

Introduction
The formation of new bones at extra skeletal sites, named heterotopic ossification (HO), is a common post-operative complication upon orthopedic surgeries (i.e., hip replacements), blast injuries, skeletal trauma and some nervous system disorders. In contrast to acquired HO, Fibrodysplasia ossificans progressiva (FOP; MIM #135100) represents the most devastating hereditary disorder involving HO. FOP affects one in two million individuals and is characterized by congenital skeletal abnormalities and postnatal progressive HO. FOP is normally manifested at birth by a characteristic skeletal malformation in the great toe (hallux valgus), and the disease progresses throughout the lifetime of the patient sequentially affecting muscle, ligaments, tendons and other connective tissues all over the body, following a predefined anatomical pattern. Noteworthy, unlike other forms of HO (i.e., progressive osseous heteroplasia, POH), HO in FOP occurs episodically, through inflammation-triggered “flare-ups”. Such a strong link between FOP and inflammation might underlie the variety of phenotypes observed in FOP patients (for example, the intensity and duration of the immune response can vary among FOP patients) (1), but also has led to the establishment of the current treatment for FOP, which consists in broad spectrum anti-inflammatory drugs (2).
Mutation in ALK2 results in altered BMP signaling
In 2006 a single point heterozygous mutation 617G-A; R206H in the glycine serine rich (GS) domain of the gene Activin receptor-like kinase 2 (ACVR1, ALK2) was found in all classically affected FOP patients (3). This ubiquitously expressed gene encodes a bone morphogenetic protein (BMP) type I receptor, which is located at the plasma membrane and is endowed with serine/threonine kinase activity. The BMPs are extracellular ligands belonging to the transforming growth factor (TGF)-β superfamily [reviewed in (4)]. BMP ligands exert their functions upon interaction with type I kinase receptors (ALK1/2/3/6). Such interaction induces the hetero-oligomerization of signaling complexes by recruiting so called type II receptors, either BMPR2, Activin receptor-like 2a (ActRIIa) or 2b (ActRIIb). These type II receptors bear constitutive serine/threonine kinase activity at their cytosolic domains, which upon ligand induced complex formation with type I receptors can phosphorylate the type I receptor at the juxtamembrane GS domain. This activates the type I receptor and the signal can be transduced intracellularly. In the case of BMPs, this consists in the phosphorylation of the Smads 1/5/8 that form heteromeric complexes with Co-Smad, i.e., Smad 4, and participate in transcriptional complexes with other factors, in order to induce the expression of genes involved in the differentiation and activation of osteoblast and chondrocyte-like cells. In contrast to BMPs, other ligands of the TGF-β family, such as TGF-βs and activins, induce the activation of the Smads 2/3 upon the formation of receptor membrane signaling complexes consisting in ALK4 (in the case of activins) or ALK5 (for TGF-βs), and ActRIIa/b. This signaling pathway does not only induce the transcription of a different subset of genes, but it also commonly counterbalances the Smad 1/5/8 pathway, for example by competing with the common mediator Smad 4. Notably, activins can bind ALK2, although this interaction fails to induce Smad 1/5/8 activation (5). Therefore, in normal circumstances, activins prevent BMP signaling by competing for ALK2 at the ligand-receptor interaction level.
The inhibitory factor FKBP12 binds the GS domain in the type I receptor and stabilizes the non-phosphorylated (or inactive) form of the receptor (6). It thereby sets up a threshold for the initiation of receptor signaling. In the case of the mutant FOP ALK2, interactions between the GS domain and FKBP12 are disturbed, thus the kinase domain remains open and accessible for Smads, which leads to hypersensitized signaling. Elevated BMP signaling has been described not only for the most common FOP ALK2 mutation (R206H), but also in several other mutant forms of ALK2 affecting either the GS or the kinase domain (described in a vast minority of FOP patients), as well as in the purely constitutive artificial ALK2 Q207D (6). This has been followed up using in vitro and in vivo models of HO, altogether demonstrating a link between an overactivation of the FOP receptor and enhanced BMP signaling, eventually leading to HO. This concept has focused the research in FOP in the subsequent years, aiming to identify novel approaches to prevent overactive ALK2 signaling by means of small molecule inhibitors targeting the kinase activity of the receptor (7-9) or anti-sense oligonucleotides, including small interference RNAs (siRNA) (10), antisense oligonucleotides (AONs) (11) and micro RNAs (miRNAs) (12) to inhibit mutant ACVR1 expression (see Figure 1).
