

Abstract
Recent studies have implicated endogenously produced H2S in the angiogenic process. On one hand, pharmacological inhibition and silencing of the enzymes involved in H2S synthesis attenuate the angiogenic properties of endothelial cells, including proliferation, migration and tube-like structure network formation. On the other hand, enhanced production of H2S by substrate supplementation or over-expression of H2S-producing enzymes leads to enhanced angiogenic responses in cultured endothelial cells. Importantly, H2S up-regulates expression of the key angiogenic factor vascular endothelial growth factor (VEGF) and contributes to the angiogenic signaling in response to VEGF. The signaling pathways mediating H2S-induced angiogenesis include mitogen-activated protein kinases, phosphoinositide-3 kinase, nitric oxide/cGMP-regulated cascades and ATP-sensitive potassium channels. Endogenously produced H2S has also been shown to facilitate neovascularization in prototypical model systems in vivo, and to contribute to wound healing, post-ischemic angiogenesis in the heart and other tissues, as well as in tumor angiogenesis. Targeting of H2S synthesizing enzymes might offer novel therapeutic opportunities for angiogenesis-related diseases.
Graphical abstract
Introduction
Angiogenesis is a physiological process through which new blood vessels are formed from the existing vasculature [1, 2]. In healthy organisms, the rate at which new blood vessels are formed varies with biological age and differs between tissues [3]. Early in life during embryonic development, organisms exhibit rapid neovascularization [4]. In contrast, during adulthood the process is tightly controlled; endothelial cells in mature vessels cells are quiescent and have a long-half life [5]. Angiogenesis in adults is observed only in the context of a limited number of physiological responses and in selected tissues. New vessel growth is restricted to the endometrium during the menstrual cycle, as an adaptive response to exercise in skeletal muscle and in wound healing [3, 6]. Dysregulation of angiogenesis is associated with many pathophysiological conditions and diseases [3, 7]. On one hand, excessive or abnormal new blood vessel growth is seen in cancer, psoriasis, arthritis, endometriosis, inflammatory bowel disease and diabetic retinopathy [3, 7, 8]. On the other hand, impaired angiogenesis or vessel regression has been linked to neurodegenerative diseases, hair loss and preeclampsia [7, 9]. Given the wide array of conditions associated with aberrant angiogenesis, there has been a considerable interest in its physiological regulatory processes and in the opportunities for pharmacological modulation. Work in this field resulted in several drug approvals, mainly for malignancies and ocular diseases [10, 11]. Further understanding of the basic mechanisms that regulate angiogenesis are expected to offer new targets with translational potential.
Hydrogen sulfide production
Hydrogen sulfide (H2S), is the newest member of the gasotransmitter family [12–14]. Once viewed exclusively as an environmental pollutant and toxicant, H2S is now recognized as an endogenous biological mediator with important roles in homeostasis, physiology and disease [15, 16]. H2S is ubiquitously present in mammalian cells and tissues and can be generated both through enzymatic and non-enzymatic pathways [16]. Three enzymes are known to be involved in catalytic reactions that yield H2S [17, 18]. Two of them, cystathionine beta synthase (CBS) [19] and cystathionine gamma lyase (CSE) [20] operate in the transsulfuration pathway, the conversion of methionine to cysteine, and require pyridoxal-5′-phosphate (PLP) as a cofactor. The third enzyme is 3-mercaptopyruvate sulfurtransferase (3-MST) and converts 3-mercaptopyruvate (3-MP) to H2S and pyruvate [21, 22]. Although 3MST itself does not need PLP, generation of 3-MP by cysteine aminotransferase (CAT) is dependent on PLP for its activity.
H2S producing enzymes exhibit widespread tissue distribution [16]. CBS is viewed as the predominant H2S-producing enzyme in the nervous system, while CSE is believed to be the main enzyme in the cardiovascular system [12, 14]. With research in the field advancing and experimental tools improving, CBS was also shown to be expressed in vascular tissues, as well as the heart [23–25]. CSE is mainly a cytosolic enzyme that can translocate to the mitochondria in response to injurious stimuli its presence in liver mitochondria increases after hypoxia [26]. CBS is found in the mitochondria of normal and tumor cells [27, 28]. 3MST appears in both the cytosolic and mitochondrial fractions of cells and is expressed in all tissues studied so far [18]. 3MST is the least studied enzyme, most likely because no pharmacological inhibitors are available [29]. All three H2S-producing enzymes have been demonstrated to be present in the endothelium [30–32]. Thus, they are well-positioned to influence the angiogenic properties of endothelial cells and modify responses to angiogenic factors. Moreover, H2S - similar to the other two gasotransmitters NO and CO - is a freely membrane permeable, diffusible molecule, not requiring transporter of carrier molecules to gain intracellular access [33]; therefore, H2S produced by neighboring cells can also act in a paracrine manner to affect angiogenesis.
In this brief review, we will focus on the role of endogenously produced H2S in the angiogenic process. H2S donors have been shown in a variety of models to promote angiogenic responses; readers interested in the effects of exogenously supplied H2S are referred to recent reviews [16, 34].
Hypoxia and H2S
Hypoxia is one of the most potent stimuli known to drive angiogenesis [35]. A drop in cellular oxygen tension stabilizes hypoxia inducible factor-1α (HIF-1α) through inhibition of prolyl-hydroxylase activity; lack of HIF-1α hydroxylation prevents recognition and ubiquitination by VHL E3 ubiquitin ligase that labels HIF-1α for degradation[35, 36]. HIF-1α can then dimerize with HIF-1β, and bind to HIF-responsive elements in the promoter region of hypoxia-regulated genes [37]; this leads to the expression of several angiogenesis-related factors, including VEGF [38].
Several observations have linked H2S bioavailability to changes in ambient oxygen concentration [39]. H2S reacts with O2, and in spite of the fact that this reaction occurs at slow rates, O2 can decrease H2S concentrations in tissues; highly oxygenated tissues might, thus, contain less H2S compared to tissues with lower O2 tensions [40–42]. Moreover, enzymatic pathways that utilize molecular O2, oxidize H2S in the mitochondria, converting it to thiosulfate and sulfate for subsequent excretion and limit its biological actions [17, 42]. Ischemia/hypoxia and the resulting intracellular acidosis have also been proposed to increase the release of H2S from acid labile pools (iron-sulfur clusters) [43]. Moreover, O2 has indirect effects on H2S; heme-containing proteins react with H2S at different rates depending on their redox status [42].
Limited information is available about the effect of hypoxia on the expression of H2S-generating enzymes in the cardiovascular system. HIF binding elements are present in the CBS promoter; hypoxia increased CBS levels in a HIF-dependent manner in cultured glioblastoma cells [44]. These results were extrapolated in vivo; rats exposed to hypobaric hypoxic exhibited increased CBS expression and activity in the cerebellum and cortex. Moreover, it was recently shown that chronic hypoxia increased tissue H2S levels and CSE activity in a mouse model of hind limb ischemia [45]. Increases in tissue H2S levels, albeit smaller, were also noted in CSE KO. In contrast, intermittent hypoxia decreased endothelial CSE expression in small mesenteric arteries [46].
Additional links between HIF-1 and H2S have been established. Flannigan and colleagues demonstrated that loss of CSE-derived H2S production decreased HIF-1α stability, indicating that endogenously generated H2S is important for HIF-1 signaling [47]. Indeed, administration of diallyl disulfide, an H2S-releasing molecule, stabilized HIF-1α expression and up-regulated hypoxia-responsive genes [47]. Similarly, in rat brain capillary endothelial cells incubation with a H2S donor, promoted an increase in HIF-1α and VEGF mRNA and protein levels, and enhanced HIF-1 DNA binding [48]. In cells treated with a H2S donor, angiogenic responses were linked to miR-640 down regulation and mediated by a VEGFR2/mTOR/HIF-1α pathway [49]. However, it should be kept in mind that under certain conditions H2S donors have been shown to suppress HIF-1 [50]. Interestingly, Kevil's group reported that H2S enhanced HIF-1α activation in hypoxic conditions and this correlated with a significantly higher stimulation of proliferation compared to normoxic cells [51]. The same group reported that expression of HIF-1α in skeletal muscle tissue was elevated under ischemia in WT but not in CSE KO mice [51]. The above observations establish multiple interactions between H2S and HIF-regulated pathways and propose that a freed forward cycle between HIF and H2S might exist in angiogenesis (Fig.1).
