

ABSTRACT
Herpes simplex virus 1 (HSV-1) remodels nuclear membranes during virus egress. Although the UL31 and UL34 proteins control nucleocapsid transit in infected cells, the molecular interactions required for their function are unclear. Here we report that the γ134.5 gene product of HSV-1 facilitates nucleocapsid release to the cytoplasm through bridging the UL31/UL34 complex, cellular p32, and protein kinase C. Unlike wild-type virus, an HSV mutant devoid of γ134.5 or its amino terminus is crippled for viral growth and release. This is attributable to a defect in virus nuclear egress. In infected cells, wild-type virus recruits protein kinase C to the nuclear membrane and triggers its activation, whereas the γ134.5 mutants fail to exert such an effect. Accordingly, the γ134.5 mutants are unable to induce phosphorylation and reorganization of lamin A/C. When expressed in host cells γ134.5 targets p32 and protein kinase C. Meanwhile, it communicates with the UL31/UL34 complex through UL31. Deletion of the amino terminus from γ134.5 disrupts its activity. These results suggest that disintegration of the nuclear lamina mediated by γ134.5 promotes HSV replication.
IMPORTANCE HSV nuclear egress is a key step that determines the outcome of viral infection. While the nuclear egress complex mediates capsid transit across the nuclear membrane, the regulatory components are not clearly defined in virus-infected cells. We report that the γ134.5 gene product, a virulence factor of HSV-1, facilitates nuclear egress cooperatively with cellular p32, protein kinase C, and the nuclear egress complex. This work highlights a viral mechanism that may contribute to the pathogenesis of HSV infection.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) replicates and packages its DNA in the cell nucleus. Once assembled, the nucleocapsids traverse the nucleoplasm and cross the nuclear lamina. The capsids bud through the nuclear membranes in a two-step process called envelopment and de-envelopment (1). In this process, the nuclear egress complex, consisting of UL31 and UL34, mediates vesiculation of the inner nuclear membrane and results in enveloped virions in the perinuclear space. Primary virions fuse with the outer nuclear membrane, which releases the capsids to the cytoplasm for further maturation (2). Accumulating evidence suggests that additional proteins, including Us3, ICP22, UL47, gB, and gH, coordinate with the UL31/34 complex to facilitate nuclear egress in infected cells (3–6).
The nuclear lamina is a dense meshwork underlying the inner nuclear membrane (7). It is composed primarily of type V intermediate filament proteins, lamin A/C and lamin B. Besides providing structural support to the nucleus, the nuclear lamina potentially presents a barrier to the transit of virus capsids. A number of studies suggest that herpesviruses alter the nuclear lamina to promote nuclear egress (8–11). For example, HSV-1 activates protein kinase C (PKC) isoforms and induces phosphorylation of lamin B, which is dependent on the UL31/UL34 complex (12). UL31 and UL34 also bind to lamin A/C and lamin B, which interrupts lamin-lamin interaction and perforates the lamina (8, 10). On the other hand, Us3, a serine/threonine kinase of HSV-1, phosphorylates lamin A/C to dissolve the nuclear lamina (3). Remarkably, isoforms of PKC also participate in nuclear envelope budding or breakdown of host cells that occurs in ribonucleoprotein export, mitosis, and apoptosis (13–18). These observations illustrate that the remodeling of the nuclear envelope is an evolutionarily conserved event. Nevertheless, the regulatory network remains largely unclear.
Previous studies suggest that the γ134.5 protein of HSV-1 facilitates nuclear egress (19). Deletion of the γ134.5 gene results in an accumulation of nucleocapsids and subsequent reduction in infectious virus. The γ134.5 gene encodes a virulence factor with an amino-terminal domain, linker (ATP) repeats, and a carboxyl-terminal domain (20, 21). When expressed, the γ134.5 protein shuttles between the nucleus and cytoplasm, presumably to perform distinct functions (22, 23). It is well established that γ134.5 acts as a regulatory subunit of protein phosphatase 1 to promote protein synthesis in HSV-infected cells (24, 25). Moreover, γ134.5 negatively modulates TANK binding kinase 1 and I-κB kinase, which inhibits the expression of cytokines, and dendritic cell maturation (26–29). HSV γ134.5 also inhibits autophagy through binding to beclin-1 (30). Additionally, the γ134.5 protein mediates nuclear egress independently of the interferon response (31). This involves the host protein p32, also known as gC1qR, which promotes HSV nuclear egress (32, 33).
This study was undertaken to investigate the mechanism of γ134.5 action. Here we report that the γ134.5 protein facilitates HSV nuclear egress through its amino-terminal domain. We show that this functional module is crucial to reorganize the nuclear lamina, translocate PKC to the nuclear membrane, and activate its activity. Furthermore, we provide evidence that while γ134.5 binds p32 and PKC, it also interacts with the UL31/UL34 complex in infected cells. These results suggest that regulation of lamin phosphorylation by γ134.5 is a mechanism to promote HSV replication.
MATERIALS AND METHODS
Cells and viruses.
HeLa and Vero cells were originally obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS). HSV-1(F) is a prototype HSV-1 strain used in this study (34). In recombinant virus R3616, a 1-kb fragment from the coding region of the γ134.5 gene was deleted (21). In H1001, the sequences of the γ134.5 gene encoding amino acids 1 to 146 were deleted, and in H1002 the deleted region was repaired with wild-type γ134.5 (29).
Reagents.
The plasmids FLAG-γ134.5, FLAG-Δ146, and FLAG-N159 were described previously (35). The FLAG-UL31 plasmid was constructed by inserting a PCR-amplified fragment into the BamHI and EcoRI sites of pCMV-tag2B. The FLAG-UL34 plasmid was constructed by inserting a PCR-amplified fragment into the EcoRV and NotI sites of pCDH-FLAG. Mouse anti-human p32 antibody (sc-271200), anti-p32 antibody (sc-23885), rabbit anti-human p32 antibody (sc-48795), mouse anti-porcine lamin A/C antibody (sc-7292), rabbit anti-human PKCδ (C-20) antibody (sc-937), anti-phospho-PKCδ (A-8) antibody (sc-377560), anti-phospho-PKCδ (H-9) antibody (sc-374613), agarose conjugated with protein A/G (sc-2003), donkey anti-rabbit IgG-FITC (sc-2090), donkey anti-rabbit IgG-R (sc-2095), donkey anti-mouse IgG–fluorescein isothiocyanate (FITC) (sc-2099), bovine anti-chicken IgY-TR antibody (sc-3914), bovine anti-chicken IgY–horseradish peroxidase (HRP) (sc-2497), goat anti-mouse IgG–HRP (sc-2005), goat anti-rabbit IgG–HRP (sc-2004), rottlerin, and bisindolylmaleimide I (BIM I) were purchased from Santa Cruz Biotechnology. Anti-phospho-lamin A/C (Ser22) antibody (CST2026), and anti-histone H3 (3H1) antibody (CST9717) were purchased from Cell Signaling Technology. Anti-γ-tubulin and anti-FLAG–HRP antibodies were purchased from Sigma. Anti-UL31 rabbit antibody and anti-UL34 hen antibody were described previously (36).
