

ABSTRACT
Cystic fibrosis (CF) is a fatal, genetic disorder that critically affects the lungs and is directly caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, resulting in defective CFTR function. Macroautophagy/autophagy is a highly regulated biological process that provides energy during periods of stress and starvation. Autophagy clears pathogens and dysfunctional protein aggregates within macrophages. However, this process is impaired in CF patients and CF mice, as their macrophages exhibit limited autophagy activity. The study of microRNAs (Mirs), and other noncoding RNAs, continues to offer new therapeutic targets. The objective of this study was to elucidate the role of Mirs in dysregulated autophagy-related genes in CF macrophages, and then target them to restore this host-defense function and improve CFTR channel function. We identified the Mirc1/Mir17-92 cluster as a potential negative regulator of autophagy as CF macrophages exhibit decreased autophagy protein expression and increased cluster expression when compared to wild-type (WT) counterparts. The absence or reduced expression of the cluster increases autophagy protein expression, suggesting the canonical inverse relationship between Mirc1/Mir17-92 and autophagy gene expression. An in silico study for targets of Mirs that comprise the cluster suggested that the majority of the Mirs target autophagy mRNAs. Those targets were validated by luciferase assays. Notably, the ability of macrophages expressing mutant F508del CFTR to transport halide through their membranes is compromised and can be restored by downregulation of these inherently elevated Mirs, via restoration of autophagy. In vivo, downregulation of Mir17 and Mir20a partially restored autophagy expression and hence improved the clearance of Burkholderia cenocepacia. Thus, these data advance our understanding of mechanisms underlying the pathobiology of CF and provide a new therapeutic platform for restoring CFTR function and autophagy in patients with CF.
KEYWORDS: autophagy, Burkholderia cenocepacia, CFTR function, cystic fibrosis, macrophages, Mirc1/Mir17-92 cluster
Introduction
Production of functional proteins requires multiple steps, including gene transcription and posttranslational processing. MicroRNAs (Mirs) can regulate individual stages of these processes.1 Mirs are an evolutionarily, conserved class of small (∼21–24 nucleotides) noncoding RNAs that play key roles in the transcriptional and posttranscriptional regulation of gene expression.1 Specific Mirs have been identified to regulate Cftr (cystic fibrosis transmembrane conductance regulator) gene expression, but their actions in the context of both autophagy and CF have not been investigated. Autophagy is a highly regulated biological process that provides energy during periods of stress and starvation. This process is impaired in CF patients and CF mice, as their macrophages exhibit limited autophagy activity.
CF is an autosomal recessive disease caused by mutations in the Cftr gene that result in defective CFTR function in many organs; however, the majority of CF-associated morbidity and mortality arises from pulmonary infection and inflammation.2 The Cftr gene encodes a transmembrane chloride channel that is regulated by ATP hydrolysis and expressed in various cell types including epithelial cells and macrophages.3 In epithelial cells, the CFTR channel conducts anions and plays a critical role in regulating the volume and composition of airway surface liquid,4 a thin layer of aqueous fluid and mucus covering the airway surface whose properties include facilitating mucociliary clearance, bacterial killing, and epithelial cell homeostasis.5 The function of the CFTR channel in macrophages remains unclear although our recent work demonstrates that defective CFTR function is accompanied by an impaired innate immune response to specific infections.6-11 In CF patients with the most common mutation, F508del, the mutant form of the protein fails to traffic properly to the plasma membrane. This leads to a critical lack of fluid exchange across the membrane. Should the F508del-CFTR traffic to the cell membrane in response to therapy, the mutant protein then regains partial channel function in epithelial cells.12,13 Heterozygote humans and mice do not suffer pathological symptoms despite the fact that their cells exhibit only 50% functional activity of the CFTR channel.14 Thus, small improvement of CFTR channel function in F508del homozygotes is accompanied by a significant improvement in ensuing symptoms as reported by several clinical trials.15,16
Recent studies have implicated reduced autophagy activity in a number of physiological and pathophysiological processes such as aging, cancer, neurodegenerative diseases, innate immunity, and CF.10,11,17-19 Autophagy functions to yield energy and nutrients during stress or starvation of the cell.20 In addition, autophagy can also restrict specific pathogens within macrophages and improve clearance of misfolded protein aggregates that cannot be managed by proteasomes.21 The process of autophagy involves formation of double-membrane compartments (phagophores) that engulf nonfunctional organelles and cytoplasm. These phagophores then mature into autophagosomes that fuse with lysosomes to form autolysosomes, within which the autophagic cargo is degraded and recycled for protein and ATP synthesis via degradative enzymes from the lysosome.22,23 Autophagosome formation is mediated by a series of autophagy-promoting molecules including Atg5, Atg12, Atg16, Atg7, Atg8 (mammalian MAP1LC3/LC3 and GABARAP) and Vps30/Atg6 (BECN1/Beclin1).22 Thus, the absence or reduction in expression of one factor can markedly impair the autophagy process.
Burkholderia (B.) cenocepacia is notorious for infecting CF patients and is resistant to the majority of antibiotics. In healthy macrophages, B. cenocepacia is cleared by autophagy. However, macrophages from CF humans and mice fail to control B. cenocepacia due to impaired autophagy activity.10,11,24 CF mice allow B. cenocepacia to establish infection in their lungs, which triggers an intense, and often lethal, inflammatory response.10,11,24 Therefore, clearance of B. cenocepacia from the lungs of CF mice reflects the amount of autophagy activity.
Reports have demonstrated the extent to which Mirs regulate CFTR expression and function16,25,26 yet their role in the context of autophagy has yet to be investigated. Thus, considering the strong implications of Mirs, both in autophagy and CF, and given the lack of current evidence linking these 2 processes, we investigated specific Mirs that regulate autophagy-related genes whose expression is altered in CF patients.
In this report, we used in silico approaches to identify the importance of the Mirc1/Mir17-92 cluster which generates a single polycistronic transcript that yields 6 mature Mirs: Mir17, Mir18a, Mir19a, Mir20a, Mir19b, and Mir92.27 The polycistronic Mirc1/Mir17-92 cluster was initially linked to tumorigenesis as published by our group and others.27-34 The role of the Mirc1/Mir17-92 cluster in CF has not been investigated. We demonstrate that members of the Mirc1/Mir17-92 cluster target multiple essential autophagy factors. In addition, we find that several specific Mirs comprising the Mir17HG/Mir17-92 cluster are overexpressed in CF human and murine macrophages with corresponding reduced expression of their predicted autophagy-targeted genes. Mirs comprising the Mir17HG/Mir17-92 cluster exhibit a trend toward upregulation in CF cells, especially Mir17 and Mir20a, which are significantly increased in murine macrophages. Importantly, Mir17 is also significantly upregulated in macrophages derived from CF patients. Luciferase assays validated that both Mir17 and Mir20a target Atg7 and Atg16l1. Notably, reducing the inherently elevated expression of Mir17 and Mir20a improves ATG7 and ATG16L1 expression both in vitro and in vivo. In addition, reducing Mir17 and Mir20a expression improves CFTR function by restoring autophagy expression. In this regard, targeting Mir17 was more efficient. Accordingly, B. cenocepacia clearance is improved in CF mice after intra-tracheal treatment with antagomirs to Mir17 and Mir20a. Containment of B. cenocepacia upon administration of these specific antagomirs is accompanied by improved expression of targeted autophagy proteins. Our study advances our understanding of the mechanism underlying defective autophagy in CF and provides a novel therapeutic approach for restoring CFTR function and autophagy.
Results
Expression of autophagy proteins is reduced in primary CF (F508del) macrophages
CF macrophages exert weak autophagic activity10,11,18 yet the underlying mechanisms behind this reduction remain unknown. To investigate the potential mechanism for compromised autophagy activity in CF cells, we examined the expression of autophagy-related proteins in wild-type (WT) C57/BL6J and CF murine macrophages by western blot using specific antibodies. Notably, the expression of members of the ATG12–ATG5 protein complex and ATG7 was decreased in CF macrophages when compared to WT cells (Fig. 1A). However, ATG12 protein and mRNA levels were comparable in WT and F508del macrophages (Figs. 1A and S1A).10 To evaluate if the low expression level of autophagy family member proteins reduces the basal autophagy activity in resting WT and CF macrophages, the number of macrophages exhibiting more than 5 LC3 labeled-autophagosomes (puncta) was quantified using confocal microscopy. LC3 is a cytosolic autophagy protein that is recruited to phagophores; the subsequent autophagosomes appear as donut-shaped structures called puncta. Significantly fewer resting CF macrophages had more than 5 puncta when compared to their WT counterparts (Fig. 1B and C). In addition, to determine autophagy activity in response to stimulation, macrophages were starved and those expressing more than 5 puncta were quantified. CF macrophages failed to increase their puncta content in response to starvation in contrast to WT macrophages (Fig. 1B and C). Therefore, CF macrophages fail to mount an autophagic flux response upon starvation. Together, these data provide evidence that CF macrophages exhibit lower expression of essential ATG proteins and are characterized by weak autophagic activity.