Figure 1. Proposed interactions between VEGF and H2S.

VEGF binding to VEGFR2 causes enhanced H2S production presumably by activating CSE in a calcium-dependent manner. Nucleophilic attack of the disulfide bond between Cys 1045-1024 by H2S leads to a disulfide reduction and boosts VEGFR2 tyrosine kinase activity. H2S generated by CBS increases the stability and transcriptional activity of Sp1, enhancing VEGFR2 transcription. H2S causes an increase in HIF-1α levels, DNA binding and transcriptional activity. VEGFR2 (vascular endothelial growth factor receptor 2); VEGF (vascular endothelial growth factor); CSE (cystathionine-γ lyase); CBS (cystathionine-β synthase); HIF (hypoxia inducible factor); HRE (hypoxia response element); Sp1 (specificity protein 1); H2S (hydrogen sulfide)
Endothelial cell metabolism, bioenergetics and H2S
Mammalian cells under normal conditions generate ATP in an oxygen-dependent manner. We recently showed that endogenously produced H2S by CBS or 3MST can be used as an inorganic energy source that complements and balances the bioenergetic role of Krebs cycle-derived electron donors [27, 52]. Interestingly, the normally cytosolic enzyme CSE can translocate from the cytosol to mitochondria during hypoxia, where L-cysteine levels are 3 times higher than those of the cytosol, under stress-related conditions [26]. Moreover, CBS in the mitochondrial matrix is degraded by Lon protease in normoxic conditions [28]; lowering of O2 tension prevents CBS degradation, allowing CBS accumulation. H2S could be used to maintain electron flow and to sustain cellular bioenergetics during hypoxia (but not anoxia as oxygen has to be present to serve as the final elector acceptor) to meet the demands of rapidly dividing endothelial cells during angiogenesis. In comparison to other respiratory chain substrates sulfide's electron yield is low: two molecules of sulfide are needed to provide a pair of electrons [53]. Moreover, it is costly in terms of oxygen, due to the involvement of the sulfide oxidation unit; sulfide oxidation needs three times more oxygen for the same electron transfer through complexes III and IV [54]. Therefore, the energy yield per oxygen atom consumed for sulfide is low compared to NADH or FADH2. However, sulfide is still an attractive alternative fuel as H2S is freely diffusible across membranes and does not require activation or conversion.
Endothelial cells exhibit considerable plasticity and can emerge from prolonged periods of quiescence to assume a rapidly proliferating phenotype during angiogenesis [5]. Although oxidative phosphorylation is a markedly more efficient mode of ATP generation than glycolysis, it has been recently proposed that endothelial cells heavily rely on glycolysis to cover their ATP needs [55]. Cultured endothelial cells have glycolytic rates that exceed glucose oxidation rates and fatty acid oxidation flux [56]. Silencing of the glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) decreased endothelial cell proliferation and migration, while over expression of PFKFB3 accelerated the angiogenic properties of endothelial cells [57]. Moreover, knockdown or pharmacologic inhibition of PFKFB3 reduced vessel branching and outgrowth in vivo [55, 58]. Since endothelial cells appear to depend on glycolysis during neovascularization, it has been proposed that inhibition of PFKFB3 will only transiently and partially reduce glycolysis in vivo, inhibiting pathological angiogenesis, without affecting blood vessels throughout the body [58]. In line with its angiogenic effects and the link between glycolysis and angiogenesis, endogenously produced H2S was shown to stimulate the activity of another glycolytic enzyme, glyceraldehyde 3-phosphate dehydrogenase (GAPDH); GAPDH undergoes S-sulfhydration on the active site cysteine 150 and stimulates glycolytic flux [59, 60]. Although evidence for the contrary (inhibition of GAPDH by H2S/persulfidation) has been presented [61], most studies have shown that H2S enhances the efficiency of glycolysis in cells [62, 63]. Future studies should aim to quantify the contribution of glycolysis enhancement to the angiogenic actions of H2S.
Effect of endogenous H2S on angiogenic properties of endothelial cells in vitro
Blood vessels can grow via sprouting, splitting of pre-existing structures through intussusception or by incorporation of circulating precursor cells [5, 64, 65]. During sprouting angiogenesis, pro-angiogenic growth factors stimulate the motility of endothelial cells. The endothelial cells at the leading edge of vascular sprouts are termed tip cells [66] and are characterized by long filopodia and high migratory activity [5, 67, 68]. Tip cells integrate environmental cues, guide the growth of the budding vessel and make new connections to secure functional integration of into a vascular network. Tip cells are followed by endothelial stalk cells that exhibit high proliferation rates, establish adherent/tight junctions to provide stability for the new sprout and forming its lumen [5]. Up to now, there are no reports investigating the expression pattern of H2S-producing enzymes in tip and stalk cells, or the contribution, if any, of H2S in tip cell selection.
When studying the angiogenic potential of pharmacological agents and endogenous molecules in vitro, typically the effects on endothelial proliferation, migration and network formation are being examined, as these are viewed as “discrete” phases of the angiogenic response. Most angiogenic factors with the notable exception of transforming growth factor-β1, are endothelial mitogens [69]. Several lines of evidence support the growth-promoting properties of endogenously generated H2S in endothelial cells. Supplementation of L-cysteine, the CBS/CSE substrate for H2S generation, increases endothelial cell growth in mouse microvascular endothelial cells [70]. The growth promoting effect of L-cysteine is abolished by propargyl glycine or CSE knockdown, suggesting that the effects of L-cysteine are CSE-dependent. Moreover, treatment of human endothelial cells with the dual CSE/CBS inhibitor aminooxyacetic acid [71] reduces their proliferation rate [31].
In line with the observations that the H2S substrate L-cysteine exerts mitogenic effects in endothelial cells, over expression of CSE in human umbilical vein endothelial cells resulted in an increase in 5-bromo-2'-deoxyuridine incorporation [72]. As has been described for other agents that enhance endothelial cell proliferation, H2S was reported to inhibit smooth muscle cell growth [73]. Similarly to what was seen with L-cysteine, endothelial cells grown in the presence of 3-MP, the 3MST substrate, exhibit higher growth rates [74]. On the other hand, silencing of 3-MST via short hairpin RNA (shRNA) lentiviral particles, resulted in a significant inhibition of ECs proliferation rate [74]. The above findings taken together provide strong evidence that H2S is an endogenous endothelial cell mitogen and indicate that the growth-promoting properties of H2S are independent of the enzymatic source of the gasotransmitter.
Endogenously generated H2S has also been demonstrated to drive endothelial cell migration in Boyden chamber-type and in scratch wound healing assays. 3-MP enhances endothelial migration and wound healing of cultured endothelial cells providing evidence for the pro-angiogenic role of 3MST. In addition, L-cysteine promotes endothelial cell migration in a propargylglycine (PAG)-reversible manner, suggesting CSE involvement in this response [70, 74]. In scratch wound healing assays, HUVEC in which CBS expression was knocked down exhibited decreased migration and wound closure rates [31]. Moreover, pharmacological inhibition of H2S production (with PAG or β-cyanoalanine) reduced VEGF-induced migration in both human umbilical vein and tumor-derived endothelial cells [30, 75]. Similarly, knockdown experiments utilizing a small interfering RNA (siRNA) against CSE, resulted in an attenuated basal migratory response of endothelial cells, as well as reduced VEGF-stimulated motility [30, 70]. Hypoxia-stimulated migration was also found to be dependent on CSE as silencing its expression reduce migration [76].
The Matrigel® assay and the ring angiogenesis assay represent two of the most widespread models used to evaluate the ability of endothelial cells to form tube-like structures and networks, in vitro. Saha and colleagues showed that cells in which CBS expression was reduced by siRNA, formed less extensive networks on Matrigel® [31]. Aortic rings from CSE KO mice and aortic rings from rats in which CSE was silenced generated fewer tube-like structures when embedded in fibrin or collagen gels in vitro [70, 72]. In addition, adenovirus-mediated CSE gene transfer resulted in increased basal and VEGF-stimulated vascular outgrowths in vitro [70]. Similarly to what was observed with proliferation and migration, incubation of cells with 3-MP [74] or L-cysteine [70] increased the neovessel growth in the ring angiogenesis assay in vitro.