Immunoblotting.
Cells were harvested, washed with phosphate-buffered saline (PBS), and lysed with ice-cold immune precipitation assay buffer (50 mM Tries-HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA, 1.0% Triton X-100, and protease inhibitor cocktail) for 30 min on ice. After centrifugation, supernatants were mixed with disruption buffer (50 mM Tris-HCl [pH 6.8], 2% [wt/vol] SDS, 0.1% bromophenol blue, 10% glycerol, and 100 nM β-mercaptoethanol) and boiled. The proteins were separated by 12% or 10% SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, and incubated with primary and secondary antibodies. Protein bands were detected by enhanced chemiluminescence (26).
Immunoprecipitation analysis.
To examine protein interactions, transfected or infected HeLa cells were harvested and lysed with ice-cold immune precipitation assay buffer on ice. After centrifugation, cell extracts were incubated with the indicated antibodies and agarose conjugated with protein A/G at 4°C. The immobilized protein beads were subjected to immunoblotting analysis (32).
Virus growth assay.
HeLa cells were infected with different viruses at the indicated multiplicity of infection. After adsorption for 2 h, the monolayers were overlaid with DMEM (plus 1% FBS) and incubated at 37°C. Cells were harvested at different hours postinfection, and viruses, released by three cycles of freezing and thawing, were titrated on Vero cells (31).
Electron microscopy analysis.
HeLa cells were infected with viruses at 5 PFU per cell. At 16 h postinfection, samples were fixed in 2.5% glutaraldehyde with 100 mM PBS (pH 7.4), treated with 1% osmium tetroxide in phosphate buffer, dehydrated in ethanol, and embedded in LX112 resin (Ladd Research Industries). Samples were removed from the petri dishes and remounted on aluminum stubs. Ultrathin sections were cut with a Leica Ultracut UCT, placed on 200-mesh copper grids, and stained with uranyl acetate and lead citrate. Grids were viewed with a Jeol 1220 transmission electron microscope at 80 kV. Images were taken at various magnifications with a digital charge-coupled device (CCD) camera (Gigital Micrograph software; Gatan Inc.).
Confocal microscopy.
Cells were washed with PBS, fixed with ice-cold 4% paraformaldehyde for 30 min, permeabilized with 0.5% Triton X-100 in PBS, and incubated with primary antibodies at 4°C overnight. Cells were then reacted with fluorescence-conjugated secondary antibodies for 2 h at room temperature. After washing with PBS, cells were mounted and visualized with a Zeiss LSM 710 Meta confocal microscope.
RESULTS
The amino-terminal domain of γ134.5 is important to viral growth.
To better define the function of γ134.5, we analyzed the kinetics of viral growth in HeLa cells infected with γ134.5 variants. As illustrated in Fig. 1A, wild-type HSV-1(F) replicated efficiently. As infection progressed, the viral yield increased to a titer of 1 × 106 PFU/ml at 48 h after infection. Under these conditions, R3616, the γ134.5 null mutant, barely replicated. H1001, which lacks amino acids 1 to 146, replicated modestly, reaching a titer of 1 × 104 PFU/ml. This mutant replicated to levels about 100-fold lower than those for the wild-type virus but 10-fold higher than those for the γ134.5 null mutant. We assayed for cell-associated and cell-free viruses in infected cells. Figure 1B shows that HSV-1(F) and repaired mutant H1002 produced comparable levels of cell-associated and cell-free viruses. Compared to HSV-1(F), R3616 or H1001 exhibited about a 10-fold reduction in cell free viruses. These phenotypes were also seen in mouse 3T6 cells (data not shown).
FIG 1.
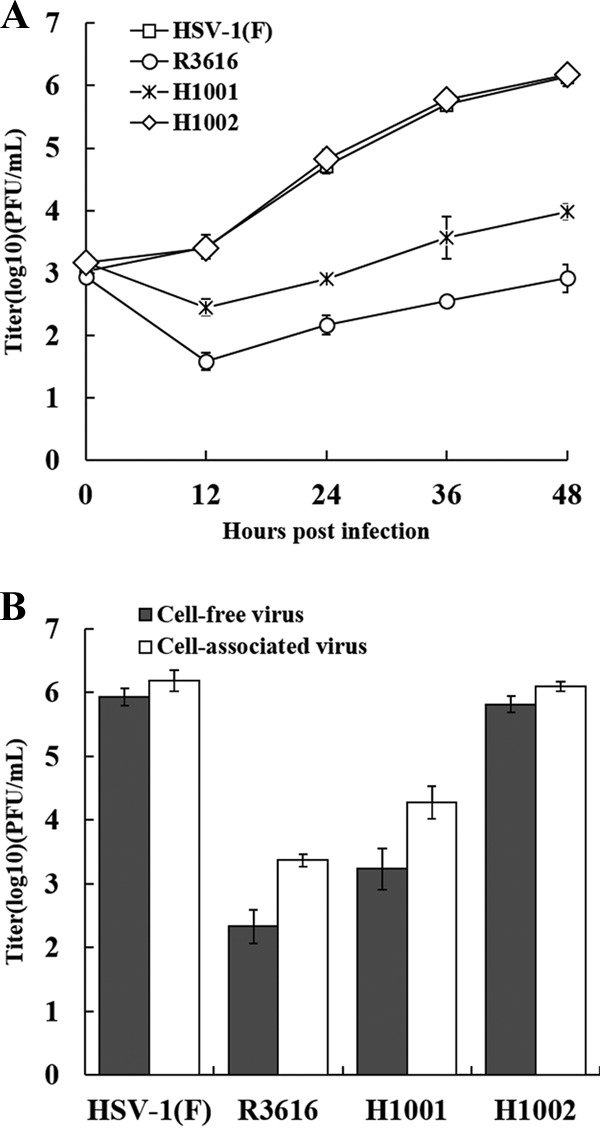
Viral replication is dependent on the amino terminus of γ134.5. (A) Monolayers of HeLa cells were infected with wild-type HSV-1(F), R3616 (the γ134.5 null mutant), H1001(the γ134.5 mutant devoid of amino acids 1 to 146), or H1002 (the repaired virus with wild-type γ134.5) at 0.05 PFU per cell and incubated at 37°C. At the indicated time points postinfection, the total virus yields were determined on Vero cells. (B) HeLa cells were infected as described above. At 48 h postinfection, cell-associated viruses and viruses in the supernatant were collected separately and titrated on Vero cells. Data represent the mean values from three independent experiments with the standard deviations.
