ###, p < 0.001 for 25 mM glucose vs. 2DG or glucose-free medium in vehicle-treated groups; n.s., not significant for 25 mM glucose vs. 2DG or glucose-free medium in compound C-treated groups; N41: n = 7 or 8, N43/5: n = 6-8). (C) Prkaa1 and Prkaa2 mRNA expression levels in hypothalamic neuronal cells were measured by quantitative real-time PCR. Cells were infected with lentiviruses containing control (shCont) or both Prkaa1 and Prkaa2 (shAmpk) shRNAs (###, p < 0.001 for shCont vs. shAmpk; N41: n = 6, N43/5: n = 8). (D) Npy and Pomc mRNA expression levels in hypothalamic neuronal cells measured by quantitative real-time PCR. Cells were infected as in (C) and then 2DG or glucose-free medium was given for 4 h (**, p < 0.01; ***, p < 0.001 for shCont vs. shAmpk; ##, p < 0.01; ###, p < 0.001 for 25 mM glucose vs. 2DG or glucose-free medium in the shCont group; ††, p < 0.01; †††, p < 0.001; n.s. for 25 mM glucose vs. 2DG or glucose-free medium in the shAmpk group; N41: n = 6, N43/5: n = 4–6).
To investigate the effects of AMPK inhibition on NPY and POMC expression, N41 and N43/5 cells were exposed to 2DG or glucose-free medium with or without compound C (Fig. 3B). 2DG or glucose-free medium increased Npy mRNA levels in N41 cells and decreased Pomc mRNA levels in N43/5 cells. More importantly, the levels of Npy mRNA expression elevated in response to these treatments were significantly decreased when compound C was added. Compound C also abolished the reduction of Pomc expression by 2DG and glucose-free medium. These results indicate that blocking AMPK activity reverses the changes in Npy and Pomc mRNA expression induced by low glucose availability.
To corroborate the effects of pharmacological activation and inhibition of AMPK on the levels of Npy and Pomc mRNA, we genetically knocked down AMPK using lentiviruses containing Prkaa1 and Prkaa2 shRNAs. The levels of both Prkaa1 and Prkaa2 transcripts were significantly reduced by 66.2% and 83.3%, respectively, in N41 cells, and by 64.1% and 83.0% in N43/5 cells compared to those in control lentivirus-infected cells (Fig. 3C). In AMPK knockdown cells, upregulated Npy expression induced by 2DG and glucose-free medium was attenuated, and the reduction of Pomc expression triggered by 2DG and glucose-free medium was abolished (Fig. 3D).
These results suggest that AMPK positively regulates Npy expression in N41 cells and negatively regulates Pomc expression in N43/5 cells, and modulation of neuropeptide expression by glucoprivation or glucose deprivation requires AMPK activity in both cell lines.
AMPK-induced autophagy regulates Npy and Pomc expression under low glucose availability
We next ascertained whether autophagy mediates the regulation of Npy and Pomc expression by activated AMPK under low glucose availability in hypothalamic cells. We knocked down the essential autophagy gene Atg5 using small interfering RNAs (siRNAs) and confirmed that Atg5 knockdown inhibited the induction of autophagy by glucoprivation and glucose deprivation (Fig. 4A). In both N41 and N43/5 cells, Atg5 knockdown had no effect on the extent of AMPK phosphorylation but attenuated the increase in LC3-II in response to these cues (Fig. 4A). Autophagy inhibition by Atg5 knockdown in both cells attenuated the changes in Npy and Pomc induced by 2DG or glucose-free medium compared to the control siRNAs (Fig. 4B). In addition, autophagy inhibition by Atg5 knockdown reduced Npy expression and induced Pomc to some degree even at 25 mM glucose, suggesting that autophagy may also be responsible for basal expression of Npy and Pomc.
Figure 4.
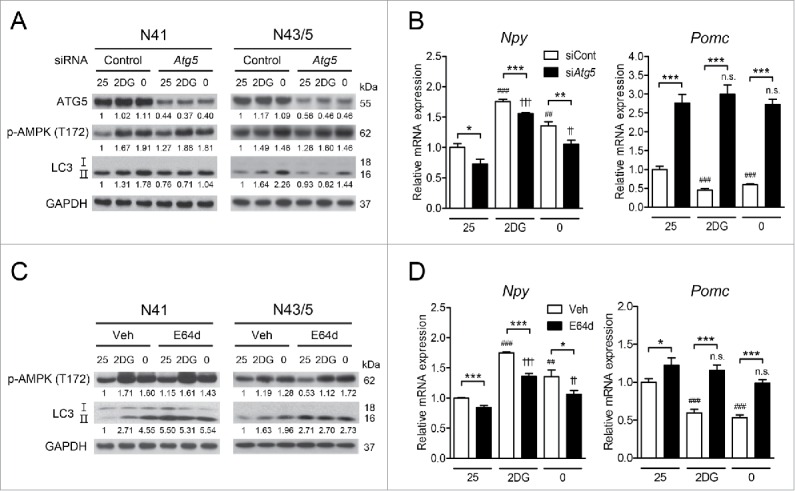
Inhibition of autophagy impedes AMPK-dependent regulation of neuropeptide expression. (A) ATG5, p-AMPK (Thr172), LC3, and GAPDH were detected by immunoblotting. (B) Atg5 was knocked down using siRNAs and the levels of Npy and Pomc mRNA in N41 and N43/5 cells exposed to 2DG (20 mM) or glucose-free medium (0 mM glucose) for 4 h were measured by quantitative real-time PCR (*, p < 0.05; **, p < 0.01; ***, p < 0.001 for control siRNAs (siCont) vs. Atg5 siRNAs (siAtg5); ##, p < 0.01; ###, p < 0.001 for 25 mM glucose vs. 2DG or glucose-free medium in the control group; ††, p < 0.01; †††, p < 0.001, n.s. for 25 mM glucose vs. 2DG or glucose-free medium in the Atg5 knockdown group; N41: n = 6, N43/5: n = 4-6). (C) The levels of p-AMPK (Thr172), LC3, and GAPDH were determined by immunoblotting. (D) The levels of Npy and Pomc mRNA in N41 and N43/5 cells after E64d (20 μg/ml) treatment in medium with 2DG or glucose-free medium for 4 h were measured by quantitative real-time PCR (*, p< 0.05; ***, p< 0.001 for vehicle [Veh] vs. E64d; ##, p < 0.01; ###, p < 0.001 for 25 mM glucose vs. 2DG or glucose-free medium in vehicle-treated groups; ††, p < 0.01; †††, p < 0.001; n.s. for 25 mM glucose vs. 2DG or glucose-free medium in E64d-treated groups; N41: n = 6, N43/5: n = 5 or 6). Protein levels were normalized to GAPDH. Numbers under blots indicate relative quantitative mean values of independent replicates (A and C: n = 3).
We also pharmacologically inhibited autophagy using the cysteine protease inhibitor E64d (20 μg/ml). E64d treatment led to LC3-II accumulation due to inhibition of autophagy; however, AMPK activation in response to 2DG and glucose-free medium remained unaffected (Fig. 4C). Similar to Atg5 knockdown, E64d reduced Npy expression and induced Pomc expression at 25 mM glucose (Fig. 4D). We also found that the effects of low glucose utilization on neuropeptide expression were attenuated when E64d treatment was used together with 2DG or glucose-free medium (Fig. 4D). Taken together, our data show that autophagy is required for low glucose availability-induced modulation of Npy and Pomc expression.