Based on the above, H2S derived from CSE, CBS or 3MST promote the expressing of an angiogenic phenotype by endothelial cells. The overlap in function of these three enzymes might be due to diffusible nature of their product (H2S), along with their similar pattern of sub-cellular distribution. Reports are begging to emerge on the role of H2S on progenitor cell function. t CSE inhibition or silencing reduces tube length of endothelial progenitor cells (EPC) on Matrigel® and inhibits EPC adhesion to the matrix [77]. Moreover, the number of circulating and homed EPCs were significantly reduced in CSE KO mice following an ischemic challenge, proving further evidence for a role of H2S in EPC biology [45]. Clearly, additional studies are needed to fully elucidate the impact of H2S on EPC behavior.
Interplay between H2S and angiogenic factors
VEGF plays a crucial role in angiogenic responses, both under normal and pathophysiological conditions [3, 8, 9]. As already mentioned in the previous section, VEGF-stimulated angiogenic properties of endothelial cells can be inhibited by the CSE inhibitor PAG or CSE silencing [30, 70, 78]. This observation suggests that incubation of endothelial cells with VEGF promotes H2S synthesis. Indeed, incubation of human EC with VEGF leads to increase output of H2S [30]. Although the mechanism through which this occurs has not been studied in detail, it was proposed to be mediated by a calcium/calmodulin-dependent activation of CSE [70] (Fig.1). However, the reverse effect has also been observed. Adenovirus-mediated triple gene transfer of CBS, CSE and 3MST was found to increase VEGF expression and to diminish the levels of the anti-angiogenic factor endostatin [79]. These findings (i.e. enhanced VEGF production after H2S stimulation) have also been reproduced using various classes of H2S donors [80–82]. The above data suggest that H2S is a downstream effector of VEGF signaling, but can also exist upstream of VEGF, depending on the conditions/model studied.
VEGF binding to VEGFR2 causes it to homodimerize, leading to transautophosphorylation of a series of tyrosine residues, including Tyr951, Tyr996, Tyr1054, Tyr1059, Tyr1175, and Tyr1214 [83]. Tao and colleagues identified a disulfide bond between Cys1045 and Cys1024 of VEGFR2 that alters the active conformation of the receptor, inhibiting its activity [76] (Fig.2). Nucleophilic attack of the disulfide bond by H2S leads to a disulfide reduction and boosts VEGFR2 tyrosine kinase activity [76] (Fig.1). Although the authors reported that CSE co-localized at sub-cellular areas at the membrane of vascular endothelial cells with VEGFR2, reduction of the Cys1045-Cys1024 disulfide bond has only been demonstrated with H2S donors.
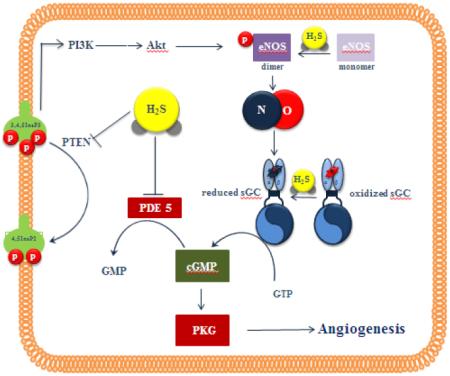
Figure 2. Interactions of H2S and NO in angiogenesis.

NO activates sGC to increase cGMP levels, while H2S both facilitates cGMP production (by activating eNOS and keeping the sGC in a reduced NO-responsive state) and prevents cGMP breakdown by inhibiting PDE. This allows cGMP to reach the threshold levels required to trigger PKG angiogenic signaling. H2S also promotes eNOS dimerization through eNOS persulfidation. PI3K (phosphoinositide 3-kinase); Akt (protein kinase B); PTEN (phosphatase and tensin homolog); 3,4,5 InsP3 (phosphatidylinositol-3,4,5-trisphosphate); 4,5 InsP2 (phosphatidylinositol-4,5-bisphosphate); eNOS (endothelial nitric oxide synthase); sGC (soluble guanylate cyclase); cGMP (cyclic guanosine monophosphate); GMP (guanosine monophosphate); PDE (phosphodiesterase); PKG (cGMP-dependent protein kinase), NO (nitric oxide); H2S (hydrogen sulfide)
In a recent study, silencing CBS in endothelial cells compromised the phenotypic and signaling responses to VEGF that were due to decreased transcription of VEGFR2 and neuropilin (NRP)-1 (Fig.1). Silencing of CBS resulted in transcriptional down-regulation of VEGFR2 and NRP-1 that was mediated by a decreased stability of the transcription factor specificity protein 1 (Sp1) [31]. Sp1 reduced stability was triggered by lack of sulfhydration on Cys68 and Cys755 that occurred in cells in which CBS had been silenced. H2S donor administration in CBS-silenced ECs restored the levels of Sp1 and its binding to the VEGFR2 promoter, as well as functional VEGF responses (proliferation, and migration phenotypes). Thus, maintaining endogenous H2S levels is crucial for preserving VEGF-responsiveness.
To summarize the interactions between H2S and VEGF evidence has been presented to show that i) H2S increases after acute exposure of EC to VEGF, ii) prolonged exposure to increased H2S concentrations up regulates VEGF expression iii) H2S aids VEGFR2 in expressing increased activity by assuming a more activity conformation and maintains its expression levels.
Considerably less information is available regarding the interaction between H2S and other growth factors. Basic fibroblast growth factor (bFGF) is another factor that is crucial for new blood vessels to form [2]. Inhibition of H2S by PAG did not reduce FGF-stimulated migration, suggesting that the actions of this angiogenic factor are H2S-independent [30, 70]. Although H2S might not be required for bFGF angiogenic signaling, H2S up regulates bFGF levels in vivo. bFGF along with VEGF and interleukin-16 were increased following hind limb ischemia in wild-type mice; this response was reduced in CSE KO animals [45]. Recent reports showed that H2S reduced angiopoietin-2 production but none of them studied this effect in the context of angiogenesis [84, 85]. The potential role of H2S in angiogenic signaling of other key growth factors, including the angiopoietins and Notch remains to be tested in future studies.
H2S signaling in angiogenesis: downstream mediators
Several studies have investigated the cellular signaling pathways, which regulate the proangiogenic effect of H2S. Exogenously added H2S promotes the phosphorylation of Akt, ERK1/2 and p38 [30, 72, 86]. The contribution of endogenous H2S in the activation of MAPK cascades was demonstrated using VEGF; stimulation of cells with VEGF promoted ERK1/2 and p38 phosphorylation that could be blocked by pharmacological inhibitors or silencing of CSE [30]. ATP-sensitive potassium channels (KATP), a major mediator of the effects of H2S, were shown to lie upstream of p38 [30]. In line with these findings, direct KATP, channel openers also promote angiogenesis [30, 87].
Treatment of tissues or cells with inhibitors of H2S synthesis, CSE gene deletion or silencing results in decreased cGMP levels [88, 89], while over expression of CSE increases cGMP [88]. The ability of H2S to up regulate cGMP levels is, at least in part, due to the direct inhibition of PDE activity by H2S [70, 88] (Fig.2). Moreover, H2S converts sGC to its ferrous, NO-responsive form and further increases cGMP [90]. Furthermore, H2S production also activates the phosphoinositide 3-kinase (PI3K)/serine/threonine kinase (Akt) pathway, leading to endothelial nitric oxide synthase (eNOS) phosphorylation and eNOS activation [70, 72, 91]. PI3-K/Akt activation might result from the ability of H2S to inhibit the lipid phosphatase and tensin homolog (PTEN)[92]. H2S has also been shown to increase eNOS activity by promoting its dimerization [93] (Fig.2). The increase in NO production, along with the inhibition of cGMP breakdown allows for significant elevations in intracellular cGMP in vascular endothelium [70]. As expected, endothelial cells incubated with L-cysteine exhibit enhanced VASP phosphorylation on Ser239, proving proof that endogenously generated H2S activates the cGMP/PKG axis [70]. cGMP accumulation, in turn, activates protein kinase G (PKG) stimulating the angiogenic properties of endothelial cells [70] (Fig.2).