Deletion of the amino-terminal domain from γ134.5 impairs nuclear egress.
Next, we examined nuclear egress by transmission electron microscopy. As presented in Fig. 2, HSV-1(F) infection produced a large fraction of virions in the cytoplasm and on the cell surface (Fig. 2A) in addition to nucleocapsids. R3616 infection produced virus particles that were mainly in the nucleus (Fig. 2B). Only a few were present in the cytoplasm or extracellular space. H1001 exhibited a similar phenotype, with viral particles predominantly confined to the nucleus. On the other hand, H1002 behaved more like wild-type virus. This pattern was evident when enumerated from 10 cells infected with each virus, as summarized in Table 1. In HSV-1(F)-infected cells, 44% of virus particles were in the nucleus, 3% in the perinuclear region, and 52% in the cytoplasm. In R3616-infected cells, 88% of virus particles were in the nucleus, 3% in the perinuclear region, and 9% in the cytoplasm. Similarly, in cells infected with H1001, 64% of virus particles were in the nucleus, 7% in the perinuclear region, and only 29% in the cytoplasm. In H1002-infected cells, viral particles were distributed evenly. These data suggest that γ134.5 mediates nuclear egress, which relies on the amino-terminal domain.
FIG 2.

Effect of γ134.5 variants on nuclear egress. Confluent monolayers of HeLa cell were infected with HSV-1(F), R3616, H1001, or H1002 at 5 PFU per cell. At 16 h postinfection, cells were harvested and processed for electron microscopy analysis as described in Materials and Methods. Abbreviations: Nuc, nucleus; Cyt, cytoplasm; NM, nuclear membrane. Scale bars are shown in each panel.
TABLE 1.
Subcellular localization of viral particles in infected cells
| Virus | No. (%) of viral particles in: |
Total particles counted/10 cellsa | ||
|---|---|---|---|---|
| Nucleus | Perinuclear space | Cytoplasm | ||
| HSV-1(F) | 422 (44) | 30 (3) | 497 (52) | 949 |
| R3616 | 102 (88) | 3 (3) | 11 (9) | 116 |
| H1001 | 206 (64) | 20 (7) | 94 (29) | 320 |
| H1002 | 431 (47) | 41 (4) | 445 (49) | 917 |
The number of viral particles in the nucleus, perinuclear space, and cytoplasm were counted in electron micrographs of 10 randomly sampled HeLa cells infected with the indicated virus.
The γ134.5 protein mediates distortion of the nuclear lamina, which involves the amino terminus.
HSV nuclear egress disrupts the nuclear lamina, which results from phosphorylation of lamin A/C and lamin B (8, 37). To investigate the role of γ134.5, HeLa cells were mock infected or infected with γ134.5 variants. Cells were stained with anti-lamin A/C antibody and visualized by confocal microscopy. As indicated in Fig. 3A, lamin A/C densely lined along with the nuclear rim in mock-infected cells. Infection with HSV-1(F) changed the appearance of lamin A/C, with a fraction dispersed into the nucleoplasm. However, in cells infected with the mutant devoid of γ134.5 (R3616) or its amino terminus (H1001), such alteration was not seen. As expected, infection with H1002, with a repaired amino terminus of γ134.5, induced reorganization of lamin A/C.
FIG 3.
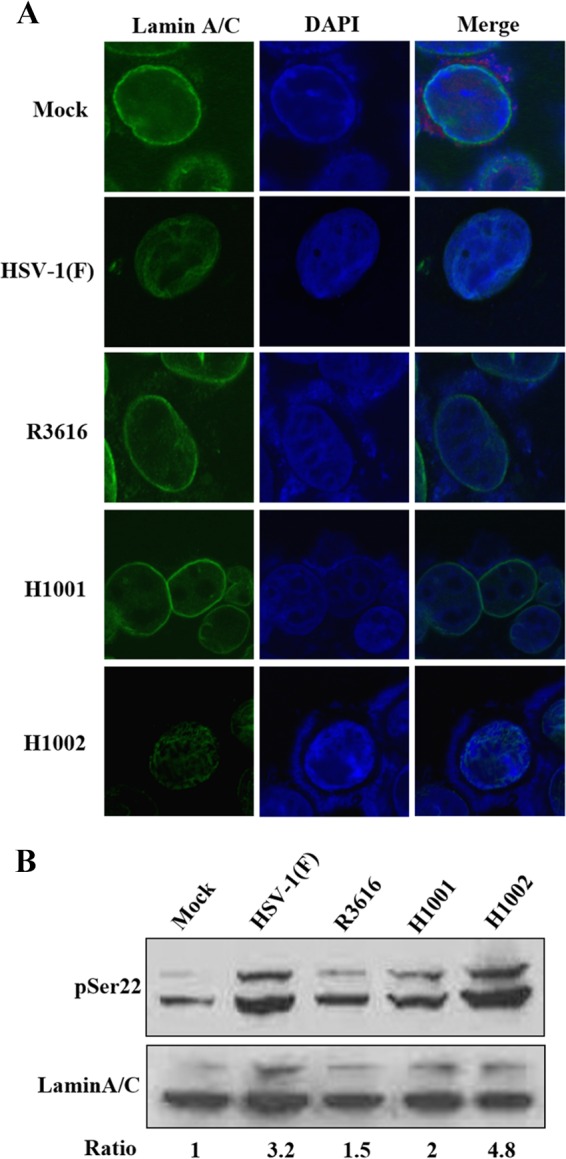
(A) Effect of γ134.5 variants on reorganization of lamin A/C. HeLa cells were mock infected or infected with HSV-1(F), R3616, H1001, or H1002 (5 PFU/cell). At 16 h postinfection, cells were processed and stained with DAPI (4′,6′-diamidino-2-phenylindole) and anti-lamin A/C antibody. Cells were visualized, and images were captured with confocal microscopy. (B) Effect of γ134.5 variants on phosphorylation of lamin A/C. HeLa cells were infected as for panel A. Lysates of cells were prepared and processed for Western blot analysis with antibodies against lamin A/C and phosphorylated lamin A/C (serine 22), respectively. Phosphorylated lamin A/C (serine 22) and lamin A/C were quantitated using Image J software. Phosphorylation of lamin A/C is presented as a ratio to lamin A/C, with normalization to mock infection.