Figure 1.
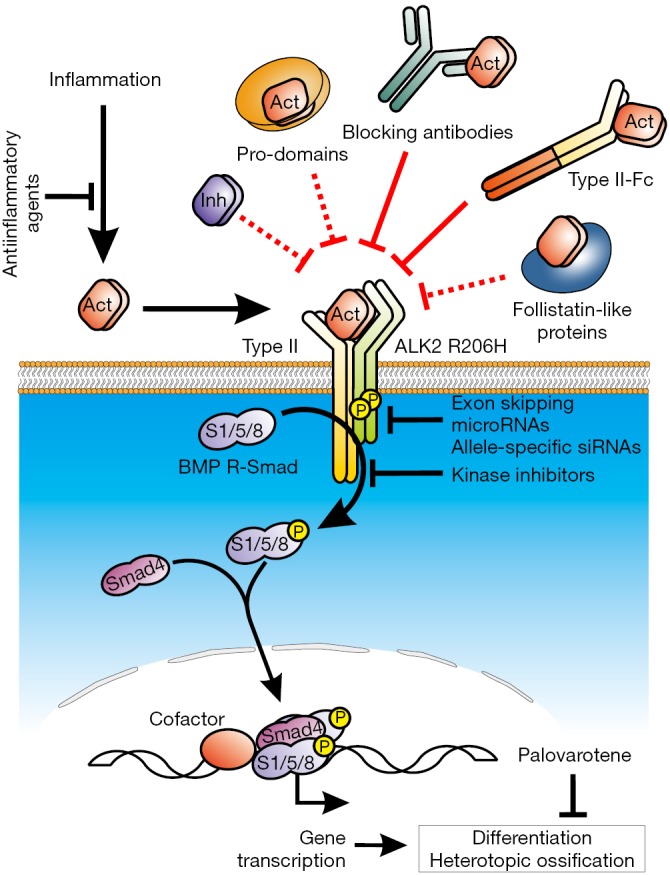
Inflammation-induced activin A is able to interact with receptor complexes incorporating the mutant ALK2 in order to induce downstream phosphorylation and activation of intracellular Smads 1/5/8, also called Receptor Smads (or R-Smads). They regulate the expression of genes involved in osteoblast differentiation and HO, as part of transcriptional complexes with other cellular factors. Normalization of the over-active mutant receptor has been intended by downregulating the expression of the receptor (using micro RNAs, exon-skipping or allele specific siRNA-mediated knock-down) or blocking its kinase activity using small molecule kinase inhibitors, which interfere with both the wild type and mutant allele. Hatsell et al. demonstrate the efficacy of type II-Fc constructs (sequestering several BMP and activin ligands) and an anti-activin A specific antibody to prevent ALK2-FOP induced Smad 1/5/8 signaling. These new extracellular strategies (depicted in red) may be complemented with inhibins and activin pro-domains (not tested thus far), or follistatins. Finally, a RAR-γ agonist that inhibits osteoblast differentiation (Palovarotene, Clementia Inc.) is currently being evaluated in Phase II clinical trials (NCT02279095, NCT02521792). Altogether, it remains to be determined whether activin-based therapies are effective in humans, which may benefit of combined treatments with anti-inflammatory drugs.
