

ABSTRACT
Autophagy, a catabolic pathway of lysosomal degradation, acts not only as an efficient recycle and survival mechanism during cellular stress, but also as an anti-infective machinery. The human pathogen Staphylococcus aureus (S. aureus) was originally considered solely as an extracellular bacterium, but is now recognized additionally to invade host cells, which might be crucial for persistence. However, the intracellular fate of S. aureus is incompletely understood. Here, we show for the first time induction of selective autophagy by S. aureus infection, its escape from autophagosomes and proliferation in the cytoplasm using live cell imaging. After invasion, S. aureus becomes ubiquitinated and recognized by receptor proteins such as SQSTM1/p62 leading to phagophore recruitment. Yet, S. aureus evades phagophores and prevents further degradation by a MAPK14/p38α MAP kinase-mediated blockade of autophagy. Our study demonstrates a novel bacterial strategy to block autophagy and secure survival inside the host cell.
KEYWORDS: ATG5, MAP kinase14, S. aureus, selective autophagy, ubiquitin
Introduction
Staphylococcus aureus is a highly adaptable human pathogen, which is able to survive as a commensal organism in the anterior nares and on human skin. A third of the human population are nasal carriers and two-thirds are intermittent carriers, forming a large reservoir for infections.1 This pathogen is the leading cause of hospital-acquired infections and its multidrug resistance makes successful treatment more and more difficult.2-4 Inside the host, S. aureus is able to cause a wide range of diseases, from minor skin infections such as impetigo and wound infections to severe systemic diseases such as bacteremia, septic arthritis and endocarditis.3 Residing inside the host cells, S. aureus must evade the intracellular defense mechanisms to survive. However, the intracellular fate of S. aureus is an ongoing controversial discussion.5,6
A fundamental process in eukaryotic cells is macroautophagy (hereafter referred to as autophagy), a catabolic pathway, that degrades damaged or unnecessary cytosolic components to supply metabolic pathways with nutrients and to maintain ATP production and macromolecular synthesis.7 Autophagy is evolutionarily conserved in all eukaryotic cells and controlled by essential key regulators, known as autophagy-related (ATG) proteins.8 For instance, the autophagic machinery is driven by 2 ubiquitin-like conjugation systems; first the ATG12–ATG5 system and second the MAP1LC3/LC3 (mammalian ortholog family of yeast Atg8) system.9 The ATG5 protein is covalently linked to ATG12 and together with ATG16L1 forms a complex, which conjugates to the phagophore and lipidates LC3.10 Lipidated LC3 and its homologs and paralogs are involved in membrane elongation and fusion resulting in a closed double-membrane vesicle, the autophagosome, which is the main morphological characteristic of autophagy.11,12
Besides its important function supplying the cell with nutrients during starvation, autophagy is also required to deliver microbial antigens to the adaptive immune response, therefore representing an essential mechanism for the cell to respond and defend to intracellular pathogens.13,14 In addition, selective autophagy via ubiquitination and recruitment of autophagy receptor proteins such as SQSTM1, OPTN and CALCOCO2 can act as a direct antimicrobial mechanism.15 However, diverse bacteria such as Listeria monocytogenes, Shigella flexneri and Francisella tularensis have evolved strategies to escape from the autophagic machinery.16-19 Nevertheless, mechanisms of autophagic escape are still relatively unknown.6,8 S. aureus has been connected to autophagy before, but the molecular mechanisms for autophagosome formation, autophagosomal escape and intracellular survival of S. aureus are controversial. For instance Schnaith et al. report that S. aureus use autophagosomes as a replicative niche, whereas Mestre et al. observe replication of S. aureus in the cytosol.5,20 In the present study, we elucidate the autophagic response of nonprofessional phagocytes to infection. We show for the first time that intracellular S. aureus is targeted by selective autophagy involving ubiquitination and autophagy receptors such as SQSTM1 in mouse fibroblasts. S. aureus activates a strong autophagic response and is able to escape lysosomal degradation via a novel MAPK14-mediated mechanism.
Results
Induction of the autophagic response during S. aureus infection
To investigate the effect of S. aureus on nonprofessional phagocytes we infected NIH/3T3 fibroblasts following the protocol depicted schematically in Fig. 1A. Briefly, the bacteria were added to the eukaryotic cells for 90 min (T = −102 min). S. aureus are nonmotile bacteria and were not centrifuged onto the eukaryotic cells to keep its stress levels as low as possible. Therefore, certain variations in the time course dependent on how soon the bacteria reach the host cells and how quickly the invasion process takes place. After incubation (T = −12 min), all extracellular bacteria were killed by the addition of lysostaphin for 12 min at 37°C. Finally, fresh DMEM was added to the infected cells (T = 0 → 0 hpi). We monitor the S. aureus-dependent induction of autophagosome formation by using NIH/3T3 fibroblasts stably expressing the autophagosomal marker GFP-MAP1LC3B, hereafter short GFP-LC3B.11,21 Cells were infected with the RFP-expressing S. aureus strain SH1000-RFP and subsequently analyzed via imaging over a defined time period. The appearance of an increasing number of GFP-LC3B-positive puncta in the cytosol of the host cells can be an indication of enhanced autophagosomal formation. As a control, cells were either cultured for 3 h in HBSS to induce autophagy or left untreated.
Figure 1.

Enclosure of intracellular S. aureus into GFP-LC3B-positive compartments. (A) Schematic representation of our experimental design. Eukaryotic cells were incubated with S. aureus for 90 min at 37°C. Afterwards, all extracellular bacteria were killed by the addition of lysostaphin. After 12 min incubation at 37°C the media was changed and recording of the experimental process started with time point 0 h postinfection (hpi). (B) NIH/3T3 cells stably expressing GFP-LC3B were either starved 3 h in HBSS, left untreated (no stimulation) or infected with RFP-expressing S. aureus for 1 hpi. Subsequently, cells were fixed and analyzed by confocal microscopy for GFP-LC3B expression as well as RFP-expressing S. aureus. Scale bars: 12 µm. (C) Wild-type MEF cells were infected with wild-type SH1000 for the indicated time periods. Uninfected cells and cells starved for 3 h in HBSS (with and without 100 nM Baf A1 for the last 2 h of culturing to prevent LC3-II degradation) served as negative and as positive controls, respectively. Cellular lysates were prepared and analyzed by immunoblotting using indicated antibodies. Normalization of ACTB/actin in comparison to LC3A/B-II expression was analyzed by ImageJ. (D) NIH/3T3 GFP-LC3B cells were infected with SH1000-RFP and recorded via live-cell imaging (recording 3 time points/min), shown are 8 time points. Arrowheads show the appearance of LC3B-positive puncta to intracellular S. aureus. Scale bars: 11 µm. (E) Quantification of the experiment shown in (D) for 1, 2, 3 and 4 hpi. The percentage of intracellular S. aureus colocalizing with GFP-LC3B is shown. 50 cells per time point were analyzed. Data are represented as mean ± SEM of 2 independent experiments.
Indeed, starved as well as S. aureus-infected NIH/3T3 fibroblasts exhibited increased GFP-LC3B puncta (Fig. 1B). To check whether enhanced autophagosomal formation is an indication for enhanced autophagic activity, we analyzed the autophagic flux by immunoblotting for endogenous LC3A/B. Again, as control cells were either cultured for 3 h in HBSS to induce autophagy in the presence or absence of the lysosomal inhibitor bafilomycin A1 (Baf A1) or left untreated. After infection of wild-type mouse embryonic fibroblasts (MEFs) with wild-type SH1000, we observed an increased amount of the lipidated LC3-II form at 1 to 3 hpi in comparison to uninfected cells (Fig. 1C). The amounts of LC3A/B-II upon infection were similar to the levels obtained by starvation (lane 3 HBSS + Baf A1). Therefore, we conclude that autophagy is initiated after infection of fibroblasts with S. aureus.
Enclosure of intracellular S. aureus by autophagy
To investigate whether S. aureus simply induces autophagy or whether intracellular bacteria become enclosed by phagophores, we employed live cell imaging to detect both, intracellular SH1000-RFP and the cellular autophagic machinery. The latter was visualized using GFP-LC3B as reporter. Live cell imaging revealed that less than 2 hpi intracellular S. aureus become rapidly enclosed (within ∼30 min) by GFP-LC3B-positive structures, i.e. phagophores presumably formed around the intracellular S. aureus (Fig. 1D, Movie S1). About 62% ± 2% of intracellular bacteria colocalized with GFP-LC3B at early time points of infection while the percentage dropped to 41% ± 1% at 4 hpi (Fig. 1E). Since LC3A/B is involved in processes other than canonical autophagy,22 we subsequently employed transmission electron microscopy. We detected multilamellar membranes fusing with S. aureus-containing phagosomes (Fig. 2B, panels iii to v). In addition, we observed membranes forming autophagic structures nearby the phagosome (Fig. 2B, panels iv to ix). These autophagic structures were in close vicinity to intracellular S. aureus (Fig. 2B, panels ix and x and Fig. S4). In agreement with the multilamellar membranes being new material for a growing phagophore, we observed dynamic transport of LC3B-positive membrane structures to or from LC3B-positive structures containing intracellular S. aureus via live-cell imaging (Fig. 2C, Movie S2). More detailed 3D analysis has to be done in the future to elucidate the origin of this membrane stack.
Figure 2.