Autophagy induction by MTOR inhibition regulates Npy and Pomc expression
Because AMPK phosphorylated ULK1 (Ser555) and RPTOR (Ser792), we further confirmed whether AMPK downstream targets in the autophagy pathway are involved in neuropeptide expression. The MTOR inhibitor rapamycin (20 nM) inhibited MTOR, as evidenced by reduced phosphorylation of RPS6KB1/S6K1 (ribosomal protein S6 kinase, polypeptide 1) at Thr389 (Fig. 5A), which is a MTOR substrate.63 Rapamycin reduced MTOR activity leading to autophagy induction verified by decreased phosphorylation of ULK1 at Ser757 (Fig. 5A), which is the MTOR inhibitory phosphorylation site.31 We also assessed lipidation of GABARAP (gamma-aminobutyric acid receptor associated protein) to verify autophagy induced by rapamycin, because lipidated GABARAP (GABARAP-II) also acts at a late stage of autophagosome biogenesis and is indicative of autophagy activity.64,65 The levels of LC3-II and GABARAP-II were elevated in response to rapamycin, indicating autophagy induction (Fig. 5A). Rapamycin-induced autophagy increased Npy expression and decreased Pomc expression (Fig. 5B).
Figure 5.

Atg5 knockdown reduces rapamycin-induced neuropeptide mRNA levels. (A) N41 and N43/5 cells were treated with vehicle (Veh) for 4 h or with rapamycin (Rapa: 20 nM) for 1 h. The levels of p-RPS6KB1 (Thr389), total RPS6KB1, p-ULK1 (Ser757), total ULK1, LC3, GABARAP, and GAPDH were examined by immunoblotting. (B) The levels of Npy and Pomc mRNA in N41 cells treated with rapamycin for 4 h and in N43/5 cells treated for 1 h (*, p < 0.05; ***, p < 0.001 for Veh vs. Rapa; N41: n = 6, N43/5: n = 6 or 7). (C) Atg5 was knocked down using siRNAs and cells were treated with rapamycin for 4 h (N41) or 1h (N43/5). The levels of ATG5, p-SRP6KB1 (Thr389), total RPS6KB1, p-ULK1 (Ser757), total ULK1, LC3, GABARAP, and GAPDH were examined by immunoblotting. (D) The levels of Npy and Pomc mRNA in cells treated with rapamycin (N41, 4 h; N43/5, 1 h) after Atg5 knockdown using siRNAs (**, p < 0.01; ***, p < 0.001 for control siRNAs [siCont] vs. Atg5 siRNAs [siAtg5]; ##, p < 0.01; ###, p < 0.001 for Veh vs. Rapa in control siRNA groups; †††, p < 0.001; n.s. for Veh vs. Rapa in Atg5 siRNA groups; N41: n = 6, N43/5: n = 6). p-RPS6KB1 and p-ULK1 were normalized to corresponding total proteins. Other protein levels were normalized to GAPDH. Numbers under blots indicate relative quantitative mean values of independent replicates (A and C: n = 3). n/a: not applicable.
Atg5 knockdown together with rapamycin treatment confirmed the role of autophagy induction in neuropeptide expression (Fig. 5C and D). In Atg5 knockdown cells, rapamycin failed to increase LC3-II and GABARAP-II. Atg5 knockdown attenuated the increase in Npy and decrease in Pomc induced by rapamycin (Fig. 5D). Taken together, these data suggest that autophagy induction by AMPK downstream signaling mediates alteration of Npy and Pomc expression.
Mice with AMPK knockdown in the ARC show decreased autophagy, food intake, and body weight
To assess the role of AMPK in the regulation of food intake in vivo, we injected lentiviruses carrying Prkaa1 and Prkaa2 shRNAs stereotaxically into the ARC of mice (shAmpk group). Lentiviruses were successfully targeted to the ARC (Fig. 6A). On d 6 and 7 after viral injection, food intake of the shAmpk group substantially decreased in comparison with that of the control shRNAs (shCont) group when fed a normal diet (ND). Due to a strong reduction in food intake on d 6, body weight of the shAmpk group was also significantly lower than that of the shCont group from d 6.
Figure 6.
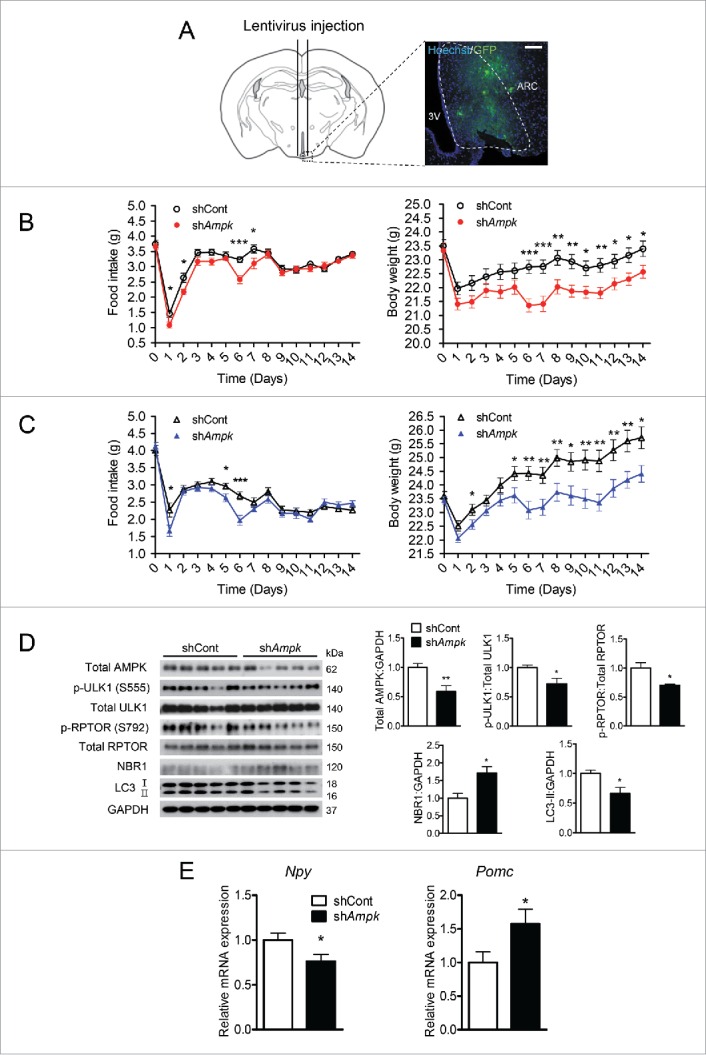
Knockdown of AMPK in the ARC decreases autophagy, food intake and body weight in mice fed ND or HFD. (A) Schematic illustration of the surgical coordinates and expression of eGFP delivered by lentiviruses targeting the arcuate nucleus (ARC). Scale bar: 200 μm. Daily food consumption and body weight changes in male mice fed (B) a normal diet (ND) and (C) a high-fat diet (HFD) after lentivirus injection for the indicated days (*, p < 0.05; **, p < 0.01; ***, p < 0.001 for the control shRNA group [shCont] vs. AMPK shRNA [shAmpk] group; ND: n = 20; HFD: shCont group, n = 18; shAmpk group, n = 23). (D) Relative levels of total AMPK, p-ULK1 (Ser555), total ULk1, p-RPTOR (Ser792), total RPTOR, NBR1, LC3, and GAPDH in the ARC tissue from the shCont and shAmpk groups fed a HFD were examined by immunoblotting. p-ULK1 and p-RPTOR were normalized to corresponding total proteins. Other protein levels were normalized to GAPDH. Protein levels in the control shRNA group were arbitrarily set to 1.0 (*, p < 0.05; **, p < 0.01 for shCont vs. shAmpk; n = 5). (E) Npy and Pomc mRNA expression levels in the ARC from shCont and shAmpk groups fed a HFD measured by quantitative real-time PCR. (*p < 0.05 for shCont group vs. shAmpk group; n = 8).
