

Abstract
Hematopoietic stem cells (HSCs), which sustain production of all blood cell lineages,1 rely on glycolysis for ATP production,2,3 yet little attention has been paid to the role of mitochondria. We show here that the short isoform of a critical regulator of HSCs, Prdm16,4,5 induces mitofusin 2 (Mfn2), a protein involved in mitochondrial fusion and in tethering of mitochondria to the endoplasmic reticulum (ER). Overexpression and deletion studies, including single cell transplantation assays, revealed that Mfn2 is specifically required for the maintenance of HSCs with extensive lymphoid potential, but not, or less so for the maintenance of myeloid-dominant HSCs. Mfn2 increased buffering of Cai2+, an effect mediated through its ER-mitochondria tethering activity,6,7 thereby negatively regulating nuclear translocation and transcriptional activity of Nuclear Factor of Activated T cells (NFAT). NFAT inhibition rescued the effects of Mfn2 deletion in HSCs, demonstrating that negative regulation of NFAT is the prime downstream mechanism of Mfn2 in the maintenance of HSCs with extensive lymphoid potential. Mitochondria therefore play an important role in HSCs. These findings provide a mechanism underlying clonal heterogeneity among HSCs8–11 and may lead to the design of approaches to bias HSC differentiation into desired lineages after transplantation.
Within the hematopoietic system, the transcriptional co-regulator, Prdm16, is expressed selectively in HSCs and its deletion severely impairs HSC maintenance.4,5 However, its molecular targets remain unknown. We observed that in Prdm16−/− HSCs (Lin−Sca1+Kit+CD48−CD150+Flt3−, Extended Data Fig. 1) and mouse embryonic fibroblasts (MEFs) mitochondria were fragmented (Fig. 1A. Extended Data Fig. 2A). Mitochondria undergo dynamic fusion and fission.12,13 Fusion is driven by the outer membrane GTPases, Mitofusin (Mfn) 1 and 2, and by the inner membrane protein Optic Atrophy 1 (Opa1), while fission requires Dynamin-Related Protein (Drp1).12,13 Culture of Prdm16−/− MEFs in the presence of the Drp1 inhibitor, mDivi1,14 restored mitochondrial length (Extended Data Fig. 2B,C), suggesting a fusion defect, which was further documented in a mitochondrial fusion assay (Extended Data Fig. 2D). Expression of Mfn2 protein and of Mfn2, but not Mfn1, mRNA was lower in Prdm16-deficient MEFs and HSCs compared to Wt (Fig. 1B–D, Fig. Extended Data Fig. 2E). Furthermore, mitochondria were similarly fragmented in Prdm16−/− and in Mfn2−/− MEFs (Extended Data Fig. 2F) and mitochondrial length was restored after lentiviral expression of Mfn2 in Prdm16−/− MEFs (Fig. 1E,F, Extended Data Fig. 2G), suggesting that Mfn2 is a target of Prdm16.
Figure 1. Prdm16 induces Mfn2.

(A) Mitochondrial morphology of MTR-stained Wt and Prdm16−/− fetal liver HSCs (Lin−Sca1+kit+CD48−CD150+) (scale bar 5µm) and MEFs (scale bar 20µm, inset 5µm). (B) Relative mRNA expression of Prdm16, Mfn1 and Mfn2 in Wt and Prdm16−/− MEFs. n=3 independent experiments; *P<0.05; two-tailed student’s t test. (C) Relative mRNA expression of Prdm16, Mfn1 and Mfn2 in Wt and Prdm16+/− adult HSCs. n=3 biological replicates; *P<0.05; two-tailed student’s t test. (D) Representative immunofluorescence for Mfn2 and Tomm20 in Wt and Prdm16−/− HSCs (scale bar 5µm) and Mfn2 quantification normalized to Tomm20. Bars, mean±s.e.m.; n≥10 fields of cells from two biological replicates; *P<0.05; two-tailed student’s t test. (E) Mitochondrial morphology in Prdm16−/− MEFs co-transduced with Mito-dsRed and Mfn2 or control lentivirus (EV) (scale bar 20µm). (F) Mitochondrial length profiles in Wt and Prdm16−/− MEFs transduced with EV or Mfn2 (≥12 cells and ≥80 mitochondria from 2 biological replicates). Note that Mfn2 overexpression in Wt MEFs causes aggregation of mitochondria and apparent shortening, as reported previously.30 (G) Relative mRNA expression in Prdm16−/− MEFs of Prdm16, Mfn1 and Mfn2 72h after retroviral expression of sPrdm16 and flPrdm16. n=3 biological replicates; *P<0.05; one-way ANOVA with Dunnett’s post hoc test.
Prdm16 exists in two isoforms arising from distinct transcription start sites, full length (fl) and short (s) Prdm16, which lacks the N-terminal PR-domain Extended Data Fig. 3A).15,16 Only sPrdm16, but not flPrdm16, activated a Mfn2 promoter luciferase reporter Extended Data Fig. 3B,C), and induced Mfn2 mRNA in Prdm16−/− MEFs (Fig. 1G). Consistent with these findings, chromatin immunoprecipitation in MEFs using FLAG-tagged isoforms of Prdm16 showed binding of sPrdm16, but not of flPrdm16, to the Mfn2 promoter Extended Data Fig. 3D).17 Mfn2 is therefore a direct target of sPrdm16. Although Prdm16-deletion did not affect Mfn1 (Fig. 1B,C), transduction of sPrdm16 did increase Mfn1 mRNA expression (Fig. 1G). Mfn1 is therefore susceptible to regulation by sPrdm16, but with a higher and likely unphysiological threshold. Lentiviral transduction of Mfn2 did not rescue the competitive repopulation defect of Prdm16+/− HSCs, however Extended Data Fig. 3E), indicating that multiple components of the sPrdm16 and flPrdm16 transcriptional program are required for HSC maintenance.
Induction of Mfn2 by Prdm16, a critical regulator of HSCs, suggests a role for Mfn2 in HSC function. We therefore assessed mitochondrial length and Mfn2 expression in hematopoietic cells. HSCs display clonal heterogeneity in their differentiation potential ranging from rare lymphoid-biased HSCs, to balanced myeloid/lymphoid and myeloid-dominant HSCs with low lymphoid potential.8–11 Though the underlying mechanism is unknown and neither functional nor phenotypic classifications are absolute, myeloid-dominant HSCs are enriched in the CD150hi, while HSCs with extensive lymphoid potential are enriched in the CD150lo fraction.18,19 HSCs expressed more Mfn2 mRNA (Fig. 2A) and protein (Fig. 2B) than more mature populations. Within the HSC compartment, CD150lo HSCs expressed more Mfn2 mRNA (Fig 2A) and protein (Fig. 2C) than did CD150hi HSCs. In contrast, Mfn1 did not show HSC-selective expression, and its expression in CD150lo HSCs was tenfold lower than that of Mfn2 (Fig. 2A). In accordance with Mfn2 induction by sPrdm16, sPrdm16 was the predominant Prdm16 isoform in CD150lo but not in CD150hi HSCs (Fig. 2D). Using mice expressing a mitochondrially targeted Dendra2 fluorescent protein (Pham mice),20 we observed longer mitochondria in HSCs compared to other hematopoietic populations, and within the HSC compartment, in CD150lo than in CD150hi cells (Fig. 2E and Extended Data Fig. 4A,B). Mitochondrial length therefore paralleled Mfn2 expression.
Figure 2. Mitochondrial morphology and Mfn2 in HSCs.

(A) Expression of Mfn2 and Mfn1 mRNA in HSCs, progenitors (MPP: multipotential progenitors, Lin−Sca1+kit+CD150−Flt3+; CMP: common myeloid progenitors, Lin−Sca1−Kit+; CLP: common lymphoid progenitor, Lin−Sca1lokitloFlt3+IL7Rα+ and Lineage+ cells. n=3 biological replicates; *P<0.05; one-way ANOVA with Dunnett’s post hoc test. (B) Mfn2 immunofluorescence in Pham-reporter+ HSCs and CMPs (Pham mice express a mitochondrially targeted Dendra2 fluorescent protein)20 (scale bars 5µm). (C) Immunofluorescence staining for Mfn2 and CD150 in CD150hi and CD150lo LSKCD48− HSCs. (D) Quantification of flPrdm16 and sPrdm16 mRNA in HSCs, progenitors and Lineage+ cells. n=3 biological replicates; *P<0.05; one-way ANOVA with Dunnett’s post hoc test. (E) Mitochondrial length frequency profile in Pham-reporter+ HSCs, progenitors and Lin+ cells (n≥15 fields from three biological replicates).