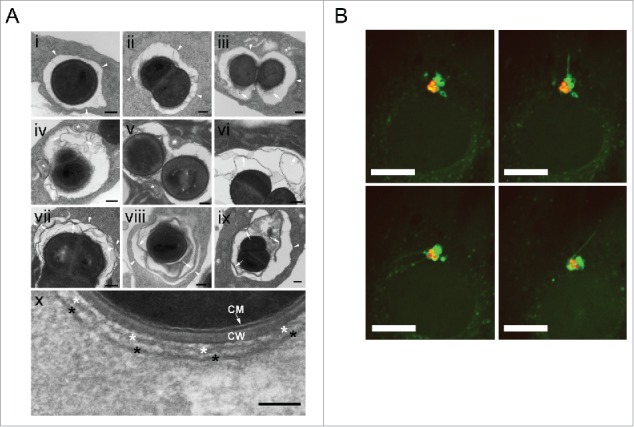
S. aureus becomes enclosed into a double-membrane structure. (A) NIH/3T3 cells were infected with SH1000-RFP, fixed 2 and 3 hpi and analyzed by transmission electron microscopy. Arrowheads depict the phagosomal or vacuolar membranes. * depicts multilamellar membranes next to phagosomes or vacuoles in panels i to ix and the double membrane in close vicinity to S. aureus in panel x. Arrows depict multilamellar and autophagosomal membranes from the host. CM, bacterial cytoplasmic membrane, CW bacterial cell wall. Scale bars: 200 nm. (B) NIH/3T3 GFP-LC3B cells were infected with SH1000-RFP and recorded via live-cell imaging (recording 3 time points/min). The images show the dynamic of GFP-LC3B-positive membrane structures from or to the existing autophagosomal membrane enclosing the intracellular S. aureus. Arrowheads show the actual way of recruitment. Scale bars: 13 µm.
Selective autophagy due to ubiquitination of the intracellular bacteria
Autophagy can proceed in bulk or in a selective manner, i.e., that cargo is specifically marked for autophagic degradation. One characteristic event of certain types of selective autophagy is ubiquitination, thus tagging the cytosolic cargo.23 We therefore examined whether ubiquitination also plays a role in the recruitment of S. aureus to phagophores using the specific anti-ubiquitin FK2 monoclonal antibody and confocal microscopy. This antibody recognizes both mono- and polyubiquitinated proteins but not free ubiquitin.24 Uninfected cells show a diffuse distribution of ubiquitinated proteins in the cytosol (data not shown). In contrast, in S. aureus-infected cells we observed ubiquitinated proteins surrounding cytosolic bacteria (Fig. 3A). Since S. aureus expresses protein A, an immunoglobulin-binding protein encoded by the spa gene, we tested the specificity of our immunofluorescence analysis using a spa mutant strain. S. aureus spaΔ colocalized with ubiquitinated proteins in a manner comparable to spa-proficient bacteria (Fig. S1). In addition, no staining of bacteria alone was observed, further confirming the specificity of the staining procedure (Fig. S2 and S3). During the time course of infection, the proportion of intracellular S. aureus SH1000-RFP associated with ubiquitin gradually increased from 26% ± 2% at 1 hpi to 63% ± 10% at 4 hpi (Fig. 3B). In summary, these results demonstrate that intracellular S. aureus becomes ubiquitinated by the host cell shortly after invasion.
Figure 3.
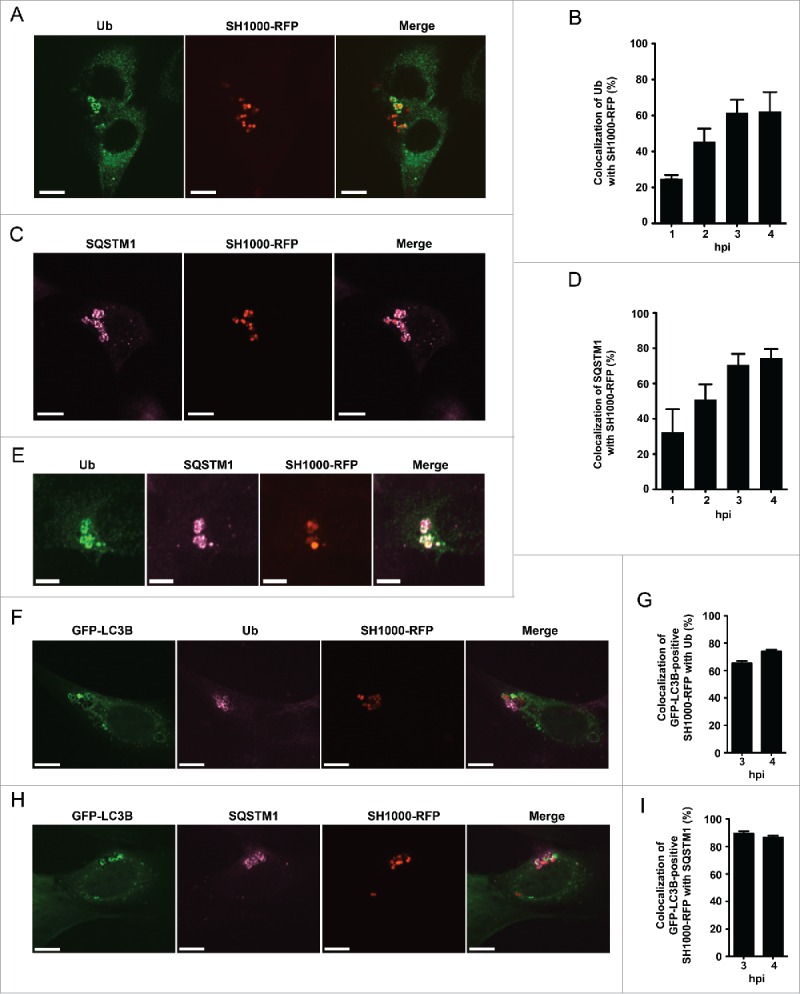
Intracellular S. aureus induces selective autophagy. (A) NIH/3T3 cells were infected with SH1000-RFP. Three hpi cells were fixed, stained with anti-ubiquitin and analyzed by confocal microscopy. Scale bars: 10 µm. (B) Quantification of the experiment shown in (A) for 1, 2, 3 and 4 hpi. The percentage of intracellular S. aureus colocalizing with ubiquitin is depicted. 100 cells per time point were analyzed. Data are represented as mean ± SEM of 3 independent experiments. (C) NIH/3T3 cells were infected with SH1000-RFP. Two hpi cells were fixed, stained with anti-SQSTM1 and analyzed by confocal microscopy. Scale bars: 12 µm. (D) Quantification of the experiment shown in (C) for 1, 2, 3 and 4 hpi. The percentage of intracellular S. aureus colocalizing with SQSTM1 is shown. 100 cells per time point were analyzed. Data are represented as mean ± SEM of 3 independent experiments. (E) NIH/3T3 cells were infected with SH1000-RFP. Two hpi cells were fixed, stained with anti-ubiquitin as well as anti-SQSTM1 and analyzed by confocal microscopy. Scale bars: 6 µm. (F) NIH/3T3 GFP-LC3B cells were infected with SH1000-RFP. Three and 4 hpi cells were fixed, stained with anti-ubiquitin and analyzed by confocal microscopy. Scale bars: 10 µm. (G) Quantification of the experiment shown in (F), visualized is the colocalization of GFP-LC3B-positive S. aureus with ubiquitin 3 and 4 hpi. 50 cells per time point were analyzed. Data are represented as mean ± SEM of 3 independent experiments. (H) NIH/3T3 GFP-LC3B cells were infected with SH1000-RFP. Three and 4 hpi cells were fixed, stained with anti-SQSTM1 and analyzed by confocal microscopy. Scale bars: 10 µm. (I) Quantification of the experiment shown in (H). Shown is the colocalization of GFP-positive S. aureus with SQSTM1 3 and 4 hpi. 50 cells per time point were analyzed. Data are represented as mean ± SEM of 3 independent experiments.
Interaction of ubiquitinated S. aureus with autophagy receptor proteins
Selective autophagy is mediated by receptor molecules, which deliver ubiquitinated cargo to the autophagosomal membrane, such as the proteins SQSTM1, OPTN and CALCOCO2.25-29 We therefore investigated if these proteins play a role in targeting S. aureus to the autophagosomal membrane. Like ubiquitin, SQSTM1 also colocalized with intracellular S. aureus by immunofluorescence microscopy (Fig. 3C). Subsequent experiments revealed that colocalization of SQSTM1 with S. aureus increases upon infection time, thus again resembling the situation found for ubiquitin colocalizing with S. aureus. The percentage of SQSTM1 colocalizing with intracellular S. aureus increased from 34% ± 21% at 1 hpi to 74% ± 7% at 4 hpi (Fig. 3D). To validate the data, we next checked for colocalization of ubiquitin and SQSTM1 in S. aureus-infected cells. As expected, colocalization of both proteins at intracellular S. aureus was found (Fig. 3E). In order to verify that ubiquitin and SQSTM1 are not only associated with S. aureus but involved in S. aureus-induced autophagy we further analyzed the localization of ubiquitin and SQSTM1 in relation to GFP-LC3B. As expected GFP-LC3B-positive S. aureus colocalized to ubiquitin as well as to SQSTM1 (Fig. 3F and 3H). Quantification of the colocalization with ubiquitin revealed 65% ± 2% for 3 hpi and 74% ± 1% for 4 hpi, respectively (Fig. 3G). GFP-LC3B-positive S. aureus colocalized to 89% ± 2% (3 hpi) and 87% ± 1% (4 hpi) with SQSTM1 (Fig. 3I).
To test whether other autophagy receptor proteins also recognize S. aureus-associated ubiquitin, we transfected a Flag-tagged human CALCOCO2 construct, which was previously shown to interact with Salmonella-associated ubiquitin, into NIH/3T3 cells and infected them with SH1000-RFP.27,28 Indeed, CALCOCO2 was enriched at sites of intracellular S. aureus (Fig. S5A). Similar findings were obtained with GFP-tagged OPTN as 62% ± 2% of intracellular S. aureus colocalized with this autophagy receptor (Fig. S5B). Again, the specificity of the staining was confirmed as similar findings were obtained using a spaΔ S. aureus mutant and no staining of bacteria alone was observed (Fig. S1 to S3). In conclusion, intracellular S. aureus becomes rapidly ubiquitinated by the host cell and the receptor proteins SQSTM1, OPTN and CALCOCO2 associate with the ubiquitinated bacteria indicating the process of selective autophagy.