To evaluate the effects of AMPK knockdown on food intake and body weight gain under acute metabolic stress, we fed mice a HFD for 2 wk after virus injection. In accordance with the ND experiment, a significant reduction in food intake was observed 6 d after viral injection when fed a HFD (Fig. 6C). Body weight of the shAmpk group became significantly lower than that of the shCont group from d 6 (Fig. 6C). In both ND and HFD experiments, a dramatic decrease in body weight was observed when food intake was reduced in the shAmpk groups. The difference in body weight between the 2 groups (shCont groups vs. shAmpk groups in both ND and HFD) was sustained after d 7, probably because both groups consumed comparable amounts of food after d 7, suggesting that changes in body weight were caused by alteration of food consumption (Fig. 6B and C).
The expression of AMPK in the ARC in the shAmpk group dropped by 41.0% of the shCont group fed a HFD. AMPK-mediated phosphorylation of ULK1 (Ser555) and RPTOR (Ser792) was significantly reduced (Fig. 6D). In addition, the accumulation of NBR1 and the reduction in LC3-II levels suggested that the levels of autophagy in the ARC were lower in the shAmpk group than in the shCont group (Fig. 6D). Consistent with the in vitro data, AMPK knockdown reduced the levels of Npy mRNA and increased the levels of Pomc mRNA in comparison with those in the shCont group (Fig. 6E).
AMPK knockdown in the ARC prevents 2DG-induced hyperphagia
Based on the in vitro finding that AMPK inhibition attenuates changes in Npy and Pomc expression induced by 2DG, we tested whether these changes in neuropeptide expression indeed drive alteration of food intake in vivo. Mice were given an ip injection of 2DG (500 mg/kg of body weight), which was previously reported to activate hypothalamic AMPK.66,67 In ND-fed mice, 2DG increased food intake 2 h after injection (Fig. 7A). Intriguingly, the 2DG-induced hyperphagic effects were greater in HFD-fed mice than in ND-fed mice (Fig. 7B). In HFD-fed mice, the cumulative food intake was significantly increased 1 h and 2 h after 2DG injection and remained elevated until 4 h in comparison with the saline-injected group.
Figure 7.

Hypothalamic AMPK knockdown attenuates autophagy and neuropeptide expression, and suppresses 2DG-induced hyperphagia. Cumulative food consumption after ip 2DG injection (500 mg/kg of body weight) in mice fed (A) a normal diet (ND) or (B) a high-fat diet (HFD) for the indicated time (**, p < 0.01; ***, p < 0.001; n.s. for saline vs. 2DG-injected group; n = 8). (C) Food consumption of mice that were sacrificed 1 h after 2DG injection. Mice were fed a HFD for 6 d after lentivirus injection, and 2DG was administered on d 6 (**, p < 0.01; ***, p < 0.001 for saline vs. 2DG in the control shRNA [shCont] group or Prkaa1+Prkaa2 shRNA [shAmpk] group; ###, p < 0.001 for shCont vs. shAmpk in the 2DG-injected group; n = 8). (D) Relative levels of total AMPK in the ARC and lateral hypothalamus (LH) (**, p < 0.01 for shCont vs. shAmpk; ARC n = 8, LH n = 8). (E) Relative levels of p-AMPK (Thr172), p-ULK1 (Ser555), and p-RPTOR (Ser792) in the ARC from the shCont group; mice were sacrificed during the light or dark cycle (*, p < 0.05; **, p < 0.01 for light cycle vs. dark cycle; n = 3). (F) Relative levels of p-AMPK (Thr172), p-ULK1 (Ser555), p-RPTOR (Ser792), NBR1, and LC3-II in the ARC of shCont and shAmpk mice that were sacrificed 1 h after 2DG injection. p-ULK1 and p-RPTOR were normalized to corresponding total proteins. Other protein levels were normalized to GAPDH. Protein levels in the shCont group were arbitrarily set to 1.0 (*, p < 0.05; **, p < 0.01 for saline vs. 2DG in the shCont group; n = 4). (G) Npy and Pomc mRNA expression levels in the ARC from shCont and shAmpk mice that were sacrificed 1 h after 2DG injection. Mice were fed a HFD for 6 d after lentivirus injection, and 2DG was administered on d 6 (*, p < 0.05 for saline vs. 2DG in shCont; n = 6 or 7).
Since 2DG showed greater hyperphagic effects in HFD-fed mice than in ND-fed mice, we evaluated its effects on food intake in the control and AMPK knockdown HFD-fed groups on d 6 after virus injection (when AMPK was effectively knocked down). In the shCont group, 2DG-injected mice consumed a greater amount of food than did saline-injected mice. Interestingly, AMPK knockdown significantly blocked 2DG-induced hyperphagia (Fig. 7C). The expression of AMPK was significantly decreased in the ARC but not in the lateral hypothalamus (LH) of the shAmpk group, which verifies specific AMPK knockdown in the ARC (Fig. 7D). Notably, 2DG increased phosphorylation of AMPK (Thr172), ULK1 (Ser555), and RPTOR (Ser792) in the ARC of the shCont group. In addition, a decrease in NBR1 and an increase in LC3-II in the 2DG-injected shCont group suggest that 2DG induced autophagy in the ARC. Interestingly, AMPK knockdown in the ARC diminished these changes induced by 2DG (Fig. 7F). Because hypothalamic AMPK activity peaks during the dark cycle,68 we observed lower phosphorylation of AMPK, ULK1, and RPTOR during the light cycle than the dark cycle (Fig. 7E). Unexpectedly, there was no further decrease in p-AMPK levels in the saline-treated shAmpk group compared with the saline-treated shCont group (Fig. 7F). That result might be due to a compensatory increase in p-AMPK levels of remaining AMPK under knockdown conditions in vivo. However, 2DG treatment failed to increase p-AMPK levels in the shAmpk group, consistent with attenuated changes in AMPK downstream targets and LC3-II following AMPK knockdown in the ARC (Fig 7F). 2DG elevated Npy and inhibited Pomc expression in the shCont group, whereas these changes were strongly abolished in the shAmpk group (Fig. 7G). Taken together, these results suggest that AMPK knockdown in the ARC suppresses 2DG-induced autophagy and hyperphagia by altering the expression of Npy and Pomc.
Discussion
In this study, we hypothesized that autophagy mediates the effects of hypothalamic AMPK on modulation of neuropeptides that play a critical role in regulation of food intake. Our in vitro study using hypothalamic cell lines shows that low glucose utilization activates AMPK, which phosphorylates ULK1 and RPTOR, induces autophagy and modulates Npy and Pomc mRNA expression. Low glucose availability-induced effects of AMPK on the neuropeptides were abolished by inhibition of autophagy. Furthermore, changes in autophagy and neuropeptide expression induced by ARC-specific AMPK knockdown in mice were attributed to the attenuation of glucoprivation-induced hyperphagia.
It remains controversial whether autophagy in the brain responds to nutritional status such as starvation. It has been suggested that autophagy in the brain is unlikely to undergo drastic changes under starvation conditions,69,70 because peripheral tissues continue to provide nutrients to the brain.71 In contrast, it has been revealed that brain autophagy protects neurons under fasting conditions or during nutrient deprivation.72-74 In line with these findings, our study demonstrates that low glucose availability induces autophagy by activating AMPK, which phosphorylates ULK1 and RPTOR in hypothalamic neuronal cells, supporting the evidence that autophagy in the brain sensitively responds to nutritional status.
For many years, scientists have strived to figure out the role of autophagy in the hypothalamus regarding food intake and body metabolism.25-30 Nevertheless, the roles of hypothalamic autophagy on neuropeptide expression related to appetite are not completely understood. Here, we found that inhibition of autophagy by siRNAs or a protease inhibitor decreases Npy mRNA expression and increases that of Pomc in normal conditions without glucoprivation or glucose deprivation. We also found that changes in neuropeptide expression induced by low glucose utilization are attenuated when autophagy is inhibited, suggesting that hypothalamic autophagy mediates neuropeptide regulation. Furthermore, our experiments that used rapamycin treatment together with Atg5 knockdown support the conclusion that autophagy plays a role in neuropeptide expression when MTORC1 is inhibited by either AMPK-mediated RPTOR inhibition or rapamycin treatment. Altogether, our data demonstrate that autophagy is required for normal neuropeptide expression not only at the basal state but also in response to low glucose availability.