As these findings suggested a subpopulation-specific role for Mfn2 in HSCs, we examined mice with conditional deletion of Mfn221 in the hematopoietic system (Mfn2fl/fl-Vav-Cre). The frequency of progenitors in the bone marrow (BM) and thymus and of mature populations in blood and spleen were similar in Mfn2fl/fl-Vav-Cre mice and Mfn2fl/fl littermates (Extended Data Table 1). The Lin−Sca1+Kit+CD48−CD150+ HSC compartment in Mfn2fl/fl-Vav-Cre mice showed mitochondrial fragmentation Extended Data Fig. 5A,B), was smaller (Extended Data Table 1) and expressed more CD150 Extended Data Fig. 5C) compared to that of Mfn2fl/fl mice, indicating a loss primarily of CD150lo HSCs. Competitive repopulation studies22 showed a further increase in CD150 expression within the donor HSC compartment Extended Data Fig. 5D,E) and a defect in long-term lymphoid repopulation in recipients of Mfn2fl/fl-Vav-Cre adult BM (Fig. 3A) and fetal liver cells Extended Data Fig. 5F). A decrease in myeloid repopulation was noted, but did not reach statistical significance (Fig. 3A, Extended Data Fig. 5F). Lentiviral overexpression of Mfn2 in Wt HSCs yielded reciprocal results (Extended Data Fig. 6A–I). As these phenotypic analyses and transplantation experiments suggested selective requirement for Mfn2 in the maintenance of HSCs with extensive lymphoid potential, we performed competitive single HSC transplantation studies to rigorously determine clonal variation in differentiation potential. Although out of >100 recipients too few mice were reconstituted to statistically assess HSC frequency, among recipients with >0.1% donor contribution most Mfn2fl/fl-Vav-Cre HSCs were myeloid-dominant, whereas most Mfn2fl/fl HSCs were balanced or lymphoid 8 weeks after transplantation. In mice that still showed repopulation after 13 weeks, only myeloid-dominant HSCs were detected recipients of Mfn2fl/fl-Vav-Cre, while most donor HSCs had extensive lymphoid potential in recipients of Mfn2fl/fl cells (Fig. 3B). To more accurately determine HSC frequencies we performed limiting dilution experiments.22 Among Mfn2fl/fl-Vav-Cre HSCs overall repopulating HSC frequency was decreased fourfold compared to Mfn2fl/fl HSC (Fig. 3C, Extended Data Table 2). The frequency of HSCs capable of >1% long-term lymphoid reconstitution was also approximately fourfold lower. However, the decrease in the frequency of HSCs capable of >1% myeloid reconstitution did not reach statistical significance (Fig. 3C, Extended Data Table 2). Taken together, these results indicate that Mfn2 is required for the maintenance of HSCs with extensive lymphoid potential.
Figure 3. Role of Mfn2 in HSC function.

(A) Donor (CD45.2) chimerism 15 weeks after competitive transplantation of 2×105 Mfn2fl/fl or Mfn2fl/fl.Vav-Cre (Mfn2−/−) adult BM cells together with 2×105 CD45.1+ competitor BM cells into CD45.1+CD45.2+ recipients. Plots, mean±s.e.m.; n=7–8 recipients from two independent transplants; *P<0.05; two-tailed student’s t test. (B) Donor GM/B+T reconstitution ratios 8 (left) and 13 (right) weeks after transplantation of single HSCs from Mfn2fl/fl or Mfn2fl/fl.Vav-Cre (Mfn2−/−) mice. Plots, mean±s.e.m.; n≥8 recipients; *P<0.05; student’s T test. Colors represent myeloid-biased (blue), balanced (pink) and lymphoid dominant (green) as defined in ref. 11. (C) Limiting dilution assay (see Methods) with Mfn2fl/fl or Mfn2fl/fl.Vav-Cre (Mfn2−/−) adult BM HSCs co-transplanted with CD45.1+ competitor BM cells analyzed for total, myeloid or lymphoid potential 15 weeks after transplantation. Plots, log % negative responders; n=2 independent experiments with 4–5 recipients each per cell dose; frequencies calculated using ELDA Poisson distribution; P<0.05; Pearson’s chi-square test.
Next, we identified the mechanism of action of Mfn2. Apoptosis and ROS production were similar in Mfn2fl/fl-Vav-Cre and Mfn2fl/fl HSCs, but Mfn2fl/fl-Vav-Cre HSCs and progenitors displayed increased expression of Grp78, a marker of ER stress, which has been shown to be associated with Mfn2-deletion23 (Extended Data Fig. 7A–C). Mfn2, but not Mfn1, also tethers mitochondria to the ER, thereby enhancing intracellular calcium (Cai2+) buffering.6 Indeed, Cai2+ was increased in Mfn2fl/fl-Vav-Cre compared to Mfn2fl/fl HSCs (Fig. 4A), in CD150hi compared to CD150lo HSCs (Fig. 4B) and in Prdm16−/− compared to Wt LSK cells (Fig. 4C), but was decreased after lentiviral transduction of Wt HSCs (Extended Data Fig. 7D). However, Mfn2 did not affect ATP- or SDF1-induced Cai2+ transients (Fig. 4A–C, Extended Data Fig. 7D). At variance with these data and with a previous report showing only delayed removal of Cai2+,6 we found both lower baseline Cai2+ and lower amplitude of ATP-induced calcium flux in Mfn2−/− MEFs (Extended Data Fig. 7E). Thus, despite cell type-specific differences in calcium homeostasis, Mfn2 negatively regulates Cai2+.
Figure 4. Mechanism of action of Mfn2.

(A-C) Calcium flux trace (left) and baseline Cai2+ (right) in (A), Mfn2fl/fl or Mfn2fl/fl.Vav-Cre (Mfn2−/−) HSCs (B), CD150hi and CD150lo HSCs (C), and Wt and Prdm16−/− HSCs. Plots, mean±s.e.m.; n=3 biological replicates; *P<0.05; two-tailed student’s t test. (D) NFAT1 staining (left, scale bars 5µm) and fraction of nuclear NFAT1 (right, *P<0.05) in Mfn2fl/fl or Mfn2fl/fl.Vav-Cre (Mfn2−/−) HSCs. Plot, mean±s.e.m.; n≥7 fields of cells from two biological replicates; *P<0.05; two-tailed student’s t test. (E) Subcellular fractionation followed by WB for NFAT1 in Wt and Mfn2−/− MEFs (left, Supplementary Fig. 1 for full scan) and relative quantification of nuclear and cytoplasmic fraction (right). Bars, mean±s.d.; n=3 biological replicates; *P<0.05; two-tailed student’s t test. (F,G) NFAT1 staining (left, scale bars = 5µm) and fraction of nuclear NFAT1 (right, *P<0.05) in CD150hi and CD150lo HSCs (F), and Wt and Prdm16−/− fetal liver HSCs (G). Plots, mean±s.e.m.; n≥7 fields of cells pooled from three biological replicates; *P<0.05; two-tailed student’s t test. (H) Luciferase activity of 3xNFAT gene reporter in 3T3 cells transfected with constructs on Y-axis (EV: empty vector). Data normalized to β-Gal activity and relative to EV. Bars, mean±s.d.; n=7 biological replicates; *P<0.05; one-way ANOVA with Dunnett’s post hoc test. (I) Lymphoid/myeloid ratio 15 weeks after competitive transplantation of Mfn2fl/fl or Mfn2fl/fl.Vav-Cre (Mfn2−/−) HSCs transduced with IRES-GFP (EV) or VIVIT-GFP. Plots, mean±s.e.m.; n>8 recipients pooled from three independent transplants; *P<0.05; one-way ANOVA with Dunnett’s post hoc test. (J) Schematic representation of the mechanism of action of Mfn2.
Sustained increase in Cai2+ activates calcineurin which dephosphorylates four isoforms of Nuclear Factor of Activated T cells (NFAT) and promotes their translocation to the nucleus,24 where they orchestrate multiple processes.25,26 We therefore examined whether Mfn2 inhibits NFAT. As measured by immunofluorescence, NFAT1 nuclear localization was decreased after lentiviral transduction of Wt HSCs (Extended Data Fig. 7F), and increased in Mfn2fl/fl-Vav-Cre compared to Mfn2fl/fl HSCs (Fig. 4D) and in Mfn2−/− compared to Wt MEFs (Extended Data Fig. 7G), which was confirmed by cellular fractionation followed by Western Blot (Fig. 4E). Consistent with inhibition of NFAT nuclear localization by Mfn2, the fraction of nuclear NFAT was also higher in CD150hi compared to CD150lo HSCs (Fig. 4F) and in Prdm16−/− compared to Wt HSCs (Fig. 4G). We next assessed the effect of Mfn2 on NFAT transcriptional activity. Transfection of Mfn2−/− MEFs with a NFAT-responsive luciferase reporter revealed increased NFAT transcriptional activity compared to Wt MEFs (Extended Data Fig. 7H). Conversely, overexpression of Mfn2 in NIH-3T3 fibroblasts reduced NFAT-responsive luciferase reporter activity to the same extent as VIVIT, a membrane-permeable peptide that specifically inhibits the interaction between calcineurin and NFAT (Fig. 4H).27 Mfn1 and a dominant negative DRP1 mutant (Extended Data Fig. 7I) had no effect however (Fig. 4H). Unlike Mfn2, Mfn1 and Drp1 only regulate mitochondrial fusion and fission. These findings therefore suggest that Mfn2 inhibits NFAT through its ER-mitochondria tethering activity rather than through mitochondrial fusion. Finally, and consistent with Mfn2 induction by sPrdm16, lentiviral transduction of Prdm16−/− HSCs with sPrdm16, but not with flPrdm16, reduced NFAT nuclear localization (Extended Data Fig. 7J), while NFAT transcriptional activation was increased in Prdm16−/− MEFs and normalized by transfection of either sPrdm16 or Mfn2, but not flPrdm16 (Extended Data Fig. 7K). We conclude that sPrdm16-mediated induction of Mfn2 inhibits NFAT activity.