ALK2 becomes promiscuous
The recent publication by Hatsell et al. introduces a paradigm shift in FOP (13). Using a new mouse model fully recapitulating the phenotype of the disease, that is, spontaneous induced HO mediated by altered BMP signaling, the authors demonstrated that activin A is capable of inducing Smad 1/5/8 phosphorylation and downstream transcriptional activation exclusively in the presence of the mutant ALK2 receptor. This finding was confirmed shortly thereafter by an independent group (14). Although this message probably summarizes the major translational finding, there is other relevant information provided within the manuscript. As such, the authors showed how HEK293 cells over-expressing the mutant ALK2 R206H receptor displayed enhanced BMP transcriptional response to the osteogenic ligands BMP-2, -4, -7, -9 and -10, although no significant changes were observed for BMP-6, which signals in a very similar manner as BMP-2 and BMP-4. Moreover, cells expressing ALK2 R206H not only acquired responsiveness to activins, but also to BMP-15, a ligand that signals via ALK6 and BMPR2/ActRIIa (15). Interestingly, the later manuscript by Hino et al. using iPSCs-derived mesenchymal stem cells, found that exclusively activin A induced a significantly stronger BMP transcriptional response in cells expressing the FOP ALK2 receptor, after screening a number of TGF-β family ligands (14). Finally, a very recent manuscript by Hsiao’s lab could not reproduce the induction of Smad 1/5/8 phosphorylation by activin A in FOP-iPSCs derived endothelial cells, suggesting that such induction requires an additional cell specific factor, or a particular receptor expression level that might be cell type dependent (16). For example, it would be of interest to check the expression level of the mutant allele in FOP cells lacking the activin pro-osteogenic response. In fact, the molecular mechanisms driving this gain of function upon the mutation of the receptor remain unknown. In receptor over-expressing cells, Hino et al. have shown that binding of radiolabeled activin A to ActRIIa/b seems to be enhanced in the presence of FOP ALK2. Moreover, enhanced chondrogenesis displayed by FOP iPSCs derived MSCs was blocked by incubation with either a BMP (DMH1, targeting ALK1/2/3/6 kinase activity) or a TGF-(greek beta) type I receptor inhibitor (SB-431542, targeting ALK4/5/7). It seems unlikely that a mutation in the cytosolic domain of ALK2, as R206H, affects the binding affinity of an extracellular ligand per se, thus pointing at additional factors, like other (co)receptors. Indeed, Hatsell et al. showed that blockade of FKBP12 binding to ALK2 does not result in increased activin A responsiveness. Interestingly, in a previous manuscript, Medici et al. described a mechanism by which the FOP mutation primed endothelial cells to undergo a process named endothelial-to-mesenchymal transition, eventually resulting in osteoblast or chondrocyte-like cells, that may be responsible for up to 50% of the heterotopic bone. Such transition was prevented in the presence of SB-431542, but also upon knock-down of ALK5 (17). These results suggest that the FOP mutation allows the formation of abnormal receptor complexes, that enable Smad 1/5/8 activation upon binding to activin A. Deciphering such mechanisms will contribute to develop novel drugs specifically targeting ALK2 FOP-Activin A interaction.
A new mouse model for FOP
In this sense, the group from Economides developed a novel mouse model that facilitates the testing of new alternatives to treat FOP (13). In order to circumvent the perinatal lethally observed in previously generated FOP transgenic mice (18), the authors made use of the so-called COIN technology to develop 9 to 15 weeks old mice ubiquitously expressing ALK2 R206H in response to tamoxifen. This model offers the possibility to investigate the effects of the expression of the mutant receptor at different time points and, upon combination with cell type specific Cre-drivers, the origin and relative contribution of different cells involved in HO. These mice develop progressive HO lesions spontaneously in a similar manner as humans: predisposed anatomical sites (i.e., sternum, vertebrae and hip joints) and comparable histology (muscle destruction followed by inflammatory infiltrate and endochondral ossification) (13). It remains to be shown whether these mice also recapitulate other particular aspects of the human disease, such as the higher incidence of osteochondromas, or the connections between the endogenous and the ectopic skeletal plaques.
Anti-activin based therapies for FOP
As a way to test the relevance of activin in FOP, Hatsell et al. treated the above described FOP transgenic mice with ActRIIa-Fc and ActRIIb-Fc ligand traps, alone or in combination, systemically. These constructs efficiently reduced HO in tamoxifen injected mice. Nevertheless, to exclude the possibility that the effect was due to a massive depletion in the osteogenic ligands available (in fact, ActRIIa-Fc and ActRIIb-Fc binds to both BMPs and activins), activin A was specifically blocked using an anti-activin A specific monoclonal antibody. Such treatment showed a similar effect as ActRIIa-Fc, successfully inhibiting HO. It remains to be investigated whether anti-activin treatments can have any effect on the existing bone, for example, by modulating osteoclasts differentiation and activity, thereby affecting bone resorption.