Figure 1.
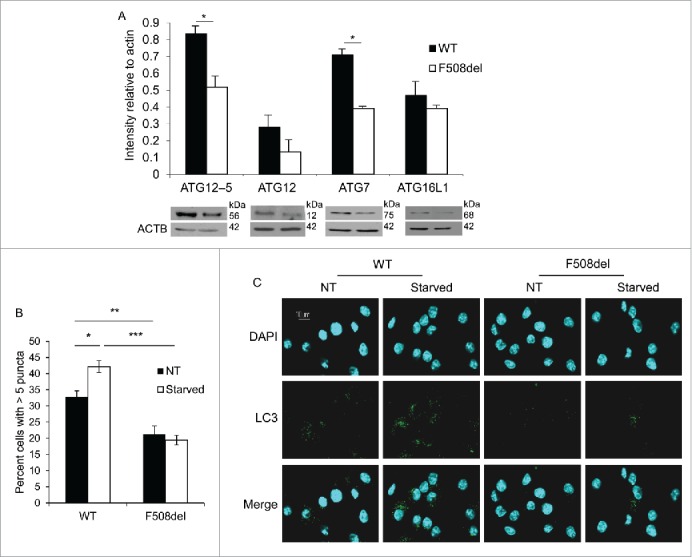
Primary F508del (CF) murine macrophages exhibit weak autophagic flux during resting and starvation conditions. (A) Western blot for basal level autophagy proteins in WT (black bars) and F508del primary mouse macrophages (white bars). Densitometry analyses of western blot bands were normalized to their respective loading control bands using ImageJ software to control for loading. Panels shown represent 3 independent experiments displaying similar results. (B) Scoring of the percentage of macrophages harboring more than 5 puncta in WT and F508del macrophages before (NT, black) and after 2 h of starvation (starved, white). Data are representative of scoring the means ± SEM of 900 WT macrophages and 700 F508del macrophages. (C) Confocal microscopy representing autophagy activity of resting and starved WT and F508del macrophages. Blue (DAPI) stain indicates nuclei and green stain indicates LC3 puncta (autophagosome formation). Asterisks (*) indicates p ≤ 0.05; **, p ≤ 0.01; and ***, p ≤ 0.001.
Expression of the Mirc1/Mir17-92 cluster is elevated in primary CF macrophages resulting in the reduction of several autophagy molecules
Given the lack of current evidence regarding autophagy regulation in CF, we investigated predicted Mirs that may play a role in modulating the autophagy process and whether their expression is altered in CF cells. Using 2 online web servers that predict targets of specific Mirs, TargetScan and MirBase, we determined that Mir101 and the Mirc1/Mir17-92 cluster are predicted to target autophagy-related mRNAs. Mir101 was reported to target STMN1, RAB5A, and ATG4D, each of which is an important autophagic regulator.35 The Mirc1/Mir17-92 cluster is predicted to target Atg4, Atg5, Becn1, Atg7, Atg12, Atg16l1 and Lc3 (Table S1). To determine whether the expression of Mir101 and the Mirc1/Mir17-92 cluster are altered in CF macrophages, we performed quantitative real-time PCR (qRT-PCR) on RNA lysates from murine WT and CF macrophages. Expression of Mir101 was not statistically different between WT and CF macrophages (Fig. S1B), whereas the Mirc1/Mir17-92 cluster expression was significantly elevated in CF macrophages (Fig. 2A). While members of the cluster exhibit a trend toward upregulation in murine CF macrophages, Mir17 and Mir20a were significantly increased (Fig. 2A). To determine whether these findings are consistent with human CF pathology, expression of Mirs comprising the cluster was assessed in macrophages derived from blood monocytes of CF patients. Several members of the cluster were upregulated, but only Mir17 was significantly upregulated in both human and mouse samples (Fig. 2B). These data suggest that the inherently elevated expression level of Mir17HG/Mir17-92 cluster members in CF macrophages contributes at least in part to the reduced expression of several autophagy-related genes that potentially impair autophagy activity in CF macrophages.
Figure 2.

Mirc1/Mir17-92 cluster expression is elevated in primary homozygous F508del macrophages and targets several autophagy molecules. (A) Quantitative real-time PCR (qPCR) representing the expression of Mirc1/Mir17-92 cluster members in resting WT (black bars) and F508del (white bars) murine macrophages. Student 2-tailed t test was used to determine significance at p ≤ 0.5. WT macrophage cluster expression compared to F508del macrophage cluster expression was also significant as calculated by 2-way ANOVA p ≤ 0.05. WT macrophages n = 5 and F508del macrophages n = 4 for Mirc1/Mir17-92 analysis. Graphs are representative of compiling means ± SD. (B) qPCR representing the expression of Mir17HG/Mir17-92 cluster in human blood monocyte-derived macrophages from 6 non-CF (black bars) and 6 CF (white bars) patients. Data shown represent the means ± SD. Student 2-tailed t test was used to assess significance. (C) Western blot for autophagy proteins in WT (black bars) and Mirc1/Mir17-92−/− macrophages (gray bars). Densitometric analyses of bands were normalized to their respective actin loading control bands using imageJ software. Each blot is representative of 3 independent experiments displaying similar results. (D) qPCR results for autophagy-regulating genes in WT (black bars) and Mirc1/Mir17-92−/− macrophages (gray bars). Data are presented as fold change compared to WT normalized to 1 and are presented as the means ± SD, n = 3. Student 2-tailed t test was used to determine significance at p ≤ 0.05. (E) Western blot for autophagy proteins in Mirc1/Mir17-92−/− macrophages untreated (NT, black bars), transfected with scramble control nucleotides (gray bars), or Mir17 and Mir20a mimics (white bars). Protein levels were normalized to their respective actin levels and quantified by ImageJ software, n = 3. * indicates p ≤ 0.05 for differences between transfected scrambled controls and mimics.
To determine the extent to which the elevated Mirc1/Mir17-92 cluster contributed to the reduced expression of autophagy molecules, their expression was examined in the absence of cluster expression. Macrophages lacking the Mirc1/Mir17-92 cluster were obtained from Mirc1/Mir17-92−/− floxed mice.36 Since a complete knockout of the cluster is embryonic lethal, these mice were obtained by breeding a Lyz2/LysM-cre mouse with a Mirc1/Mir17-92−/− floxed mouse, thus, knocking out the Mirc1/Mir17-92 cluster specifically in the myeloid lineage.27 The protein lysates of WT and Mirc1/Mir17-92−/− macrophages were analyzed by western blot using specific autophagy antibodies. ATG12–ATG5, ATG7, and ATG16L1 protein levels were substantially elevated in the absence of the Mirc1/Mir17-92 cluster, but not ATG12, (Fig. 2C) as were their corresponding mRNA expression (Fig. 2D). To determine if the elevated levels of ATG proteins in Mir17-92−/− macrophages are directly associated with low levels of the cluster, we transfected Mir17-92−/− macrophages with Mir17 and Mir20a mimics to restore their expression and assessed the autophagy protein expression profile. Overexpression of Mir17 and Mir20a led to a reduction in ATG12–ATG5 and ATG7 expression when compared to their scrambled controls (Fig. 2E). Collectively, these data provide strong evidence that the Mir17-92 cluster modulates the expression of essential autophagy-related genes in macrophages.