To directly examine the role of NFAT downstream of Mfn2, we inhibited its function in HSCs. Culture of Wt HSCs in the presence of VIVIT peptide increased expression of CD150lo HSC-associated lymphoid commitment markers, Il7r and Sox418 (Extended Data Fig. 8A), and increased lymphoid/myeloid repopulation ratio after subsequent competitive transplantation (Extended Data Fig. 8B). Most importantly, lentiviral VIVIT-GFP27 transduction fully rescued the long-term lymphoid reconstitution defect of Mfn2fl/fl-Vav-Cre HSCs (Fig. 4I) and, similar to Mfn2 transduction, increased lymphoid repopulation of Wt HSCs (Extended Data Fig. 8C). NFAT inhibition is therefore the prime mechanism downstream of Mfn2.
The observation that Mfn2, induced by sPrdm16, maintains HSCs with extensive lymphoid potential by negatively regulating calcineurin/NFAT activity through enhanced Cai2+ buffering (Fig. 4L) identifies such HSCs, which decline with age,28,29 as a mechanistically defined subset and provides a mechanism underpinning clonal heterogeneity in HSCs. These findings may lead to the design of approaches to bias HSC differentiation into desired lineages after transplantation.
Methods
Animals
C57BL/6J mice (CD45.2) and B6.SJL-PtprcaPep3b/BoyJ (CD45.1) were purchased from The Jackson Laboratory (Bar Harbor, ME). Prdm16Gt(OST67423)Lex knockout mice31 were obtained from Lexicon Genetics. Conditional Mito-Dendra2 transgenic (Pham) mice32 (B6;129S–Gt(ROSA)26Sortm1(CAG-COX8A/Dendra2)Dcc/J) and E2A–Cre mice33 (B6.FVB-Tg(EIIa-cre)C5379Lmgd/J) were purchased from Jackson Laboratory. Pham mice contain a mitochondrially targeted Dendra preceeded by a stoplox sequence in the Rosa locus. These mice were crossed with E2A–Cre mice to effect ubiquitous induction of the MitoDendra2 reporter. Conditional Mfn2 knockout mice34 (B6/129SF1Mfn2tm3Dcc/Mmucd) were obtained from MMRRC and crossed to Vav-Cre transgenic mice35 (B6.Cg-Tg(Vav1-Cre)A2Kio/J) to obtain a homozygous floxed allele Mfn2 allele which generated a B6.Cg-Tg(Vav1-Cre)A2Kio/J;B6/126SF1 Mfn2tm3Dcc/Mmucd mixed mouse strain. All mouse strains were rederived by in vitro fertilization at the Jackson Laboratory. Animals were housed in a specific pathogen-free facility. Experiments and animal care were performed in accordance with the Columbia University Institutional Animal Care and Use Committee. All mice were used at age 8–12 weeks, except in experiments involved fetal liver cells, when E14.4 embryos were used. Both sexes were used for experiments. Results were analyzed in non-blinded fashion. In all experiments, randomly chosen wild type and littermates were used.
MEF Isolation and Cell Lines
MEFs were established from approximately 14.5 dpc embryos as previously described36 from Prdm16+/− breeder pairs. Briefly, dissected embryo trunks were minced into 1–2mm fragments, resuspended in 3mL 0.25% trypsin/EDTA (Gibco, Carlsbad, CA) and passed 20–30 times through a 16ga needle. Cell suspensions were incubated at 37 °C for 1h with frequent agitation. Erythrocytes were lysed with ACK buffer, washed and cells were plated for 3h in 10% FBS/DMEM. Cells remaining in suspension were aspirated and adherent cells were cultured with fresh media. MEFs were passaged 1:3 every 3 days and cells between passage 2 and 5 were used for all experiments. 293 cells and NIH-3T3 cells were purchased from ATCC (Manassas, VA) and sub-cultured in 10% FBS/DMEM or 10% calf serum/DMEM, respectively. Wt and Mfn2−/− MEFs were a kind gift from Dr. Eric Schon (Columbia University). All lines are tested yearly for Mycoplasma contamination and found negative.
Plasmids
Prdm16 constructs were generated by subcloning the murine full length (flPrdm16) or truncated (sPrdm16) cDNA into the XhoI/EcoRI sites of the pMSCV-IRES-GFP retroviral expression plasmid. The Mito-dsRed construct was purchased from Addgene (Cambridge, MA) (plasmid 11151). Mfn2 constructs were generated by subcloning the murine Mfn2 cDNA into the EcoRI/BamHI sites of the pLVX-EF1α-IRES-GFP or pLVX-EF1α-IRES-mCherry lentiviral expression plasmid (Clontech). The pGreenFire-NFAT & pGreenFire-CMV gene reporter construct was purchased from System Biosciences (San Jose, CA) and contained three canonical NFAT response elements (5’-GGAAAAN-3’) driving the expression of copGFP and luciferase reporters. The DNDrp1-pcDNA3.1 construct was purchased from Addgene (#45161) and subcloned using the BamHI/EcoRI restriction sites into the pLVX-IRES-GFP vector. Lentiviral 2nd generation packaging construct ΔR8.2 (8455) and pDM2.6 (12259) were purchased from Addgene. The −950/+22 murine MFN2 promoter was constructed by PCR amplification of the RP23–458J18 BAC clone (CHORI, Oakland, CA) and subcloned into the pGL4 luciferase reporter vector (Promega, Madison, WI). All cloning was carried out using KOD hot-start polymerase (Novagen, Billerica, MA) and subcloned for screening and sequencing into the pCR2.1 shuttle vector (Invitrogen, Carlsbad, CA).
FACS Sorting and Analysis
For peripheral blood analyses, erythrocytes were lysed twice with ACK lysis buffer and nucleated cells were stained with antibody cocktail (Supplementary Table 1) in FACS buffer for 15min on ice, washed and analyzed on a BD FACSCantoII flow cytometer (Becton Dickinson, Mountain View, CA). For bone marrow analyses, cells were isolated using the crushing method and erythrocytes were lysed with ACK lysis buffer followed by 40µm filtration. BM cells were stained with antibody cocktail in FACS buffer for 30min on ice, washed and analyzed on a BD LSRII flow cytometer (Becton Dickinson, Mountain View, CA). Dead cells were excluded from analyses by gating out 7AAD-positive cells. To isolate purified hematopoietic populations, BM cells were isolated, stained and sorted using a BD Influx cell sorter (Becton Dickinson, Mountain View, CA) into complete media. Data were analyzed using FlowJo9.6 (TreeStar Inc., Ashland, OR).
Hematopoietic Stem Cell Transplantation
Mfn2fl/fl-Vav-Cre fetal liver cells, BM cells or purified LT-HSCs (Lin−cKit+Sca1+CD48−Flt3−CD150+) were transplanted into lethally irradiated (two doses of 478 cGy over 3 hours using a Rad Source RS-2000 X-ray irradiator (Brentwood, TN)) recipients together with 2×105 competitor cells. As Mfn2fl/fl-Vav-Cre mice were not fully backcrossed onto the C57BL/6 background, recipient mice and competitor BM cells were from the B6.Cg-Tg(Vav1-Cre)A2Kio/J;B6/126SF1 Mfn2tm3Dcc/Mmucd mixed background mouse strain crossed to B6.SJL-Ptprca Pep3b/BoyJ (CD45.1) to generate a CD45.1+CD45.2+ mixed background mouse. Competitor cells were T-cell depleted using MACS beads. For all competitive transplantation experiments, at least two independent transplants, each with at least 4 recipients per condition of genotype were performed, and result of all recipients pooled for statistical analysis. Power calculation was based on results of the first experiment. In limiting dilution assays, cohorts of recipients received 20 or 50 HSCs together with 2×105 competitor cells, allowing calculation of HSC frequency based on the number of non-repopulated mice (<1% donor contribution) using Poisson statistics 15 weeks after reconstitution. For Mfn2 KO single cell transplantation, LT-HSCs were sorted directly into complete media (StemPro34, 100ng/mL SCF, 100ng/mL TPO, 50ng/mL IL-6) and single cells were visually confirmed. Positive single cell wells were combined with 2×105 CD45.1 competitor BM cells and transplanted into lethally irradiated CD45.1 recipient mice. Recipients showing ≥0.1% CD45.2 donor contribution were considered positive and GM/(B+T) ratios were calculated as previously described for characterizing heterogeneous HSC phenotypes.37 In transplantations using Wt or Prdm16−/− HSCs (Lin−cKit+Sca1+CD48−Flt3−CD150+) B6.CD45.2 cells were mixed with 2×105 freshly isolated B6.CD45.1 BM cells and injected via tail vein into lethally irradiated (two doses of 478 cGy over 3 hours using a Rad Source RS-2000 X-ray irradiator (Brentwood, TN)) B6.CD45.1+CD45.2+ F1 hybrid recipients. After 8 to 15 weeks, peripheral blood (PB) and bone marrow (BM) were analyzed.