S. aureus escapes from LC3B-positive structures
Subsequent to the initiation of autophagy, enclosed pathogens can be degraded.15 However, some pathogens have developed avoidance strategies.18,19,30-32 Using live cell imaging, we tested whether or not S. aureus is able to escape from the autophagic machinery. Indeed, escape from the autophagosomal compartment due to rupturing the membrane within 30 to 45 min after enclosure was observed (Fig. 4A; Movie S3). In addition, we checked for colocalization of intracellular S. aureus with degradative compartments via LysoTracker Deep Red dye, revealing no substantial acidification of S. aureus-containing compartments or autophagosomes (Fig. 4B) with only about 15% ± 7% of GFP-LC3B-associated bacteria colocalizing with LysoTracker Deep Red at 3 and 4 hpi (Fig. 4C). Since GFP can lose its fluorescence in acidic compartments, we also quantified the colocalization of total intracellular S. aureus with LysoTracker Deep Red. Again, only a minor fraction (24% ± 3% for the 3 h time point and 27% ± 2% at 4 h) of the intracellular bacteria was associated with acidic compartments (Fig. 4D). Consistent with the finding that S. aureus escapes from the autophagosomes, we analyzed the cellular amount of receptor protein SQSTM1. A correlation between levels of endogenous SQSTM1 and the inhibition of autophagy-lysosomal protein degradation has been described earlier.11,33,34 Of note, after an initial degradation the level of endogenous SQSTM1 increased to baseline again at 3 hpi, suggesting the inhibition of autolysosome formation and consequently the degradation of the receptor protein SQSTM1 (Fig. 4E). We therefore conclude that S. aureus can escape from autophagosomes and prevents the formation of autolysosomes, thus avoiding its own degradation.
Figure 4.

Autophagosomal escape of S. aureus. (A) NIH/3T3 GFP-LC3B cells were infected with SH1000-RFP and recorded via live-cell imaging (recording 3 time points/min) shown are 12 time points. Scale bar, 13 µm. (B) NIH/3T3 GFP-LC3B cells were infected with SH1000-RFP and incubated with LysoTracker Deep Red, 3 hpi. Scale bars: 11 µm. (C) Quantification of the experiment shown in (B) for 3 and 4 hpi. The percentage of GFP-LC3B-positive intracellular S. aureus colocalizing with LysoTracker Deep Red is shown. 50 cells per time point were analyzed. Data are represented as mean ± SEM of 3 independent experiments. (D) Quantification of the experiment shown in (B) for 3 and 4 hpi. The percentage of total intracellular S. aureus colocalizing with LysoTracker Deep Red is shown. 50 cells per time point were analyzed. Data are represented as mean ± SEM of 3 independent experiments. (E) Wild-type MEF cells were infected with wild-type SH1000. As a negative control uninfected cells were included and as positive control cells were incubated for 3 h in HBSS with and without Baf A1. Cellular lysates were prepared and analyzed by immunoblotting using indicated antibodies. Normalization of ACTB/actin in comparison to SQSTM1 expression was analyzed with ImageJ.
Intracellular replication of S. aureus is independent from autophagy
Previous studies report that replication of S. aureus inside host cells highly depends on autophagy induction.20 However, whether replication of intracellular S. aureus takes place inside autophagosomes remained controversial.5,20 To check in our experimental system whether or not induction of autophagy is essential for S. aureus replication in nonprofessional phagocytes, we used atg5 knockout (KO) MEFs for infection as well as control cells that were stably reconstituted with wild-type ATG5 (Fig. 5A). As expected, ATG5-proficient cells exhibited enhanced autophagic flux upon starvation while atg5 KO cells were defective in LC3 conversion (Fig. 5A). Analyzing the bacterial load inside the living (i.e. adherent) infected fibroblasts, we observed that more S. aureus survived inside atg5 KO than ATG5-reconstitued MEFs, indicating that autophagy is restricting intracellular S. aureus and is not essential for intracellular proliferation (Fig. 5B). This became even more evident, when the total bacterial load was analyzed in the cultures, i.e., including bacteria within dead cells and ones that evaded from the host cells (Fig. 5C). The higher replication of S. aureus in atg5 KO MEFs suggested that bacterial replication inside the host cells can occur without autophagosomes. Indeed, we demonstrated the ability of S. aureus to replicate inside the cytosol using live cell imaging (Fig. 5D; Movie S4). Consistent with these data, transmission electron microscopy of ultrathin sections revealed infected NIH/3T3 cells containing dividing bacteria residing in the cytoplasm without a surrounding membrane (Fig. 5E). Cell division of the cytoplasmic S. aureus is clearly indicated by its septum (Fig. 5E, white asterisk). The septum is made of peptidoglycan in the division plane of S. aureus and ring shaped.35-37 Therefore we conclude, that replication of S. aureus inside nonprofessional phagocytes is independent from autophagy induction.
Figure 5.
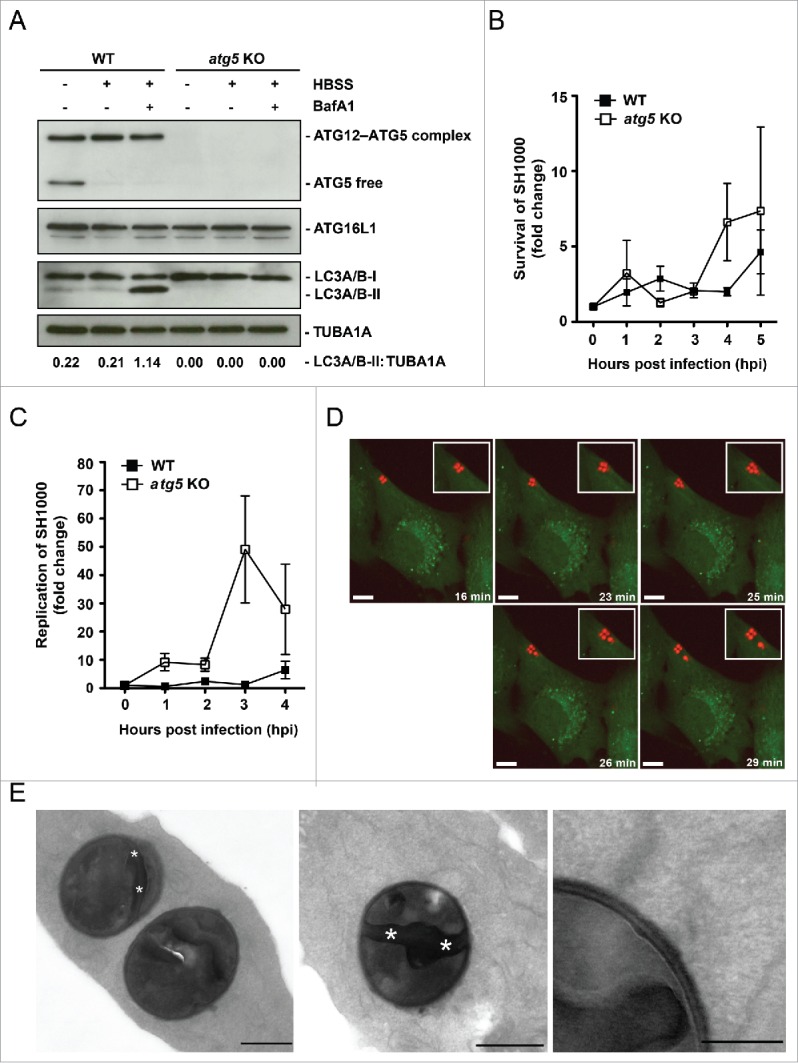
Cytosolic replication of S. aureus. (A) Cellular lysates of atg5 KO and ATG5-reconstituted MEFs were prepared and analyzed by immunoblotting using the indicated antibodies. (B) MEFs reconstituted with wild-type ATG5 (ATG5 WT) and atg5 KO MEFs were infected with wild-type SH1000. Survival of intracellular S. aureus was measured hourly. (C) Cells were infected as in (B). Bacterial load was measured hourly in the whole culture, i.e., including dead cells and the supernatant. (D) NIH/3T3 GFP-LC3B cells were infected with SH1000-RFP and recorded via live-cell imaging (recording 3 time points/min) shown are 5 time points. Scale bars: 8 µm. (E) NIH/3T3 cells were infected with SH1000-RFP. Two hpi cells were fixed and cut in ultrathin sections. Dividing bacteria are characterized by the septum, the dividing zone within bacteria (marked by asterisks). They are detectable in the cytoplasm with no visible membrane surrounding the bacteria. Scale bars: 500 nm in the left and middle image and 200 nm in the right image.
Activation of MAPK14 due to S. aureus invasion
Recently, we have reported that active MAPK14 can block autophagosome maturation after the initiation process.21 We therefore explored whether phosphorylation of MAPK14 plays a role during staphylococcal infection. NIH/3T3 fibroblasts were infected with either S. aureus or Salmonella typhi and protein expression during infection was analyzed via immunoblotting. Of note, S. aureus infection strongly induced MAPK14 phosphorylation over time, whereas no MAPK14 activation was observed during Salmonella typhi infection (Fig. 6A). We further employed immunofluorescence for intracellular phospho-MAPK14 localization. As shown in Figure S6 we did not detect any phospho-MAPK14 signal in uninfected cells, whereas a strong signal was observed 1 hpi using S. aureus (Fig. 6B and Fig. S6). Importantly, we could further show, that MAPK14 was phosphorylated in the vicinity of intracellular S. aureus 3 hpi (Fig. 6B). The majority of the phospho-MAPK14-positive signal near bacteria was also positive for GFP-LC3B (Fig. 6C), ubiquitin (Fig. 6D) and SQSTM1 (Fig. 6E) suggesting that autophagosome-associated intracellular S. aureus activated the kinase. Interestingly, survival of S. aureus inside mapk14 KO MEFs is reduced compared to wild-type cells, indicating that activation of MAPK14 plays a role in survival and proliferation of intracellular S. aureus (Fig. 7A). Moreover, colocalization of intracellular S. aureus with autophagosomes, as analyzed by CytoID staining, was increased in the absence of MAPK14 (Fig. 7B, C).