Mir17 and Mir20a target the 3′-untranslated region of Atg7 and Atg16l1
We have shown that several microRNAs comprising the cluster are elevated in human and murine CF macrophages; however, Mir17 and Mir20a exhibit the most significantly increased expression in mice, and Mir17 is elevated in human CF macrophages (Fig. 2A and B). Therefore, we focused on further characterizing Mir17 and Mir20a in CF mice. Targeting only 2 Mirs is a more feasible therapeutic approach to take in the future, as it would potentially reduce off-target effects. To confirm the binding of Mir17 and Mir20a to the 3′ untranslated (3′-UTR) regions of Atg7 and Atg16l1, NIH3T3 cells were transiently transfected with a luciferase reporter construct containing the full length 3′-UTR of Atg7, Atg16l1 or an empty vector luciferase reporter. Transfection with Mir17 or Mir20a mimics reduced luciferase activity from cells transfected with either Atg7-3′-UTR-luc or Atg16l1-3′-UTR-luc constructs (Fig. 3A and B). Notably, mutations in the 3′-UTR of Atg7 (M-Atg7) or Atg16l1 (M-Atg16l1) eliminated the Mir17 and Mir20a mimic-mediated reduction of luciferase activity (Fig. 3A and B). To further verify the relationship between Mir17 and Mir20a and the expression of their respective targets Atg7 and Atg16l1, we inhibited the expression of these Mirs in CF macrophages using specific antagomirs. CF macrophages transfected with either antagomir17 or antagomir20a elicited a nonsignificant increase in ATG7 and ATG16L1 protein expression (Fig. S1C). However when transfected together, antagomir17 and antagomir20a significantly increased the expression of ATG7 and ATG16L1 when compared to scramble-transfected cells (Fig. 3C and D). Therefore, transfection with specific antagomir17 and antagomir20a improved the expression of ATG7 and ATG16L1, whereas transfection with scramble antagomirs did not (Fig. 3A and B). Taken together, these data demonstrate that Mir17 and Mir20a target the Atg7 and Atg16l1 genes and modulate their expression in CF macrophages.
Figure 3.
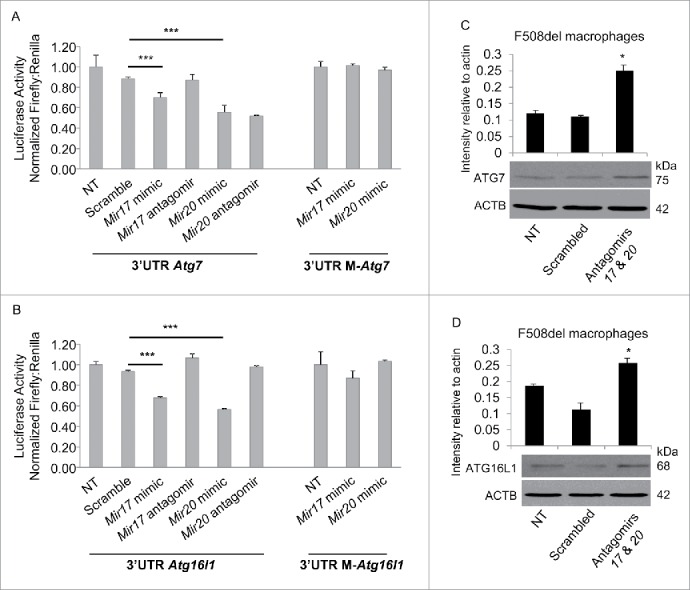
Mir17 and Mir20a target the 3′UTRs of Atg7 and Atg16l1. (A and B) NIH3T3 cells transfected with luciferase reporter constructs containing the 3′UTR of murine Atg7 (A) or Atg16l1 (B), or their mutated 3′UTR (M-Atg7, M-Atg16l1), were not treated (NT) or treated with scramble control nucleotides (A and B), or Mir17 or Mir20a mimics or their corresponding antagomirs. Antagomir17 and antagomir20a were transfected to reduce corresponding endogenous miRNA levels. Luciferase expression was normalized to the level of Renilla measured. Data shown are representative of the average luciferase production conducted in quadruplicate ± SD. n = 3 and asterisks indicate significant differences by one-way ANOVA. (C and D) Western blot for (C) ATG7 or (D) ATG16L1 in F508del macrophages transfected with antagomir17 and antagomir20a. The blot is representative of 3 independent experiments displaying similar results and protein bands were normalized to their respective ACTB bands and quantified by ImageJ software. * indicates p ≤ 0.05; and ***, p ≤ 0.001.
Pulmonary delivery of antagomir17 and antagomir20a improves the expression of targeted autophagy genes and autophagy activity in CF mice
To determine whether reducing the elevated expression of Mir17 and Mir20a in vivo improves the expression of targeted autophagy genes, we delivered antagomirs against Mir17 and Mir20a or scrambled control intratracheally, once a day for 3 d, to mice. The lungs were harvested, homogenized and analyzed for the expression of Mir17 and Mir20a by qRT-PCR. We found that this regimen effectively lowered the expression of elevated Mir17 and Mir20a in the lungs of CF mice and did not alter the expression of other members of the cluster (Fig. 4A). To determine the extent to which decreasing the expression of Mir17 and Mir20a rescues the expression of targeted autophagy genes in vivo, lung homogenates were analyzed for the mRNA levels of autophagy genes Atg5, Atg7 and Atg16l1 by qRT-PCR and western blot. Delivery of Mir17 and Mir20a antagomirs to the lungs of CF mice resulted in a statistically significant increase of mRNA levels of Atg7 and protein levels of ATG7 and ATG16L1 (Fig. 4B and C).
Figure 4.
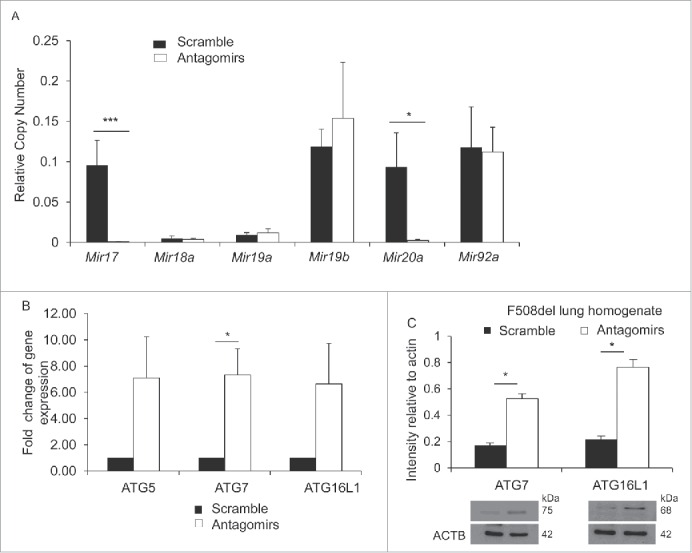
Decreasing the expression of Mir17 and Mir20a in live F508del mice improves the expression of autophagy molecules. (A) Expression of members of the Mirc1/Mir17-92 cluster in F508del mice intratracheally treated with scrambled control (black bars) or antagomirs to Mir17 and Mir20a (white bars) as measured by qPCR. Data are presented as the mean ± SD and are representative of n = 6. Student 2-tailed t test was used to determine significance for Mir17 and Mir20a. Significance assessed at p ≤ 0.05 (B) Expression of autophagy-regulating genes in F508del murine lung homogenate after intra-tracheal treatment with scramble (black bars) or antagomir17 and antagomir20a (white bars) assessed by qPCR. Data shown are representative of the average expression analyzed in duplicate and presented as fold-change compared to WT normalized to one ± SD (n = 3). Student 2-tailed t test was performed and asterisks indicate significant differences at p ≤ 0.05. (C) Western blot for autophagy proteins, ATG7 and ATG16L1, in F508del lung homogenates post-intra-tracheal administration of scrambled control (black bars) or antagomir17 and anatgoMir20a (white bars). The blots shown are representative of 3 independent experiments displaying similar results and protein production bands were normalized to their respective ACTB bands and quantified by ImageJ software. * indicates p ≤ 0.05; and ***, p ≤ 0.001.
B. cenocepacia is primarily cleared by autophagy in the lungs of healthy mice and represents an often fatal infection in CF patients who suffer from reduced autophagy activity. Stimulation of autophagy by rapamycin in live mice and their macrophages contains the infection.10 Thus, we examined bacterial loads in the lungs of antagomir-treated CF mice to determine the functional consequences of targeted reduction of Mir17 and Mir20a. Notably, CF mice treated with Mir17 and Mir20a antagomirs improved clearance of B. cenocepacia infection compared to their counterparts treated with scrambled control antagomirs (Fig. 5A). Both groups harbored similar bacterial loads at 4 h (data not shown).15 Improved autophagy activity as indicated by improved bacterial clearance, was accompanied by significant reductions in the expression of Mir17 and Mir20a in lung tissues of mice treated with the corresponding antagomirs even in the presence of B. cenocepacia (Fig. 5B). Therefore, targeting Mir17 and Mir20a in vivo improves autophagy activity in live CF mice.
Figure 5.