As reorganization of lamin is accompanied by phosphorylation (11–13), we assessed the effect of γ134.5 variants on lamin A/C in infected HeLa cells. Figure 3B shows that in mock-infected cells, lamin A/C was minimally phosphorylated at serine 22. At 16 h postinfection, HSV-1(F) or H1002 induced two prominent bands, representing phosphorylated lamin A/C. The relative phosphorylation increased by 3- to 4-fold compared to that in mock infection. Nevertheless, phosphorylated bands were modestly induced upon infection with R3616 or H1001, with the ratio of phosphorylation increased by 1- to 2-fold. This weaker phosphorylation suggests that the amino terminus of γ134.5 is required. We conclude that lamin reorganization and its phosphorylation rely on the amino terminus of γ134.5.
The γ134.5 protein directs translocation and activation of PKC.
Protein kinase C (PKC) mediates the remodeling of nuclear membranes (11–13). Accordingly, treatment of HeLa cells with rottlerin and bisindolylmaleimide I (BIM I), inhibitors of protein kinase C, precluded phosphorylation of lamin A/C induced by wild-type virus (Fig. 4A). This paralleled with a 20- to 80-fold reduction in viral production (Fig. 4B), although cell viability was unaffected (data not shown). To investigate whether γ134.5 regulates protein kinase C, we focused on subcellular localization of PKCδ in HeLa cells mock infected or infected with viruses (Fig. 5A). Cells were reacted with anti-PKCδ antibody and analyzed by confocal microscopy. In mock-infected cells, PKCδ localized to the cytoplasm, with faint punctates distributed diffusely. Infection with wild-type HSV-1(F) or H1002 resulted in translocation, where PKCδ concentrated at the nuclear rim. However, in cells infected with R3616 or H1001, PKCδ remained dispersed in the cytoplasm. A similar pattern was seen with PKCα (data not shown), suggesting that γ134.5 recruits PKC to the nuclear membrane, which requires its amino-terminal domain.
FIG 4.
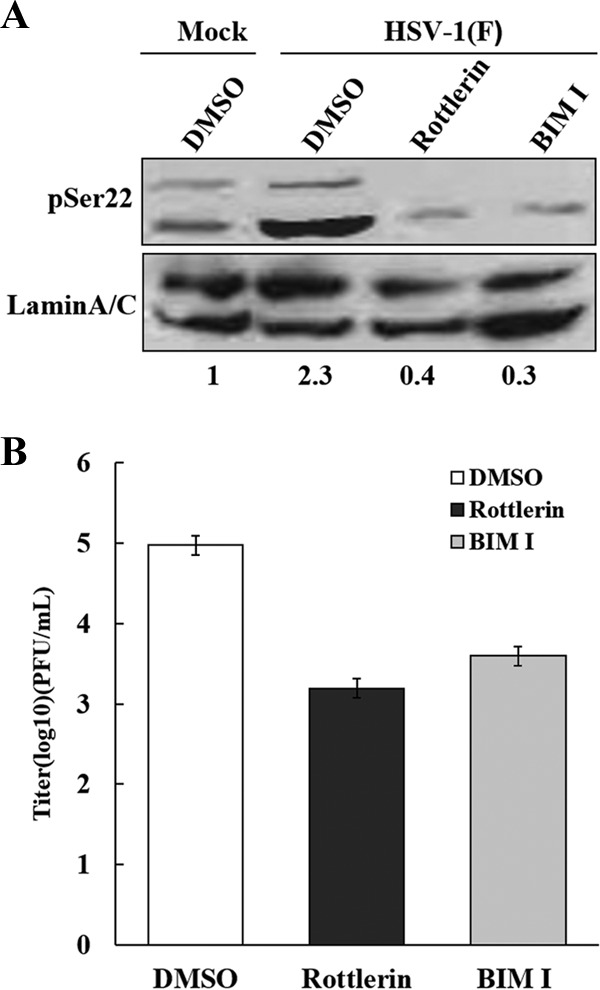
PKC activity is required for virus replication and lamin phosphorylation. (A) HeLa cells were mock infected or infected with HSV-1(F) (5 PFU/cell). Cells, treated with dimethyl sulfoxide (DMSO), rottlerin, and BIM I, were harvested at 16 h postinfection and subjected to Western blot analysis with antibodies against phosphorylated lamin A/C (serine 22) and lamin A/C. Phosphorylation of lamin A/C (serine 22) is presented as a ratio to lamin A/C, with normalization to mock infection. (B) HeLa cells were infected with HSV-1(F) at 0.05 PFU per cell. At 5 h postinfection, cells were treated with DMSO, rottlerin (10 μM), and BIM I (10 μM), and viral yields were determined at 24 h postinfection. Data represent the mean values from three independent experiments with the standard deviations.
FIG 5.