Figure 6.
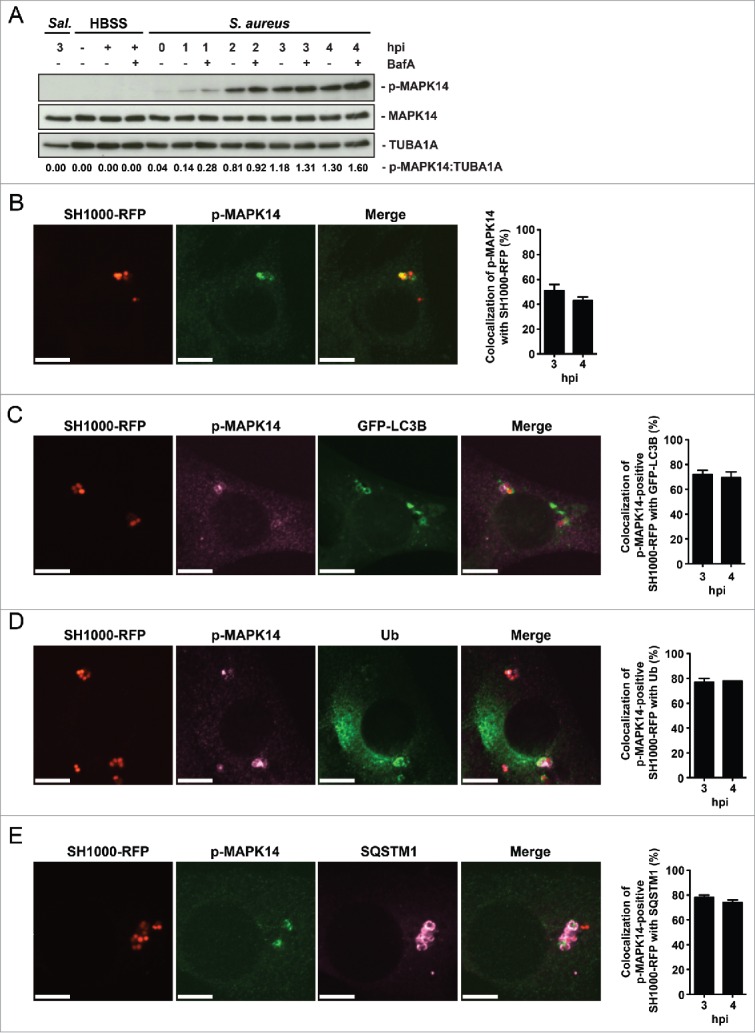
Activation of MAPK14 during S. aureus infection. (A) NIH/3T3 cells were infected with S. aureus strain SH1000. As a negative control uninfected cells were included and as positive control cells were incubated for 3 h in HBSS. Time points were performed in the presence of 100 nM Baf A1 for the last 2 h of culturing or without Baf A1. As further control NIH/3T3 cells were infected with Salmonella thypi (Sal.) for 1 h at an MOI of 100. Cellular lysates were prepared and 30 µg of protein were analyzed by immunoblotting using indicated antibodies. Normalization of tubulin in comparison to p-MAPK14 expression was analyzed by ImageJ. (B) NIH/3T3 cells were infected with SH1000-RFP. Cells were fixed, stained with anti-p-MAPK14 and analyzed by confocal microscopy, 3 hpi. Scale bars: 10 µm. The quantification of the experiment is shown in the right panel for 3 and 4 hpi. The percentage of intracellular S. aureus colocalizing with p-MAPK14 is depicted. 50 cells per time point were analyzed. Data are represented as mean ± SEM of 2 independent experiments. (C) NIH/3T3 GFP-LC3B cells were infected with SH1000-RFP. Cells were fixed, stained with anti-p-MAPK14 and analyzed by confocal microscopy, 3 hpi. Scale bars: 10 µm. The quantification of the experiment is shown in the right panel for 3 and 4 hpi. The percentage of intracellular S. aureus colocalizing with p-MAPK14 and GFP-LC3B is shown. 50 cells per time point were analyzed. Data are represented as mean ± SEM of 3 independent experiments. (D) NIH/3T3 cells were infected with SH1000-RFP. Cells were fixed, stained with anti-p-MAPK14 and anti-ubiquitin and analyzed by confocal microscopy, 3 hpi. Scale bars: 10 µm. The quantification of the experiment is shown in the right panel for 3 and 4 hpi. The percentage of intracellular S. aureus colocalizing with p-MAPK14 and ubiquitin is depicted. 50 cells per time point were analyzed. Data are represented as mean ± SEM of 2 independent experiments. (E) NIH/3T3 cells were infected with SH1000-RFP. Cells were fixed, stained with anti-p-MAPK14 and anti-SQSTM1 and analyzed by confocal microscopy, 3 hpi. Scale bars: 10 µm. The quantification of the experiment is shown in the right panel for 3 and 4 hpi. The percentage of intracellular S. aureus colocalizing with p-MAPK14 and SQSTM1 is shown. 50 cells per time point were analyzed. Data are represented as mean ± SEM of 2 independent experiments.
Figure 7.
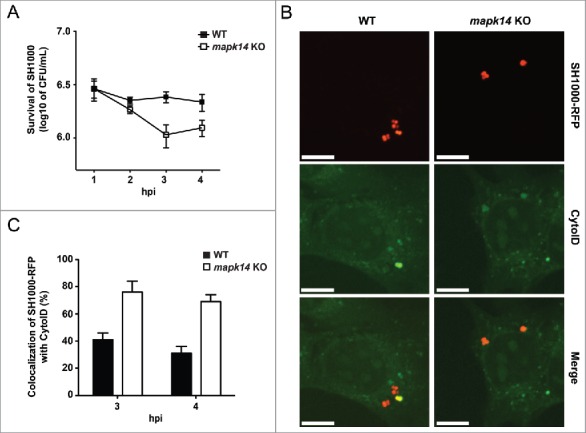
The importance of MAPK14 for S. aureus evading autophagy. (A) mapk14 KO and wild-type MEFs were infected with wild-type SH1000. Survival of intracellular S. aureus was measured hourly. Data are represented as mean ± SEM of 3 independent experiments. (B) mapk14 KO and wild-type MEFs were infected with SH1000-RFP and incubated with CytoID. Cells were fixed and analyzed by confocal microscopy, 3 hpi. Scale bars: 10 µm. (C) Quantification of the experiment shown in (B) for 3 and 4 hpi. The percentage of intracellular S. aureus colocalizing with CytoID is depicted. 50 cells per time point were analyzed. Data are represented as mean ± SEM of 2 independent experiments.
To check if MAPK14 plays a role in phosphorylation of ATG5 and therefore blocking the autophagic pathway, we used atg5 KO MEFs, which were reconstituted with wild-type or a T75A mutant of ATG5.21 These cells were infected with SH1000-RFP and colocalization of intracellular S. aureus with the lysosomal marker LAMP2 was analyzed by immunofluorescence. While S. aureus colocalized strongly with LAMP2 in ATG5T75A mutant cells, this was less frequently observed in wild-type ATG5-expressing cells (Fig. 8A). We observed 45% ± 3% colocalization of LAMP2 with intracellular S. aureus in wild-type ATG5-expressing MEFs and 68% colocalization of LAMP2 with intracellular S. aureus in ATG5T75A mutant cells at 4 hpi (Fig. 8B). Surprisingly, this did not result in targeting S. aureus more frequently to acidified compartments since colocalization of bacteria and LysoTracker Deep Red was equally low in wild-type ATG5 and ATG5T75A-expressing cells (Fig. 8C and D) suggesting that S. aureus has additional MAPK14-mediated mechanisms to block autophagy next to targeting ATG5.
Figure 8.
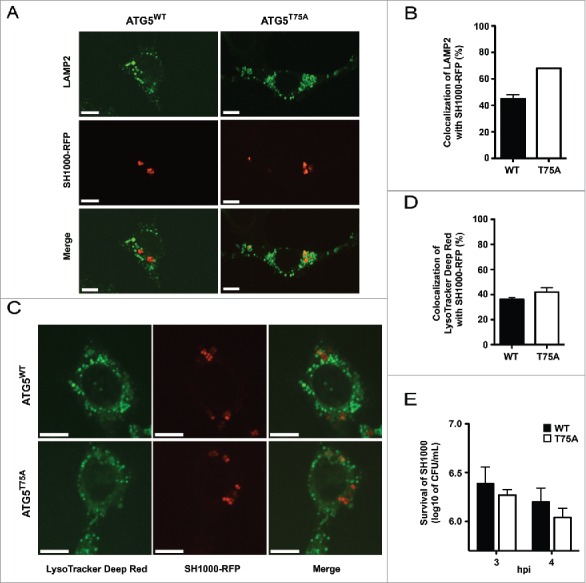
Inhibition of autolysosomal fusion. (A) atg5 KO cells reconstituted with either wild-type ATG5 (WT) or ATG5T75A (T75A), in which threonine (T75) has been mutated to alanine, were infected with SH1000-RFP, fixed, stained with anti-LAMP2 and analyzed by confocal microscopy. Images were taken 4 hpi. Scale bars: 11 µm. (B) Quantification of the experiment shown in (A). The percentage of intracellular S. aureus colocalizing with LAMP2 is shown. 50 cells per condition were analyzed. Data are represented as mean ± SEM of 2 independent experiments. (C) ATG5 WT or ATG5T75A cells were infected with SH1000-RFP, fixed, stained with LysoTracker Deep Red and analyzed by confocal microscopy. Images were taken 4 hpi. Scale bars: 10 µm. (D) Quantification of the experiment shown in (C). The percentage of intracellular S. aureus colocalizing with LysoTracker Deep Red is depicted. 50 cells per condition were analyzed. Data are represented as mean ± SEM of 4 independent experiments. (E) MEF cells reconstituted with wild-type ATG5 (ATG5 WT) and atg5 KO MEFs were infected with SH1000 wild-type. Survival of intracellular S. aureus was measured hourly.