Targeting Mir17 and Mir20a in F508del mice increases autophagy-mediated clearance of Burkholderia cenocepacia. F508del mice were intratracheally treated with scrambled control (black bars) or antagomir17 and antagomir20a (white bars), then infected with B. cenocepacia. (A) After 48 h of infection, lungs were homogenized and plated for colony forming units (CFUs). Data shown represent the average CFU/gram of lung ± SD, n = 4 per condition. Students 2-tailed t test was used to determine significance (*) at p ≤ 0.05. (B) qPCR of individual Mirs within the Mirc1/Mir17-92 cluster in lung homogenates. Data represent the mean expression ± SD, n = 4 per condition. Student 2-tailed t test was used to determine significance. * indicates p ≤ 0.05; **, p ≤ 0.01.
CFTR function is impaired in primary F508del macrophages and is improved by reducing the levels of Mir17 and Mir20a via improved autophagy
As demonstrated above, correcting the inherently elevated levels of Mir17 and Mir20a in CF macrophages in vitro and in vivo improved autophagy activity; however, the impact of these Mirs on CFTR function remains unknown. To determine the effect of reducing Mir17 and Mir20a on the function of the F508del-CFTR channel, primary CF macrophages were transfected with antagomirs against Mir17 and Mir20a or a scramble control. After 48 h, CFTR function was assessed using the fluorescent and halide-sensitive compound 6-methoxy-N-(3-sulfopropyl)-quinolinium (SPQ) after modification of a protocol used to measure CFTR function in airway epithelial cells.37,38 CFTR was activated using a “cocktail” containing forskolin, cpt-cAMP and 3-isobutyl-1-methylxanthine/IBMX to increase cAMP, since CFTR is a cAMP-activated channel. Results demonstrated that resting CF macrophages exhibited significantly reduced CFTR activity compared to WT cells. Forty-eight h after transfection of combined antagomirs against Mir17 and Mir20a, an increase in fluorescence was detected in CF macrophages (Fig. 6). These results indicate that reducing the expression of inherently elevated Mir17 and Mir20a in CF macrophages improves F508del-CFTR function. Furthermore, to determine if this improvement in CFTR function was mediated by increased autophagy activity in antagomir-treated cells, CF macrophages were transfected with antagomir17 and antagomir20a in the presence or absence of the autophagy inhibitor 3-methyladenine (3-MA). The function of CFTR was determined by the SPQ assay as described above. Notably, inhibition of autophagy activity by 3-MA abolished the beneficial effect on CFTR function in CF macrophages achieved via decreasing Mir17 and Mir20a expression (Fig. 6). Collectively, these findings provide evidence that restoration of CFTR function upon reduction of Mir17 and Mir20a is mediated by autophagy. Together, our data provide evidence that correcting elevated levels of Mir17 and Mir20a in CF improves autophagy and CFTR function.
Figure 6.
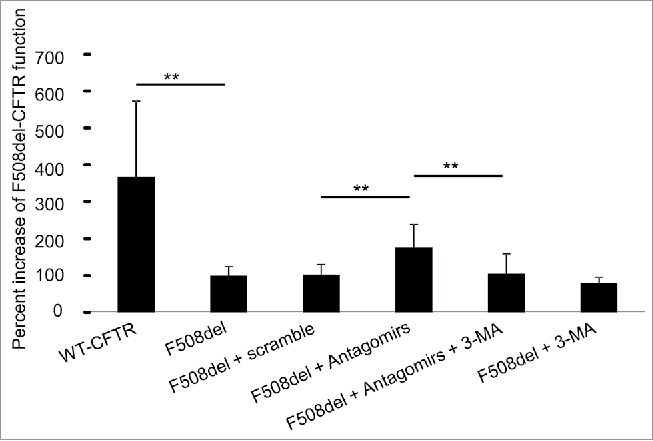
CFTR function is reduced in F508del murine macrophages and partially restored by decreasing Mir17 and Mir20a expression levels via autophagy. F508del macrophages were untransfected, transfected with scramble control or antagomir17 and −20a (Antagomirs) for 48 h, and 3-methyladenine (3-MA) for 6 h to inhibit autophagic flux. Cells were loaded with SPQ dye, quenched using an iodide buffer, and fluorescence output was measured. Data are presented as means of percent increase of F508del-CFTR ± SD from 5 independent experiments. Student 2-tailed t test was used to determine significance of antagomir transfection, and 3-MA inhibition. ** indicates p ≤ 0 .01.
Discussion
CF symptoms have been attributed to defective CFTR function in epithelial cells where impairment of CFTR function reduces chloride secretion, thereby impairing mucociliary clearance due to dehydration.15 Our data support the notion that the pathobiology of CF is multi-factorial, including functional impairment of autophagy in macrophages, as indicated by increased susceptibility to specific bacterial infections and inflammation.39 Macrophages expressing mutant F508del-CFTR on both alleles inherently exert reduced autophagy activity, which is associated with exacerbated inflammatory responses.10,11,19,40 In this regard, autophagy controls bacterial infection and IL-1β production in macrophages.41 The link between the CFTR mutation and the autophagy impairment has established macrophages as major players in CF pathology.10,11,42 Emerging studies also investigate neutrophil function in CF.43,44 Thus, CF pathobiology encompasses several immune cells, in addition to epithelial cells, recognizing that CF is a newly identified immune deficiency disorder.
All previous work in CF macrophages has focused on various phenomena such as impairment of bacterial clearance or exacerbated IL-1β production.24,25 Here, we examine CFTR function in macrophages using a modified SPQ assay initially used for epithelial cells. This is the first report to directly demonstrate that halide transport is impaired in F508del macrophages. Therefore, it is plausible to conclude that impairment of CFTR function in macrophages is responsible for innate immune deficiency in CF immune cells.
Defective autophagy plays a critical role in the pathology of several pulmonary diseases including chronic obstructive pulmonary disease, pulmonary hypertension, idiopathic lung fibrosis, and CF.22,45 We have previously demonstrated in a CF mouse model, that treatment with rapamycin, an autophagy-inducing drug, improves autophagy activity and reduces the lung inflammatory response both in vitro and in vivo.10,11,24,46,47 Rapamycin treatment also improves CFTR function in airway epithelial cells.19,40,48 The use of rapamycin reinforces the concept that restoration of autophagy activity is beneficial for CF patients. However, rapamycin elicits severe side-effects and thus cannot be used for the young CF population. Therefore, alternative approaches to improve autophagy in CF are needed.39,49
Despite the rapidly growing literature describing defective autophagy in CF, the underlying mechanisms remain to be fully elucidated. To characterize the impaired autophagy activity and inherently reduced expression of autophagy proteins observed in CF macrophages, we examined the expression of several Mirs predicted to target autophagy. Mir101 and the Mirc1/Mir17-92 cluster were identified. However, only the Mirc1/Mir17-92 cluster was overexpressed in both human and mouse CF macrophages. Within this cluster, only Mir17 was significantly elevated in both human and mouse samples. Downregulation of inherently elevated Mir17 and Mir20a in murine macrophages results in improved CFTR channel function. Other Mirs within the cluster were unregulated only to a small extent. Thus, it appears that correcting Mir17 and Mir20a is sufficient to restore significant autophagy activity in mice. Since only Mir17 is significantly elevated in human macrophages, it is possible that reducing its expression alone will be sufficient to improve CFTR functions and bacterial clearance in CF patients.
We observe that both Mir17 and Mir20a mimics were able to downregulate Atg7 and Atg16l1; however, the Mir20a antagomir failed to elevate Atg7 expression, suggesting that Mir17 has more specificity toward Atg7 than Mir20a. Notably, the seed sequences of Mir17 and Mir20a are the same but have different duplex structures causing different silencing efficacies for their targets. Online computational prediction data showed that the free energy release between Mir17 and Atg7 or Atg16l1 is higher than between Mir20a and Atg7 or Atg16l1 suggesting that Mir17 binds to the 3′UTR of Atg7 or Atg16l1 more stably and complementary than Mir20a. Thus, we propose that targeting Mir17 can be used in conjunction with CFTR potentiators that are demonstrating partial efficacy in clinical trials. Our findings are corroborated by the recent clinical trials demonstrating the restoration of defective autophagy in CF airways can be achieved by proteostasis regulators, such as cystamine and its reduced form, cysteamine.50 As suggested above, these agents also rescue and stabilize F508del-CFTR at the plasma membrane.48,51
Improved autophagy activity may be a result of the release of sequestered essential autophagy proteins, such as ATG7, from SQSTM1/p62 collections within F508del aggregates.10,18,19,39-41 The disassembly of these aggregates would allow for the autophagy molecules to traffic and localize to the phagophore membrane, and also permit folded F508del-CFTR protein to reach the plasma membrane and facilitate chloride transport. This mechanism was suggested by the Luciani group and ours.10,19,39
Another potential regulatory element of autophagy is the existence of 2 paralogs of the cluster, Mir106a-363 and Mir106b-25. Each paralog shares high sequence similarity with one another and intersects the predicted targets. The role of autophagy regulation by the paralogs is unknown, although their expression is dispensable early in life, whereas expression of the Mirc1/Mir17-92 cluster is required for normal development.52
In addition to the Mirc1/Mir17-92a cluster, other Mirs also play a role in the function of the CFTR protein.25,26,53 Mir138 was identified to regulate CFTR expression through its interaction with the transcriptional regulatory protein SIN3A.25 Mir384, Mir494 and Mir1246 are involved in the post-transcriptional regulation of CFTR channel synthesis.25 Individuals carrying the F508del mutation exhibit increased expression of Mir145, Mir223, and Mir494 in bronchial epithelium that correlates with decreased CFTR expression.16 Therefore, these findings suggest that overexpression of a variety of Mirs cooperate to disrupt several functions in the CF cell.