The proangiogenic effect of 3-MP/3MST/H2S pathway is also associated with the activation of Akt and PKG. 3-MP stimulates Akt phosphorylation on its activating site (Ser473). Similarly, 3-MST silencing results in lower levels of Akt and vasodilator-stimulated phosphoprotein phosphorylation [74]. Based on the above observations, the concerted action of NO and H2S is believed to be crucial for the angiogenic response. Inhibition of NO blocks the proliferation, migration and tube-structure formation [70, 72], while inhibiting H2S abolishes the NO-stimulated angiogenesis [70, 72].
Pharmacological agents that promote angiogenesis through endogenous H2S
In this section we will refer to pharmacologically active compounds that modify angiogenic responses by modulating endogenous H2S. Zofenopril, an SH-containing inhibitor of the angiotensin converting enzyme, was recently demonstrated to up regulate CSE in the cardiovascular system without affecting CBS and 3MST levels [23]. In a follow up study, Terzuoli et al reported that the zofenoprilat-induced angiogenic properties of endothelial cells were attenuated by CSE inhibition or silencing [94]. Similarly, PAG administration reduced neovascularization in the Matrigel® plug assay in vivo [94]. The molecular mechanisms underlying zofenopril-induced angiogenesis were dependent on Akt, eNOS and ERK1/2 cascades, in line with what is known about mediators of H2S-triggered angiogenic signaling.
S-propargyl-l-cysteine (SPRC) is a H2S donor that also increases CSE levels [95]. Recent findings show that SPRC increased HUVECs proliferation and in vitro angiogenesis in a PAG-inhibitable manner [96]. Results from in vivo studies using SPRC provided additional evidence for its CSE-mediated effects, as PAG inhibited angiogenesis in an Matrigel® plug model [96]. Since other H2S donors have been shown to increase CSE expression under certain conditions [97], it is possible that endogenous H2S contributes to the angiogenic effects of some H2S donors. It should also be mentioned that NO donors require the presence of endogenous H2S to exert their angiogenic effects. CSE silencing resulted in the blockade of NO-induced proliferation, migration and in in vitro aortic ring angiogenesis [70]. Based on what is known about the mechanism of action of H2S, we predict that additional pharmacological agents that target cGMP-regulated pathways and promote angiogenesis (e.g. PDE inhibitors, sGC activators/stimulators, natriuretic peptides) could interact with endogenously produced H2S to regulate new blood vessel formation.
Effect of endogenous H2S on angiogenic properties of endothelial cells in vivo
CSE KO mice do not display a major cardiovascular phenotype, apart from a mild hypertension [98]. Although extensively used in the literature for almost a decade, there are no reports describing developmental angiogenic defects or alterations in vascular structure or density in the organs of these animals. However, as discussed above, vessels isolated from CSE KO animals show reduced responsiveness to VEGF in in vitro assays. On the other hand, very limited information on the 3MST KO mice is available, with no studies evaluating their cardiovascular phenotype [99]. In contrast, CBS KO mice exhibit a severe phenotype that results in reduced lifespan [100]. CBS heterozygotes are used in most studies; loss of just one CBS allele results in mild hypehomocysteinemia causing endothelial dysfunction [101] and vascular complications [102]. In a hind limb ischemia model, Bosch-Marcé and colleagues observed that arteriogenesis/angiogenesis was reduced in CBS+/− animals; angiogenesis was inversely related to plasma levels of homocysteine [103]. The observed impairment, was not dependent on reduced expression of VEGF or impaired phosphorylation of its receptor Flk-1, but was rather attributed to impaired Akt phosphorylation associated with hyperhomocysteinemia.
The first in vivo model in which the role of CSE-derived H2S was investigated was the chicken chorioallantoic membrane (CAM). Treatment of CAMs with the H2S synthesis inhibitors PAG and β-Cyano-L-alanine (BCA) lead to reduced vessel length and branching [30] (Fig.3). The effect of PAG and BCA was reversed following addition of NaHS. Although the CAM is a widely used model and in most cases accurately predicts the angiogenic potential of the tested agents, it is a developmental model of angiogenesis in a non-mammalian organism. To further investigate the contribution of endogenous H2S in new blood vessel formation and to extend the in vitro findings that H2S is required for VEGF-stimulated proliferation, migration and network formation, we employed the Matrigel® plug assay. In these experiments reduced Matrigel® plugs were implanted into mice that received water for injection or PAG for 15days. Some of the xenografted plugs contained VEGF to drive the angiogenic response, while others contained vehicle. At the end of the experiment plugs were removed and angiogenesis was assessed by measuring hemoglobin content, as an index of vascularization (Fig.4). We observed that VEGF stimulates vessel ingrowth in the Matrigel® plugs and that PAG administration reduced both basal and VEGF-stimulated angiogenesis. In agreement with the angiogenic properties of CSE-derived H2S, 3-MP administration to enhance production of H2S by 3MST lead to an increase in neovascularization of Matrigel ®plugs implanted in mice [74].
Figure 3.
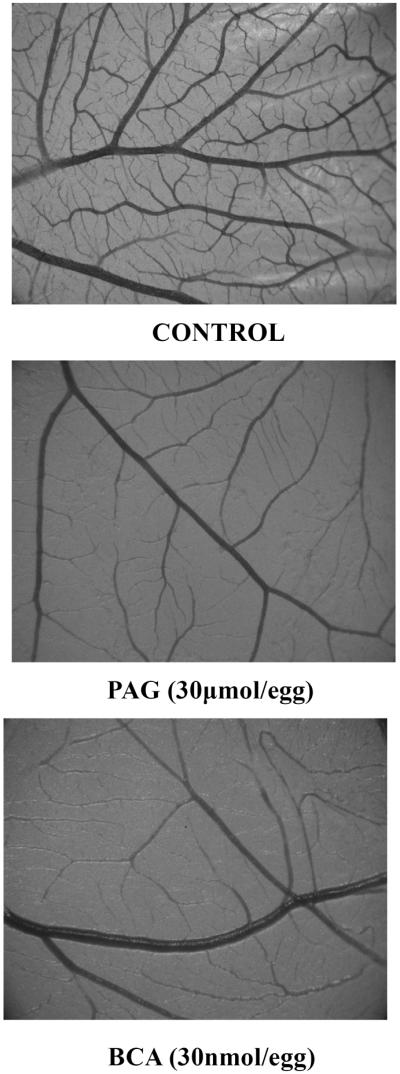
Representative photomicrographs of chorioallantoic membranes incubated with vehicle (control), PAG or BCA. A clear reduction in the vascular network is seen when CSE is inhibited (magnification × 2,5).
Figure 4. PAG inhibits VEGF-stimulated angiogenesis in vivo.

C57BL/6 male mice 10–12 weeks old mice were randomized in 4 groups (4 mice/group): 1. CONTROL: 100μl water for injection (WFI) intra-peritoneally (i.p.) for 15 days, 2. PAG: 100μl of 50mg/kg in WFI PAG i.p. for 15 days, 3. VEGF:100μl WFI i.p. for 15 days and 4.PAG+VEGF: 100μl of 50mg/kg in WFI PAG i.p. for 15 days. The first day, mice were anesthetized by a 1–2 min exposure to isoflurane and 400μl 50:50 Matrigel® Matrix: 1xPBS with or without VEGF(100ng/ml) was infused subcutaneously. Mice were sacrificed 16 days after Matrigel® injection. (A) Macroscopic appearance and vessel infiltration in Matrigel plugs (B) Hemoglobin content in μg/mg *p<0.05 vs Control, ***p<0.01 vs Control, #p<0.05 vs VEGF. (C) Immunostaining for the expression of endothelial cell marker CD31 staining in 5μm sections of Matrigel® plugs. 40X magnification.