According to our in vitro results, there were differences in the regulation of the expression levels of Npy and Pomc by autophagy under low glucose availability and rapamycin treatment. Atg5 knockdown abolished the low glucose-induced reduction in Pomc expression, whereas the increase in Npy expression was still observed. Modulation of autophagy may not be the only mechanism of the regulation of neuropeptide gene expression, since NPY is also regulated by other mechanisms such as the ACAC-malonyl-CoA-CPT1/carnitine palmitoyltransferase 1 pathway.75,76
It has been reported that intracerebral infusion of rapamycin enhances neural projection and excitability of POMC neurons in old (12-mo old) mice.77 Interestingly, effects of rapamycin on increasing POMC activity were not observed in young (2-mo old) mice. These observations suggest that MTOR signaling in hypothalamic POMC neurons is relatively lower in young mice than old mice. As Pomc mRNA levels were not measured in their study, it is difficult to conclude that a change in POMC activity induced by rapamycin is directly correlated with Pomc expression levels. In contrast, another study has shown that intracerebral infusion of rapamycin rather decreases Pomc mRNA expression in young (7-10-wk old) mice.78 Consistent with this finding, we observed decreased Pomc mRNA expression following rapamycin treatment in hypothalamic POMC-expressing cells. It is unclear whether this discrepancy comes from different levels of MTOR signaling depending on age, as well as different doses and duration of rapamycin treatment. Therefore, the effect of rapamycin on Pomc expression should be further investigated and interpreted in the context of various experimental conditions.
Our in vitro data suggest a mechanism by which AMPK regulates feeding behavior. Whether hypothalamic AMPK affects feeding behavior by regulating neuropeptide mRNA expression has been intensively investigated.20,21,24 However, the exact mechanism of this regulation has not been fully understood. We hypothesized that glucoprivation-induced AMPK activation regulates neuropeptide levels via autophagy and increases food intake in mice. It is assumed that peripheral administration of 2DG leads to hyperphagia by inhibiting glucose utilization in the hypothalamus and upregulating NPY,79-82 because 2DG penetrates the blood-brain barrier through a glucose transporter.83,84 PRKAA1 alone also regulates autophagy in mice85 and human monocytes.86 Selective Prkaa2 knockout in AGRP and POMC neurons leads to lean and obese phenotypes, respectively.24 We knocked down both AMPK subunits (PRKAA1 and PRKAA2) using lentiviral shRNAs in the mouse ARC to avoid possible complications caused by knockdown of a single subunit. AMPK knockdown in the ARC attenuated 2DG-induced hyperphagia, in line with other studies that used compound C and overexpression of AMPK-DN.66,67 Our data suggest that hypothalamic AMPK mediates 2DG-induced hyperphagia by activating autophagy, which changes Npy and Pomc expression.
Although our study demonstrates that AMPK-activated autophagy controls neuropeptide expression in the hypothalamus, it is still unclear how autophagy modulates the levels of neuropeptides involved in feeding regulation. Notably, other research groups have suggested an autophagic mechanism by which the expression of hypothalamic neuropeptides is changed. Kaushik et al. have reported that intracellular free fatty acids produced by autophagy promote AGRP expression in hypothalamic neuronal cells.26 Phosphorylation of STAT3 (signal transducer and activator of transcription 3) by LEP upregulates Pomc transcription by releasing its inhibition by FOXO1 (forkhead box O1).87 It has been shown that the loss of Atg7 in POMC neurons undermines phosphorylation of STAT3 and impairs LEP sensitivity during refeeding.27 In light of these reports, autophagy induction by low glucose conditions probably modulates the intracellular metabolites derived from autophagy or activity of downstream transcription factors related to Npy or Pomc gene expression, thereby increasing food consumption. Molecular mechanisms by which AMPK-induced autophagy changes the levels of Npy and Pomc mRNA warrant further investigation.
We could not rule out that AMPK-induced autophagy in the ARC regulates appetite not only by regulating neuropeptide expression but also by modulating other factors. Considering that hypothalamic autophagy regulates body metabolism by affecting inflammation,28 lipid metabolism,26 axonal growth,29 or LEP signaling,27 it would be interesting to investigate whether regulation of food intake by AMPK-induced autophagy is mediated by its effects on some of these processes.
In summary, our study identifies the AMPK-autophagy axis in the ARC as a regulatory pathway for Npy and Pomc expression and feeding behavior under low glucose availability. We showed that hypothalamic AMPK activation in response to low glucose availability induces autophagy and is a possible pathway that upregulates Npy and downregulates Pomc expression, thereby increasing food intake. Our study may contribute to a better understanding of molecular mechanisms and physiological dynamics of feeding behavior by providing integrative insights into the role of AMPK-mediated autophagy in the hypothalamus.
Materials and methods
Antibodies and chemical reagents
Target proteins were probed with the following antibodies: phospho-AMPK (Thr172; Cell Signaling Technology [CST], 2531), PRKAA/AMPKα (CST, 2532), phospho-ACAC/ACC (Ser79; CST, 3661), ACAC/ACC (CST, 3662), phospho-ULK1 (Ser555; CST, 5869), phospho-ULK1 (Ser757; CST, 6888), ULK1 (CST, 8054), ATG5 (CST, 12994), phospho-RPTOR (Ser792; CST, 2083), RPTOR (CST, 2280), phospho-RPS6KB1/S6K1 (Thr389; CST, 9206), RPS6KB1/S6K1 (CST, 2708), GABARAP (CST, 13733), NBR1 (CST, 9891), GAPDH (CST, 2118), and LC3 (Sigma, L8918). When indicated, cells were treated with either AICAR (CST, 9944) to activate AMPK or with compound C (Calbiochem, 171260) to inhibit AMPK. For autophagy inhibition, cells were treated with E64d (Calbiochem, 330005) and pepstatin A (Calbiocam, 516481). Rapamycin (Sigma, R8781) was used to induce autophagy. 2DG (Sigma, D6134) was used to induce glucoprivation. 2DG was dissolved in DPBS (Corning, 21-031-CVR) or saline; AICAR, compound C, E64d, pepstatin A and rapamycin were dissolved in dimethyl sulfoxide (Sigma, D2650).
Cell culture
The embryonic mouse hypothalamic mHypoE-N41 (N41; Cellutions Biosystems Inc., CLU121) and mHypoE-N43/5 (N43/5; Cellutions Biosystems Inc., CLU127) cell lines were maintained in DMEM (Sigma, D5796) with 10% fetal bovine serum (Hyclone Laboratories Inc., SH30919.03) and 1% penicillin/streptomycin (Hyclone Laboratories Inc., SV30010) at 37°C. For glucoprivation (2DG) or glucose deprivation (glucose-free), 2DG was added in 25 mM glucose DMEM (Welgene, LM001-07) or 0 mM glucose DMEM (Welgene, LM001-56) was used to replace DMEM with 25 mM glucose. Lenti-X 293T (Clontech, 632180) cells were maintained in DMEM (Hyclone Laboratories Inc., SH30243) under the same conditions as hypothalamic cell lines.