We demonstrate that inherently elevated Mirc1/Mir17-92 expression contributes to decreased autophagy in CF cells and that defective CFTR protein function is improved when autophagy activity is enhanced by reducing the expression of Mir17 and Mir20a. Several reports demonstrate that partial restoration of CFTR function is sufficient for improving CF pathobiology. More than 80% of CF individuals carry at least one allele with the F508del-CFTR mutation,54 which leads to misfolding of the CFTR protein preventing its proper trafficking to the plasma membrane.3,55 These heterozygote individuals express 50% of the normal amount of CFTR protein and secrete 50% of the airway surface fluid and chloride ions compared to healthy non-CF individuals.56,57 However, heterozygote humans and mice do not exhibit pathological symptoms.58-60 Furthermore, it is important to note that increasing the expression of F508del-CFTR in only 10% of CF epithelial cells is sufficient to improve the level of CFTR-mediated chloride ion transport.61 Similarly, expression of CFTR in 25% of airway cells is sufficient to restore normal mucus transport due to proper fluid homeostasis.15 Therefore, the positive effect we found on autophagy in response to antagomir17 and antagomir20 is promising.
Importantly, cancer has emerged as an increasingly significant problem affecting the CF community as the average life expectancy for CF patients now exceeds 40 y.62,63 The fact that elevated expression of the Mirc1/Mir17-92 cluster is associated with several types of malignancies, and our findings that this cluster is elevated in CF patients, raises growing concerns about cancer predispositions in the CF population.29,64,65 Current strides in research aim to prolong and improve the life of CF patients, however CF patients are prone to different types of cancers later in life.62,65,66 Elevated expression of the Mirc1/Mir17-92 cluster in CF may contribute to this predisposition. It has also been shown that the Mirc1/Mir17-92 cluster is highly expressed in intestinal tissue in CF patients.26 Should high levels of the Mirc1/Mir17-92 cluster correlate with increased susceptibility to cancer in CF, it would be even more critical to find approaches to control expression of the Mirc1/Mir17-92 cluster throughout the patient's life in an effort to improve bacterial clearance and chronic inflammation early in life, and prevent cancer at later stages in life. However, it will be important to carefully titrate the expression of Mir17 and Mir20a to normal physiological levels, rather than abolish them, given the importance of these Mirs in regulating diverse biological pathways.
Despite advancements in CF research, this disease remains without a cure. Our data build a foundation for the development of novel therapeutics to improve autophagy and CFTR protein function in CF patients by targeting Mir17. This approach can be applied to any cell type in CF should the elevated expression of the cluster be a global phenomenon rather than a macrophage-specific one. This approach may also be used in other disease conditions characterized by weak autophagy accompanied by upregulation of the Mirc1/Mir17-92 cluster or its members.
Materials and methods
Mice and bone-marrow-derived macrophages (BMDMs)
All animal experiments were performed according to protocols approved by the Animal Care and Use Committee (IACUC) of the Ohio State University College of Medicine. Wild-type (WT) C57BL/6 mice were obtained from Jackson Laboratories. F508del mice were obtained from Case Western Reserve University. Mirc1/Mir17-92−/− floxed mice were generously donated from the lab of Dr. Clay Marsh at The Dorothy M. Davis Heart and Lung Research Institute. All mice were housed in the OSU vivarium. BMDMs were isolated as previously described.10,11
Immunoblotting
Macrophages were lysed in lysis buffer solution (10 mM HEPES, 5 mM MgCl2, 1 mM EGTA, 142 mM KCl, 1% Ige-Pal [Sigma Aldrich, I-3021]) supplemented with a protease inhibitor cocktail (Roche Applied Science, 11 836 170 001). Thirty micrograms of protein were separated by sodium dodecyl sulfate-12% PAGE and transferred to polyvinylidene difluroide membranes (Bio-Rad Laboratories, 162-0117). Membranes were probed for ATG12–ATG5, ATG12, ATG16L1, ATG7 and ACTB/actin (Sigma-Aldrich, A0856, A8731, SAB2501677, A2856 and Abcam ab8226, respectively). Protein bands were detected with secondary antibodies conjugated to horseradish peroxidase; Rabbit: GE Healthcare, NA934; mouse: ThermoFisher Scientific, A28177; goat: Santa Cruz Biotechnology, sc-2020 followed by enhanced chemiluminescence reagents (Amersham/GE Health Care-Life Sciences, RPN 2106)
Confocal microscopy
Immunofluorescence microscopy experiments were performed as previously described.10,11 Antibodies used to stain autophagosomes were rabbit anti-LC3 (Abgent, AP1805a) followed by fluorescent secondary antibodies (Molecular Probes, A11008). Nuclei were stained with the nucleic acid dye 4′,6′-diamino-2-phenylindole (DAPI). Samples were analyzed with a FluoView FV10i confocal microscope.
Quantitative real-time PCR (qRT-PCR) for expression of Mirs
Total RNA was isolated from cells that were lysed in Trizol (Invitrogen Life Technologies, 15596-026). Chloroform (Fisher Scientific, 268320010), isopropanol (Fisher Scientific, BP2618-212), and glycogen (Fisher Scientific, 10814010) were used to isolate total macrophage RNA and its concentration was measured by Nanodrop. Expression of mature Mir17, Mir18a, Mir19a, Mir19b, Mir20a, Mir92a, Mir101, and snoRNA202 as an endogenous control, were analyzed by first converting the RNA to cDNA by priming with specific primers (Applied Biosystems, Assay ID 2308, 2422, 395, 396, 580, 431, 002253, 001232, respectively) using the TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystems, 4366596). PCR was conducted according to the manufacturer's guidelines. For qRT-PCR, cDNA was primed with specific TaqMan primers listed above and assayed using TaqMan Universal PCR MasterMix (Applied Biosystems, 4304437) and Applied Biosystems ABI 7900HT real-time PCR system. Expression was calculated as relative copy numbers. Ct values of each Mir were subtracted from the average Ct of the internal control, snoRNA Snord68/MBII-202 (mouse) or SNORD48/RNU48 (human), and the resulting ΔCt was used in the equation: relative copy numbers = (2−ΔΔCt)
Quantitative real-time PCR for expression of autophagy genes
Total RNA was isolated from cells lysed in Trizol. Atg5, Atg7, and Atg16l1 mRNA expression was assessed using SYBR Green PCR Master Mix (Life Technologies, 4309155). Briefly, Ct values of each target gene were subtracted from the average Ct of the housekeeping gene, Gapdh, and the resulting ΔCt was used.