The role of endogenous H2S has also been studied in a number of pathophysiological conditions associated with angiogenesis, including wound healing, critical limb ischemia and cancer [30, 45, 70]. Burn wounds in CSE KO mice healed at a slower rate compared to those of wild-type control mice [30]. Moreover, administration of 3-MP to boost the 3MST-derived H2S facilitated wound closure in a burn wound model in rats [74]. Wound skin capillary densities were reduced in db/db animals which received the CSE inhibitor PAG; these animals exhibited reduced wound-closure rates [77]. In the only study to address the role of H2S in retinal neovascularization, mice were maintained in 70% oxygen from postnatal day P7 to P12 and room air from P12 to P17. It was observed that this treatment resulted in up regulation of CSE [104]. Moreover, CSE KO exhibited a 19% reduction in retinal neovascularization and a 31% increase in vaso-obliteration compared to heterozygotes during oxygen-induced retinopathy. Similarly, inhibition of CSE/CBS by aminooxyacetic acid (AOAA) decreased retinal retinal neovascularization, while accelerating vaso-obliteration. These results indicated that the CSE is critically involved in the development of pathological retinal neovascularization during retinal ischemia.
In a femoral artery ligation study, arteriogenesis was inhibited in the absence of CSE. Mature vessel density, angiogenic indices and blood flow were significantly reduced in CSE KO mice compared with WT mice with femoral artery ligation [45]; administration of diallyl disulfide restored these parameters. In line with this notion, patients with critical limb ischemia exhibit reduced levels of all three H2S generating enzymes and attenuated H2S bioavailability accompanied by increased oxidative stress [105]. Mechanistically, H2S-stimulated angiogenesis is heavily dependent on the presence of eNOS/NO in wound healing, ischemia-driven angiogenesis and in Matrigel® plug neovessel growth [45, 51, 70].
Tumor angiogenesis is an area with significant translational implications. It is, thus, not surprising that production of H2S has been studied by several laboratories in the context of cancer [27, 106–111]. Endogenous H2S levels were found to be increased in VHL-deficient clear cell renal cell carcinoma (ccRCC) cell lines. Inhibition of endogenous H2S production in the CAM decreased vascularization of ccRCC xenografts [112]. We have previously demonstrated that CBS, but not CSE or 3MST, is over expressed in human colon adenocarcinomas and colon cancer-derived epithelial cell lines (HCT116, HT- 29, LoVo), resulting in increased H2S production [27]. CBS silencing in HCT116 resulted in reduced tumor growth. Moreover, pharmacological inhibition of CSE/CBS by AOAA reduced the growth of patient-derived tumor xenotransplants and HCT116 tumors. Tumors from AOAA-treated mice also exhibited reduced vessel density [27], suggesting a significant role of endogenous H2S in tumor angiogenesis. CBS expression, was also found increased primary epithelial ovarian cancer and ovarian cancer cell lines; CBS down regulation via siRNA or inhibition of its activity by AOAA impairs proliferation and viability of cancer cells in vitro [106]. Silencing CBS in a cisplatin-resistant orthotopic ovarian cancer model reduced nodule formation, sensitized tumor cells to cisplatin and inhibited angiogenesis [106]. Taken together, the studies in colon and ovarian cancer cells clearly establish CBS-produced H2S as a novel, endogenous, regulator of tumor progression and angiogenesis [113].
Conclusions and perspective
The evidence presented above shows that H2S derived from CSE, CBS or 3-MST, stimulates the angiogenic properties of endothelial cells. MAPK pathways and NO are the main mediators of the H2S-induced angiogenesis. In addition, endogenously produced H2S contributes to angiogenesis during development in vivo, as well as angiogenic responses that accompany wound healing and tumor growth, pointing towards translational opportunities. In spite of the compelling evidence in the literature for the role of endogenously generated H2S in new blood vessel formation, a multitude of questions regarding the regulation and biology of H2S in the context of angiogenesis remain unanswered. In growing, maturing and quiescent vessels, what cell type and which H2S-producing enzyme is the major source of H2S? Does H2S contribute to inflammation-associated angiogenesis? How are H2S signaling pathways integrated in vivo in the different settings where angiogenesis occurs? How does H2S impact on the endothelial metabolome and how does that contribute to its angiogenic properties? Can H2S be used to stimulate therapeutic angiogenesis? In addition to cancer, what other diseases related to angiogenesis can benefit from inhibition of H2S production? These questions and many more, some of which are mentioned in the sections above, await answers. Gaining more knowledge on H2S in angiogenesis will not only satisfy scientific curiosity, but will only increase the odds of successful translation of basic science findings into clinical applications.
Acknowledgments
Funding This work has been funded by the Hellenic State Scholarship Foundation IKY-Siemens research projects of excellence (11/3056 to AP and AK), by the National Institutes of Health (R01CA175803 to CS) and co-financed by the European Union (European Social Fund – ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) - Research Funding Program: Aristeia 2011 (1436) to AP
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- [2].Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- [3].Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- [4].Flamme I, Frölich T, Risau W. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J Cell Physiol. 1997;173:206–210. doi: 10.1002/(SICI)1097-4652(199711)173:2<206::AID-JCP22>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- [5].Potente M, Gerhardt H, Carmeliet P. Basic and Therapeutic Aspects of Angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- [6].Olfert IM, Baum O, Hellsten Y, Egginton S. Advances and challenges in skeletal muscle angiogenesis. Am J Physiol Heart Circ Physiol. 2016;310:H326–H336. doi: 10.1152/ajpheart.00635.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- [8].Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- [9].Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- [10].Sennino B, McDonald DM. Controlling escape from angiogenesis inhibitors. Nat Rev Cancer. 2012;12:699–709. doi: 10.1038/nrc3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang K, Zhang L, Weinreb RN. Ophthalmic drug discovery: novel targets and mechanisms for retinal diseases and glaucoma. Nat Rev Drug Discov. 2012;11:541–559. doi: 10.1038/nrd3745. [DOI] [PubMed] [Google Scholar]
- [12].Li L, Hsu A, Moore PK. Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation — a tale of three gases! Pharmacol Ther. 2009;123:386–400. doi: 10.1016/j.pharmthera.2009.05.005. [DOI] [PubMed] [Google Scholar]
- [13].Mustafa AK, Gadalla MM, Snyder SH. Signaling by Gasotransmitters. Sci Signal. 2009;2:re2–re2. doi: 10.1126/scisignal.268re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- [15].Kimura H. The physiological role of hydrogen sulfide and beyond. Nitric Oxide. 2014;41:4–10. doi: 10.1016/j.niox.2014.01.002. [DOI] [PubMed] [Google Scholar]
- [16].Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- [17].Kabil O, Banerjee R. Enzymology of H(2)S Biogenesis, Decay and Signaling. Antioxid Redox Signal. 2014;20:770–782. doi: 10.1089/ars.2013.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid Redox Signal. 2014;20:783–793. doi: 10.1089/ars.2013.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Miles EW, Kraus JP. Cystathionine β-Synthase: Structure, Function, Regulation, and Location of Homocystinuria-causing Mutations. J Biol Chem. 2004;279:29871–29874. doi: 10.1074/jbc.R400005200. [DOI] [PubMed] [Google Scholar]