Animals
Male C57BL/6 mice were purchased from KOATECH and housed (one per cage) in individually ventilated cages under a 12-h light/dark cycle (lights on from 6:00 to 18:00) in a temperature- and humidity-controlled room with ad libitum access to water and ND (LabDiet, Inc., 38057) or HFD (60% kcal from fat; Research Diets, Inc., D12492). Food intake and body weight were measured daily just before the onset of the dark cycle. All animal studies followed the guidelines on care and use of laboratory animals as approved by the Institutional Animal Care and Use Committee at Daegu Gyeongbuk Institute of Science & Technology (DGIST; Daegu, Korea).
mRFP-GFP-LC3 plasmid transfection and confocal microscopy
N41 and N43/5 cells were seeded in 12-well plates on microscope coverslips (Marienfeld, 0111580) and were transfected with 1.2 μg of the mRFP-GFP-LC3 plasmid (a gift from Dr. Inhee Mook-Jung, Seoul National University, Korea) using Lipofectamine 3000 (Invitrogen, L3000-015) for 24 h following the manufacturer's instructions. After the treatments indicated in the figure legends cells were fixed with 4% formaldehyde in PBS and stained with Hoechst 33342 (Invitrogen, H3570) following the manufacturer's instructions. Images were obtained using an inverted confocal microscope (Carl Zeiss, LSM 700). For quantification of autophagic cells, red, green, or yellow puncta were counted from at least 30 cells. Confocal microscopy images were analyzed using ZEN 2009 and ImageJ software.
siRNA transfection
N41 and N43/5 cells were seeded in 6-well plates and transfected with ON-TARGETplus mouse siRNA composed of 4 different siRNAs. Scrambled siRNAs (100 nM; Dharmacon, D-001810-10-10-05) or Atg5 siRNAs (100 nM; Dharmacon, L-064838-00-0010) were transfected using Lipofectamine 3000 for 48 h following the manufacturer's instructions.
Lentivirus preparation
Lenti-X 293T cells were seeded on 10-cm dishes (6.0 × 106 cells per dish) and cultured overnight. To produce lentiviruses, the pLKO.3G (encoding eGFP) or pLKO.1 construct (8 μg each) carrying control, Prkaa1- or Prkaa2-targeting shRNAs were co-transfected with the psPAX2 packaging plasmid (6 μg) and pMD2.G envelope plasmid (2 μg) using TurboFect (Thermo Scientific, R0531) following the manufacturer's instructions. To avoid off-target effects, 2 different shRNA plasmids for each AMPK isoform were used (Prkaa1/AMPKα1: Sigma, TRCN0000360842 and TRCN0000360770; Prkaa2/AMPKα2: Sigma, TRCN0000360775 and TRCN0000360848). After transfection, culture medium was replaced with fresh medium to remove the plasmids, and culture medium containing viruses was harvested 48 and 72 h after medium change as described previously.88 Media containing lentiviruses were filtered through 0.45-μm syringe filters (Millipore, SLHV033RS) and ultra-centrifuged 4 times in the same ultra-clear centrifuge tubes (Beckman, 344058) at 43,000 × g for 90 min at 4°C to obtain concentrated viruses.89 After final centrifugation, pellet fractions were resuspended in saline and the lentivirus copy number was measured by using a titration kit (Clontech, 631235).
Lentiviral infection of hypothalamic cell lines
N41 and N43/5 cells were placed in 6-well plates at a density of 0.5 × 105 cells per well. After 12 h, cells were treated with lentiviruses carrying control shRNAs or shRNAs for Prkaa1 and Prkaa2 knockdown with polybrene (Sigma, H9268) at a concentration of 4 μg/ml and further cultured for 48 h. The medium was replaced with fresh medium containing puromycin (N41: 4 μg/ml; N43/5: 6 μg/ml) to select the infected cells; selected cells were used for experiments.
Stereotaxic injection of lentiviruses into the ARC
Seven-wk-old mice were acclimated for a wk and were given an anesthetic (10 ml/kg of body weight) composed of Zoletil, Rumpun, and saline 20 min before surgery. Lentiviruses were adjusted to 4.0 × 108 copies/μl (equal amounts of Prkaa1 and Prkaa2 shRNA were used), 1.5 × 108 copies/μl (control shRNAs), or 3.48 × 108 copies/μl (eGFP) with saline and injected at a speed of 0.5 μl/min (2 μl on each side) with a microliter syringe (Hamilton, 7768) using the following coordinates: 1.4 mm posterior to bregma; 6.2 mm ventral; 0.35 mm bilateral.
Fluorescence imaging of mouse brain
To visualize GFP in the brain, eGFP lentivirus-injected mice were perfused with ice-cold saline and fixed with 4% paraformaldehyde under anesthesia during the light cycle. The brains were then fixed again in 4% paraformaldehyde and incubated in 30% sucrose in PBS at 4°C overnight. After incubation, the brains were frozen and sectioned at 40 µm for imaging. To visualize nuclei, Hoechst 33342 (Invitrogen, H3570) was used following the manufacturer's instructions. Images were obtained using an inverted confocal microscope (Carl Zeiss, LSM 700) and analyzed using ZEN2009 software.
Preparation of brain tissue
For immunoblotting, mice (except the 2DG-injected group) were sacrificed and the hypothalamic tissues including ARC were quickly dissected immediately after the onset of the dark cycle (18:00) on d 7 after viral infection. For mRNA analysis, the ARC-containing hypothalamus was quickly dissected at 00:00 on d 6 after viral infection. The collected tissues were frozen in liquid nitrogen and stored at −80°C until use.
Administration of 2DG and analysis
The ip injection of 2DG (500 mg/kg of body weight) dissolved in saline was conducted during the light cycle (15:00) and mice were sacrificed at 16:00. Specific hypothalamic areas were dissected using anatomical landmarks. A small part of the hypothalamus including the ARC was dissected between the optic chiasm and mammillary bodies, including the area located within ± 0.5 mm from the 3rd ventricle to a depth of 0.5 mm. After ARC dissection, the remaining hypothalamus was harvested along its lateral border at the same depth as the ARC to obtain the LH.21,90,91
Immunoblot analysis
Cell and tissue samples were lysed in lysis buffer.92 Samples were dissolved in 50 mM Tris-HCl, pH 7.4, 250 mM sucrose (Bioshop, SUC507), 5 mM sodium pyrophosphate, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100 (Sigma, T8787), 0.1 mM benzamidine (Sigma, B6506), 1 mM DTT, 0.5 mM PMSF (Sigma, P7626), 50 mM NaF, protease inhibitor cocktail (Calbiochem, 535140), and phosphatase inhibitor cocktail (Sigma, P5726). Lysates were resolved on SDS-polyacrylamide gels and blotted onto PVDF membranes (Millipore, IPVH00010) for 35 min at 20 V in transfer buffer (25 mM Tris base, pH 7.4, 192 mM glycine, 10% methanol). The membranes were blocked with 5% skim milk for 1 h and incubated with appropriate primary antibodies for 1 h at room temperature or at 4°C overnight. After 3 washes with TBST buffer (20 mM Tris [Bioshop, TRS001], 125 mM NaCl [Bioshop, SOD001], 0.1% Tween 20 [Sigma, P1379]), the membrane was incubated with appropriate HRP-linked secondary antibody (anti-mouse: CST, 7076S; anti-rabbit: Thermo Scientific, NCI1460KR) and visualized by using ECL solutions (Thermo Scientific, NCI4080KR; Advansta, K-12045-D50) according to the manufacturer's instructions. Band intensities were measured and quantified using ImageJ software.