Induction of autophagy in human cells
S. aureus can cause severe skin infections. Therefore, human keratinocytes are a likely cell type that becomes invaded during clinically relevant infections. To show the relevance of autophagy during S. aureus infection of human cells, we infected the human keratinocyte cell line HaCaT stably expressing GFP-LC3B38 with SH1000-RFP and employed live cell imaging to detect both intracellular SH1000-RFP and the cellular autophagic machinery. As demonstrated before in NIH/3T3 cells, we observed the enclosure of S. aureus by GFP-LC3B-positive structures, i.e. autophagosomes presumably formed around the intracellular S. aureus (Fig. 9A; Movie S5). Further, we also observed the escape of S. aureus from the autophagosomal compartment due to rupturing the membrane (Fig. 9B; Movie S6) indicating that similar mechanisms apply in murine fibroblasts and human keratinocytes.
Figure 9.
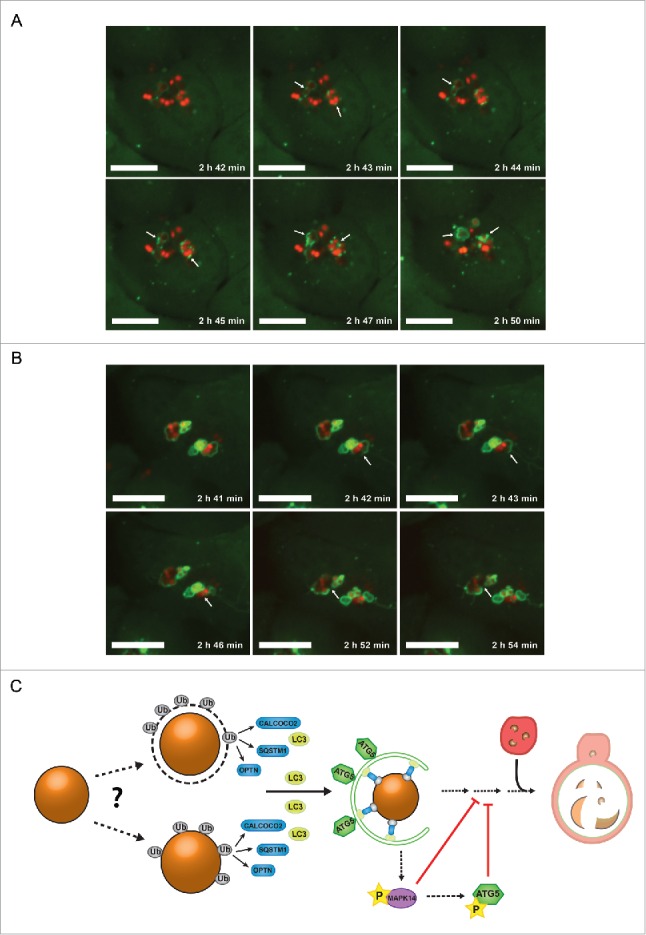
S. aureus evades autophagy in human cells. (A) Human keratinocyte cells (HaCaT) expressing GFP-LC3B were infected with SH1000-RFP and recorded via live-cell imaging. Three time points/min were recorded and 6 time points are shown. Scale bars: 11 µm. Arrowheads show forming of LC3B-positive phagophore membrane around intracellular S. aureus. (B) Human keratinocyte cells (HaCaT) expressing GFP-LC3B were infected with SH1000-RFP and recorded via live-cell imaging. Three time points/min were recorded and 6 time points are shown. Scale bars: 13 µm. Arrowheads show the disruption of the previously formed autophagosomal membrane around S. aureus. (C) Model of staphylococcal evasion from the autophagic machinery. After invasion of the host cell, S. aureus becomes directly ubiquitinated or associated with ubiquitinated proteins leading to recognition by the host cell receptor proteins SQSTM1, OPTN and CALCOCO2. The receptor proteins deliver S. aureus to LC3-containing phagophores. After autophagosomal capture, S. aureus is able to evade autophagy and blocks autophagosomal maturation via activation of MAPK14. Ub, ubiquitin-associated proteins (gray); SQSTM1, OPTN and CALCOCO2, host cell receptor proteins (blue); LC3, microtubule-associated protein 1 light chain 3 (yellow); ATG5, autophagy-related 5 (green); MAPK14, mitogen-activated protein kinase 14 (purple); P, phosphorylation (yellow star); the lysosome is depicted in red.
Discussion
In order to reduce the emergence of staphylococcal infections and diseases, it is crucial to gain detailed knowledge about the interaction between S. aureus and the host cell. It is therefore fundamental to determine the key parameters involved in invasion, intracellular survival and persistence, from the pathogenic side as well as the host cell side. Investigations of these underlying mechanisms are crucial to develop new strategies to be able to treat S. aureus infections more successfully. Importantly, autophagy can act as an antimicrobial mechanism of the host cell. However, some intracellular pathogens have developed autophagy evasion strategies to secure intracellular survival. Also S. aureus is able to survive the autophagic pathway, but details in terms of S. aureus and autophagy are controversial in literature.5,6,20 Here we show the intracellular fate of S. aureus from evasion of the autophagic machinery to intracellular replication in nonprofessional phagocytes, namely murine fibroblasts and human keratinocytes, in detail. We further show the enclosure process of intracellular S. aureus by the phagophore membrane in real time.
Our live cell imaging and immunoblot data revealed induction of autophagy and enclosure of S. aureus within LC3B-positive structures at 2 to 3 h postinfection (hpi). Since LC3 is not only a marker for canonical autophagy but also for noncanonical processes such as LC3-associated phagocytosis (LAP),22 selective autophagy or LAP could be initiated by S. aureus infection. However, several lines of evidence argue that S. aureus induced selective autophagy in MEFs. First, though RB1CC1 and the ULK1-complex have been implicated in xenophagy of Salmonella,39 it is currently unclear whether xenophagy in general requires the preinitiation complex. Second, it was shown that LAP occurs without ubiquitination and the recruitment of autophagy receptors.40 However, we were able to show for the first time colocalization of ubiquitinated proteins with intracellular S. aureus, as well as colocalization with the ubiquitin-LC3 receptor proteins SQSTM1, OPTN and CALCOCO2 strongly arguing that selective autophagy is induced. Finally, we found S. aureus closely associated with double-membrane vesicles by electron microscopy. These findings are consistent with previous results that documented S. aureus-dependent autophagosome formation in human HeLa cells.20 Therefore, we conclude that selective autophagy is induced after invasion of S. aureus into the host cell.
Colocalization of ubiquitinated proteins with other intracellular pathogens has been described before. Recently, Barnett et al. report about colocalization of group A Streptococcus strain M6JRS4 with ubiquitinated proteins.30 Further, Perrin et al. show ubiquitinated proteins that colocalized with the surface of cytosolic Salmonella typhimurium.41 Nevertheless, it is currently unknown whether bacterial proteins or only host cell factors are ubiquitinated during selective autophagy. One study has identified LRSAM1 as an E3 ligase that is relevant for ubiquitination associated with intracellular bacteria.42 However, it remains to be determined whether auto-ubiquitinated LRSAM1 associates with bacteria or whether the enzyme ubiquitinates bacterial proteins directly.
Our observation of induction of selective autophagy followed by inhibition of the autophagosomal maturation process to prevent bacterial degradation is consistent with previous reports.5,20 In contrast to other publications, we did not, however, observe the need of autophagosome formation for bacterial replication, which might be due to differences in the experimental design.5,20 We used ATG5-reconstituted KO MEFs to be as close to the original cell type and genetic background as possible. Further, for infection we used a low multiplicity of infection (MOI) of 8 instead of a high MOI of 200 as performed in a study investigating S. aureus induced cell death.20 Thus, at high bacterial burden autophagy-deficient host cells may die earlier preventing intracellular replication of S. aureus. In contrast, autophagy serves as an antibacterial mechanism by restricting intracellular S. aureus at low MOI. Thirdly, different bacterial strains were used in our study compared to previously published reports. We employed the strain SH1000, which originates from the strain NCTC8325 but is devoid of prophages and has a repaired rsbU gene resulting in normal SigB activity.43 Prophages often insert into genes affecting their expression, including virulence genes. RsbU is an important transcriptional regulator of virulence factors such as hla (α-toxin) and spa (protein A) and additionally affects expression of the important virulence regulator agr.44 The community acquired methicillin-resistant S. aureus (MRSA) strain MW2 and USA300, which are e.g. used by Schnaith et al., contain a mecA cassette conferring methicillin resistance and often also the Panton–Valentine leukocidin (PVL) pore-forming toxin.45,46 In conclusion, the different strains used in our study and others may vary in the expression of virulence factors and pore-forming toxins that probably affect the interplay between S. aureus and the host cell. Thus, a comprehensive analysis of different S. aureus strains of known genetic background is required to elucidate this interplay.
Intracellular bacterial pathogens have evolved different strategies to escape the autophagic degradation mechanism. Salmonella is able to manipulate the ubiquitin system due to delivery of the deubiquitinase SseL to the host cell cytoplasm, which further interferes with the host cell ubiquitination and finally prevents recruitment of SQSTM1 and LC3.47-49 Moreover, Legionella secretes the effector protein RavZ, which uncouples and therefore inactivates LC3-II from the autophagosomal membrane, leading to inhibition of autophagy.32 As mentioned before, M6JRS4 group A Streptococcus is targeted by autophagy, but a different strain, M1T1 group A Streptococcus, escapes the autophagic pathway due to expression of SpeB, a cysteine protease, degrading the receptor proteins SQSTM1, CALCOCO2 and NBR1.30 Shigella binds the effector protein IcsB to its surface protein IcsA/VirG to prevent the recognition of VirG by ATG5 and avoiding recruitment of ubiquitin and SQSTM1.18,26 In terms of S. aureus, the molecular mechanisms for autophagosome formation, autophagosomal escape and intracellular survival of S. aureus have not been fully addressed before.