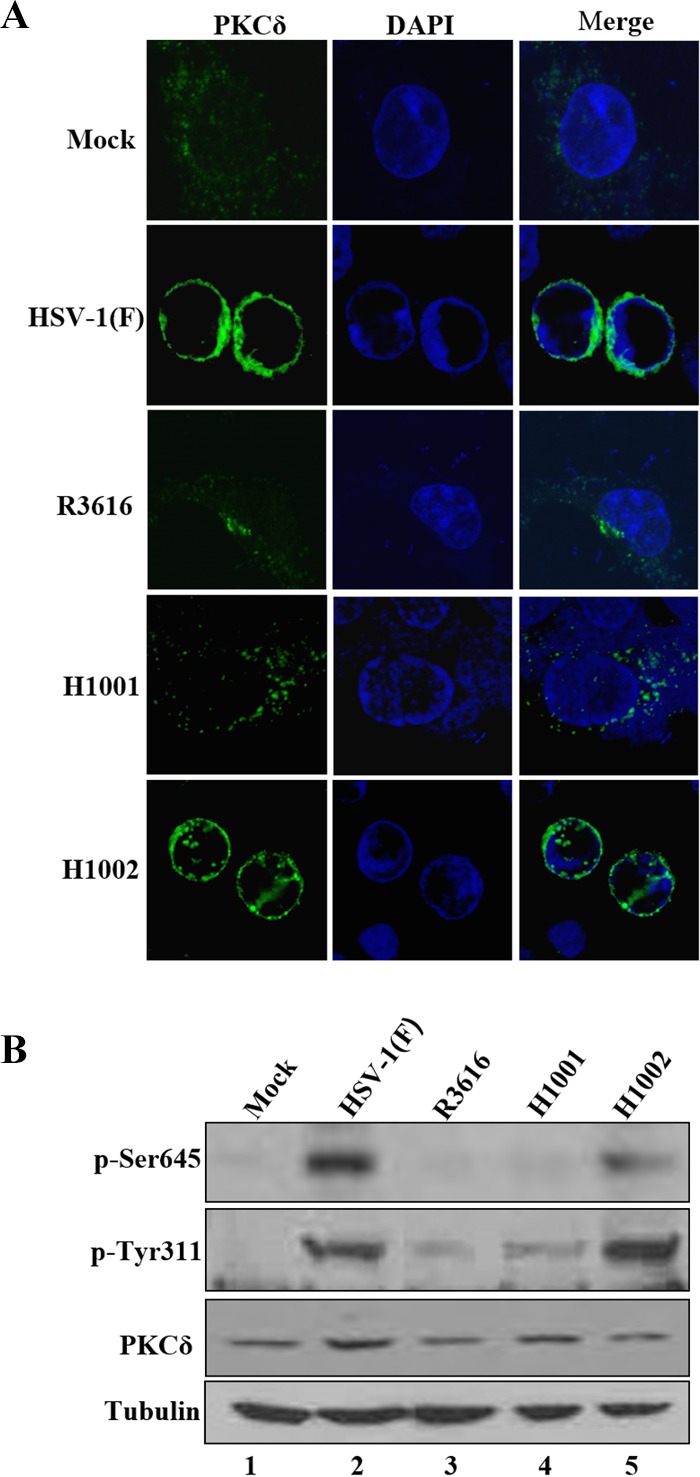
The γ134.5 protein mediates the recruitment and activation of PKCδ in infected cells. (A) HeLa cells were mock infected or infected with the indicated viruses (5 PFU/cell). At 16 h postinfection, cells were stained with DAPI and anti-PKCδ antibody. Cells are then visualized with confocal microscopy. (B) HeLa cells were mock infected or infected with indicated viruses as for panel A. Cell lysates were prepared and analyzed by Western blotting with antibodies against PKCδ, phosphorylated PKCδ (Ser645), phosphorylated PKCδ (Tyr311), and tubulin.
To evaluate the status of PKC activation, we examined its phosphorylation in response to HSV infection. HeLa cells were mock infected or infected with γ134.5 variants. At 16 h postinfection, cells were processed for immunoblotting analysis with antibodies against PKCδ and phosphorylated PKCδ. As shown in Fig. 5B, expression of PKCδ was detectable in all cells. Infection with HSV-1(F) or H1002 induced phosphorylation of serine 645 and tyrosine 311 on PKCδ. This was not observed in mock-infected cells. Similarly, little phosphorylation was seen in cells infected with R3616 or H1001. These results suggest that HSV-1 induces PKCδ activation, which is dependent on γ134.5 or its amino-terminal domain.
The γ134.5 protein associates with PKC and p32 through its amino-terminal domain.
To investigate the mechanism of γ134.5 action, we assessed the relationship of γ134.5, PKC, and cellular p32, which is linked to the reorganization of lamin A/C in HSV-1 infection (32). HeLa cells were infected with γ134.5 variants and subjected to immunoprecipitation analyses. With anti-γ134.5 antibody (Fig. 6A), γ134.5 pulled down PKCδ and p32 in cells infected with HSV-1(F) or H1002 (lanes 1 and 4). This interaction was not detectable in cells infected with R3616 or H1001 (lanes 2 and 3). In a reverse experiment with anti-p32 antibody (Fig. 6B), p32 captured PKCδ in the presence of wild-type γ134.5 (lanes 1 and 4). Deletion of γ134.5 or its amino terminus abrogated this interaction (lanes 2 and 3).
FIG 6.

The amino terminus of γ134.5 is required to form a complex with PKCδ and p32. (A) HeLa cells were infected with the indicated viruses at 5 PFU per cell. At 16 h postinfection, cells were harvested and subjected to immunoprecipitation (IP) with anti-γ134.5 antibody. Precipitated proteins and whole-cell lysates (WCL) were processed for Western blot analysis with antibodies against PKCδ, p32, γ134.5, and tubulin. Tubulin was used as a loading control. (B) HeLa cells were infected as for panel A and processed for immunoprecipitation with anti-p32 antibody, which was followed by Western blotting with antibodies against PKCδ, p32, γ134.5, and tubulin. (C) HeLa cells were transfected with a vector plasmid or a plasmid expressing wild-type γ134.5 or the γ134.5 mutant devoid of amino acids 1 to 146 (DN) or amino acids 147 to 263 (DC). At 36 h after transfection, cells were processed for immunoprecipitation with anti-γ134.5 antibody. Precipitated proteins and whole-cell lysates were probed with antibodies against p32, PKCδ, γ134.5, and tubulin.
To explore whether γ134.5 forms a complex with PKC and p32 in the absence of other viral protein, HeLa cells, transfected with plasmid vector or γ134.5 variants, were subjected to immunoprecipitation with anti-γ134.5 antibody. As shown Fig. 6C, wild-type γ134.5 bound to endogenous PKCδ and p32 (lane 2). In contrast, the mutant lacking the amino terminus lost its binding activity (lane 3). The γ134.5 mutant with deletion of the carboxyl terminus retained its ability to interact with PKCδ and p32 (lane 4), suggesting that the amino-terminal domain of γ134.5 interacts with PKCδ and p32. These experimental results are consistent with a model where γ134.5 bridges PKCδ and p32, forming a complex that mediates reorganization of the nuclear lamina.