Lentivirus Production, Transduction and Integration Verification
Lentiviral particles were produced by seeding 293 cells at 7×105/cm2, or PlatE cells (Cell Biolabs, San Diego, CA), in Ultra Culture serum-free media (Lonza, Basel, Switzerland) overnight followed by transfection of each packaging and expression construct (1:1:1) using Trans-It 293 (Mirus, Madison, WI) for 2 hours. Media were pooled after 36–48h, clarified and concentrated by ultracentrifugation (100,000 xg), resuspended in StemPro-34 media and stored at −80°C. Virus titer was calculated from transduction of NIH-3T3 fibroblasts serial dilutions of the viral preparation. Sorted LT-HSCs were transduced with ≥150 MOI lentivirus particles in the presence of 6µg/mL polybrene (Sigma) and spun at 900xg for 20min at 20°C. Supernatant was aspirated and replaced with complete media and cultured overnight. Transduction efficiency of cells was confirmed after 24h. To assess proviral copy number 15 weeks post-transplantation in vivo, splenocytes were harvested and sorted into donor (CD45.2) or competitor (CD45.1) populations and gDNA was isolated as previously described.38 Amplification of the proviral WPRE region was achieved using SYBR Green qPCR assay using the primer pair WPREFor: 5’-CCGTTGTCAGGCAACGTG-3’ and WPRERev: 5’-AGCTGACAGGTGGTGGCAAT-3’. Quantification of proviral copies was derived from the linear regression of serial dilutions of viral vector and normalized to input cell number.
Quantitative RT-PCR
Sorted or cultured cell populations (2–5×103 cells) were lysed in Trizol LS reagent (Invitrogen, Carlsbad, CA) and RNA was isolated according to manufacturer’s instructions. cDNA was synthesized using Superscript III Reverse Transcriptase (Invitrogen) and target CT values were determined using inventoried Taqman probes (Applied Biosystems, Carlsbad, CA, see Supplementary Table 2) spanning exon/exon boundaries and detected using a Viia7 Real Time PCR System (Applied Biosystems). Relative quantification was calculated using the ΔΔCT method. To estimate relative copy number of Mfn1 and Mfn2 transcripts (Fig. 4A), copy numbers were derived from the linear regression of serial dilutions of respective cDNA plasmids and normalized to GAPDH-VIC values. To estimate relative copy number of flPrdm16 transcripts (Fig. 4D), a probe was designed to span the SET methyltransferase domain of Prdm16 (exon2/3 junction) and copy number was derived from the linear regression of serial dilutions of respective cDNA plasmids. Another probe (exon 14/15 junction) was used to quantify total Prdm16 copy numbes derived from the linear regression of serial dilutions of respective cDNA plasmids. The values derived from total Prdm16 probe was subtracted from flPrdm16-specific probe to determine sPrdm16 transcript quantity. All values were normalized to relative multiplexed GAPDH-VIC values.
LT-HSC Culture
Culture of sorted LT-HSCs was carried out using StemPro34 media (Invitrogen) supplemented with 10mM HEPES and 50ng/mL of recombinant murine SCF, TPO, IL-6 (Peptrotech, Rocky Hill, NJ) and cultured in 5% O2 at 37 °C. In some experiments, LT-HSCs were cultured in the presence of 500ng/mL VIVIT (Millipore, Billerica, MA) or 30µM mDivi1 (MolPort, Riga, Latvia).
Mitochondrial PEG-1500 Fusion Assay
To demonstrate a mitochondrial fusion activity, cell fusion experiments were performed using MEFs as previously described.37 Briefly, BacMam baculovirus constructs (Invitrogen) expressing the signaling peptide from cytochrome c fused to either GFP or RFP were transduced separately into MEF cells. Sorted GFP+ and RFP+ MEFs were co-cultured for 24h and plasma membranes were fused using PEG-1500 (Roche, Basel, Switzerland. Fused cells were cultured in DMEM containing cyclohexamide (Sigma, St. Louis, MO) for 4h and analyzed for colocalization of mitochondrial labels.
Chromatin Immunoprecipitation (ChIP)
Early passage Prdm16−/− MEFs were transduced with 10 MOI retrovirus for 72h and fixed with 4% paraformaldehyde for 10min. Protein lysates were isolated and chromatin immunoprecipiation was carried out using the ChIP-IT Express Enzyme kit (Active Motif, Carlsbad, CA). Antibodies used for ChIP include anti-FLAG and anti-TF2D. Primer probes were designed to span regions of the Mfn2 promoter previously shown to regulate Mfn2 transcriptional activity (see Supplementary Table 3).39 Quantification of precipitated Mfn2 promoter regions were derived from the linear regression of serial dilutions of BM genomic DNA, normalized to input DNA concentration and quantifiable IgG detection was subtracted from sample values.
Indo-1-AM Calcium Flux
BM was freshly isolated and lineage depleted with the MACS Lineage Depletion Kit (Miltenyi Biotech, San Diego, CA). Cells were cultured for 30min in complete medium supplemented with 1µM Indo-1 prepared as stock supplemented with Pluronic-F127 and incubated at 37 °C for 30min. Cells were washed and stained for surface markers for 15min, washed and allowed to rest in for 15min PBS in PBS with Ca2+. FACS tubes were run at 37 °C in the sample port of the LSRII flow cytometer equipped with a 355nM excitation laser. Events were collected for 40 seconds prior to incubation with 25uM ATP or 1uM SDF1 to induce calcium transients. The average ratio, R, of bound/free Indo-1 (405nm/485nm emission) before simulation was used to determine baseline values. Identical samples were equilibrated in 10mM EDTA PBS w/o Ca2+ to determine Rmin or stimulated with 1uM ionomycin to determine Rmax. The Indo-1 dissociation constant (Kd) was assumed to be 237nM at 37 °C based on previous studies.40 The following equation was then used to relate Indo-1 intensity ratios to [Cai2+] levels;
Immunofluorescence
Sorted or cultured hematopoietic populations (2–5×103 cells) were collected in complete media and plated on onto MicroWell 96-well glass-bottom plates (Thermo, Waltham, MA) coated with 1ug/mL poly-D-lysine. Cells were allowed to adhere for 10min and fixed with 4% PFA for 15min. Cells were then permeabilized with 0.1% TritonX-100/PBS for 5min and blocked with 2% BSA/PBS for 1h at 4 °C. Cells were incubated with anti-NFAT1 (1:100), anti-Mfn2 (1:200), anti-tubulin (1:200), anti CD150-APC (1:100) or anti-FLAG (1:250) (see Supplementary Table 1) overnight, washed and incubated with AlexaFluor secondary antibodies (Invitrogen) for 1h. Cell nuclei were counterstained with DAPI and mounted with fluorescent mounting media (Vector Labs, Burlingame, CA). Confocal images were acquired with a Zeiss LSM 700 confocal microscope or a Leica DMI 6000B and images were deconvoluted and processed with Leica AF6000 software package.
Gene Reporter Assays
NIH-3T3, Wt or Mfn2−/− MEF cells were plated at 2×104 cells/cm2 in triplicate overnight and transfected with 500ng of pGF-NFAT, pGF-CMV or −950/+22 Mfn2-pGL4 reporter construct, 500ng of cDNA plasmids as indicated and 500ng of either pSV-βGal or pLVX-IRES-mCherry plasmids with Lipofectamine 3000 according to manufacturer’s instructions for 24 or 48h. Cells were lysed in reporter lysis buffer (Promega, Madison, WI) and analyzed for luciferase activity using BrightGlo luciferase (Promega) and detected on a Synergy H2 plate reader (BioTek, Winooski, VT). To visualize βGal activity, cell lysate was incubated in Buffer Z (1mg/mL ONPG, 0.1M phosphate, pH 7.5, 10mM KCl, 1mM βME, 1mM MgSO4) at 37C for 1h. Absorbance values were measured at 405nm and used to normalize for transfection efficiency. In Wt and Prdm16−/− MEFs, gene reporter luciferase values were normalized to mCherry excitation values.