In contrast to previously described evasion mechanisms as shown by Salmonella, Listeria or Shigella, we were able to provide evidence of a new mechanism of evasion used by S. aureus. After successful invasion S. aureus becomes recognized by the host cell and colocalizes with ubiquitin-associated proteins. Subsequently, the receptor proteins SQSTM1, OPTN and CALCOCO2 bind to the ubiquitin and deliver the bacteria to LC3-containing phagophore membranes (Fig. 9C). However, S. aureus can evade the autophagic machinery. Here, we observed the phosphorylation of MAPK14 increasing from 1 hpi, whereas no activation of MAPK14 was observed performing infections with Salmonella. Our findings are in line with a previous study that reports early MAPK14 activation upon S. aureus infection in a human macrophage cell line.50 Furthermore, we show that MAPK14 is activated upon S. aureus infection in a local manner since we observed strong phospho-MAPK14 staining only in the vicinity of S. aureus. This suggests a specific guidance of active MAPK14 to autophagosomal compartments containing S. aureus.
MAPK14 is one member of the p38 MAPK family comprising MAPK11, MAPK12, MAPK13 and MAPK14, which are highly homologous with an amino acid identity of approximately 60%.51 Since MAPK12 and MAPK13 exhibit a tissue-specific expression pattern and are not expressed in fibroblasts52 and, furthermore, MAPK11 has only a negligible activity compared to MAPK14 in fibroblasts,53 we think that we observe a MAPK14-mediated effect in our experimental system. Nevertheless, due to the high amino acid identity within the p38 MAPK family MAPK11, MAPK12 and MAPK13 might regulate autophagic flux in other cell types.
Recently, we have reported a direct role of MAPK14 in the regulation of autophagy and demonstrate that activated MAPK14 phosphorylates ATG5 and therefore inhibits the fusion of autophagosomes with lysosomes.21 This leads us to the following evasion mechanism: after enclosure of intracellular S. aureus within an autophagosome, MAPK14 becomes activated leading to inhibition of autophagic flux and therefore bacterial degradation (Fig. 9C). One pathway of MAPK14-mediated autophagy inhibition is via phosphorylation of ATG5.21 However, our data imply that MAPK14 deficiency has a more pronounced effect than the ATG5T75A mutation. Thus, additional MAPK14-dependent mechanisms have been described, e.g., availability of SUPT20H/p38IP for ATG9 trafficking,54 which might operate during S. aureus infection as well. In any case, the activation of MAPK14 by S. aureus to inhibit the autophagic pathway and therefore its own degradation is, so far, an unrecognized strategy by an intracellular pathogen to manipulate the autophagic pathway.
In conclusion, we show for the first time that intracellular S. aureus becomes targeted by selective autophagy through ubiquitination and the receptor proteins SQSTM1, OPTN and CALCOCO2. Further we show that S. aureus is able to evade autophagic degradation via a novel mechanism, involving activation of MAPK14, ATG5 phosphorylation and inhibition of fusion with lysosomes. Further understanding of the molecular mechanisms of host-pathogen interaction will yield new strategies to combat such an important pathogen.
Material and methods
Antibodies and reagents
The following antibodies were used in this study: anti-SQSTM1 (clone 2C11; Abnova, H00008878-M01), anti-ubiquitin (clone FK2; Enzo Life Science, BML-PW8810–0100), anti-ATG5 (clone D5F5U; Cell Signaling Technology, 12994), anti-ATG12 (Cell Signaling Technology, 2011), anti-ATG16L1 (clone D6D5; Cell Signaling Technology, 8089), anti-LC3A/B (Cell Signaling Technology, 4108), anti-TUBA/α-tubulin (clone DM1A; Sigma-Aldrich, T9026), anti-ACTB/β-actin (clone AC-74; Sigma-Aldrich, A2228), anti-phospho-MAPK14 (Cell Signaling Technology, 4511 for immunofluorescence, 9211 for western blotting), anti-MAPK14 (Cell Signaling Technology, 9212), anti-Flag (clone M2; Sigma-Aldrich, F7425), anti-LAMP2 (Developmental Studies Hybridoma Bank, ABL-93). Biochemicals used in this study included bafilomycin A1/Baf A1 (Enzo Life Science, BML-CM110–0100), CytoID (Enzo Life Science, ENZ-51031–0050) and LysoTracker Deep Red (Thermo Fisher Scientific, L12492).
Bacterial strains and culture conditions
The following bacterial strains were used in this study: SH1000 WT, SH1000-RFP and SH1000-spaΔ.55 Staphylococcus aureus strains were grown in tryptic soy broth (TSB) (Sigma-Aldrich, 22092). For growth on solid media, 1.5 (w/v) agar (BD Biosciences, 214010) was added. Thirty µg/ml chloramphenicol (Carl Roth, 3886.2) was added when required. Cultures were grown at 37°C and inoculated with an overnight culture to an optical density at 600 nm (OD600) of 0.05 into TSB, followed by incubation with agitation at 37°C. Bacterial growth was monitored by measuring the OD600.
Construction of bacterial RFP fluorescence reporter
A gene encoding mRFPmars was PCR-amplified from a plasmid pCR2.1TOPO::mRFPmars56 using the following primer pair (forward: ATAATAGGGCCCATAAAGGAGGTGTTTAAACGTG, reverse: TAATAGAATTCTTAGGATCCTGCACCTGTTG). The PCR product was then cloned into pTKP003-CFP57 to replace the CFP-encoding gene with mRFPmars. Inserts and the plasmid were restriction-digested using ApaI (New England Bioloabs, R0114S) and EcoRI (New England Bioloabs, R3101S) prior to ligation. The resulting plasmid, pTKP005-RFP, was introduced into S. aureus RN4220 by electroporation, resulting in transformants expressing mRFPmars (verified by fluorescence microscopy). The plasmid was then transferred into S. aureus SH1000 by Φ11 transduction and transductants were verified by fluorescence microscopy.
Cell culture and transfection
Murine fibroblasts NIH/3T3 were cultured in Dulbecco's modified Eagle's medium (DMEM high glucose; Thermo Fisher Scientific, 11965092) supplemented with 10% fetal calf serum (PAA Laboratories, A15–101). NIH/3T3 cells stably expressing GFP-LC3 were generated by transfection using JetPEI™ (Polyplus transfection™, 101–10N) reagents. mapk14 KO MEFs and their wild-type counterparts were kindly provided by Dr. Angel Nebreda (Institute for Research in Biomedicine, Barcelona, Spain).58 HaCaT cells stably expressing GFP-LC3 were kindly provided by Dr. Christian Münz (Viral Immunobiology, Institute of Experimental Immunology, University Hospital of Zürich, Switzerland).38 atg5 KO fibroblasts and its corresponding control cells were kindly provided by Dr. Noboru Mizushima (Department of Physiology and Cell Biology; Tokyo Medical and Dental University; Tokyo, Japan).59 atg5 KO fibroblasts were reconstituted either with wild-type or ATG5T75A, in which threonine 75 has been mutated to alanine, as described before.21 Transient transfection of NIH/3T3 cells was performed with JetPEI™ or JetPRIME™ (Polyplus transfection™, 114–01) according to manufacturer's instructions. Cells were transfected with Flag-tagged human CALCOCO2 or GFP-tagged human OPTN, both were kindly provided by Dr. Ivan Dikic (Frankfurt Institute for Molecular Life Sciences and Institute of Biochemistry II, Goethe University School of Medicine in Frankfurt Main, Germany).28
Intracellular survival assay
For intracellular survival assay, early stationary phase bacteria (OD600 = 1.0 to 1.5) were harvested and washed once in cold phosphate-buffered saline (PBS; Thermo Fisher Scientific, 10010–015). Bacteria diluted and added to confluent cell culture monolayers at a MOI of 8. After 1.5 h, cells were treated with 10 µg/mL lysostaphin (Sigma-Aldrich, L9043) for 12 min, to kill extracellular bacteria. After incubation, cells were washed with PBS and further incubated in 10% FCS-DMEM. Finally, cells were washed with PBS to wash out extracellular bacteria and dead cells, lysed by the addition of 0.5% Triton X-100 (Sigma-Aldrich, X100) and serial dilutions were plated on TSB agar (Sigma-Aldrich, 22092 and BD Biosciences, 214010) for bacterial enumeration.
Replication assay
For the intracellular replication assay early stationary phase bacteria (OD600 = 1.0 to 1.5) were harvested and washed once in cold PBS. Bacteria diluted and added to confluent cell culture monolayers at a MOI of 8. After 1.5 h, cells were treated with 10 µg/mL lysostaphin for 12 min, to kill extracellular bacteria. After incubation, cells were washed with PBS and further incubated in fresh 10% FCS-DMEM. Finally, the supernatant was collected and cells were lysed by the addition of 0.5% Triton X-100. Lysate was combined with the supernatant and serial dilutions were plated on TSB agar for bacterial enumeration.
Infection
For live cell imaging 1.5 × 103 NIH/3T3 GFP-LC3 cells were seeded onto an 8-well tissue culture chamber (Sarstedt, 94.6170.802). For immunofluorescence studies, 5 to 8 × 104 cells were seeded on a coverslip in a 12-well dish. After 24 h, cells were infected with S. aureus early stationary phase (OD600 = 1.0 to 1.5; MOI 8) for 90 min. Subsequently, cells were treated with 10 µg/mL lysostaphin for 12 min, to kill all extracellular bacteria (= 0 hpi).