The γ134.5 protein communicates with the nuclear egress complex in HSV-infected cells.
The UL31/UL34 complex mediates nucleocapsid docking, reorganization of the nuclear lamina, and remodeling of nuclear envelope (1). Deletion of UL31 or UL34 from HSV-1 impedes viral nuclear egress (37, 38). Because γ134.5 facilitates viral egress from the nucleus, we asked whether it associates with the UL31/UL34 complex upon viral infection. Accordingly, HeLa cells were infected with wild-type virus and γ134.5 variants. At 16 h postinfection, cells were processed for immunoprecipitation with anti-γ134.5 antibody (Fig. 7A). In cells infected with wild-type HSV-1(F), γ134.5 associated with UL31 and UL34. However, in cells infected with H001, which lacks the amino terminus of γ134.5, such interaction disappeared. Infection with H1002 restored the interaction of γ134.5 with UL31 and UL34. Further analysis showed that PCKδ and p32 were present in the UL31/UL34 complex. These results suggest that γ134.5 forms a multiprotein complex that mediates HSV nuclear egress.
FIG 7.

(A) The γ134.5 protein forms a complex with UL31, UL34, PKC, and p32 in HSV-infected cells. Monolayer HeLa cells were mock infected or infected with HSV-1(F), H1001, or H1002 (5 PFU/cell). At 16 h postinfection, lysates of cells were immunoprecipitated with anti-γ134.5 antibody. Precipitated proteins and whole-cell lysates (WCL) were processed for immunoblotting with antibodies against PKCδ, p32, UL31, UL34, γ134.5, and tubulin. (B) UL31 and UL34 localize to the nuclear membrane independently of γ134.5. HeLa cells were mock infected or infected with the indicated viruses (5 PFU/cell). At 16 h postinfection, cells were stained with DAPI and antibodies against UL31 and UL34. Samples were then visualized by confocal microscopy.
To determine whether γ134.5 affects the localization of UL31 and UL34 in viral infection, HeLa cells were mock infected or infected with viruses. Cells were stained with antibody against UL31 or UL34 and visualized by confocal microscopy (Fig. 7B). Consistent with previous findings, UL31 and UL34 localized to the nuclear rim in cells infected with wild-type HSV-1(F). In cells infected with H1001 or H1002, UL31 and UL34 similarly distributed to the nuclear membrane, suggesting that γ134.5 does not regulate subcellular localization of the UL31/UL34 complex in HSV-infected cells.
To delineate whether γ134.5 alone binds to UL31 or UL34, HeLa cells were transfected with γ134.5 along UL31 or UL34. Samples were then subjected to immunoprecipitation with anti-γ134.5 antibody. As illustrated in Fig. 8A, wild-type γ134.5 interacted with UL31 but not UL34. Furthermore, the γ134.5 mutant that lacks the carboxyl terminus bound to UL31 (Fig. 8B). However, the γ134.5 mutant devoid of its amino terminus lost its activity (Fig. 8B). Therefore, when expressed, the γ134.5 protein binds to UL31 via its amino-terminal domain.
FIG 8.

(A) The γ134.5 protein interacts with UL31. Monolayers of HeLa cells were transfected with γ134.5 along with a plasmid expressing UL31 or UL34. At 36 h after transfection, cells were processed for immunoprecipitation with anti-γ134.5 antibody. Precipitated proteins and whole-cell lysates were probed with antibodies against FLAG, γ134.5, and tubulin. (B) The amino terminus of γ134.5 interacts with UL31. HeLa cells were transfected with a vector plasmid or a plasmid expressing wild-type γ134.5 or the γ134.5 mutant devoid of amino acids 1 to 146 (DN) or amino acids 147 to 263 (DC). Cells were then processed for immunoprecipitation with anti-γ134.5 antibody. This was followed by Western blotting with antibodies against FLAG, γ134.5, and tubulin as for panel A.
DISCUSSION
Several lines of evidence suggest that nucleocapsid budding of herpesviruses alters the architecture of nuclear envelope (8, 11). Relevant to this is local disintegration of the nuclear lamina, which is necessary to engage capsids to the nuclear membrane and for subsequent release. In the present work, we show that HSV-1 mediates reorganization of lamin A/C through the amino terminus of the γ134.5 protein. This parallels with efficient release of capsids from the nucleus and viral growth. In particular, the γ134.5 protein engages with the UL31/UL34 complex and activates protein kinase C in infected cells. These observations suggest a model in which HSV γ134.5 is a mediator of nuclear egress in HSV-infected cells (Fig. 9).
FIG 9.
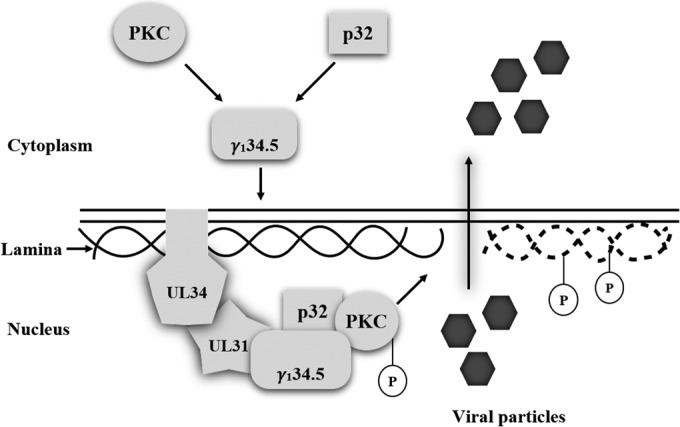
A model of γ134.5-mediated nuclear egress. Upon HSV-1 infection, the γ134.5 protein binds and redirects PKC and p32 to the nuclear membrane where the UL31/UL34 complex resides. Formation of a multiprotein complex activates PKC, which dephosphorylates and subsequently disintegrates the nuclear lamina for nucleocapsid egress.