Western Blot
For total cell lysate experiments, MEF cultures were lysed in RIPA buffer, 50mM Tris pH 7.5, 137mM NaCl, 0.1% SDS, 0.5% deoxycholate and protease inhibitors (Roche). For subcellular fractionation studies, cells were scraped, washed in PBS. Cell pellets were lysed in 5x packed cell volume (pcv) Buffer A for 10min on ice and vortexed for 15min in the presence of 1/10 volume 3% NP-40. Plasma membrane lysis was verified by trypan blue staining. Lysate was spun at 15,000xg for 10min at 4C and the cytoplasmic fraction was saved. The remaining nuclear pellet was resuspended in 2.5x pc Buffer C and incubated at 4C for 1h with rotation and spun at 15,000xg for 10min. The nuclear fraction was diluted with 2.5x volume of Nuclear Diluent Buffer and stored at −80C. To achieve even fractionation loading, equivalent percentages of nuclear and cytoplasmic fractions were loaded on each gel. All protein samples were denatured in 4x sample buffer at 95 °C and loaded onto 4–12% Bis-Tris SDS-PAGE gradient gels (Invitrogen). Gels were transferred onto 0.22µm nitrocellulose membrane and stained with Ruby Red (Molecular Probes, Carlsbad, CA) to confirm transfer. Membranes were blocked with 3% non-fat milk or BSA in 0.3%Tween-20/TBS and incubated with anti-Mfn2 (1:200), anti-βGal (1:1000), anti-NFAT1 (1:250), anti-tubulin (1:500), anti-lamin A/C (1:500) and anti-β-actin (1:5000) overnight (see Supplementary Table 1). Membranes were washed, incubated with HRPO-conjugated secondary antibodies and exposed to X-ray film (Denville) after incubation with Super Signal West Fempto ECL reagent (Pierce).
Image Quantification
For mitochondrial length measurements, confocal or deconvoluted z-stacks were collected and projected as a z-project in ImageJ (NIH, Bethesda, MD). Individual mitochondria were manually traced, binned into length categories and expressed as percent of cellular mitochondria. The mean ±SEM number of mitochondria falling into each length category collected from ≥15 fields (30–50 cells) are expressed. For NFAT nuclear localization quantification, confocal or deconvoluted z-stacks were collected and a 1µm in the center of the cell was projected as a z-project in ImageJ. Nuclear boundaries were constructed using DAPI staining. The ratio of staining within the nuclear boundary to total staining was expressed as percent of NFAT signal. The mean ±SEM for ≥10 fields (20–40 cells) are expressed. For immunofluorescence intensity measurements, confocal or deconvoluted z-stacks were collected and projected as a z-project in ImageJ. Thresholds were set based on IgG-stained negative control cells and the integrated density value of each signal per cell was recorded. The mean ±SEM for ≥15 fields (30–50 cells) are expressed.
Statistics
For statistical analysis between two groups, the unpaired Student’s t test was used. When more than two groups were compared, one-way ANOVA was used. Results are expressed as mean ± SEM. The Bonferroni and Dunnett multiple comparison tests were used for post hoc analysis to determine statistical significance between multiple groups. All statistics were calculated using Prism5 (GraphPad, La Jolla, CA) software. Differences among group means were considered significant when the probability value, p, was less than 0.05. Sample size (‘n’) always represents biological replicates. Cochran test was used for exclusion of outliers.
Extended Data
Extended Data Fig. 1. Representative sort gates for isolation of HSCs.

Flow cytometric plots showing the gates used to isolate HSCs (Lin−Sca1+kit+Flt3−CD48−) and CD150hi and CD150lo HSCs.
Extended Data Figure 2. Mitochondrial dynamics in Prdm16−/− MEFs.

(A) Frequency distribution (left) and frequency distribution profile (right) of mitochondrial length in Wt or Prdm16−/− fetal HSCs. Bars, mean±s.e.m.; n≥20 fields of cells from three biological replicates; *P<0.05 within length bins; two-tailed student’s t test within length bins. (B) Mitochondrial morphology in Prdm16−/− MEFs treated for 24hrs with the Drp1 inhibitor, mDivi (30µM), or vehicle is shown for comparison, Mitotracker Red staining, scale bar 20µm). (C) Frequency distribution (left) and frequency distribution profile (right) of mitochondrial length in Wt and Prdm16−/− MEFs treated for 24hrs with the Drp1 inhibitor, mDivi (30µM), or vehicle. Bars, mean±s.e.m.; n≥16 fields from three biological replicates; *P<0.05 within length bins; one-way ANOVA with Bonferroni’s post hoc test within length bins. (D) Fluorescence micrographs showing fusion hybrids of Wt or Prdm16−/− MEFs transduced with baculovirus expressing mitochondria-tagged GFP and RFP prior to PEG-mediated fusion (scale bar 50µm). Fused mitochondria are yellow (arrows), and were only observed in fusions of Wt cells. (E) Western blot (upper, Supplementary Fig. 1 for full scans) and quantification of Western blots for Mfn2 of Wt and Prdm16−/− MEFs. Bars, mean±s.e.m.; n=3 fields biological replicates; *P<0.05; two-tailed student’s t test. (F) Mitochondrial morphology in Wt, Prdm16−/− and Mfn2−/− MEFs visualized by Tomm20 staining (red) (scale bar 20µm). (G) Mitochondrial length in Wt and Prdm16−/− MEFs transduced with EV or Mfn2-IRES-GFP (Mfn2-GFP). Bars, mean±s.e.m.; n≥24 fields from three biological replicates; *P<0.05 compared to Prdm16−/− EV in each length bin; one-way ANOVA with Bonferroni’s post hoc test within length bins.
Extended Data Fig. 3. sPrdm16 Interacts with the Mfn2 Promoter Mfn2 transduction in adult Prdm16+/− HSCs.

(A) Schematic representation of Prdm16 protein domain structure. (B) qPCR probe amplification scheme covering the Mfn2 promoter used in chromatin immunoprecipitation assays. (C) Proximal Mfn2 promoter luciferase gene reporter assay in Wt MEFs transfected 24h prior with flPrdm16, sPrdm16 or control vectors. Bars, mean±s.e.m.; n=3 biological replicates; *P<0.05; one-way ANOVA with Dunnett’s post hoc test. (D) ChIP quantification in Prdm16−/− MEFs transduced with retroviral FLAG-Prdm16 constructs immunoprecipitated using FLAG and TF2D antibodies. TFIID positive (GAPDH) and negative (intergenic Chr 6) control probes are also shown. Quantification was performed after establishment of qPCR standard curves for all probes (see Methods). Bars, mean±s.e.m.; n=3 technical replicates representative tow biological replicates; *P<0.05; one-way ANOVA with Dunnett’s post hoc test. (E) Percent CD45.2 (donor) contribution in PB WBC in CD45.1+CD45.2+ mice reconstituted with 200 transduced (IRES-GFP or Mfn2-IRES-GFP) Prdm16+/− CD45.2+ HSCs and 2×105 CD45.1+ competitor BM cells. Plots, mean±s.e.m.; n=17 recipients pooled from four independent transplants; *P<0.05; two-tailed student’s t test.
Extended Data Fig. 4. Mitochondrial Morphology of BM Populations.
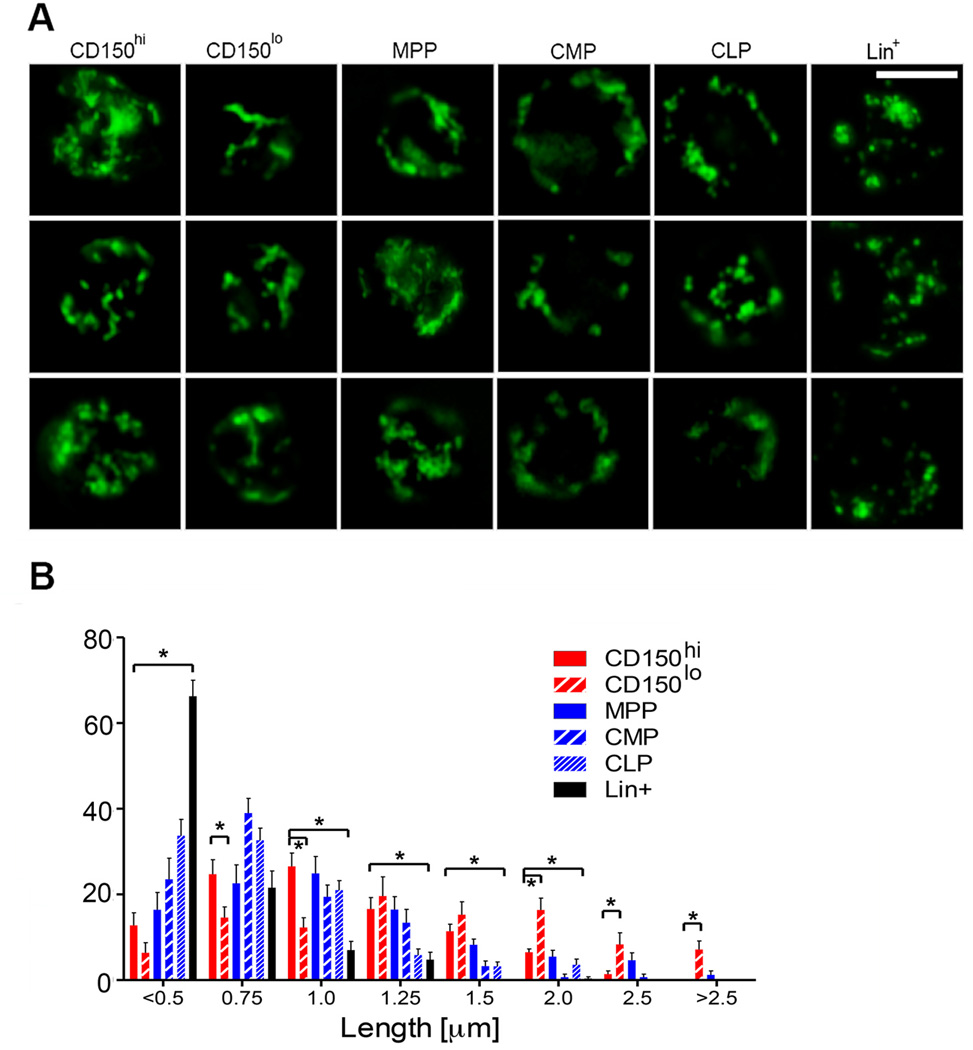
(A) Representative images of mitochondrial morphology in Pham-reporter+ CD150hi and CD150lo HSCs (lin−Sca1+kit+Flt3−CD48−), MPPs (lin−Sca1+kit+CD48+), CMPs (lin−Sca1−kit+), CLPs (lin−Sca1lokitloIL7Ra+Flt3+) and lineage+ cells (Scale bar 5µm) (B) Mitochondrial length frequency distribution in Pham-reporter+ HSCs, progenitors and Lin+ cells Bars, mean±s.e.m.; n≥14 fields from three biological replicates; *P<0.05 within length bins; one-way ANOVA with Bonferroni’s post hoc test within length bins.