- [20].Pan LL, Liu XH, Gong QH, Yang HB, Zhu YZ. Role of Cystathionine γ-Lyase/Hydrogen Sulfide Pathway in Cardiovascular Disease: A Novel Therapeutic Strategy? Antioxid Redox Signal. 2012;17:106–118. doi: 10.1089/ars.2011.4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nagahara N. Regulation of Mercaptopyruvate Sulfurtransferase Activity Via Intrasubunit and Intersubunit Redox-Sensing Switches. Antioxid Redox Signal. 2012;19:1792–1802. doi: 10.1089/ars.2012.5031. [DOI] [PubMed] [Google Scholar]
- [22].Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, Kimura H. 3-Mercaptopyruvate Sulfurtransferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid Redox Signal. 2009;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- [23].Bucci M, Vellecco V, Cantalupo A, Brancaleone V, Zhou Z, Evangelista S, Calderone V, Papapetropoulos A, Cirino G. Hydrogen sulfide accounts for the peripheral vascular effects of zofenopril independently of ACE inhibition. Cardiovasc Res. 2014;102:138–147. doi: 10.1093/cvr/cvu026. [DOI] [PubMed] [Google Scholar]
- [24].Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G, Gojon G, Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ. H(2)S Protects Against Pressure Overload Induced Heart Failure via Upregulation of Endothelial Nitric Oxide Synthase (eNOS) Circulation. 2013;127:1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Polhemus DJ, Calvert JW, Butler J, Lefer DJ. The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica (Cairo) 2014;2014:768607. doi: 10.1155/2014/768607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Hydrogen sulfide (H(2)S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci U S A. 2012;109:2943–2948. doi: 10.1073/pnas.1115634109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Szabo C, Coletta C, Chao C, Módis K, Szczesny B, Papapetropoulos A, Hellmich MR. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci U S A. 2013;110:12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Teng H, Wu B, Zhao K, Yang G, Wu L, Wang R. Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proc Natl Acad Sci U S A. 2013;110:12679–12684. doi: 10.1073/pnas.1308487110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Papapetropoulos A, Whiteman M, Cirino G. Pharmacological tools for hydrogen sulphide research: a brief, introductory guide for beginners. British Journal of Pharmacology. 2015;172:1633–1637. doi: 10.1111/bph.12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R, Szabo C. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Saha S, Chakraborty PK, Xiong X, Dwivedi SKD, Mustafi SB, Leigh NR, Ramchandran R, Mukherjee P, Bhattacharya R. Cystathionine β-synthase regulates endothelial function via protein S-sulfhydration. FASEB J. 2016;30:441–456. doi: 10.1096/fj.15-278648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, Kimura H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- [33].Mathai JC, Missner A, Kügler P, Saparov SM, Zeidel ML, Lee JK, Pohl P. No facilitator required for membrane transport of hydrogen sulfide. Proc Natl Acad Sci U S A. 2009;106:16633–16638. doi: 10.1073/pnas.0902952106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Szabó C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. Br J Pharmacol. 2011;164:853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rey S, Semenza GL. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res. 2010;86:236–242. doi: 10.1093/cvr/cvq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- [37].Ke Q, Costa M. Hypoxia-Inducible Factor-1 (HIF-1) Mol Pharmacol. 2006;70:1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- [38].Liu Y, Cox SR, Morita T, Kourembanas S. Hypoxia Regulates Vascular Endothelial Growth Factor Gene Expression in Endothelial Cells: Identification of a 5′ Enhancer. Circ Res. 1995;77:638–643. doi: 10.1161/01.res.77.3.638. [DOI] [PubMed] [Google Scholar]
- [39].Kolluru GK, Prasai PK, Kaskas AM, Letchuman V, Pattillo CB. Oxygen tension, H2S, and NO bioavailability: is there an interaction? J Appl Physiol. 2016;120:263–270. doi: 10.1152/japplphysiol.00365.2015. [DOI] [PubMed] [Google Scholar]
- [40].Olson KR, Dombkowski RA, Russell MJ, Doellman MM, Head SK, Whitfield NL, Madden JA. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J Exp Biol. 2006;209:4011–4023. doi: 10.1242/jeb.02480. [DOI] [PubMed] [Google Scholar]
- [41].Li Q, Lancaster JR., Jr Chemical foundations of hydrogen sulfide biology. Nitric Oxide. 2013;35:21–34. doi: 10.1016/j.niox.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Stein A, Bailey SM. Redox biology of hydrogen sulfide: Implications for physiology, pathophysiology, and pharmacology. Redox Biol. 2013;1:32–39. doi: 10.1016/j.redox.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yuan S, Kevil C. Nitric Oxide and Hydrogen Sulfide Regulation of Ischemic Vascular Remodeling. Microcirculation. 2016;23:134–145. doi: 10.1111/micc.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Takano N, Peng Y-J, Kumar GK, Luo W, Hu H, Shimoda LA, Suematsu M, Prabhakar NR, Semenza GL. Hypoxia-Inducible Factors Regulate Human and Rat Cystathionine β-Synthase Gene Expression. Biochem J. 2014;458:203–211. doi: 10.1042/BJ20131350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kolluru GK, Bir SC, Yuan S, Shen X, Pardue S, Wang R, Kevil CG. Cystathionine γ-lyase regulates arteriogenesis through NO-dependent monocyte recruitment. Cardiovasc Res. 2015;107:590–600. doi: 10.1093/cvr/cvv198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jackson-Weaver O, Paredes DA, Bosc LVG, Walker BR, Kanagy NL. Intermittent Hypoxia in Rats Increases Myogenic Tone Through Loss of Hydrogen Sulfide Activation of Large-Conductance Ca(2+)-Activated Potassium Channels. Circ Res. 2011;108:1439–1447. doi: 10.1161/CIRCRESAHA.110.228999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Flannigan KL, Agbor TA, Motta J-P, Ferraz JGP, Wang R, Buret AG, Wallace JL. Proresolution effects of hydrogen sulfide during colitis are mediated through hypoxia-inducible factor-1α. FASEB J. 2015;29:1591–1602. doi: 10.1096/fj.14-266015. [DOI] [PubMed] [Google Scholar]
- [48].Liu X, Pan L, Zhuo Y, Gong Q, Rose P, Zhu Y. Hypoxia-Inducible Factor-1α Is Involved in the Pro-angiogenic Effect of Hydrogen Sulfide under Hypoxic Stress. Biol Pharmac Bull. 2010;33:1550–1554. doi: 10.1248/bpb.33.1550. [DOI] [PubMed] [Google Scholar]
- [49].Zhou Y, Li X-H, Zhang C-C, Wang M-J, Xue W-L, Wu D-D, Ma F-F, Li W-W, Tao B-B, Zhu Y-C. Hydrogen sulfide promotes angiogenesis by downregulating miR-640 via the VEGFR2/mTOR pathway. Am J Physiol Cell Physiol. 2016;310:C305–C317. doi: 10.1152/ajpcell.00230.2015. [DOI] [PubMed] [Google Scholar]
- [50].Wu B, Teng H, Zhang L, Li H, Li J, Wang L, Li H. Interaction of Hydrogen Sulfide with Oxygen Sensing under Hypoxia. Oxid Med Cell Longev. 2015;2015:9. doi: 10.1155/2015/758678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bir SC, Kolluru GK, McCarthy P, Shen X, Pardue S, Pattillo CB, Kevil CG. Hydrogen Sulfide Stimulates Ischemic Vascular Remodeling Through Nitric Oxide Synthase and Nitrite Reduction Activity Regulating Hypoxia-Inducible Factor-1α and Vascular Endothelial Growth Factor-Dependent Angiogenesis. J Am Heart Assoc. 2012;1:e004093. doi: 10.1161/JAHA.112.004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Modis K, Coletta C, Erdelyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27:601–611. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- [53].Szabo C, Ransy C, Módis K, Andriamihaja M, Murghes B, Coletta C, Olah G, Yanagi K, Bouillaud F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol. 2014;171:2099–2122. doi: 10.1111/bph.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Abou-Hamdan A, Guedouari-Bounihi H, Lenoir V, Andriamihaja M, Blachier F. F. Bouillaud Chapter Twelve - Oxidation of H2S in Mammalian Cells and Mitochondria. In: Enrique C, Lester P, editors. Methods in Enzymology. Academic Press; 2015. pp. 201–228. [DOI] [PubMed] [Google Scholar]