Fluorescence microscopy
For immunofluorescence experiments, cells were washed in PBS and fixed with 3% paraformaldehyde (Sigma-Aldrich, F8775) in PBS for 10 min at 37°C after different times of infections. Afterwards, cells were washed twice in PBS and permeabilized with 0.001% saponin (Sigma-Aldrich, 47036) and 4% BSA (PAA Laboratories, K51-001) in PBS for 45 min at room temperature (RT). Then, cells were incubated with the appropriate antibody at 4°C overnight and washed several times with PBS. Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific, A21441, A21121, A21131, A21070, A21126, A21136) were used for 1 h at room temperature. Finally, coverslips were fixed using fluorescence mounting medium (DAKO, S3023). For analyzing immune fluorescence as well as performing live cell imaging, an UltraViewVox Spining Disc module (Perkin Elmer, Waltham, MA/USA) together with a Nikon Ti Eclipse inverted microscope (Nikon GmbH, Duesseldorf, Germany) need address for apparatus) was used. Images were analyzed via Volocity (Perkin Elmer) and ImageJ software.
Immunoblot assay
For protein extraction cells were lysed in TPNE buffer (1% Triton X-100, 2 mM EDTA [Carl Roth, 8043], 1 mM PMSF [Sigma, P7626] and 1 µg/mL of leupeptin [Applichem, A2183], aprotinin [Applichem, A2132], chymostatin [Applichem, A2144] and pepstatin [Applichem, A2205] each, in PBS adjusted to 300 mM NaCl [Carl Roth, 3957.1]). Protein concentration was measured via Pierce BCA protein assay kit (Thermo Fisher Scientific, 23225). A total of 20 or 30 µg protein were separated on 12 or 15% SDS polyacrylamide gels (Serva, 20765.03, Carl Roth, 3029.1) and transferred onto Amersham Hybond 0.45 PVDF membrane (GE Healthcare Life Sciences, 10600023). The membrane was blocked for 1 h in 5% nonfat dry milk (Carl Roth, T145.3) in TBS (137 mM NaCl, 2.68 mM KCl [Carl Roth, 6781.1], 24.76 mM Tris [Sigma, T1503], pH 7.4) containing 0.05% Tween 20 (Sigma-Aldrich, P1379) and incubated with a primary antibody over night at 4°C. After 4 × 10 min washing with TBS-Tween, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody (1:20,000; SouthernBiotech, 1070–05, 1080–05, 4030–05) for 1 h at RT. After washing the membranes were developed with an Amersham ECL Select Western Blotting Detection Reagent (GE Healthcare Life Sciences, RPN2235). The corresponding bands were then detected via the Fusion FX-7 camera (Vilber Lourmat, Eberhardzell, Germany) or by usage of high performance chemiluminescence films (GE Healthcare Life Sciences, 28–9068-37) and the Curix 60-System (AGFA, Mortsel, Belgium). Afterwards blots were stripped using Re-Blot mild solution (EMD Millipore, 2502) according to the manufacturer´s instructions.
Transmission electron microscopy
For visualization of intracellular membranes within S. aureus-infected NIH/3T3 fibroblasts, 2 fixation schemes were applied. O-T-O method: After fixation with 2.5% glutaraldehyde (Merck, 04239.0250) and 5% formaldehyde (Carl Roth, 0335.3) samples were treated with osmium ferrocyanide (1% osmium tetroxide (Carl Roth, 8371.3) and 1.5% ferrocyanide (Sigma-Aldrich, 31254–500G)) followed by incubation with 1% thiocarbohydrazide (Sigma, 88535). Thereafter, another fixation with osmium ferrocyanide was performed. Samples were then embedded in 1.75% agar (Difco, 0142–01), cut into small cubes and dehydrated with a graded series of acetone (J.T. Baker, 8002) and embedded into the epoxy low viscosity resin (Agar Scientific, R1078). Second fixation scheme: Samples were fixed with 1% glutaraldehyde and 5% formaldehyde in PBS. After washing with PBS samples were further treated with 1% aqueous osmium tetroxide for 1 h, washed and incubated with 4% aqueous uranyl acetate (Science Services, 22400) for 1 h. After washing with PBS samples were further processed as for the O-T-O method. Ultrathin sections were cut with a diamond knife, collected onto butvar-coated grids, and counterstained with uranyl acetate and lead citrate (Leica, Ultastain2 705530). Samples were examined in a Zeiss TEM 910 (Zeiss, Oberkochen, Germany) at an acceleration voltage of 80 kV and at calibrated magnifications. Images were recorded digitally at calibrated magnifications with a Slow-Scan CCD-Camera (ProScan, 1024 ×1024, Scheuring, Germany) with ITEM-Software (Olympus Soft Imaging Solutions, Münster, Germany). Contrast and brightness were adjusted with Adobe Photoshop CS5.
Supplementary Material
Abbreviations
- Atg
autophagy related
- ATG5T75A
autophagy-related 5; threonine 75 mutated to alanine
- Baf A1
bafilomycin A1
- CALCOCO2/NDP52
calcium binding and coiled-coil domain 2
- GFP
green fluorescent protein
- HBSS
Hank's buffered salt solution
- hpi
hours postinfection
- LC3
microtubule-associated protein 1 light chain 3
- MAPK14/p38α
mitogen-activated protein kinase 14
- MEF
mouse embryonic fibroblast
- min
minute
- MOI
multiplicity of infection
- OPTN
optineurin
- RFP
red fluorescent protein
- S. aureus
Staphylococcus aureus
- SQSTM1/p62
sequestosome 1
- SEM
standard error of the mean
- Ub
ubiquitin
Disclosure of potential conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
We thank Ina Schleicher, Sabrina Schumann and Christian Kozowsky for their excellent technical assistance. We wish to thank Drs. Tobias Kruse and Oliver Goldmann for critically reading the manuscript and Dr. Susanne Engelmann for many helpful discussions. We are grateful to Drs. Ivan Dikic, Noboru Mizushima, Christian Münz, Angel Nebreda and Björn Stork for cells and plasmids.
Funding
SAB was supported by the President's Initiative and Networking Fund of the Helmholtz Association of German Research Centers (HGF) under contract number VH-GS-202. TKP was supported by the British Infection Association (BIA) research fellowship. This work has been supported by the Deutsche Forschungsgemeinschaft (SCHM1586/3–1) and the Helmholtz portfolio program Metabolic Dysfunction.
References
- [1].Peacock SJ, de Silva I, Lowy FD. What determines nasal carriage of Staphylococcus aureus? Trends Microbiol 2001; 9:605-10; PMID:11728874; http://dx.doi.org/ 10.1016/S0966-842X(01)02254-5 [DOI] [PubMed] [Google Scholar]
- [2].Kobayashi SD, DeLeo FR. An update on community-associated MRSA virulence. Curr Opin Pharmacol 2009; 9:545-51; PMID:19726228; http://dx.doi.org/ 10.1016/j.coph.2009.07.009 [DOI] [PubMed] [Google Scholar]
- [3].Lowy FD. Staphylococcus aureus infections. N Engl J Med 1998; 339:520-32; PMID:9709046; http://dx.doi.org/ 10.1056/NEJM199808203390806 [DOI] [PubMed] [Google Scholar]
- [4].Lowy FD. Is Staphylococcus aureus an intracellular pathogen? Trends Microbiol 2000; 8:341-3; PMID:10920387; http://dx.doi.org/ 10.1016/S0966-842X(00)01803-5 [DOI] [PubMed] [Google Scholar]
- [5].Mestre MB, Fader CM, Sola C, Colombo MI. Alpha-hemolysin is required for the activation of the autophagic pathway in Staphylococcus aureus-infected cells. Autophagy 2010; 6:110-25; PMID:20110774; http://dx.doi.org/ 10.4161/auto.6.1.10698 [DOI] [PubMed] [Google Scholar]
- [6].Sinha B, Fraunholz M. Staphylococcus aureus host cell invasion and post-invasion events. Int J Med Microbiol 2010; 300:170-5; PMID:19781990; http://dx.doi.org/ 10.1016/j.ijmm.2009.08.019 [DOI] [PubMed] [Google Scholar]
- [7].Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102-9; PMID:17909521; http://dx.doi.org/ 10.1038/ncb1007-1102 [DOI] [PubMed] [Google Scholar]
- [8].Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/ 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2:211-6; PMID:11265251; http://dx.doi.org/ 10.1038/35056522 [DOI] [PubMed] [Google Scholar]
- [10].Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657-68; PMID:11266458; http://dx.doi.org/ 10.1083/jcb.152.4.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805-12; PMID:15169837; http://dx.doi.org/ 10.1242/jcs.01131 [DOI] [PubMed] [Google Scholar]
- [13].Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, et al.. Autophagy defends cells against invading group A Streptococcus. Science 2004; 306:1037-40; PMID:15528445; http://dx.doi.org/ 10.1126/science.1103966 [DOI] [PubMed] [Google Scholar]
- [14].Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol 2003; 5:455-68; PMID:12814436; http://dx.doi.org/ 10.1046/j.1462-5822.2003.00292.x [DOI] [PubMed] [Google Scholar]
- [15].Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol 2014; 12:101-14; PMID:24384599; http://dx.doi.org/ 10.1038/nrmicro3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chong A, Wehrly TD, Child R, Hansen B, Hwang S, Virgin HW, Celli J. Cytosolic clearance of replication-deficient mutants reveals Francisella tularensis interactions with the autophagic pathway. Autophagy 2012; 8:1342-56; PMID:22863802; http://dx.doi.org/ 10.4161/auto.20808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ogawa M, Yoshikawa Y, Mimuro H, Hain T, Chakraborty T, Sasakawa C. Autophagy targeting of Listeria monocytogenes and the bacterial countermeasure. Autophagy 2011; 7:310-4; PMID:21193840; http://dx.doi.org/ 10.4161/auto.7.3.14581 [DOI] [PubMed] [Google Scholar]
- [18].Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science 2005; 307:727-31; PMID:15576571; http://dx.doi.org/ 10.1126/science.1106036 [DOI] [PubMed] [Google Scholar]
- [19].Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, et al.. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol 2009; 11:1233-40; PMID:19749745; http://dx.doi.org/ 10.1038/ncb1967 [DOI] [PubMed] [Google Scholar]