Real-time PCR
Total RNA from cells or brain tissues was isolated using Trizol reagent (Invitrogen, 15596018). The RNA pellet was dissolved in nuclease-free water (Promega, P1193) and total RNA concentration was determined using a NanoDrop spectrophotometer (DeNovix, DS-11). Total RNA, reaction buffer, and GoScript Reverse Transcriptase (Promega, A5004) were mixed in a total volume of 20 μl and reverse transcription was carried out in a thermal cycler (Bio-Rad, C1000) at 25°C for 5 min, 42°C for 60 min, and 70°C for 15 min. Real-time PCR was performed with a SYBR Green PCR kit (TaKaRa Biotechnology, RR820A) in a qPCR machine (Bio-Rad, CFX96) for 40 cycles (95°C for 10 sec, 60°C for 30 sec). The following primers were synthesized by Integrated DNA Technologies: Gapdh Forward, 5′-ATCACTGCCACCCAGAAGAC-3′; Gapdh Reverse, 5′-ACACATTGGGGGTAGGAACA-3′; Npy Forward, 5′-CAGAAAACGCCCCCAGAA-3′; Npy Reverse, 5′-AAAAGTCGGGAGAACAAGTTTCATT-3′; Pomc Forward, 5′-GAACAGCCCCTGACTGAAAA-3′; Pomc Reverse, 5′-ACGTTGGGGTACACCTTCAC-3′; Prkaa1 Forward, 5′-GGAACCGGTTCCACCATGCGCAGACTCAGTTCCTGG-3′; Prkaa1 Reverse, 5′-CTGCAGAACCAATGCATGGATGCATTTACTGTGCAAGAAT-3′; Prkaa2 Forward, 5′-GGAACCGGTTCCACCATGGCTGAGAAGCAGAAG-3′; and Prkaa2 Reverse, 5′-CCGGAATTCCGGTCAACGAGCTAAAGC-3′. Relative mRNA expression of each target gene was analyzed by the delta-delta Ct method and normalized to that of Gapdh.
Statistical analysis
All data are shown as mean ± standard error of the mean (SEM). Statistical significance was determined by unpaired t test using built-in software in Graphpad Prism 5. p values of <0.05 were considered statistically significant.
Abbreviations
- 2DG
2-deoxy-d-glucose
- ACAC/ACC
acetyl-coenzme A carboxylase
- AGRP
agouti-related neuropeptide
- AICAR
5-aminoimidazole-4-carboxamide ribonucleotide
- AMPK
AMP-activated protein kinase
- ARC
arcuate nucleus
- ATG
autophagy related
- DN
dominant negative
- GABARAP
gamma-aminobutyric acid receptor associated protein
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GHRL
ghrelin
- HFD
high-fat diet
- ip
intraperitoneal
- LEP
leptin
- LH
lateral hypothalamus
- MAP1LC3A/LC3A
microtubule-associated protein 1 light chain 3 alpha
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- MTORC1
MTOR complex 1
- NBR1
neighbor of Brca1 gene 1
- ND
normal diet
- NPY
neuropeptide Y
- POMC
pro-opiomelanocortin-alpha
- RPS6KB1/S6K1
ribosomal protein S6 kinase, polypeptide 1
- RPTOR/raptor
regulatory associated protein of MTOR, complex 1
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- STAT3
signal transducer and activator of transcription 3
- ULK1
unc-51 like kinase 1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Research Foundation (2013M3C7A1056099) of South Korea.
References
- [1].Nguyen DM, El-Serag HB. The epidemiology of obesity. Gastroenterol Clin North Am 2010; 39:1-7; PMID:20202574; http://dx.doi.org/ 10.1016/j.gtc.2009.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Field AE, Coakley EH, Must A, Spadano JL, Laird N, Dietz WH, Rimm E, Colditz GA. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Int Med 2001; 161:1581-6; PMID:11434789; http://dx.doi.org/ 10.1001/archinte.161.13.1581 [DOI] [PubMed] [Google Scholar]
- [3].Anand BK, Chhina GS, Sharma KN, Dua S, Singh B. Activity of single neurons in the hypothalamic feeding centers: effect of glucose. Am J Physiol 1964; 207:1146-54; PMID:14237464 [DOI] [PubMed] [Google Scholar]
- [4].Oomura Y, Ooyama H, Sugimori M, Nakamura T, Yamada Y. Glucose inhibition of the glucose-sensitive neurone in the rat lateral hypothalamus. Nature 1974; 247:284-6; PMID:4818362; http://dx.doi.org/ 10.1038/247284a0 [DOI] [PubMed] [Google Scholar]
- [5].Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes 2004; 53:2521-8; PMID:15448079; http://dx.doi.org/ 10.2337/diabetes.53.10.2521 [DOI] [PubMed] [Google Scholar]
- [6].Garcia M, Millan C, Balmaceda-Aguilera C, Castro T, Pastor P, Montecinos H, Reinicke K, Zúñiga F, Vera JC, Oñate SA, et al.. Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J Neurochem 2003; 86:709-24; PMID:12859684; http://dx.doi.org/ 10.1046/j.1471-4159.2003.01892.x [DOI] [PubMed] [Google Scholar]
- [7].Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocrine Rev 1999; 20:68-100; PMID:10047974 [DOI] [PubMed] [Google Scholar]
- [8].Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest 1996; 98:1101-6; PMID:8787671; http://dx.doi.org/ 10.1172/JCI118891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci 2002; 5:566-72; PMID:12021765; http://dx.doi.org/ 10.1038/nn0602-861 [DOI] [PubMed] [Google Scholar]
- [10].Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, et al.. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003; 37:649-61; PMID:12597862; http://dx.doi.org/ 10.1016/S0896-6273(03)00063-1 [DOI] [PubMed] [Google Scholar]
- [11].Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci 1983; 6:269-324; PMID:6132586; http://dx.doi.org/ 10.1146/annurev.ne.06.030183.001413 [DOI] [PubMed] [Google Scholar]
- [12].Varela L, Horvath TL. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep 2012; 13:1079-86; PMID:23146889; http://dx.doi.org/ 10.1038/embor.2012.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Elias CF, Lee C, Kelly J, Aschkenasi C, Ahima RS, Couceyro PR, Kuhar MJ, Saper CB, Elmquist JK. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron 1998; 21:1375-85; PMID:9883730; http://dx.doi.org/ 10.1016/S0896-6273(00)80656-X [DOI] [PubMed] [Google Scholar]
- [14].Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci 1998; 1:271-2; PMID:10195157; http://dx.doi.org/ 10.1038/1082 [DOI] [PubMed] [Google Scholar]
- [15].Broberger C, Johansen J, Johansson C, Schalling M, Hokfelt T. The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci U S A 1998; 95:15043-8; PMID:9844012; http://dx.doi.org/ 10.1073/pnas.95.25.15043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 1999; 5:1066-70; PMID:10470087; http://dx.doi.org/ 10.1038/12506 [DOI] [PubMed] [Google Scholar]
- [17].Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, Wulff BS, Clausen JT, Jensen PB, Madsen OD, Vrang N, et al.. Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature 1998; 393:72-6; PMID:9590691; http://dx.doi.org/ 10.1038/29993 [DOI] [PubMed] [Google Scholar]
- [18].Coll AP, Farooqi IS, Challis BG, Yeo GS, O'Rahilly S. Proopiomelanocortin and energy balance: insights from human and murine genetics. J Clin Endocrinol Metab 2004; 89:2557-62; PMID:15181023; http://dx.doi.org/ 10.1210/jc.2004-0428 [DOI] [PubMed] [Google Scholar]
- [19].Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 2004; 279:12005-8; PMID:14742438; http://dx.doi.org/ 10.1074/jbc.C300557200 [DOI] [PubMed] [Google Scholar]
- [20].Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferré P, Birnbaum MJ, et al.. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004; 428:569-74; PMID:15058305; http://dx.doi.org/ 10.1038/nature02440 [DOI] [PubMed] [Google Scholar]
- [21].Kim EK, Miller I, Aja S, Landree LE, Pinn M, McFadden J, Kuhajda FP, Moran TH, Ronnett GV. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. J Biol Chem 2004; 279:19970-6; PMID:15028725; http://dx.doi.org/ 10.1074/jbc.M402165200 [DOI] [PubMed] [Google Scholar]
- [22].Han SM, Namkoong C, Jang PG, Park IS, Hong SW, Katakami H, Chun S, Kim SW, Park JY, Lee KU, et al.. Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycaemia in rats. Diabetologia 2005; 48:2170-8; PMID:16132951; http://dx.doi.org/ 10.1007/s00125-005-1913-1 [DOI] [PubMed] [Google Scholar]