- [55].De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong Brian W., Cantelmo Anna R., Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, Phng L-K, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins Russel T., Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven Paul P., Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, DeBerardinis Ralph J., Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M, Carmeliet P. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- [56].Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial Cell Metabolism in Normal and Diseased Vasculature. Circ Res. 2015;116:1231–1244. doi: 10.1161/CIRCRESAHA.116.302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Xu Y, An X, Guo X, Habtetsion TG, Wang Y, Xu X, Kandala S, Li Q, Li H, Zhang C, Caldwell RB, Fulton DJ, Su Y, Hoda MN, Zhou G, Wu C, Huo Y. Endothelial PFKFB3 Plays a Critical Role in Angiogenesis. Arterioscler Thromb Vasc Biol. 2014;34:1231–1239. doi: 10.1161/ATVBAHA.113.303041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schoors S, De Bock K, Cantelmo Anna R., Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong Brian W., Quaegebeur A, Goveia J, Bifari F, Wang X, Blanco R, Tembuyser B, Cornelissen I, Bouché A, Vinckier S, Diaz-Moralli S, Gerhardt H, Telang S, Cascante M, Chesney J, Dewerchin M, Carmeliet P. Partial and Transient Reduction of Glycolysis by PFKFB3 Blockade Reduces Pathological Angiogenesis. Cell Metabol. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- [59].Gao X-H, Krokowski D, Guan B-J, Bederman I, Majumder M, Parisien M, Diatchenko L, Kabil O, Willard B, Banerjee R, Wang B, Bebek G, Evans CR, Fox PL, Gerson SL, Hoppel CL, Liu M, Arvan P, Hatzoglou M. Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. eLife. 2015;4:e10067. doi: 10.7554/eLife.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jarosz AP, Wei W, Gauld JW, Auld J, Özcan F, Aslan M, Mutus B. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is inactivated by S-sulfuration in vitro. Free Rad Biol Med. 2015;89:512–521. doi: 10.1016/j.freeradbiomed.2015.09.007. [DOI] [PubMed] [Google Scholar]
- [62].Liang M, Jin S, Wu D-D, Wang M-J, Zhu Y-C. Hydrogen sulfide improves glucose metabolism and prevents hypertrophy in cardiomyocytes. Nitric Oxide. 2015;46:114–122. doi: 10.1016/j.niox.2014.12.007. [DOI] [PubMed] [Google Scholar]
- [63].Sanokawa-Akakura R, Ostrakhovitch EA, Akakura S, Goodwin S, Tabibzadeh S. A H(2)S-Nampt Dependent Energetic Circuit Is Critical to Survival and Cytoprotection from Damage in Cancer Cells. PLoS One. 2014;9:e108537. doi: 10.1371/journal.pone.0108537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Fang S, Salven P. Stem cells in tumor angiogenesis. J Cell Mol Cardiol. 50:290–295. doi: 10.1016/j.yjmcc.2010.10.024. [DOI] [PubMed] [Google Scholar]
- [65].Makanya AN, Hlushchuk R, Djonov VG. Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis. 2009;12:113–123. doi: 10.1007/s10456-009-9129-5. [DOI] [PubMed] [Google Scholar]
- [66].Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. The Journal of Cell Biology. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Phng LK, Gerhardt H. Angiogenesis: A Team Effort Coordinated by Notch. Dev Cell. 16:196–208. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- [69].Papapetropoulos A, Desai KM, Rudic RD, Mayer B, Zhang R, Ruiz-Torres MP, García-Cardeña G, Madri JA, Sessa WC. Nitric oxide synthase inhibitors attenuate transforming-growth-factor-beta 1-stimulated capillary organization in vitro. Am J Pathol. 1997;150:1835–1844. [PMC free article] [PubMed] [Google Scholar]
- [70].Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Modis K, Panopoulos P, Asimakopoulou A, Gero D, Sharina I, Martin E, Szabo C. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci U S A. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, Giannis A, Szabo C, Spyroulias GA, Papapetropoulos A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE) Br J Pharmacol. 2013;169:922–932. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Altaany Z, Yang G, Wang R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J Cell Mol Med. 2013;17:879–888. doi: 10.1111/jcmm.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Yang G, Wu L, Wang R. Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 2006 doi: 10.1096/fj.05-4712fje. [DOI] [PubMed] [Google Scholar]
- [74].Coletta C, Módis K, Szczesny B, Brunyánszki A, Oláh G, Rios ECS, Yanagi K, Ahmad A, Papapetropoulos A, Szabo C. Regulation of Vascular Tone, Angiogenesis and Cellular Bioenergetics by the 3-Mercaptopyruvate Sulfurtransferase/H(2)S Pathway: Functional Impairment by Hyperglycemia and Restoration by dl-α-Lipoic Acid. Mol Med. 2015;21:1–14. doi: 10.2119/molmed.2015.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Pupo E, Fiorio Pla A, Avanzato D, Moccia F, Avelino Cruz J-E, Tanzi F, Merlino A, Mancardi D, Munaron L. Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Rad Biol Med. 2011;51:1765–1773. doi: 10.1016/j.freeradbiomed.2011.08.007. [DOI] [PubMed] [Google Scholar]
- [76].Tao B-B, Liu S-Y, Zhang C-C, Fu W, Cai W-J, Wang Y, Shen Q, Wang M-J, Chen Y, Zhang L-J, Zhu Y-Z, Zhu Y-C. VEGFR2 Functions As an H(2)S-Targeting Receptor Protein Kinase with Its Novel Cys1045–Cys1024 Disulfide Bond Serving As a Specific Molecular Switch for Hydrogen Sulfide Actions in Vascular Endothelial Cells. Antioxid Redox Signal. 2013;19:448–464. doi: 10.1089/ars.2012.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Liu F, Chen D-D, Sun X, Xie H-H, Yuan H, Jia W-P, Chen AF. Hydrogen Sulfide Improves Wound Healing via Restoration of Endothelial Progenitor Cell Functions and Activation of Angiopoietin-1 in Type 2 Diabetes. Diabetes. 2014;63:1763–1778. doi: 10.2337/db13-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Potenza DM, Guerra G, Avanzato D, Poletto V, Pareek S, Guido D, Gallanti A, Rosti V, Munaron L, Tanzi F, Moccia F. Hydrogen sulphide triggers VEGF-induced intracellular Ca2+ signals in human endothelial cells but not in their immature progenitors. Cell Calcium. 2014;56:225–234. doi: 10.1016/j.ceca.2014.07.010. [DOI] [PubMed] [Google Scholar]
- [79].Sen U, Sathnur PB, Kundu S, Givvimani S, Coley DM, Mishra PK, Qipshidze N, Tyagi N, Metreveli N, Tyagi SC. Increased endogenous H(2)S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am J Physiol Cell Physiol. 2012;303:C41–C51. doi: 10.1152/ajpcell.00398.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Holwerda KM, Burke SD, Faas MM, Zsengeller Z, Stillman IE, Kang PM, van Goor H, McCurley A, Jaffe IZ, Karumanchi SA, Lely AT. Hydrogen Sulfide Attenuates sFlt1-Induced Hypertension and Renal Damage by Upregulating Vascular Endothelial Growth Factor. J Am Soc Nephrol. 2014;25:717–725. doi: 10.1681/ASN.2013030291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G, Gojon G, Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ. H2S Protects Against Pressure Overload–Induced Heart Failure via Upregulation of Endothelial Nitric Oxide Synthase. Circulation. 2013;127:1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wang M-J, Cai W-J, Li N, Ding Y-J, Chen Y, Zhu Y-C. The Hydrogen Sulfide Donor NaHS Promotes Angiogenesis in a Rat Model of Hind Limb Ischemia. Antioxid Redox Signal. 2009;12:1065–1077. doi: 10.1089/ars.2009.2945. [DOI] [PubMed] [Google Scholar]
- [83].Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19:2003–2012. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- [84].Tang B, Ma L, Yao X, Tan G, Han P, Yu T, Liu B, Sun X. Hydrogen sulfide ameliorates acute lung injury induced by infrarenal aortic cross-clamping by inhibiting inflammation and angiopoietin 2 release. J Vasc Surg. doi: 10.1016/j.jvs.2015.10.010. [DOI] [PubMed] [Google Scholar]
- [85].Faller S, Spassov S, Zimmermann K, Ryter S, Buerkle H, Loop T, Schmidt R, Strosing K, Hoetzel A. Hydrogen sulfide prevents hyperoxia-induced lung injury by downregulating reactive oxygen species formation and angiopoietin-2 release. Curr Pharm Des. 2013;19:2715–2721. doi: 10.2174/1381612811319150006. [DOI] [PubMed] [Google Scholar]