Downregulation of elevated Mir17 and Mir20a in vitro and in vivo
Antagomir17 and antagomir20a (Applied Biosystems, MH12412 and AM10057, respectively) were diluted to 100 nM in phosphate-buffered saline (Gibco, 14190-144) and transfected into macrophages using the Lipofectamine LTX and PLUS reagents (Life Technologies, 15338100) for 48 h according to the manufacturer's instructions. For in vivo studies, 2-mo-old F508del mice were anesthetized via isofluorane (Ohio State University Veterinary Hospital) and intratracheally received antagomir17 and antagomir20 or scramble control (GE Dharmacon, IH-310561-08-0020, IH-310514-07-0020, or IN-001005-01-20, respectively) at 25 µg per mouse in 24-h intervals for 72 h.67,68 The dose was derived from our previous publications.69 The antagomirs were reconstituted in siRNA buffer (GE Dharmacon, B2000UB100) and diluted in 1X phosphate-buffered saline prior to administration. After 3 treatments, the mice were sacrificed and lungs were isolated. One lobe of the lung was homogenized in Trizol for RNA isolation. The other lobe was homogenized in lysis buffer with protease inhibitor, as described previously, for protein analysis. Homogenization was accomplished using the Qiagen Tissue Lyser (Qiagen, 85600) and accompanying 5-mm stainless steel beads (Qiagen, 69989). Another set of mice was treated as described above then infected intratracheally with B. cenocepacia and colony-forming units (CFUs) were quantified from homogenized lungs as previously described.10
Luciferase reporter assays
The NIH3T3 cell line was co-transfected with a plasmid containing either the Mir 3′UTR target clone for murine Atg7, Atg16l1 or control vector (GeneCopoeia, MmiT035820-MT01, MmiT036321-MT01 or CmiT000001, respectively) and Mir mimics to Mir17 and Mir20a (Applied Biosystems, 4464066 or AM17101, respectively) using Lipofectamine 2000 according to the manufacturer's protocol. Antagomir17 and antagomir20a were also transfected separately to downregulate endogenous Mir17 or Mir20a expression within the NIH3T3 cells. Firefly and Renilla Luciferase activities were measured consecutively by using the Dual-Luciferase Reporter Assay system (Promega, E1910) 24 h after transfection. The seed regions (5′ nucleotides 2 through 8) of mature Mir17 and Mir20a/b are predicted to bind with perfect complementarity to the sequence 5′-GCACUUU-3′, which is present in the Atg7 and Atg16l1 3′UTR. Site-directed mutagenesis was used to introduce 3 point mutations within these 7 nucleotides of the Atg7 and Atg16l1 3′UTR luciferase reporters to further test Mir17 and Mir20a/b binding specificity. Primers for introducing point mutations were designed using the QuikChange Primer Design online tool to be compatible with the QuikChange Lightning Multi Site-Directed Mutagenesis kit (Agilent Technologies, 210514). A list of mutagenesis primers can be found in Table S2.
CFTR function
CFTR channels can transport chloride as well as iodide. Therefore, CFTR function was assessed by measuring iodide efflux using the fluorescent and halide-sensitive dye 6-methoxy-N-(3-sulfopropyl) quinolinium (SPQ) as previously described by our group.70 Macrophages were briefly plated in a 96-well plate. Cells were loaded with SPQ (Molecular Probes, M-440) using hypotonic shock and were incubated with 10 mM SPQ in Opti-MEM:water (1:1) for 15 min at 37°C. Cells were then washed and incubated twice for 10 min with fluorescence quenching NaI buffer (130 mM NaI, 5 mM KNO3, 2.5 mM Ca[NO3]2, 2.5 mM Mg[NO3]2, 10 mM D-glucose, 10 mM N-[2-hydroxyethyl] piperazine-N′-[2-ethanesulfonic] acid/HEPES, pH 7.4). Subsequently, cells were switched to a dequenching isotonic NaNO3 buffer (identical to NaI buffer except that 130 mM NaI was replaced with 130 mM NaNO3) in the presence of 20 µM forskolin (Abcam, ab120058) and 100 µM 8-(4-chlorophenylthio)-c-AMP (Sigma-Aldrich, c3912) to activate CFTR. Nonspecific increase in fluorescence was measured by incubating the cells with the activation cocktail and the specific CFTR inhibitor GlyH101 (10 µM; Calbiochem, 219671. Fluorescence was measured using the plate reader VICTOR X3 (Perkin Elmer) with excitation wavelength at 350 nm and DAPI emission filter.
Statistical analysis
All experiments were performed at least 3 independent times unless stated otherwise and yielded similar results. Comparisons of groups for statistical difference were done using 2-tailed Student t test or ANOVA when described. P-value ≤ 0.05 was considered significant. * indicates p ≤ 0.05; **, p ≤ 0.01; and ***, p ≤ 0.001.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- BMDMs
bone marrow-derived macrophages
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- CFUs
colony-forming units
- Mirs
microRNAs
- qRT-PCR
quantitative real-time PCR
- SPQ
6-methoxy-N-(3-sulfopropyl)-quinolinium
- UTR
untranslated region
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Mary Severin for her technical help.
Funding
Studies in Dr. Amer's laboratory are supported by the Cystic Fibrosis Research Grant from the Cystic Fibrosis Foundation and Ohio State University Center for Clinical and Translational Science Longitudinal Pilot Award (CCTS), R21 AI113477, and R01 HL094586. MT is supported by the Cystic Fibrosis Foundation Student Traineeship Grant, the American Association of Immunologists (AAI) Careers in Immunology Fellowship, Center for Microbial Interface Biology Training Grant supported by The Ohio State University College of Medicine, Ohio State University Graduate School, and the Ohio State Sigma Xi Chapter Grants-In-Aid of Research Fund. KC is supported by Cystic Fibrosis Foundation Postdoctoral Researcher Fellowship. HK is supported by Egyptian Science and Technology Development Fund (STDF) through project ID 6117. KK is supported by Deutsche Forschungsgemeinschaft (DFG - German Research Foundation).
References
- [1].Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and function. Thromb Haemost 2012; 107:605-10; PMID:22318703; http://dx.doi.org/ 10.1160/TH11-12-0836 [DOI] [PubMed] [Google Scholar]
- [2].Jiang Q, Engelhardt JF. Cellular heterogeneity of CFTR expression and function in the lung: implications for gene therapy of cystic fibrosis. Eur J Hum Genet 1998; 6:12-31; PMID:9781011; http://dx.doi.org/ 10.1038/sj.ejhg.5200158 [DOI] [PubMed] [Google Scholar]
- [3].Welsh MJ, Denning GM, Ostedgaard LS, Anderson MP. Dysfunction of CFTR bearing the delta F508 mutation. J Cell Sci Suppl 1993; 17:235-9; PMID:7511616; http://dx.doi.org/ 10.1242/jcs.1993.Supplement_17.33 [DOI] [PubMed] [Google Scholar]
- [4].Coakley RD, Grubb BR, Paradiso AM, Gatzy JT, Johnson LG, Kreda SM, O'Neal WK, Boucher RC. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci U S A 2003; 100:16083-8; PMID:14668433; http://dx.doi.org/ 10.1073/pnas.2634339100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Blouquit S, Regnier A, Dannhoffer L, Fermanian C, Naline E, Boucher R, Chinet T. Ion and fluid transport properties of small airways in cystic fibrosis. Am J Respir Crit Care Med 2006; 174:299-305; PMID:16645176; http://dx.doi.org/ 10.1164/rccm.200506-987OC [DOI] [PubMed] [Google Scholar]
- [6].Bruscia EM, Bonfield TL. Innate and Adaptive Immunity in Cystic Fibrosis. Clin Chest Med 2016; 37:17-29; PMID:26857765; http://dx.doi.org/ 10.1016/j.ccm.2015.11.010 [DOI] [PubMed] [Google Scholar]
- [7].Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, Greene C, McElvaney G, Mall M, Döring G. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros 2012; 11:363-82; PMID:22917571; http://dx.doi.org/ 10.1016/j.jcf.2012.07.003 [DOI] [PubMed] [Google Scholar]
- [8].Doring G, Gulbins E. Cystic fibrosis and innate immunity: how chloride channel mutations provoke lung disease. Cell Microbiol 2009; 11:208-16; PMID:19068098; http://dx.doi.org/ 10.1111/j.1462-5822.2008.01271.x [DOI] [PubMed] [Google Scholar]