- [20].Schnaith A, Kashkar H, Leggio SA, Addicks K, Kronke M, Krut O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem 2007; 282:2695-706; PMID:17135247; http://dx.doi.org/ 10.1074/jbc.M609784200 [DOI] [PubMed] [Google Scholar]
- [21].Keil E, Hocker R, Schuster M, Essmann F, Ueffing N, Hoffman B, Liebermann DA, Pfeffer K, Schulze-Osthoff K, Schmitz I. Phosphorylation of Atg5 by the Gadd45beta-MEKK4-p38 pathway inhibits autophagy. Cell Death Differ 2013; 20:321-32; PMID:23059785; http://dx.doi.org/ 10.1038/cdd.2012.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al.. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450:1253-7; PMID:18097414; http://dx.doi.org/ 10.1038/nature06421 [DOI] [PubMed] [Google Scholar]
- [23].Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ 2013; 20:21-30; PMID:22722335; http://dx.doi.org/ 10.1038/cdd.2012.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fujimuro M, Sawada H, Yokosawa H. Production and characterization of monoclonal antibodies specific to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett 1994; 349:173-80; PMID:7519568; http://dx.doi.org/ 10.1016/0014-5793(94)00647-4 [DOI] [PubMed] [Google Scholar]
- [25].Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009; 5:732-3; PMID:19398892; http://dx.doi.org/ 10.4161/auto.5.5.8566 [DOI] [PubMed] [Google Scholar]
- [26].Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, Cossart P. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem 2011; 286:26987-95; PMID:21646350; http://dx.doi.org/ 10.1074/jbc.M111.223610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 2009; 10:1215-21; PMID:19820708; http://dx.doi.org/ 10.1038/ni.1800 [DOI] [PubMed] [Google Scholar]
- [28].Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al.. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011; 333:228-33; PMID:21617041; http://dx.doi.org/ 10.1126/science.1205405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol 2009; 183:5909-16; PMID:19812211; http://dx.doi.org/ 10.4049/jimmunol.0900441 [DOI] [PubMed] [Google Scholar]
- [30].Barnett TC, Liebl D, Seymour LM, Gillen CM, Lim JY, Larock CN, Davies MR, Schulz BL, Nizet V, Teasdale RD, et al.. The globally disseminated M1T1 clone of group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe 2013; 14:675-82; PMID:24331465; http://dx.doi.org/ 10.1016/j.chom.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dortet L, Mostowy S, Samba-Louaka A, Gouin E, Nahori MA, Wiemer EA, Dussurget O, Cossart P. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog 2011; 7:e1002168; PMID:21829365; http://dx.doi.org/ 10.1371/journal.ppat.1002168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Choy A, Dancourt J, Mugo B, O'Connor TJ, Isberg RR, Melia TJ, Roy CR. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 2012; 338:1072-6; PMID:23112293; http://dx.doi.org/ 10.1126/science.1227026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/ 10.1091/mbc.E03-09-0704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res 2011; 109:296-308; PMID:21659648; http://dx.doi.org/ 10.1161/CIRCRESAHA.111.244707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pinho MG, Errington J. Dispersed mode of Staphylococcus aureus cell wall synthesis in the absence of the division machinery. Mol Microbiol 2003; 50:871-81; PMID:14617148; http://dx.doi.org/ 10.1046/j.1365-2958.2003.03719.x [DOI] [PubMed] [Google Scholar]
- [36].Steele VR, Bottomley AL, Garcia-Lara J, Kasturiarachchi J, Foster SJ. Multiple essential roles for EzrA in cell division of Staphylococcus aureus. Mol Microbiol 2011; 80:542-55; PMID:21401734; http://dx.doi.org/ 10.1111/j.1365-2958.2011.07591.x [DOI] [PubMed] [Google Scholar]
- [37].Turner RD, Ratcliffe EC, Wheeler R, Golestanian R, Hobbs JK, Foster SJ. Peptidoglycan architecture can specify division planes in Staphylococcus aureus. Nat Commun 2010; 1:26; PMID:20975691; http://dx.doi.org/ 10.1038/ncomms1025 [DOI] [PubMed] [Google Scholar]
- [38].Gannage M, Dormann D, Albrecht R, Dengjel J, Torossi T, Ramer PC, Lee M, Strowig T, Arrey F, Conenello G, et al.. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009; 6:367-80; PMID:19837376; http://dx.doi.org/ 10.1016/j.chom.2009.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kageyama S, Omori H, Saitoh T, Sone T, Guan JL, Akira S, Imamoto F, Noda T, Yoshimori T. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol Biol Cell 2011; 22:2290-300; PMID:21525242; http://dx.doi.org/ 10.1091/mbc.E10-11-0893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lam GY, Cemma M, Muise AM, Higgins DE, Brumell JH. Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy 2013; 9:985-95; PMID:23584039; http://dx.doi.org/ 10.4161/auto.24406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Perrin AJ, Jiang X, Birmingham CL, So NS, Brumell JH. Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr Biol 2004; 14:806-11; PMID:15120074; http://dx.doi.org/ 10.1016/j.cub.2004.04.033 [DOI] [PubMed] [Google Scholar]
- [42].Huett A, Heath RJ, Begun J, Sassi SO, Baxt LA, Vyas JM, Goldberg MB, Xavier RJ. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe 2012; 12:778-90; PMID:23245322; http://dx.doi.org/ 10.1016/j.chom.2012.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Giachino P, Engelmann S, Bischoff M. Sigma(B) activity depends on RsbU in Staphylococcus aureus. Journal of bacteriology 2001; 183:1843-52; PMID:11222581; http://dx.doi.org/ 10.1128/JB.183.6.1843-1852.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ. sigmaB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. Journal of bacteriology 2002; 184:5457-67; PMID:12218034; http://dx.doi.org/ 10.1128/JB.184.19.5457-5467.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, Nagai Y, Iwama N, Asano K, Naimi T, et al.. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 2002; 359:1819-27; PMID:12044378; http://dx.doi.org/ 10.1016/S0140-6736(02)08713-5 [DOI] [PubMed] [Google Scholar]
- [46].Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, et al.. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 2006; 367:731-9; PMID:16517273; http://dx.doi.org/ 10.1016/S0140-6736(06)68231-7 [DOI] [PubMed] [Google Scholar]
- [47].Hicks SW, Galan JE. Hijacking the host ubiquitin pathway: structural strategies of bacterial E3 ubiquitin ligases. Curr Opin Microbiol 2010; 13:41-6; PMID:20036613; http://dx.doi.org/ 10.1016/j.mib.2009.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mesquita FS, Thomas M, Sachse M, Santos AJ, Figueira R, Holden DW. The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog 2012; 8:e1002743; PMID:22719249; http://dx.doi.org/ 10.1371/journal.ppat.1002743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rytkonen A, Poh J, Garmendia J, Boyle C, Thompson A, Liu M, Freemont P, Hinton JC, Holden DW. SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc Natl Acad Sci U S A 2007; 104:3502-7; PMID:17360673; http://dx.doi.org/ 10.1073/pnas.0610095104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Miller M, Dreisbach A, Otto A, Becher D, Bernhardt J, Hecker M, Peppelenbosch MP, van Dijl JM. Mapping of interactions between human macrophages and Staphylococcus aureus reveals an involvement of MAP kinase signaling in the host defense. J Proteome Res 2011; 10:4018-32; PMID:21736355; http://dx.doi.org/ 10.1021/pr200224x [DOI] [PubMed] [Google Scholar]
- [51].Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell research 2005; 15:11-8; PMID:15686620; http://dx.doi.org/ 10.1038/sj.cr.7290257 [DOI] [PubMed] [Google Scholar]
- [52].Ono K, Han J. The p38 signal transduction pathway: activation and function. Cellular signalling 2000; 12:1-13; PMID:10676842; http://dx.doi.org/ 10.1016/S0898-6568(99)00071-6 [DOI] [PubMed] [Google Scholar]
- [53].Tamura K, Sudo T, Senftleben U, Dadak AM, Johnson R, Karin M. Requirement for p38alpha in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell 2000; 102:221-31; PMID:10943842; http://dx.doi.org/ 10.1016/S0092-8674(00)00027-1 [DOI] [PubMed] [Google Scholar]
- [54].Webber JL, Tooze SA. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J 2010; 29:27-40; PMID:19893488; http://dx.doi.org/ 10.1038/emboj.2009.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sibbald MJ, Yang XM, Tsompanidou E, Qu D, Hecker M, Becher D, Buist G, van Dijl JM. Partially overlapping substrate specificities of staphylococcal group A sortases. Proteomics 2012; 12:3049-62; PMID:22930668; http://dx.doi.org/ 10.1002/pmic.201200144 [DOI] [PubMed] [Google Scholar]
- [56].Paprotka K, Giese B, Fraunholz MJ. Codon-improved fluorescent proteins in investigation of Staphylococcus aureus host pathogen interactions. J Microbiol Methods 2010; 83:82-6; PMID:20708040; http://dx.doi.org/ 10.1016/j.mimet.2010.07.022 [DOI] [PubMed] [Google Scholar]
- [57].Prajsnar TK, Hamilton R, Garcia-Lara J, McVicker G, Williams A, Boots M, Foster SJ, Renshaw SA. A privileged intraphagocyte niche is responsible for disseminated infection of Staphylococcus aureus in a zebrafish model. Cell Microbiol 2012; 14:1600-19; PMID:22694745; http://dx.doi.org/ 10.1111/j.1462-5822.2012.01826.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Porras A, Zuluaga S, Black E, Valladares A, Alvarez AM, Ambrosino C, Benito M, Nebreda AR. P38 α mitogen-activated protein kinase sensitizes cells to apoptosis induced by different stimuli. Mol Biol Cell 2004; 15:922-33; PMID:14617800; http://dx.doi.org/ 10.1091/mbc.E03-08-0592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; http://dx.doi.org/ 10.1038/nature03029 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.