Extended Data Fig. 5. Phenotype and function of Mfn2−/− HSCs.
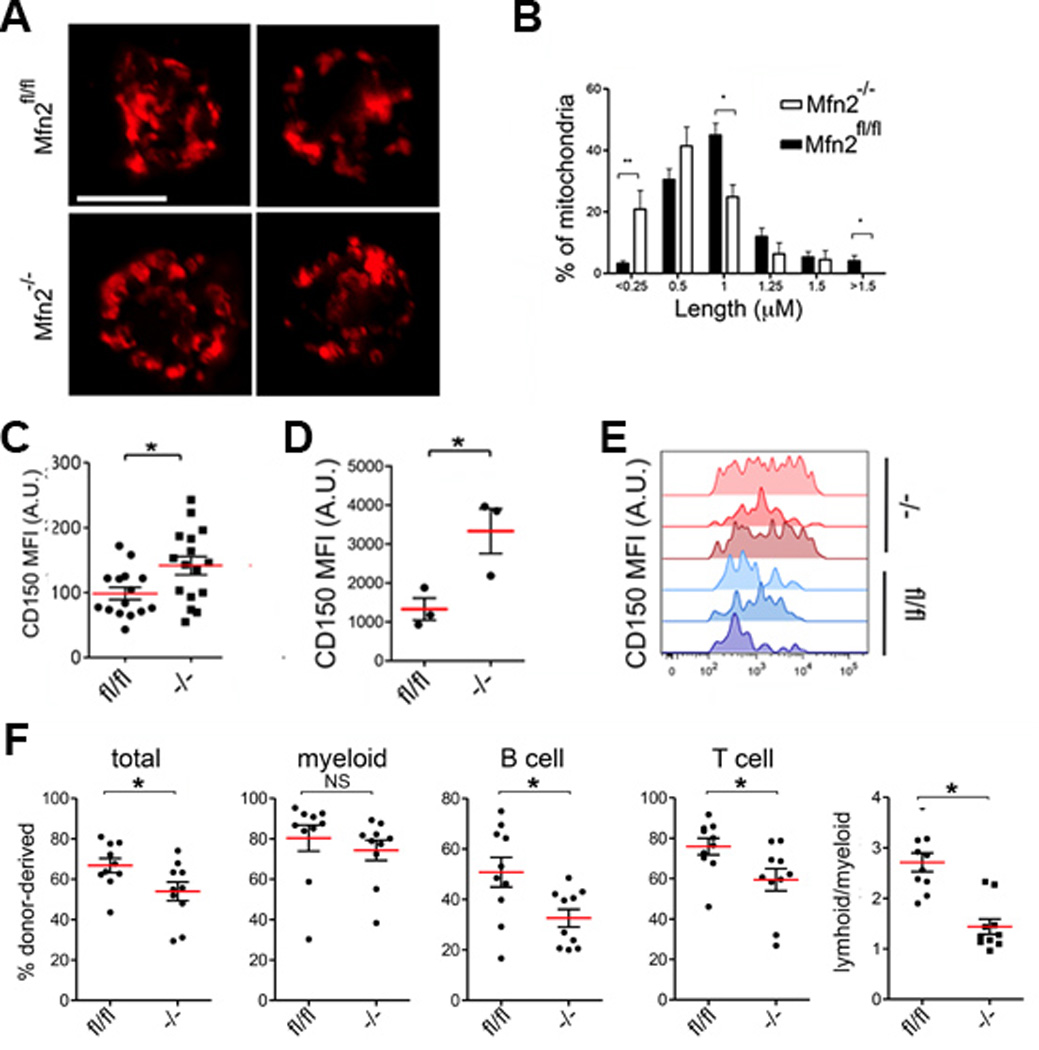
(A) Mitochondrial morphology HSCs from Mfn2fl/fl or Mfn2fl/fl-Vav-Cre mice visualized with immunostaining for Tomm20. (B) Frequency distribution of mitochondrial lengths (right panel, compared to wt in each length bin). Bars, mean±s.e.m.; n≥7 fields from two biological replicates; *P<0.05 within length bins; two-tailed student’s t test within length bins. (C) CD150 surface staining mean fluorescence intensity (MFI) of HSCs (Lin−Sca1+kit+CD48−) in 8–12 week old Mfn2fl/fl or Mfn2fl/fl.Vav-Cre (Mfn2−/−) mice. n=25 biological replicates, *P<0.05, two-tailed student’s t test. (D,E) CD150 MFI of Mfn2fl/fl or Mfn2fl/fl.Vav-Cre (Mfn2−/−) donor HSCs 15 weeks after competitive transplantation. Plots in (D), mean±s.e.m.; n≥15 mice from three biological replicates. (F); *P<0.05; two-tailed student’s t test. (F) Donor (CD45.2) chimerism 15 weeks after competitive transplantation of 2×105 Mfn2fl/fl or Mfn2fl/fl.Vav-Cre (Mfn2−/−) fetal liver cells together with 2×105 CD45.1+ competitor BM cells into CD45.1+CD45.2+ recipients Plots, mean±s.e.m.; n=10 mice pooled from two independent transplants; *P<0.05; two-tailed student’s t test.
Extended Data Fig. 6. Analysis of competitive repopulation experiments and lentiviral vector transduction.

(A) Schematic representation of lentiviral constructs used. (B) Representative example of transduction efficiency of purified HSCs with IRES-GFP and IRES-mCherry vectors 24–48h post-transduction. (C) Schematic representation of transduction and subsequent competitive repopulation experiments. (D) Representative flow cytometric plots of the gates used to analyze recipients of competitive repopulation experiments. The CD45.1/CD45.2 gates were also applied to the myeloid, B and T cell gates to determine donor contribution to individual lineages. In this example, HSCs had been transduced with an IRES-GPF lentiviral vectors (efficiency 80%, see (B)). Although donor repopulation was high in the periphery (upper left panel, 71.8%) no GFP was detected (lower left panel), suggesting silencing of the vectors. (E) Quantification of proviral copy number in donor-derived (CD45.2+) 15 weeks after transduction and competitive transplantation of HSCs. Despite silencing of the vector (see (D)), approximately 1 proviral copy was present per donor HSC-derived cell. n=3 biological replicates; *P<0.05; two-tailed student’s t test. (F) Representative example of transduction efficiency of purified HSCs with IRES-GFP and Mfn2-IRES-GFP lentiviral vectors. (G) Expression of Mfn2 mRNA in CD150hi and CD150lo HSCs 24h post-transduction relative to EV control (left panel) and in donor-derived HSCs, CMPs and lineage+ cells 15 weeks after competitive transplantation of 200 transduced HSCs together with 2×105 CD45.1+ competitor cells (right panel). The data indicate partial silencing of the vector in HSCs, and complete silencing in the progenitors and more mature cells. n=3 biological replicates; *P<0.05; two-way analysis of variance (ANOVA) with Bonferroni’s post hoc test. (H) Donor (CD45.2+) chimerism 15 weeks after transplantation of 200 CD45.2+ HSCs transduced for 24h with SIN LTR constructs pLVX-IRES-GFP or pLVX-Mfn2-IRES-GFP together with 2×105 CD45.1+ competitor fetal liver cells into lethally irradiated CD45.1+CD45.2+ recipients mice. Plots, mean±s.e.m.; n≥19 recipients from four transplantation experiments; *P<0.05; two-tailed student’s t test. (I) Same experiments as in (H), but using the non-SIN LTR vector, pHR. Plots, mean±s.e.m.; n=5 recipients; *P<0.05; two-tailed student’s t test.
Extended Data Fig. 7. NFAT activity in Mfn2−/− and Prdm16−/− MEFs.