The γ134.5 protein is essential to promote HSV virulence (21). In addition to inhibition of autophagy and interferon responses, it facilitates viral nuclear egress (19, 26, 30). We noted that an HSV mutant lacking the amino terminus of γ134.5 was severely impaired in viral growth compared to wild-type virus. While this phenotype may reflect a compounding effect, it is partly attributable to aberrant nuclear egress. Restoration of wild-type γ134.5 reversed the phenotype, suggesting that the region encompassing amino acids 1 to 146 is functionally important. Two lines of evidence support this interpretation. First, in cells infected with the amino-terminal mutant of γ134.5, the relative yield of cell-free virus was reduced. Second, electron microscopy examination suggests that this γ134.5 mutant was confined mainly to the nucleus upon infection. A simple explanation is that a block or reduction in the rate of nucleocapsid transit may account for defective nuclear egress. Therefore, concerted activities of γ134.5 likely contribute to efficient viral replication. We favor the notion that the amino terminus of γ134.5 may act as a functional module that mediates the budding machinery in HSV infection.
Our work suggests a role of γ134.5 in remodeling the nuclear envelope in virus-infected cells. Several studies indicate that herpesvirus infection dismantles the nuclear lamina through phosphorylation of lamin A/C and lamin B (3, 11, 12), which permits capsid access for primary envelopment. The data presented in this work suggest that such changes are linked to the amino terminus of γ134.5. In its absence, neither reorganization nor phosphorylation of lamin A/C occurred. In HSV-infected cells, protein kinase C is believed to phosphorylate lamin A/C and lamin B (12, 32). In addition, Us3 phosphorylates lamin A/C (3). Therefore, the γ134.5 protein may regulate a kinase(s) that phosphorylates lamin A/C and locally dissolves the lamina. Indeed, the γ134.5 protein mediated the activation of PKCδ in infected cells. Deletion of γ134.5 or its amino terminus precluded the translocation of protein kinase Cδ to the nuclear membrane and its activation. Previous studies suggest that chemical inhibition of PKC impairs nuclear egress (39). In line with this, we observed that bisindolylmaleimide I or rottlerin inhibited phosphorylation of lamin A/C. Given that BIM I is a pan-PKC small-molecule inhibitor (40), multiple kinases may contribute to lamin phosphorylation. Although it was not tested, our results do not exclude the possibility that γ134.5 may target an additional kinase(s).
In HSV-infected cells, the γ134.5 protein forms complex with p32 and protein kinase C through its amino-terminal domain. Deletion of the amino terminus disrupted the interaction of p32 and PKCδ. p32, a mitochondrial protein, travels to nuclear membrane upon HSV infection (32). While incompletely characterized, p32 is required for nuclear egress of HSV-1 and human cytomegalovirus (HCMV) (32, 33, 41). It has been shown that knockdown of p32 prevents HSV-induced reorganization of lamin A/C (32). Given a link of PKC to the nuclear envelope budding, it is logical to postulate that the γ134.5 complex regulates reorganization of the nuclear lamina. At this point, the precise role of γ134.5 is unknown. A plausible hypothesis is that γ134.5 bridges p32 and PKC via its amino-terminal domain, where p32 may modulate PKC or a distinct kinase(s). Alternatively, HSV γ134.5 may simply work in the cytoplasm as a trafficking protein that delivers p32 and PKC to the nuclear membrane. In this special context, it is noteworthy that γ134.5 shuttles between the nucleus and cytoplasm (22, 23).
In addition, the γ134.5 protein interacts with the UL31/UL34 complex in HSV-infected cells. Removal of the amino terminus of γ134.5 abrogated such interaction and crippled viral nuclear egress. These experimental data suggest a model in which γ134.5 may facilitate nuclear egress through cooperation with UL31 and UL34. The UL31/UL34 complex is the workhorse that mediates nuclear egress (1). In infected cells, UL31 and UL34 colocalize to the nuclear rim and bind to lamin A/C and lamin B (8, 36). This is suggested to modulate disintegration of the nuclear lamina. Although the UL31/UL34 complex is necessary to induce the recruitment of PKC isoforms to the nuclear membrane, a direct interaction is not detectable (12). In this respect, we suspect that the UL31/UL34 complex may function in a γ134.5-dependent mechanism. An attractive possibility is that γ134.5 recruits PKC in the cytoplasm, which is directed to the UL31/UL34 complex in the nuclear membrane. In this regard, UL31 probably serves as a contact site. Together with p32, γ134.5 may activate PKC, which leads to lamina disassembly and subsequent nuclear egress.
ACKNOWLEDGMENTS
We thank Yu Wang for assistance.
This work was supported by grants from the National Institute of Allergy and Infectious Diseases (AI112755 to B.H.), the National Natural Science Foundation of China (31370862 to Y.C.), the Ministry of Science and Technology of China (BE091561 to Y.C.), and the Program of Introducing Talents of Discipline to Universities (B08011). S.W. was supported in part by a fellowship from the China Scholarship Council.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 2.Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bauerlein FJ, Cheleski J, Werner S, Guttmann P, Rehbein S, Henzler K, Demmerle J, Adler B, Koszinowski U, Schermelleh L, Schneider G, Enquist LW, Plitzko JM, Mettenleiter TC, Grunewald K. 2015. Structural basis of vesicle formation at the inner nuclear membrane. Cell 163:1692–1701. doi: 10.1016/j.cell.2015.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mou F, Forest T, Baines JD. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J Virol 81:6459–6470. doi: 10.1128/JVI.00380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maruzuru Y, Shindo K, Liu Z, Oyama M, Kozuka-Hata H, Arii J, Kato A, Kawaguchi Y. 2014. Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J Virol 88:7445–7454. doi: 10.1128/JVI.01057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 2014. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88:4657–4667. doi: 10.1128/JVI.00137-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci U S A 104:10187–10192. doi: 10.1073/pnas.0703790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burke B, Stewart CL. 2013. The nuclear lamins: flexibility in function. Nat Rev Mol Cell Biol 14:13–24. doi: 10.1038/nrm3488. [DOI] [PubMed] [Google Scholar]