- [86].Cai W-J, Wang M-J, Moore PK, Jin H-M, Yao T, Zhu Y-C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76:29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- [87].Umaru B, Pyriochou A, Kotsikoris V, Papapetropoulos A, Topouzis S. ATP-Sensitive Potassium Channel Activation Induces Angiogenesis In Vitro and In Vivo. J Pharmacol Exp Ther. 2015;354:79–87. doi: 10.1124/jpet.114.222000. [DOI] [PubMed] [Google Scholar]
- [88].Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, Roviezzo F, Brancaleone V, Cirino G. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- [89].Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Zaid A, Giannogonas P, Cantalupo A, Dhayade S, Karalis KP, Wang R, Feil R, Cirino G. cGMP-dependent protein kinase contributes to hydrogen sulfide-stimulated vasorelaxation. PLoS One. 2012;7:e53319. doi: 10.1371/journal.pone.0053319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhou Z, Martin E, Sharina I, Esposito I, Szabo C, Bucci M, Cirino G, Papapetropoulos A. Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide. Pharmacol Res. 2016;111:556–562. doi: 10.1016/j.phrs.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao Y-X, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc Natl Acad Sci U S A. 2014;111:3182–3187. doi: 10.1073/pnas.1321871111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Greiner R, Pálinkás Z, Bäsell K, Becher D, Antelmann H, Nagy P, Dick TP. Polysulfides Link H(2)S to Protein Thiol Oxidation. Antioxid Redox Signal. 2013;19:1749–1765. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Altaany Z, Ju Y, Yang G, Wang R. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci Signal. 2014;7:ra87–ra87. doi: 10.1126/scisignal.2005478. [DOI] [PubMed] [Google Scholar]
- [94].Terzuoli E, Monti M, Vellecco V, Bucci M, Cirino G, Ziche M, Morbidelli L. Characterization of zofenoprilat as an inducer of functional angiogenesis through increased H2S availability. Br J Pharmacol. 2015;172:2961–2973. doi: 10.1111/bph.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ma K, Liu Y, Zhu Q, Liu C.-h., Duan J-L, Tan BKH, Zhu YZ. H(2)S Donor, S-Propargyl-Cysteine, Increases CSE in SGC-7901 and Cancer-Induced Mice: Evidence for a Novel Anti-Cancer Effect of Endogenous H(2)S? PLoS One. 2011;6:e20525. doi: 10.1371/journal.pone.0020525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kan J, Guo W, Huang C, Bao G, Zhu Y, Zhu YZ. S-Propargyl-Cysteine, a Novel Water-Soluble Modulator of Endogenous Hydrogen Sulfide, Promotes Angiogenesis Through Activation of Signal Transducer and Activator of Transcription 3. Antioxid Redox Signal. 2013;20:2303–2316. doi: 10.1089/ars.2013.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Tsai C-Y, Wen S-Y, Shibu MA, Yang Y-C, Peng H, Wang B, Wei Y-M, Chang H-Y, Lee C-Y, Huang C-Y, Kuo W-W. Diallyl trisulfide protects against high glucose-induced cardiac apoptosis by stimulating the production of cystathionine gamma-lyase-derived hydrogen sulfide. Int J Cardiol. 2015;195:300–310. doi: 10.1016/j.ijcard.2015.05.111. [DOI] [PubMed] [Google Scholar]
- [98].Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine γ-Lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Nagahara N, Nagano M, Ito T, Shimamura K, Akimoto T, Suzuki H. Antioxidant enzyme, 3-mercaptopyruvate sulfurtransferase-knockout mice exhibit increased anxiety-like behaviors: a model for human mercaptolactate-cysteine disulfiduria. Sci Reports. 2013;3:1986. doi: 10.1038/srep01986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, Maeda N. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Eberhardt RT, Forgione MA, Cap A, Leopold JA, Rudd MA, Trolliet M, Heydrick S, Stark R, Klings ES, Moldovan NI, Yaghoubi M, Goldschmidt-Clermont PJ, Farber HW, Cohen R, Loscalzo J. Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia. J Clin Invest. 106:483–491. doi: 10.1172/JCI8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Beard RJ, Bearden S. Vascular complications of cystathionine β synthase deficiency: future directions for homocysteine-to-hydrogen sulfide research. Am J Physiol Heart Circ Physiol. 2011;300:H13–H26. doi: 10.1152/ajpheart.00598.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bosch-Marcé M, Pola R, Wecker AB, Silver M, Weber A, Luedemann C, Curry C, Murayama T, Kearney M, Yoon Y.-s., Malinow MR, Asahara T, Isner JM, Losordo DW. Hyperhomocyst(e)inemia impairs angiogenesis in a murine model of limb ischemia. Vasc Med. 2005;10:15–22. doi: 10.1191/1358863x05vm585oa. [DOI] [PubMed] [Google Scholar]
- [104].Gersztenkorn D, Coletta C, Zhu S, Ha Y, Liu H, Tie H, Zhou J, Szabo C, Zhang W, Motamedi M. Hydrogen Sulfide Contributes to Retinal Neovascularization in Ischemia-Induced RetinopathyH2S Contributes to Retinal Neovascularization. Invest Ophthalmol Vis Sci. 2016;57:3002–3009. doi: 10.1167/iovs.15-18555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Islam KN, Polhemus DJ, Donnarumma E, Brewster LP, Lefer DJ. Hydrogen Sulfide Levels and Nuclear Factor-Erythroid 2-Related Factor 2 (NRF2) Activity Are Attenuated in the Setting of Critical Limb Ischemia (CLI) J Am Heart Assoc. 2015;4:e001986. doi: 10.1161/JAHA.115.001986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB, Rodriguez-Aguayo C, Lopez-Berestein G, Basal E, Weaver AL, Visscher DW, Cliby W, Sood AK, Bhattacharya R, Mukherjee P. Cystathionine Beta-Synthase (CBS) Contributes to Advanced Ovarian Cancer Progression and Drug Resistance. PLoS One. 2013;8:e79167. doi: 10.1371/journal.pone.0079167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Gai J-W, Qin W, Liu M, Wang H-F, Zhang M, Li M, Zhou W-H, Ma Q-T, Liu G-M, Song W-H, Jin J, Ma H-S. Expression profile of hydrogen sulfide and its synthases correlates with tumor stage and grade in urothelial cell carcinoma of bladder. Urol Oncol Seminars Original Invest. 2016;34:166.e115–166.e120. doi: 10.1016/j.urolonc.2015.06.020. [DOI] [PubMed] [Google Scholar]
- [108].Guo H, Gai J-W, Wang Y, Jin H-F, Du J-B, Jin J. Characterization of Hydrogen Sulfide and Its Synthases, Cystathionine β-Synthase and Cystathionine γ-Lyase, in Human Prostatic Tissue and Cells. Urology. 2012;79:483.e481–483.e485. doi: 10.1016/j.urology.2011.10.013. [DOI] [PubMed] [Google Scholar]
- [109].Pan Y, Ye S, Yuan D, Zhang J, Bai Y, Shao C. Hydrogen sulfide (H2S)/cystathionine γ-lyase (CSE) pathway contributes to the proliferation of hepatoma cells. Mutat Res. 2014;763–764:10–18. doi: 10.1016/j.mrfmmm.2014.03.002. [DOI] [PubMed] [Google Scholar]
- [110].Panza E, De Cicco P, Armogida C, Scognamiglio G, Gigantino V, Botti G, Germano D, Napolitano M, Papapetropoulos A, Bucci M, Cirino G, Ianaro A. Role of the cystathionine γ lyase/hydrogen sulfide pathway in human melanoma progression. Pigment Cell Melanoma Res. 2015;28:61–72. doi: 10.1111/pcmr.12312. [DOI] [PubMed] [Google Scholar]
- [111].Sen S, Kawahara B, Gupta D, Tsai R, Khachatryan M, Roy-Chowdhuri S, Bose S, Yoon A, Faull K, Farias-Eisner R, Chaudhuri G. Role of cystathionine β-synthase in human breast Cancer. Free Rad Biol Med. 2015;86:228–238. doi: 10.1016/j.freeradbiomed.2015.05.024. [DOI] [PubMed] [Google Scholar]
- [112].Sonke E, Verrydt M, Postenka CO, Pardhan S, Willie CJ, Mazzola CR, Hammers MD, Pluth MD, Lobb I, Power NE, Chambers AF, Leong HS, Sener A. Inhibition of endogenous hydrogen sulfide production in clear-cell renal cell carcinoma cell lines and xenografts restricts their growth, survival and angiogenic potential. Nitric Oxide. 2015;49:26–39. doi: 10.1016/j.niox.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Hellmich MR, Coletta C, Chao C, Szabo C. The Therapeutic Potential of Cystathionine β-Synthetase/Hydrogen Sulfide Inhibition in Cancer. Antioxid Redox Signal. 2015;22:424–448. doi: 10.1089/ars.2014.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]