(A) Mfn2fl/fl (blue) or Mfn2fl/fl-Vav-Cre (red) HSCs were stained with DCFDA to visualize intracellular ROS levels. Cells were treated with oligomcyin for 15min as a positive control. (B) HSC apoptosis levels were analyzed by surface staining of Annexin V. (C) Intracellular staining for the ER stress response chaperone GRP78 in Mfn2fl/fl or Mfn2fl/fl-Vav-Cre HSCs (red colors) MPPs (green colors) and CMPs (blue colors). Histograms; representative of three biological replicates. (D) Calcium flux trace (left) and baseline Cai2+ (right) in Wt LSK cells transduced with IRES-GFP or Mfn2-IRES-GFP lentiviral vectors (E) and with Wt or Mfn2−/− MEFs stained with Indo1. Bars, mean±s.e.m.; n=3 biological replicates; *P<0.05; two-tailed student’s t test. (F) NFAT1 staining (left, scale bars 5µm) and fraction of nuclear NFAT1 (right, *P<0.05) in HSCs transduced with IRES-GFP or Mfn2-IRES-GFP lentiviral vector. Bars, mean±s.e.m.; n≥10 from two biological replicates; *P<0.05; two-tailed student’s t test. (G) NFAT1 staining (left, scale bars = 20 µm) and fraction of nuclear NFAT1 (right, *P<0.05) in Wt or Mfn2−/− MEFs. Bars, mean±s.d.; n≥20 fields from three independent experiments; *P<0.05; two-tailed student’s t test. (H) NFAT luciferase gene reporter activity in Wt or Mfn2−/− MEFs. Bars, mean±s.d.; n=3 experiments; *P<0.05; two-tailed student’s t test. (I) Mitochondrial morphology after transfection of dominant negative Drp1 into HeLa cells (scale bar 5 µM). Note extreme elongation of mitochondria (arrow), confirming that this vector is functional. (J) NFAT1 staining (left, scale bars 5µm) and fraction of nuclear NFAT1 (right) in HSCs lenitvirally transduced with sPRrdm16 and flPRdm16.. Plots, mean±s.e.m.; n≥14 fields of cells pooled from two biological replicates; *P<0.05; one-way ANOVA with Dunnett’s post hoc test. (K) NFAT luciferase gene reporter activity in Wt and in Prdm16−/− MEFs transfected with Mfn2, flPrdm16 or sPrdm16 constructs Bars, mean±s.d.; n=3 experiments; *P<0.05; one-way ANOVA with Dunnett’s post hoc test.
Extended Data Fig. 8. Effect of VIVIT on Wt HSCs.

(A) Expression of lymphoid (Sox4 and Il7r) and myeloid/platelet (Vwf and Fli1) genes in HSCs treated with VIVIT or DMSO for 24h. n=3 biological replicates; *P<0.05; student’s T test. (B) Lymphoid/myeloid ratio of CD45.2+ HSCs cultured for 4 days in DMSO and 500nM VIVIT, transplanted with 2×105 CD45.1+.CD45.2+ competitor BM cells into CD45.1+ recipients and analyzed 15 weeks post-transplant for lymphoid/myeloid ratio of donor compartment. Plots, mean±s.e.m.; n=5–9 recipients from two transplant experiments; *P<0.05; two-tailed student’s t test. (C) Donor chimerism analysis 15 weeks after transplantation of CD45.2+ HSCs transduced with IRES-GFP or VIVIT-GFP with 2×105 CD45.1+.CD45.2+ competitor BM cells into CD45.1+ recipients Plots, mean±s.e.m.; n=9–12 recipients from two independent transplant experiments; *P<0.05; two-tailed student’s t test. Lower panel shows lymphoid and myeloid donor contribution in each individual recipient.
Extended Data Table 1. Phenotypic analysis of the hematopoietic system in Mfn2fl/fl-Vav-Cre mice.
Analysis of 8-10 week-old Mfn2fl/fl or Mfn2fl/fl-Vav-Cre mice for (A) frequency of LSK cells and HSCs, (B) peripheral blood counts, (C) BM progenitor populations (MPPs (lin−Sca1+kit+CD48+), CMPs (lin−Sca1−kit+), CLPs (lin−Sca1lokitloIL7Ra+Flt3+), MEPs (Lin−Kit+Sca1−CD16/32lowCD34−)), (D) myeloid (Gr1+/Mac1+), T (CD3+) and B (CD19+) cells in the spleen, (E) myeloid (Gr1+/Mac1+), T (CD3+) and B (CD19+) cells in peripheral blood mononuclear cells, and (F) thymocyte subpopulations (DP: CD4+CD8+, DN: CD4−CD8−, DN1: CD4−CD8−CD44+CD25−, DN2: CD4−CD8−CD44+CD25+, DN3: CD4−CD8−CD44−CD25+, DN4: CD4−CD8−CD44−CD25−) Tables, mean±s.e.m.; n=3 mice; *P<0.05; two-tailed student’s t test.
| A | ||
|---|---|---|
| Hematopoietic Stem Cell Compartments | ||
| LSK | LSK CD48- Flt3- CD150+ |
|
| Mfn2 f/f | ||
| 0.277% | 0.053% | |
| 0.319% | 0.082% | |
| 0.346% | 0.058% | |
| Mean | 0.314% ± 0.03 | 0.064% ± 0.02* |
| Mfn2 f/fVav-Cre | ||
| 0.213% | 0.021% | |
| 0.575% | 0.047% | |
| 0.220% | 0.039% | |
| Mean | 0.336% ± 0.20 | 0.036% ± 0.01* |
| B | ||
|---|---|---|
| Peripheral blood counts | ||
| WT | KO | |
| N= | 8 | 4 |
| WBC | 7.3 ± 2.6 | 8.7 ± 1.7 |
| Neutrophil% | 15.4 ± 9.2 | 15.4 ± 12.0 |
| Lymphocytes% | 73.0 ± 12.8 | 78.2 ± 12.6 |
| Monocytes% | 6.2 ± 2.8 | 5.9 ± 3.2 |
| Eosinophiles% | 0.4 ± 0.3 | 1.3 ± 1.4 |
| Basophiles% | 0.1 ± 0.1 | 0.4 ± 0.4 |
| NRBC% | ± | ± |
| Hematocrit | 45.1 ± 4.2 | 45.3 ± 6.7 |
| RBC | 8.7 ± 0.7 | 8.9 ± 1.2 |
| HB | 12.3 ± 1.2 | 12.8 ± 1.6 |
| MCV | 51.5 ± 1.7 | 50.7 ± 1.8 |
| MCH | 14.1 ± 0.5 | 14.4 ± 0.5 |
| MCHC | 27.3 ± 0.8 | 28.4 ± 0.7 |
| RDW | 15.2 ± 1.7 | 15.5 ± 1.4 |
| RSD | 7.9 ± 0.9 | 7.9 ± 0.9 |
| RETIC# | 47.4 ± 29.9 | 49.1 ± 42.0 |
| RETIC% | 0.6 ± 0.4 | 0.6 ± 0.5 |
| Platelet | 642.8 ± 146.4 | 653.0 ± 66.7 |
| C | |||||
|---|---|---|---|---|---|
| Bone Marrow Progenitor Compartments | |||||
| MPP | GMP | CMP | MEP | CLP | |
| Mfn2 f/f | |||||
| 0.00272% | 0.00184% | 0.00281% | 0.00049% | ||
| 0.00841% | 0.00469% | 0.00958% | 0.00067% | ||
| 0.00149% | 0.00164% | 0.00135% | 0.00409% | ||
| Mean | 0.314% ± 0.03 | 0.0042% ± 4e-5 | 0.0027% ± 2e-5 | 0.0046% ± 4e-5 | 0.00175% ± 2e-5 |
| Mfn2 f/f Vav-Cre | |||||
| 0.00321% | 0.00224% | 0.00369% | 0.00108% | ||
| 0.00844% | 0.00550% | 0.00589% | 0.00041% | ||
| 0.00136% | 0.00110% | 0.00142% | 0.00916% | ||
| Mean | 0.336% ± 0.20 | 0.0043% ± 4e-5 | 0.0029% ± 2e-5 | 0.0037% ± 2e-5 | 0.00355% ± 5e-5 |
| D | |||
|---|---|---|---|
| Splenocyte Lineages | |||
| Mac + or GR1 + | CD3+ | CD19+ | |
| Mfn2 f/f | |||
| 6.840% | 17.60000% | 59.70000% | |
| 9.910% | 27.80000% | 63.70000% | |
| 8.375% | 22.700% | 61.700% | |
| Mean | 8.37% ± 0.02 | 22.70% ± 0.05 | 61.70% ± 0.02 |
| Mfn2 f/f Vav-Cre | |||
| 11.800% | 13.79000% | 62.59000% | |
| 9.070% | 27.50000% | 63.20000% | |
| 10.435% | 20.645% | 62.895% | |
| Mean | 10.43% ± 0.01 | 20.65% ± 0.07 | 62.90% ± 0.01 |
| E | |||
|---|---|---|---|
| Peripheral Blood Lineages | |||
| Mac1+ or GR1 + | CD3+ | CD19+ | |
| Mfn2 f/f | |||
| 26.300% | 9.75280% | 54.75000% | |
| 23.100% | 26.800% | 42.135% | |
| 19.900% | 43.84800% | 29.51910% | |
| Mean | 23.1% ± 0.03 | 26.8% ± 17 | 42.1% ± 13 |
| Mfn2 f/f Vav-Cre | |||
| 15.200% | 15.63765% | 63.22500% | |
| 17.900% | 35.944% | 41.143% | |
| 20.600% | 56.24940% | 19.06100% | |
| Mean | 17.9% ± 0.02 | 35.9% ± 20 | 62.9% ± 22 |
| F | ||||||||
|---|---|---|---|---|---|---|---|---|
| Thymus Lineages | ||||||||
| CD4 | CD8 | DP | DN | DN1 | DN2 | DN3 | DN4 | |
| Mfn2 f/f | ||||||||
| 10.50% | 2.95% | 81.60% | 2.11% | 0.03% | 0.10% | 0.54% | 0.19% | |
| 11.10% | 2.10% | 81.30% | 3.10% | 0.04% | 0.15% | 0.81% | 0.17% | |
| 5.95% | 3.01% | 83.20% | 1.48% | 0.03% | 0.10% | 0.41% | 0.06% | |
| Mean | 9.2% ± 2.8 | 2.7% ± 0.5 | 82.7% ± 1.0 | 2.2% ± 0.8 | 0.33% ± 0.006 | 0.117% ± 0.03 | 0.587% ± 0.20 | 0.14% ± 0.07 |
| Mfn2 f/f Vav-Cre | ||||||||
| 9.42% | 2.56% | 83.00% | 1.86% | 0.03% | 0.13% | 0.54% | 0.20% | |
| 9.25% | 2.55% | 84.50% | 2.09% | 0.06% | 0.13% | 0.81% | 0.20% | |
| 11.30% | 3.44% | 80% | 2.74% | 0.03% | 0.09% | 0.57% | 0.21% | |
| Mean | 10.0% ± 1.1 | 2.8% ± 0.5 | 82.5% ± 2.3 | 2.2% ± 0.5 | 0.040% ± 0.02 | 0.117% ± 0.02 | 0.64% ± 0.15 | 0.203% ± 0.01 |
Extended Data Table 2.