Noteworthy, clinical trials performed with ActRII-Fc ligand traps have revealed a number of considerable side effects (19,20), which likely would turn out to be devastating in the case of FOP. In this sense, administration of an activin A specific antibody may result to be more tolerable for patients. Importantly, activins are released in response to inflammatory stimulation, which coincides with the progression of HO in FOP through episodic “flare-ups”. This might allow to adjust the drug regimen to episodes of inflammation (such as soft tissue trauma and viral infections), or in the case of undergoing surgery to remove ectopic bone. Such restrictions might minimize the undesired collateral effects of blocking activins function. To investigate this, Regeneron Pharmaceuticals has recently initiated in Belgium a Phase I clinical trial (NCT02870400) to evaluate the safety and pharmacodynamics of REGN2477, a humanized anti-Activin A antibody. The company expects to collect enough data before the end of the year, in order to initiate studies in FOP patients.
A number of alternatives may be also considered. For example, activins are synthetized as non-mature proteins that require further processing to remove a pro-domain to become active. Certainly, pro-domains in the TGF-β family can be synthetized independently and retain their ability to interact with the mature peptides to reconstitute the original non-mature inactive peptides (21). Inhibins have been investigated in the context of the reproductive system, where they have shown to block activins activity by preventing the formation of the signaling receptor complexes at the membrane competing with the ActRII receptors (22). Several decades ago, follistatins were identified as the natural antagonists of activins. These are secreted antagonists characterized by an activin binding site, although they can also interact with other ligands of the TGF-β family (23). Indeed, Hino et al. tested a number of follistatin-like proteins to prevent activin A induced chondrogenesis in FOP iPSCs-derived MSCs (14). Finally, mutual antagonism is usually observed between members of the TGF-β family, by means of competing for the same receptors at the membrane or intracellular regulators that mediate gene transcription (24). Therefore, antagonism of activin A-ALK2 FOP activity could be theoretically achieved by up-regulating the expression of a factor (or a combination of them) that makes use of any member of the activin A-ALK2 FOP signaling cascade, without inducing a pro-osteogenic transcriptional program. All these possibilities need to be studied and tested carefully in preclinical models of the disease, since blockade of activins function can lead to skeletal abnormalities, reproductive failure and even postnatal death (25), as shown in animal models. Perhaps a combined treatment of low dose novel anti-activin agents with anti-inflammatory agents/drugs may be the most efficient and less toxic approach to treat FOP.
Acknowledgements
Our research on FOP is supported by the LeDucq Foundation. GSD is supported by a Postdoctoral Fellowship from AFM-Telethon.
Footnotes
Provenance: This is a Guest Editorial commissioned by Section Editor Bingrong Zhou, MD, PhD (Department of Dermatology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China).