- [9].Pier GB. Role of the cystic fibrosis transmembrane conductance regulator in innate immunity to Pseudomonas aeruginosa infections. Proc Natl Acad Sci U S A 2000; 97:8822-8; PMID:10922041; http://dx.doi.org/ 10.1073/pnas.97.16.8822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DH, Newland C, Rosales-Reyes R, Kopp B, McCoy K, et al.. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy 2011; 7:1359-70; PMID:21997369; http://dx.doi.org/ 10.4161/auto.7.11.17660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Abdulrahman BA, Khweek AA, Akhter A, Caution K, Tazi M, Hassan H, Zhang Y, Rowland PD, Malhotra S, Aeffner F, et al.. Depletion of the ubiquitin-binding adaptor molecule SQSTM1/p62 from macrophages harboring cftr DeltaF508 mutation improves the delivery of Burkholderia cenocepacia to the autophagic machinery. J Biol Chem 2013; 288:2049-58; PMID:23148214; http://dx.doi.org/ 10.1074/jbc.M112.411728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].French PJ, van Doorninck JH, Peters RH, Verbeek E, Ameen NA, Marino CR, de Jonge HR, Bijman J, Scholte BJ. A delta F508 mutation in mouse cystic fibrosis transmembrane conductance regulator results in a temperature-sensitive processing defect in vivo. J Clin Invest 1996; 98:1304-12; PMID:8823295; http://dx.doi.org/ 10.1172/JCI118917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gomes-Alves P, Neves S, Coelho AV, Penque D. Low temperature restoring effect on F508del-CFTR misprocessing: A proteomic approach. J Proteomics 2009; 73:218-30; PMID:19775599; http://dx.doi.org/ 10.1016/j.jprot.2009.09.001 [DOI] [PubMed] [Google Scholar]
- [14].Hogenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, Rosenblatt RL, Prestidge CB, Fordtran JS. Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion. Am J Hum Genet 2000; 67:1422-7; PMID:11055897; http://dx.doi.org/ 10.1086/316911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang L, Button B, Gabriel SE, Burkett S, Yan Y, Skiadopoulos MH, Dang YL, Vogel LN, McKay T, Mengos A, et al.. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol 2009; 7:e1000155; PMID:19621064; http://dx.doi.org/ 10.1371/journal.pbio.1000155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Oglesby IK, Chotirmall SH, McElvaney NG, Greene CM. Regulation of cystic fibrosis transmembrane conductance regulator by microRNA-145, −223, and −494 is altered in DeltaF508 cystic fibrosis airway epithelium. J Immunol 2013; 190:3354-62; PMID:23436935; http://dx.doi.org/ 10.4049/jimmunol.1202960 [DOI] [PubMed] [Google Scholar]
- [17].Klionsky DJ. The molecular machinery of autophagy and its role in physiology and disease. Semin Cell Dev Biol 2010; 21:663; PMID:20430106; http://dx.doi.org/ 10.1016/j.semcdb.2010.04.005 [DOI] [PubMed] [Google Scholar]
- [18].Assani K, Tazi MF, Amer AO, Kopp BT. IFN-gamma stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS One 2014; 9:e96681; PMID:24798083; http://dx.doi.org/ 10.1371/journal.pone.0096681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina DL, Settembre C, Gavina M, Raia V, Ballabio A, Maiuri L. Cystic fibrosis: a disorder with defective autophagy. Autophagy 2011; 7:104-6; PMID:21048426; http://dx.doi.org/ 10.4161/auto.7.1.13987 [DOI] [PubMed] [Google Scholar]
- [20].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/ 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bjorkoy G, Lamark T, Johansen T. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy 2006; 2:138-9; PMID:16874037; http://dx.doi.org/ 10.4161/auto.2.2.2405 [DOI] [PubMed] [Google Scholar]
- [22].Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature 1998; 395:395-8; PMID:9759731; http://dx.doi.org/ 10.1038/26506 [DOI] [PubMed] [Google Scholar]
- [23].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717-21; PMID:11099404; http://dx.doi.org/ 10.1126/science.290.5497.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Abdelaziz HAD, Khalil H, Cormet-Boyaka E, Amer A. The cooperation between the autophagy machinery and the inflammasome to implement an appropriate innate immune response: Do they regulate each other? Immunological Reviews 2015; 265(1):194-204; PMID:25879294; http://dx.doi.org/10.1111/imr.12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ramachandran S, Karp PH, Jiang P, Ostedgaard LS, Walz AE, Fisher JT, Keshavjee S, Lennox KA, Jacobi AM, Rose SD, et al.. A microRNA network regulates expression and biosynthesis of wild-type and DeltaF508 mutant cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci U S A 2012; 109:13362-7; PMID:22853952; http://dx.doi.org/ 10.1073/pnas.1210906109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bazett M, Paun A, Haston CK. MicroRNA profiling of cystic fibrosis intestinal disease in mice. Mol Genet Metab 2011; 103:38-43; PMID:21333573; http://dx.doi.org/ 10.1016/j.ymgme.2011.01.012 [DOI] [PubMed] [Google Scholar]
- [27].Mendell JT. Miriad roles for the Mir17-92 cluster in development and disease. Cell 2008; 133:217-22; PMID:18423194; http://dx.doi.org/ 10.1016/j.cell.2008.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dakhlallah D, Batte K, Wang Y, CanteMirStone CZ, Yan P, Nuovo G, Mikhail A, Hitchcock CL, Wright VP, Nana-Sinkam SP, et al.. Epigenetic Regulation of Mir17∼92 Contributes to the Pathogenesis of Pulmonary Fibrosis. Am J Respir Crit Care Med 2013; 187:397-405; PMID:23306545; http://dx.doi.org/ 10.1164/rccm.201205-0888OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Aguda BD, Kim Y, Piper-Hunter MG, Friedman A, Marsh CB. MicroRNA regulation of a cancer network: consequences of the feedback loops involving Mir17-92, E2F, and Myc. Proc Natl Acad Sci U S A 2008; 105:19678-83; PMID:19066217; http://dx.doi.org/ 10.1073/pnas.0811166106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nana-Sinkam SP, Karsies T, Riscili B, Ezzie M, Piper M. Lung microRNA: from development to disease. Expert Rev Respir Med 2009; 3:373-85; PMID:20477329; http://dx.doi.org/ 10.1586/ers.09.30 [DOI] [PubMed] [Google Scholar]
- [31].Bonauer A, Dimmeler S. The microRNA-17-92 cluster: still a Miracle? Cell Cycle 2009; 8:3866-73; PMID:19887902; http://dx.doi.org/ 10.4161/cc.8.23.9994 [DOI] [PubMed] [Google Scholar]
- [32].Chen L, Li C, Zhang R, Gao X, Qu X, Zhao M, Qiao C, Xu J, Li J. Mir17-92 cluster microRNAs confers tumorigenicity in multiple myeloma. Cancer Lett 2011; 309:62-70; PMID:21664042; http://dx.doi.org/ 10.1016/j.canlet.2011.05.017 [DOI] [PubMed] [Google Scholar]
- [33].Grillari J, Hackl M, Grillari-Voglauer R. Mir17-92 cluster: ups and downs in cancer and aging. Biogerontology 2010; 11:501-6; PMID:20437201; http://dx.doi.org/ 10.1007/s10522-010-9272-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y, Takahashi T. A polycistronic microRNA cluster, Mir17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005; 65:9628-32; PMID:16266980; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-2352 [DOI] [PubMed] [Google Scholar]
- [35].Frankel LB, Wen J, Lees M, Hoyer-Hansen M, Farkas T, Krogh A, Jäättelä M, Lund AH. microRNA-101 is a potent inhibitor of autophagy. EMBO J 2011; 30:4628-41; PMID:21915098; http://dx.doi.org/ 10.1038/emboj.2011.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chakraborty S, Mehtab S, Patwardhan A, Krishnan Y. Pri-Mir17-92a transcript folds into a tertiary structure and autoregulates its processing. RNA 2012; 18:1014-28; PMID:22450760; http://dx.doi.org/ 10.1261/rna.031039.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sermet-Gaudelus I, Vallee B, Urbin I, Torossi T, Marianovski R, Fajac A, Feuillet MN, Bresson JL, Lenoir G, Bernaudin JF, et al.. Normal function of the cystic fibrosis conductance regulator protein can be associated with homozygous (Delta)F508 mutation. Pediatr Res 2002; 52:628-35; PMID:12409506 [DOI] [PubMed] [Google Scholar]
- [38].Xie J, Drumm ML, Ma J, Davis PB. Intracellular loop between transmembrane segments IV and V of cystic fibrosis transmembrane conductance regulator is involved in regulation of chloride channel conductance state. J Biol Chem 1995; 270:28084-91; PMID:7499295; http://dx.doi.org/ 10.1074/jbc.270.47.28084 [DOI] [PubMed] [Google Scholar]
- [39].Junkins RD, McCormick C, Lin TJ. The emerging potential of autophagy-based therapies in the treatment of cystic fibrosis lung infections. Autophagy 2014; 10:538-47; PMID:24434788; http://dx.doi.org/ 10.4161/auto.27750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello-Mantovani M, et al.. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol 2010; 12:863-75; PMID:20711182; http://dx.doi.org/ 10.1038/ncb2090 [DOI] [PubMed] [Google Scholar]