- 8.Reynolds AE, Liang L, Baines JD. 2004. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes U(L)31 and U(L)34. J Virol 78:5564–5575. doi: 10.1128/JVI.78.11.5564-5575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leach N, Bjerke SL, Christensen DK, Bouchard JM, Mou F, Park R, Baines J, Haraguchi T, Roller RJ. 2007. Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and US3. J Virol 81:10792–10803. doi: 10.1128/JVI.00196-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mou F, Wills EG, Park R, Baines JD. 2008. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus U(L)34-encoded protein to the inner nuclear membrane. J Virol 82:8094–8104. doi: 10.1128/JVI.00874-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857. doi: 10.1126/science.1071506. [DOI] [PubMed] [Google Scholar]
- 12.Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 80:494–504. doi: 10.1128/JVI.80.1.494-504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, Moore MJ, Budnik V. 2012. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 149:832–846. doi: 10.1016/j.cell.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hocevar BA, Burns DJ, Fields AP. 1993. Identification of protein kinase C (PKC) phosphorylation sites on human lamin B. Potential role of PKC in nuclear lamina structural dynamics. J Biol Chem 268:7545–7552. [PubMed] [Google Scholar]
- 15.Goss VL, Hocevar BA, Thompson LJ, Stratton CA, Burns DJ, Fields AP. 1994. Identification of nuclear beta II protein kinase C as a mitotic lamin kinase. J Biol Chem 269:19074–19080. [PubMed] [Google Scholar]
- 16.Thompson LJ, Fields AP. 1996. BetaII protein kinase C is required for the G2/M phase transition of cell cycle. J Biol Chem 271:15045–15053. doi: 10.1074/jbc.271.25.15045. [DOI] [PubMed] [Google Scholar]
- 17.Collas P. 1999. Sequential PKC- and Cdc2-mediated phosphorylation events elicit zebrafish nuclear envelope disassembly. J Cell Sci 112:977–987. [DOI] [PubMed] [Google Scholar]
- 18.Cross T, Griffiths G, Deacon E, Sallis R, Gough M, Watters D, Lord JM. 2000. PKC-delta is an apoptotic lamin kinase. Oncogene 19:2331–2337. doi: 10.1038/sj.onc.1203555. [DOI] [PubMed] [Google Scholar]
- 19.Brown SM, MacLean AR, Aitken JD, Harland J. 1994. ICP34.5 influences herpes simplex virus type 1 maturation and egress from infected cells in vitro. J Gen Virol 75:3679–3686. doi: 10.1099/0022-1317-75-12-3679. [DOI] [PubMed] [Google Scholar]
- 20.Chou J, Roizman B. 1990. The herpes simplex virus 1 gene for ICP34.5, which maps in inverted repeats, is conserved in several limited-passage isolates but not in strain 17syn+. J Virol 64:1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to γ134.5, a gene nonessential for growth in culture. Science 250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 22.Cheng G, Brett ME, He B. 2002. Signals that dictate nuclear, nucleolar, and cytoplasmic shuttling of the γ134.5 protein of herpes simplex virus type 1. J Virol 76:9434–9445. doi: 10.1128/JVI.76.18.9434-9445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao H, Rosenthal KS. 2002. An N-terminal arginine rich cluster and a proline-alanine-threonine repeat region determines the cellular localization of the herpes simplex virus type-1 ICP34.5 protein and Its ligand, protein phosphatase 1. J Biol Chem 11:11. [DOI] [PubMed] [Google Scholar]
- 24.He B, Gross M, Roizman B. 1997. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He B, Gross M, Roizman B. 1998. The γ134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J Biol Chem 273:20737–20743. doi: 10.1074/jbc.273.33.20737. [DOI] [PubMed] [Google Scholar]
- 26.Verpooten D, Ma Y, Hou S, Yan Z, He B. 2009. Control of TANK-binding kinase 1-mediated signaling by the γ134.5 protein of herpes simplex virus 1. J Biol Chem 284:1097–1105. doi: 10.1074/jbc.M805905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin H, Ma Y, Prabhakar BS, Feng Z, Valyi-Nagy T, Yan Z, Verpooten D, Zhang C, Cao Y, He B. 2009. The γ134.5 protein of herpes simplex virus 1 is required to interfere with dendritic cell maturation during productive infection. J Virol 83:4984–4994. doi: 10.1128/JVI.02535-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin H, Ma Y, Yan Z, Prabhakar BS, He B. 2012. Activation of NF-kappaB in CD8+ dendritic cells ex vivo by the γ134.5 null mutant correlates with immunity against herpes simplex virus 1. J Virol 86:1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Y, Jin H, Valyi-Nagy T, Cao Y, Yan Z, He B. 2012. Inhibition of tank binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J Virol 86:2188–2196. doi: 10.1128/JVI.05376-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Jing X, Cerveny M, Yang K, He B. 2004. Replication of herpes simplex virus 1 depends on the γ134.5 functions that facilitate virus response to interferon and egress in the different stages of productive infection. J Virol 78:7653–7666. doi: 10.1128/JVI.78.14.7653-7666.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Yang Y, Wu S, Pan S, Zhou C, Ma Y, Ru Y, Dong S, He B, Zhang C, Cao Y. 2014. p32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J Biol Chem 289:35795–35805. doi: 10.1074/jbc.M114.603845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z, Kato A, Oyama M, Kozuka-Hata H, Arii J, Kawaguchi Y. 2015. Role of host cell p32 in herpes simplex virus 1 de-envelopment during viral nuclear egress. J Virol 89:8982–8998. doi: 10.1128/JVI.01220-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 35.Verpooten D, Feng Z, Valyi-Nagy T, Ma Y, Jin H, Yan Z, Zhang C, Cao Y, He B. 2009. Dephosphorylation of eIF2alpha mediated by the γ134.5 protein of herpes simplex virus 1 facilitates viral neuroinvasion. J Virol 83:12626–12630. doi: 10.1128/JVI.01431-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. 2000. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J Virol 74:117–129. doi: 10.1128/JVI.74.1.117-129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leach NR, Roller RJ. 2010. Significance of host cell kinases in herpes simplex virus type 1 egress and lamin-associated protein disassembly from the nuclear lamina. Virology 406:127–137. doi: 10.1016/j.virol.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamelll L, Charon D, Kirilovsky J. 1991. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem 266:15771–15781. [PubMed] [Google Scholar]
- 41.Milbradt J, Kraut A, Hutterer C, Sonntag E, Schmeiser C, Ferro M, Wagner S, Lenac T, Claus C, Pinkert S, Hamilton ST, Rawlinson WD, Sticht H, Coute Y, Marschall M. 2014. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol Cell Proteomics 13:2132–2146. doi: 10.1074/mcp.M113.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]