Statistical analysis of limiting dilution transplantation studies
| total reconstitution | lymphoid reconstitution | myeloid reconstitution | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | Lower | Estimate | Upper | Group | Lower | Estimate | Upper | Group | Lower | Estimate | Upper |
| KO | 108.1 | 55.6 | 28.72 | KO | 407.6 | 153.2 | 57.8 | KO | 35.8 | 20.23 | 11.5 |
| WT | 30.3 | 17 | 9.59 | WT | 69.8 | 38.2 | 21 | WT | 18.5 | 8.96 | 4.5 |
| Chisq | DF | P.value | Chisq | DF | P.value | Chisq | DF | P.value | |||
| 7.64 | 1 | 0.00571 | 6.71 | 1 | 0.00957 | 3.1 | 1 | 0.0785 | |||
Supplementary Material
Acknowledgments
The pHR-IRES-GFP SIN vector was a generous gift from Dr. Remi Creusot (Columbia University). Mfn2−/− MEFs were a kind gift of Dr. David Chan (Caltech). We thank Dr. Siu-Hong Ho (CCTI Flow Core) and Dr. Eric A. Schon (Columbia University) for assistance and discussion. This work was supported by grant NIH RO1 CA167286 and RO1 AG029262 (HWS), the Druckenmiller Fellowship from the New York Stem Cell Foundation (LLL), the Ruth L. Kirschstein F31 CA196045 fellowship (DJC), the NIH National Center for Research Resources (NCRR) award 1S10RR027050-01 and the NIH Office of the Director (OD) award 1S10OD020056-01.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Authors Contributions
LLL designed and performed most experiments, contributed to the concept, and co-wrote the manuscript with HWS. MJA generated the data in Fig. 3, 4F. MM cloned the DN Drp1 construct and verified its function (Extended Data Fig. 7D). DJC generated the Mfn2 promoter construct (Extended Data Fig. 3C). HWS provided concept and guidance, and co-wrote with LLL.
The authors have no conflicts of interest to declare.
References
- 1.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simsek T, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takubo K, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12:49–61. doi: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguilo F, et al. Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood. 2011;117:5057–5066. doi: 10.1182/blood-2010-08-300145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nature cell biology. 2010;12:999–1006. doi: 10.1038/ncb2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 7.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors regulators of calcium signalling. Nature reviews. Molecular cell biology. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 8.Muller-Sieburg CE, Cho RH, Thoman M, Adkins B, Sieburg HB. Deterministic regulation of hematopoietic stem cell self-renewal and differentiation. Blood. 2002;100:1302–1309. [PubMed] [Google Scholar]
- 9.Sanjuan-Pla A, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 2013;502:232–236. doi: 10.1038/nature12495. [DOI] [PubMed] [Google Scholar]
- 10.Muller-Sieburg CE, Sieburg HB. Clonal diversity of the stem cell compartment. Current opinion in hematology. 2006;13:243–248. doi: 10.1097/01.moh.0000231421.00407.65. [DOI] [PubMed] [Google Scholar]
- 11.Dykstra B, et al. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell. 2007;1:218–229. doi: 10.1016/j.stem.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 12.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annual review of genetics. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 13.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka A, Youle RJ. A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Molecular cell. 2008;29:409–410. doi: 10.1016/j.molcel.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Shing DC, et al. Overexpression of sPRDM16 coupled with loss of p53 induces myeloid leukemias in mice. J Clin Invest. 2007;117:3696–3707. doi: 10.1172/JCI32390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida M, et al. Aberrant expression of the MEL1S gene identified in association with hypomethylation in adult T-cell leukemia cells. Blood. 2004;103:2753–2760. doi: 10.1182/blood-2003-07-2482. [DOI] [PubMed] [Google Scholar]
- 17.Sorianello E, et al. The promoter activity of human Mfn2 depends on Sp1 in vascular smooth muscle cells. Cardiovasc Res. 2012;94:38–47. doi: 10.1093/cvr/cvs006. [DOI] [PubMed] [Google Scholar]
- 18.Beerman I, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A. 2010;107:5465–5470. doi: 10.1073/pnas.1000834107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weksberg DC, Chambers SM, Boles NC, Goodell MA. CD150- side population cells represent a functionally distinct population of long-term hematopoietic stem cells. Blood. 2008;111:2444–2451. doi: 10.1182/blood-2007-09-115006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pham AH, McCaffery JM, Chan DC. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis. 2012;50:833–843. doi: 10.1002/dvg.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 22.Szilvassy SJ, Humphries RK, Lansdorp PM, Eaves AC, Eaves CJ. Quantitative assay for totipotent reconstituting hematopoietic stem cells by a competitive repopulation strategy. Proc Natl Acad Sci U S A. 1990;87:8736–8740. doi: 10.1073/pnas.87.22.8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngoh GA, Papanicolaou KN, Walsh K. Loss of mitofusin 2 promotes endoplasmic reticulum stress. The Journal of biological chemistry. 2012;287:20321–20332. doi: 10.1074/jbc.M112.359174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Rao A, Hogan PG. Interaction of calcineurin with substrates and targeting proteins. Trends in cell biology. 2011;21:91–103. doi: 10.1016/j.tcb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller MR, et al. Requirement for balanced Ca/NFAT signaling in hematopoietic and embryonic development. Proc Natl Acad Sci U S A. 2009;106:7034–7039. doi: 10.1073/pnas.0813296106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu H, Peisley A, Graef IA, Crabtree GR. NFAT signaling and the invention of vertebrates. Trends in cell biology. 2007;17:251–260. doi: 10.1016/j.tcb.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 27.Aramburu J, et al. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- 28.Benz C, et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell. 2012;10:273–283. doi: 10.1016/j.stem.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. The Journal of experimental medicine. 2011;208:2691–2703. doi: 10.1084/jem.20111490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen H, et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. The Journal of cell biology. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
References for Methods
- 31.Zambrowicz BP, et al. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci U S A. 2003 Nov 25;100:14109. doi: 10.1073/pnas.2336103100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pham AH, McCaffery JM, Chan DC. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis. 2012 Nov;50:833. doi: 10.1002/dvg.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lakso M, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996 Jun 11;93:5860. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007 Aug 10;130:548. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 35.Shimshek DR, et al. Codon-improved Cre recombinase (iCre) expression in the mouse. Genesis. 2002 Jan;32:19. doi: 10.1002/gene.10023. [DOI] [PubMed] [Google Scholar]
- 36.Ausubel FM. Current Protocols in Molecular Biology. Brooklyn, N.Y: Greene Pub. Associates ; J. Wiley and Sons. Inc; 1987. [Google Scholar]
- 37.Benz C, et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell. 2012 Mar 2;10:273–283. doi: 10.1016/j.stem.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 38.Chen H, et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003 Jan 20;160:189. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorianello E, et al. The promoter activity of human Mfn2 depends on Sp1 in vascular smooth muscle cells. Cardiovasc Res. 2012 Apr 1;94:38. doi: 10.1093/cvr/cvs006. [DOI] [PubMed] [Google Scholar]
- 40.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985 Mar 25;260 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.