- [41].Mayer ML, Blohmke CJ, Falsafi R, Fjell CD, Madera L, Turvey SE, Hancock RE. Rescue of dysfunctional autophagy attenuates hyperinflammatory responses from cystic fibrosis cells. J Immunol 2013; 190:1227-38; PMID:23264659; http://dx.doi.org/ 10.4049/jimmunol.1201404 [DOI] [PubMed] [Google Scholar]
- [42].Kopp BT, Abdulrahman BA, Khweek AA, Kumar SB, Akhter A, Montione R, Tazi MF, Caution K, McCoy K, Amer AO. Exaggerated inflammatory responses mediated by Burkholderia cenocepacia in human macrophages derived from Cystic fibrosis patients. Biochem Biophys Res Commun 2012; 424:221-7; PMID:22728038; http://dx.doi.org/ 10.1016/j.bbrc.2012.06.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Witko-Sarsat V, Sermet-Gaudelus I, Lenoir G, Descamps-Latscha B. Inflammation and CFTR: might neutrophils be the key in cystic fibrosis? Mediators Inflamm 1999; 8:7-11; PMID:10704083; http://dx.doi.org/ 10.1080/09629359990658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhou Y, Song K, Painter RG, Aiken M, Reiser J, Stanton BA, Nauseef WM, Wang G. Cystic fibrosis transmembrane conductance regulator recruitment to phagosomes in neutrophils. J Innate Immun 2013; 5:219-30; PMID:23486169; http://dx.doi.org/ 10.1159/000346568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med 2003; 9:65-76; PMID:12865942 [PMC free article] [PubMed] [Google Scholar]
- [46].Amer AO, Swanson MS. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol 2005; 7:765-78; PMID:15888080; http://dx.doi.org/ 10.1111/j.1462-5822.2005.00509.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Khalil H, Tazi M, Caution K, Ahmed A, Kanneganti A, Assani K, Kopp B, Marsh C, Dakhlallah D, Amer AO. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics 2016; 11:381-8; PMID:26909551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Luciani A, Villella VR, Esposito S, Gavina M, Russo I, Silano M, Guido S, Pettoello-Mantovani M, Carnuccio R, Scholte B, et al.. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on DeltaF508 cystic fibrosis transmembrane conductance regulator. Autophagy 2012; 8:1657-72; PMID:22874563; http://dx.doi.org/ 10.4161/auto.21483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Huang S, Houghton PJ. Inhibitors of mammalian target of rapamycin as novel antitumor agents: from bench to clinic. Curr Opin Investig Drugs 2002; 3:295-304; PMID:12020063 [PubMed] [Google Scholar]
- [50].De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, De Gregorio F, Salvadori L, Grassia R, Leone CA, De Rosa G, et al.. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014; 10:2053-74; PMID:25350163; http://dx.doi.org/ 10.4161/15548627.2014.973737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Villella VR, Esposito S, Maiuri MC, Raia V, Kroemer G, Maiuri L. Towards a rational combination therapy of cystic fibrosis: How cystamine restores the stability of mutant CFTR. Autophagy 2013; 9:1431-4; PMID:23800975; http://dx.doi.org/ 10.4161/auto.25517 [DOI] [PubMed] [Google Scholar]
- [52].Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, et al.. Targeted deletion reveals essential and overlapping functions of the Mir17 through 92 family of MirNA clusters. Cell 2008; 132:875-86; PMID:18329372; http://dx.doi.org/ 10.1016/j.cell.2008.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gillen AE, Gosalia N, Leir SH, Harris A. MicroRNA regulation of expression of the cystic fibrosis transmembrane conductance regulator gene. Biochem J 2011; 438:25-32; PMID:21689072; http://dx.doi.org/ 10.1042/BJ20110672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bowling FG, McGill JJ, Shepherd RW, Danks DM. Screening for cystic fibrosis: use of delta F508 mutation. Lancet 1990; 335:925-6; PMID:1970022; http://dx.doi.org/ 10.1016/0140-6736(90)90534-C [DOI] [PubMed] [Google Scholar]
- [55].Morris MR, Pereira MM, Hallett MB, McPherson MA, Dormer RL. Cellular localisation of the most common mutant form of the CF gene protein, delta F508-CFTR. Biochem Soc Trans 1998; 26:S293; PMID:9766012; http://dx.doi.org/ 10.1042/bst026s293 [DOI] [PubMed] [Google Scholar]
- [56].Patel S, Sinha IP, Dwan K, Echevarria C, Schechter M, Southern KW. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database Syst Rev 2015; 3:CD009841; PMID:25811419 [DOI] [PubMed] [Google Scholar]
- [57].Kuk K, Taylor-Cousar JL. Lumacaftor and ivacaftor in the management of patients with cystic fibrosis: current evidence and future prospects. Ther Adv Respir Dis 2015; 9:313-26; PMID:26416827; http://dx.doi.org/ 10.1177/1753465815601934 [DOI] [PubMed] [Google Scholar]
- [58].Cohen JC, Lundblad LK, Bates JH, Levitzky M, Larson JE. The “Goldilocks effect” in cystic fibrosis: identification of a lung phenotype in the cftr knockout and heterozygous mouse. BMC Genet 2004; 5:21; PMID:15279681; http://dx.doi.org/ 10.1186/1471-2156-5-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 1994; 266:107-9; PMID:7524148; http://dx.doi.org/ 10.1126/science.7524148 [DOI] [PubMed] [Google Scholar]
- [60].Guilbault C, Saeed Z, Downey GP, Radzioch D. Cystic fibrosis mouse models. Am J Respir Cell Mol Biol 2007; 36:1-7; PMID:16888286; http://dx.doi.org/ 10.1165/rcmb.2006-0184TR [DOI] [PubMed] [Google Scholar]
- [61].Johnson LG, Olsen JC, Sarkadi B, Moore KL, Swanstrom R, Boucher RC. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet 1992; 2:21-5; PMID:1284642; http://dx.doi.org/ 10.1038/ng0992-21 [DOI] [PubMed] [Google Scholar]
- [62].Maisonneuve P, Marshall BC, Knapp EA, Lowenfels AB. Cancer risk in cystic fibrosis: a 20-year nationwide study from the United States. J Natl Cancer Inst 2013; 105:122-9; PMID:23178438; http://dx.doi.org/ 10.1093/jnci/djs481 [DOI] [PubMed] [Google Scholar]
- [63].Maisonneuve P, Marshall BC, Lowenfels AB. Risk of pancreatic cancer in patients with cystic fibrosis. Gut 2007; 56:1327-8; PMID:17698876; http://dx.doi.org/ 10.1136/gut.2007.125278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Olive V, Jiang I, He L. Mir17-92, a cluster of MirNAs in the midst of the cancer network. Int J Biochem Cell Biol 2010; 42:1348-54; PMID:20227518; http://dx.doi.org/ 10.1016/j.biocel.2010.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Padua RA, Warren N, Grimshaw D, Smith M, Lewis C, Whittaker J, Laidler P, Wright P, Douglas-Jones A, Fenaux P, et al.. The cystic fibrosis delta F508 gene mutation and cancer. Hum Mutat 1997; 10:45-8; PMID:9222759; http://dx.doi.org/ 10.1002/(SICI)1098-1004(1997)10:1%3c45::AID-HUMU6%3e3.0.CO;2-L [DOI] [PubMed] [Google Scholar]
- [66].Neglia JP, FitzSimmons SC, Maisonneuve P, Schoni MH, Schoni-Affolter F, Corey M, Lowenfels AB. The risk of cancer among patients with cystic fibrosis. Cystic Fibrosis and Cancer Study Group. N Engl J Med 1995; 332:494-9; PMID:7830730; http://dx.doi.org/ 10.1056/NEJM199502233320803 [DOI] [PubMed] [Google Scholar]
- [67].Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with 'antagomirs'. Nature 2005; 438:685-9; PMID:16258535; http://dx.doi.org/ 10.1038/nature04303 [DOI] [PubMed] [Google Scholar]
- [68].Wahlquist C, Jeong D, Rojas-Munoz A, Kho C, Lee A, Mitsuyama S, van Mil A, Park WJ, Sluijter JP, Doevendans PA, et al.. Inhibition of Mir25 improves cardiac contractility in the failing heart. Nature 2014; 508:531-5; PMID:24670661; http://dx.doi.org/ 10.1038/nature13073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lovett-Racke AE, Rocchini AE, Choy J, Northrop SC, Hussain RZ, Ratts RB, Sikder D, Racke MK. Silencing T-bet defines a critical role in the differentiation of autoreactive T lymphocytes. Immunity 2004; 21:719-31; PMID:15539157; http://dx.doi.org/ 10.1016/j.immuni.2004.09.010 [DOI] [PubMed] [Google Scholar]
- [70].Cormet-Boyaka E, Jablonsky M, Naren AP, Jackson PL, Muccio DD, Kirk KL. Rescuing cystic fibrosis transmembrane conductance regulator (CFTR)-processing mutants by transcomplementation. Proc Natl Acad Sci U S A 2004; 101:8221-6; PMID:15141088; http://dx.doi.org/ 10.1073/pnas.0400459101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.