- [23].McCrimmon RJ, Fan X, Ding Y, Zhu W, Jacob RJ, Sherwin RS. Potential role for AMP-activated protein kinase in hypoglycemia sensing in the ventromedial hypothalamus. Diabetes 2004; 53:1953-8; PMID:15277372; http://dx.doi.org/ 10.2337/diabetes.53.8.1953 [DOI] [PubMed] [Google Scholar]
- [24].Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, et al.. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 2007; 117:2325-36; PMID:17671657; http://dx.doi.org/ 10.1172/JCI31516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kaushik S, Arias E, Kwon H, Lopez NM, Athonvarangkul D, Sahu S, Schwartz GJ, Pessin JE, Singh R. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep 2012; 13:258-65; PMID:22249165; http://dx.doi.org/ 10.1038/embor.2011.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kaushik S, Rodriguez-Navarro JA, Arias E, Kiffin R, Sahu S, Schwartz GJ, Cuervo AM, Singh R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab 2011; 14:173-83; PMID:21803288; http://dx.doi.org/ 10.1016/j.cmet.2011.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Quan W, Kim HK, Moon EY, Kim SS, Choi CS, Komatsu M, Jeong YT, Lee MK, Kim KW, Kim MS, et al.. Role of hypothalamic proopiomelanocortin neuron autophagy in the control of appetite and leptin response. Endocrinology 2012; 153:1817-26; PMID:22334718; http://dx.doi.org/ 10.1210/en.2011-1882 [DOI] [PubMed] [Google Scholar]
- [28].Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem 2011; 286:32324-32; PMID:21784844; http://dx.doi.org/ 10.1074/jbc.M111.254417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Coupe B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG. Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab 2012; 15:247-55; PMID:22285542; http://dx.doi.org/ 10.1016/j.cmet.2011.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Malhotra R, Warne JP, Salas E, Xu AW, Debnath J. Loss of Atg12, but not Atg5, in pro-opiomelanocortin neurons exacerbates diet-induced obesity. Autophagy 2015; 11:145-54; PMID:25585051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992-2003; PMID:19225151; http://dx.doi.org/ 10.1091/mbc.E08-12-1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181:497-510; PMID:18443221; http://dx.doi.org/ 10.1083/jcb.200712064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297-305; PMID:19258318; http://dx.doi.org/ 10.1074/jbc.M900573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214-26; PMID:18439900; http://dx.doi.org/ 10.1016/j.molcel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011; 13:1016-23; PMID:21892142; http://dx.doi.org/ 10.1038/ncb2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PloS One 2010; 5:e15394; PMID:21072212; http://dx.doi.org/ 10.1371/journal.pone.0015394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673; http://dx.doi.org/ 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Weikel KA, Cacicedo JM, Ruderman NB, Ido Y. Glucose and palmitate uncouple AMPK from autophagy in human aortic endothelial cells. Am J Physiol Cell Physiol 2015; 308:C249-63; PMID:25354528; http://dx.doi.org/ 10.1152/ajpcell.00265.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Moruno F, Perez-Jimenez E, Knecht E. Regulation of autophagy by glucose in Mammalian cells. Cells 2012; 1:372-95; PMID:24710481; http://dx.doi.org/ 10.3390/cells1030372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Watanabe T, Takemura G, Kanamori H, Goto K, Tsujimoto A, Okada H, Kawamura I, Ogino A, Takeyama T, Kawaguchi T, et al.. Restriction of food intake prevents postinfarction heart failure by enhancing autophagy in the surviving cardiomyocytes. Am J Pathol 2014; 184:1384-94; PMID:24641899; http://dx.doi.org/ 10.1016/j.ajpath.2014.01.011 [DOI] [PubMed] [Google Scholar]
- [42].Di Nardo A, Wertz MH, Kwiatkowski E, Tsai PT, Leech JD, Greene-Colozzi E, Goto J, Dilsiz P, Talos DM, Clish CB, et al.. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum Mol Genet 2014; 23:3865-74; PMID:24599401; http://dx.doi.org/ 10.1093/hmg/ddu101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 2011; 25:1895-908; PMID:21937710; http://dx.doi.org/ 10.1101/gad.17420111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007; 100:914-22; PMID:17332429; http://dx.doi.org/ 10.1161/01.RES.0000261924.76669.36 [DOI] [PubMed] [Google Scholar]
- [45].Duan X, Ponomareva L, Veeranki S, Choubey D. IFI16 induction by glucose restriction in human fibroblasts contributes to autophagy through activation of the ATM/AMPK/p53 pathway. PloS One 2011; 6:e19532; PMID:21573174; http://dx.doi.org/ 10.1371/journal.pone.0019532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yin L, Kharbanda S, Kufe D. MUC1 oncoprotein promotes autophagy in a survival response to glucose deprivation. Int J Oncol 2009; 34:1691-9; PMID:19424588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 1996; 271:27879-87; PMID:8910387; http://dx.doi.org/ 10.1074/jbc.271.44.27879 [DOI] [PubMed] [Google Scholar]
- [48].Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 2005; 1:15-25; PMID:16054041; http://dx.doi.org/ 10.1016/j.cmet.2004.12.003 [DOI] [PubMed] [Google Scholar]
- [49].Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002; 415:339-43; PMID:11797013; http://dx.doi.org/ 10.1038/415339a [DOI] [PubMed] [Google Scholar]
- [50].Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett 1987; 223:217-22; PMID:2889619; http://dx.doi.org/ 10.1016/0014-5793(87)80292-2 [DOI] [PubMed] [Google Scholar]
- [51].Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al.. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/ 10.1126/science.1196371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, et al.. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505-16; PMID:19250911; http://dx.doi.org/ 10.1016/j.molcel.2009.01.020 [DOI] [PubMed] [Google Scholar]
- [53].Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12:836-41; PMID:20811356; http://dx.doi.org/ 10.1038/ncb0910-836 [DOI] [PubMed] [Google Scholar]
- [54].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- [55].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/ 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/ 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- [58].Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 2011; 7:188-204; PMID:21107021; http://dx.doi.org/ 10.4161/auto.7.2.14181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu X, Chhipa RR, Nakano I, Dasgupta B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol Cancer Ther 2014; 13:596-605; PMID:24419061; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Vucicevic L, Misirkic M, Janjetovic K, Vilimanovich U, Sudar E, Isenovic E, Prica M, Harhaji-Trajkovic L, Kravic-Stevovic T, Bumbasirevic V, et al.. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy 2011; 7:40-50; PMID:20980833; http://dx.doi.org/ 10.4161/auto.7.1.13883 [DOI] [PubMed] [Google Scholar]
- [61].Lieberthal W, Tang M, Zhang L, Viollet B, Patel V, Levine JS. Susceptibility to ATP depletion of primary proximal tubular cell cultures derived from mice lacking either the alpha1 or the alpha2 isoform of the catalytic domain of AMPK. BMC Nephrol 2013; 14:251; PMID:24228806; http://dx.doi.org/ 10.1186/1471-2369-14-251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem 2004; 279:1070-9; PMID:14573616; http://dx.doi.org/ 10.1074/jbc.M306205200 [DOI] [PubMed] [Google Scholar]