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Al Kaissi A, Kenis V, Ben Ghachem M, et al. The Diversity of the Clinical Phenotypes in Patients With Fibrodysplasia Ossificans Progressiva. J Clin Med Res 2016;8:246-53. 10.14740/jocmr2465w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glaser DL, Kaplan FS. Treatment considerations for the management of fibrodysplasia ossificans progressiva. Clinical Reviews in Bone and Mineral Metabolism 2005;3:243-50. 10.1385/BMM:3:3-4:243 [DOI] [Google Scholar]
- 3.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006;38:525-7. 10.1038/ng1783 [DOI] [PubMed] [Google Scholar]
- 4.Salazar VS, Gamer LW, Rosen V. BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol 2016;12:203-21. 10.1038/nrendo.2016.12 [DOI] [PubMed] [Google Scholar]
- 5.ten Dijke P, Yamashita H, Ichijo H, et al. Characterization of type I receptors for transforming growth factor-greek beta and activin. Science 1994;264:101-4. 10.1126/science.8140412 [DOI] [PubMed] [Google Scholar]
- 6.Chaikuad A, Alfano I, Kerr G, et al. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem 2012;287:36990-8. 10.1074/jbc.M112.365932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanvitale CE, Kerr G, Chaikuad A, et al. A new class of small molecule inhibitor of BMP signaling. PLoS One 2013;8:e62721. 10.1371/journal.pone.0062721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohedas AH, Xing X, Armstrong KA, et al. Development of an ALK2-biased BMP type I receptor kinase inhibitor. ACS Chem Biol 2013;8:1291-302. 10.1021/cb300655w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerr G, Sheldon H, Chaikuad A, et al. A small molecule targeting ALK1 prevents Notch cooperativity and inhibits functional angiogenesis. Angiogenesis 2015;18:209-17. 10.1007/s10456-014-9457-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaplan J, Kaplan FS, Shore EM. Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele-specific targeting. Gene Ther 2012;19:786-90. 10.1038/gt.2011.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi S, Cai J, de Gorter DJ, et al. Antisense-oligonucleotide mediated exon skipping in activin-receptor-like kinase 2: inhibiting the receptor that is overactive in fibrodysplasia ossificans progressiva. PLoS One 2013;8:e69096. 10.1371/journal.pone.0069096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mura M, Cappato S, Giacopelli F, et al. The role of the 3'UTR region in the regulation of the ACVR1/Alk-2 gene expression. PLoS One 2012;7:e50958. 10.1371/journal.pone.0050958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatsell SJ, Idone V, Wolken DM, et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med 2015;7:303ra137. 10.1126/scitranslmed.aac4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hino K, Ikeya M, Horigome K, et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci U S A 2015;112:15438-43. 10.1073/pnas.1510540112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore RK, Otsuka F, Shimasaki S. Molecular basis of bone morphogenetic protein-15 signaling in granulosa cells. J Biol Chem 2003;278:304-10. 10.1074/jbc.M207362200 [DOI] [PubMed] [Google Scholar]
- 16.Barruet E, Morales BM, Lwin W, et al. The ACVR1 R206H mutation found in fibrodysplasia ossificans progressiva increases human induced pluripotent stem cell-derived endothelial cell formation and collagen production through BMP-mediated SMAD1/5/8 signaling. Stem Cell Res Ther 2016;7:115. 10.1186/s13287-016-0372-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medici D, Shore EM, Lounev VY, et al. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 2010;16:1400-6. 10.1038/nm.2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakkalakal SA, Zhang D, Culbert AL, et al. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res 2012;27:1746-56. 10.1002/jbmr.1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruckle J, Jacobs M, Kramer W, et al. Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J Bone Miner Res 2009;24:744-52. 10.1359/jbmr.081208 [DOI] [PubMed] [Google Scholar]
- 20.Relizani K, Mouisel E, Giannesini B, et al. Blockade of ActRIIB signaling triggers muscle fatigability and metabolic myopathy. Mol Ther 2014;22:1423-33. 10.1038/mt.2014.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Böttinger EP, Factor VM, Tsang ML, et al. The recombinant proregion of transforming growth factor greek beta1 (latency-associated peptide) inhibits active transforming growth factor beta1 in transgenic mice. Proc Natl Acad Sci U S A 1996;93:5877-82. 10.1073/pnas.93.12.5877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vale W, Wiater E, Gray P, et al. Activins and inhibins and their signaling. Ann N Y Acad Sci 2004;1038:142-7. 10.1196/annals.1315.023 [DOI] [PubMed] [Google Scholar]
- 23.Hedger MP, de Kretser DM. The activins and their binding protein, follistatin-Diagnostic and therapeutic targets in inflammatory disease and fibrosis. Cytokine Growth Factor Rev 2013;24:285-95. 10.1016/j.cytogfr.2013.03.003 [DOI] [PubMed] [Google Scholar]
- 24.Aykul S, Martinez-Hackert E. Transforming growth factor-β family ligands can function as antagonists by competing for type II receptor binding. J Biol Chem 2016;291:10792-804. 10.1074/jbc.M115.713487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matzuk MM, Kumar TR, Bradley A. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature 1995;374:356-60. 10.1038/374356a0 [DOI] [PubMed] [Google Scholar]