- [63].Pearson RB, Dennis PB, Han JW, Williamson NA, Kozma SC, Wettenhall RE, Thomas G. The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J 1995; 14:5279-87; PMID:7489717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010; 29:1792-802; PMID:20418806; http://dx.doi.org/ 10.1038/emboj.2010.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805-12; PMID:15169837; http://dx.doi.org/ 10.1242/jcs.01131 [DOI] [PubMed] [Google Scholar]
- [66].Kim MS, Park JY, Namkoong C, Jang PG, Ryu JW, Song HS, Yun JY, Namgoong IS, Ha J, Park IS, et al.. Anti-obesity effects of alpha-lipoic acid mediated by suppression of hypothalamic AMP-activated protein kinase. Nat Med 2004; 10:727-33; PMID:15195087; http://dx.doi.org/ 10.1038/nm1061 [DOI] [PubMed] [Google Scholar]
- [67].Ropelle ER, Pauli JR, Fernandes MF, Rocco SA, Marin RM, Morari J, Souza KK, Dias MM, Gomes-Marcondes MC, Gontijo JA, et al.. A central role for neuronal AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) in high-protein diet-induced weight loss. Diabetes 2008; 57:594-605; PMID:18057094; http://dx.doi.org/ 10.2337/db07-0573 [DOI] [PubMed] [Google Scholar]
- [68].Um JH, Pendergast JS, Springer DA, Foretz M, Viollet B, Brown A, Kim MK, Yamazaki S, Chung JH. AMPK regulates circadian rhythms in a tissue- and isoform-specific manner. PloS One 2011; 6:e18450; PMID:21483791; http://dx.doi.org/ 10.1371/journal.pone.0018450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/ 10.1091/mbc.E03-09-0704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci 2006; 29:528-35; PMID:16859759; http://dx.doi.org/ 10.1016/j.tins.2006.07.003 [DOI] [PubMed] [Google Scholar]
- [71].Goodman MN, Lowell B, Belur E, Ruderman NB. Sites of protein conservation and loss during starvation: influence of adiposity. Am J Physiol 1984; 246:E383-90; PMID:6720943 [DOI] [PubMed] [Google Scholar]
- [72].Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, Kiosses WB. Short-term fasting induces profound neuronal autophagy. Autophagy 2010; 6:702-10; PMID:20534972; http://dx.doi.org/ 10.4161/auto.6.6.12376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, Kochanek PM, Jenkins LW, Ren J, Gibson G, et al.. Starving neurons show sex difference in autophagy. J Biol Chem 2009; 284:2383-96; PMID:19036730; http://dx.doi.org/ 10.1074/jbc.M804396200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Young JE, Martinez RA, La Spada AR. Nutrient deprivation induces neuronal autophagy and implicates reduced insulin signaling in neuroprotective autophagy activation. J Biol Chem 2009; 284:2363-73; PMID:19017649; http://dx.doi.org/ 10.1074/jbc.M806088200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 2000; 288:2379-81; PMID:10875926; http://dx.doi.org/ 10.1126/science.288.5475.2379 [DOI] [PubMed] [Google Scholar]
- [76].Wolfgang MJ, Lane MD. Hypothalamic malonyl-coenzyme A and the control of energy balance. Mol Endocrinol 2008; 22:2012-20; PMID:18356287; http://dx.doi.org/ 10.1210/me.2007-0538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Yang SB, Tien AC, Boddupalli G, Xu AW, Jan YN, Jan LY. Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron 2012; 75:425-36; PMID:22884327; http://dx.doi.org/ 10.1016/j.neuron.2012.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yue Y, Wang Y, Li D, Song Z, Jiao H, Lin H. A central role for the mammalian target of rapamycin in LPS-induced anorexia in mice. J Endocrinol 2015; 224:37-47; PMID:25349249; http://dx.doi.org/ 10.1530/JOE-14-0523 [DOI] [PubMed] [Google Scholar]
- [79].Sergeyev V, Broberger C, Gorbatyuk O, Hokfelt T. Effect of 2-mercaptoacetate and 2-deoxy-D-glucose administration on the expression of NPY, AGRP, POMC, MCH and hypocretin/orexin in the rat hypothalamus. Neuroreport 2000; 11:117-21; PMID:10683841; http://dx.doi.org/ 10.1097/00001756-200001170-00023 [DOI] [PubMed] [Google Scholar]
- [80].He B, White BD, Edwards GL, Martin RJ. Neuropeptide Y antibody attenuates 2-deoxy-D-glucose induced feeding in rats. Brain Res 1998; 781:348-50; PMID:9507187; http://dx.doi.org/ 10.1016/S0006-8993(97)01310-3 [DOI] [PubMed] [Google Scholar]
- [81].Giraudo SQ, Kim EM, Grace MK, Billington CJ, Levine AS. Effect of peripheral 2-DG on opioid and neuropeptide Y gene expression. Brain Res 1998; 792:136-40; PMID:9593862; http://dx.doi.org/ 10.1016/S0006-8993(98)00197-8 [DOI] [PubMed] [Google Scholar]
- [82].Ozawa Y, Arima H, Watanabe M, Shimizu H, Ito Y, Banno R, Sugimura Y, Ozaki N, Nagasaki H, Oiso Y. Repeated glucoprivation delayed hyperphagic responses while activating neuropeptide Y neurons in rats. Peptides 2011; 32:763-9; PMID:21184790; http://dx.doi.org/ 10.1016/j.peptides.2010.12.009 [DOI] [PubMed] [Google Scholar]
- [83].Lundgaard I, Li B, Xie L, Kang H, Sanggaard S, Haswell JD, Sun W, Goldman S, Blekot S, Nielsen M, et al.. Direct neuronal glucose uptake Heralds activity-dependent increases in cerebral metabolism. Nat Commun 2015; 6:6807; PMID:25904018; http://dx.doi.org/ 10.1038/ncomms7807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bruckner BA, Ammini CV, Otal MP, Raizada MK, Stacpoole PW. Regulation of brain glucose transporters by glucose and oxygen deprivation. Metabolism 1999; 48:422-31; PMID:10206432; http://dx.doi.org/ 10.1016/S0026-0495(99)90098-7 [DOI] [PubMed] [Google Scholar]
- [85].Zhu H, Foretz M, Xie Z, Zhang M, Zhu Z, Xing J, Leclerc J, Gaudry M, Viollet B, Zou MH. PRKAA1/AMPKalpha1 is required for autophagy-dependent mitochondrial clearance during erythrocyte maturation. Autophagy 2014; 10:1522-34; PMID:24988326; http://dx.doi.org/ 10.4161/auto.29197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Obba S, Hizir Z, Boyer L, Selimoglu-Buet D, Pfeifer A, Michel G, Hamouda MA, Gonçalvès D, Cerezo M, Marchetti S, et al.. The PRKAA1/AMPKalpha1 pathway triggers autophagy during CSF1-induced human monocyte differentiation and is a potential target in CMML. Autophagy 2015; 11:1114-29; PMID:26029847; http://dx.doi.org/ 10.1080/15548627.2015.1034406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Plum L, Belgardt BF, Bruning JC. Central insulin action in energy and glucose homeostasis. J Clin Investigat 2006; 116:1761-6; PMID:16823473; http://dx.doi.org/ 10.1172/JCI29063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc 2006; 1:241-5; PMID:17406239; http://dx.doi.org/ 10.1038/nprot.2006.37 [DOI] [PubMed] [Google Scholar]
- [89].Ichim CV, Wells RA. Generation of high-titer viral preparations by concentration using successive rounds of ultracentrifugation. J Transl Med 2011; 9:137; PMID:21849073; http://dx.doi.org/ 10.1186/1479-5876-9-137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008; 135:61-73; PMID:18854155; http://dx.doi.org/ 10.1016/j.cell.2008.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Imbernon M, Sanchez-Rebordelo E, Gallego R, Gandara M, Lear P, Lopez M, Dieguez C, Nogueiras R. Hypothalamic KLF4 mediates leptin's effects on food intake via AgRP. Mol Metab 2014; 3:441-51; PMID:24944903; http://dx.doi.org/ 10.1016/j.molmet.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lee J, Kim K, Yu SW, Kim EK. Wnt3a upregulates brain-derived insulin by increasing NeuroD1 via Wnt/beta-catenin signaling in the hypothalamus. Mol Brain 2016; 9:24; PMID:26956881; http://dx.doi.org/ 10.1186/s13041-016-0207-5 [DOI] [PMC free article] [PubMed] [Google Scholar]