

Abstract
The integrity of the nuclear envelope barrier relies on membrane remodeling by the ESCRTs, which seal nuclear envelope holes and contribute to the quality control of nuclear pore complexes (NPCs); whether these processes are mechanistically related remains poorly defined. Here, we show that the ESCRT‐II/III chimera, Chm7, is recruited to a nuclear envelope subdomain that expands upon inhibition of NPC assembly and is required for the formation of the storage of improperly assembled NPCs (SINC) compartment. Recruitment to sites of NPC assembly is mediated by its ESCRT‐II domain and the LAP2‐emerin‐MAN1 (LEM) family of integral inner nuclear membrane proteins, Heh1 and Heh2. We establish direct binding between Heh2 and the “open” forms of both Chm7 and the ESCRT‐III, Snf7, and between Chm7 and Snf7. Interestingly, Chm7 is required for the viability of yeast strains where double membrane seals have been observed over defective NPCs; deletion of CHM7 in these strains leads to a loss of nuclear compartmentalization suggesting that the sealing of defective NPCs and nuclear envelope ruptures could proceed through similar mechanisms.
Keywords: ESCRT, LEM domain, nuclear envelope, nuclear transport, quality control
Subject Categories: Membrane & Intracellular Transport
Introduction
It is well established that the nuclear envelope (NE) in multicellular eukaryotes undergoes a dramatic breakdown and reformation during cell division (Wandke & Kutay, 2013), while emerging work suggests that the two membranes of the NE undergo extensive remodeling during interphase as well (Hatch & Hetzer, 2014; King & Lusk, 2016). Classic examples are the insertion of massive protein assemblies like nuclear pore complexes (NPCs)(Rothballer & Kutay, 2013) and the yeast centrosome (spindle pole body; SPB) into the NE, but now extend to the nuclear egress of “mega” ribonucleoprotein particles through a vesicular intermediate in the NE lumen/perinuclear space (Speese et al, 2012; Jokhi et al, 2013) and the degradative clearance of nuclear/NE contents by autophagy pathways that act specifically at the NE (Roberts et al, 2003; Dou et al, 2015; Mochida et al, 2015). Understanding the molecular mechanisms that drive the membrane remodeling necessary for NE homeostasis is a critical goal for the field, particularly with the ever‐growing links between disruptions in nuclear compartmentalization and human disease (Burke & Stewart, 2014; Hatch & Hetzer, 2014; Webster & Lusk, 2016).
There is a particular lack of clarity regarding the fundamental mechanism of de novo NPC assembly. To build the massive ~50 MD yeast or ~100 MD human NPC requires the assembly of ~30 proteins (nucleoporins(nups)) in multiple copies such that an individual NPC requires upwards of 500 proteins in yeast (Alber et al, 2007) and perhaps twice as many in human cells (Bui et al, 2013; von Appen et al, 2015). This requires remarkable spatiotemporal control over hundreds of proteins that converge at a NE domain competent for NPC assembly; what defines a biogenesis site remains unclear, but it might require local changes in Ran‐GTP levels (Ryan et al, 2003; Walther et al, 2003; D'Angelo et al, 2006), nup binding to integral inner nuclear membrane (INM) proteins (Talamas & Hetzer, 2011; Yewdell et al, 2011; Chen et al, 2014), or to chromatin (Franz et al, 2007; Rasala et al, 2008; Rotem et al, 2009; Doucet et al, 2010), or changes in the properties of the membrane itself, perhaps by altering lipid composition (Schneiter et al, 1996; Scarcelli et al, 2007; Hodge et al, 2010; Lone et al, 2015). Such changes to lipid composition might provide a more permissive environment for the protein‐mediated membrane remodeling thought to be required for INM and outer nuclear membrane (ONM) fusion to form a pore.
While no single dedicated membrane bending or fusion machinery has been identified that drives nuclear pore formation, an emerging theme is the cooperative action of nups containing amphipathic helices or reticulon domains capable of recognizing and/or generating membrane curvature (Marelli et al, 2001; Drin et al, 2007; Dawson et al, 2009; Chadrin et al, 2010; Doucet et al, 2010; Vollmer et al, 2012, 2015; von Appen et al, 2015; Casey et al, 2015; Floch et al, 2015; Mészáros et al, 2015). Interestingly, budding yeast requires the membrane bending and scission endosomal sorting complexes required for transport (ESCRT)‐III proteins to ensure formation of functional NPCs, raising the possibility that an established multifunctional membrane remodeler that acts during pore biogenesis may yet be identified (Webster et al, 2014; Webster & Lusk, 2015). Consistent with this idea, genetic ablation of ESCRT‐III components and the AAA‐ATPase Vps4 leads to the accumulation of defective NPCs in a compartment called the storage of improperly assembled NPCs compartment (SINC), which is subsequently retained in mother cells (Webster et al, 2014). This example of spatial quality control supports a model in which ESCRTs surveil early steps in nuclear pore biogenesis to prevent SINC formation, but the mechanisms of SINC formation and NPC assembly surveillance remain to be fully determined.
Any potential mechanism for how ESCRTs contribute to NPC biogenesis and/or quality control must be reconciled with the well‐established role for ESCRT‐III as a membrane scission machinery that acts to fuse membranes to form continuous bilayers (McCullough et al, 2013; Hurley, 2015). For example, recent work suggests that ESCRT‐III is recruited to the NE during post‐mitotic NE reformation and participates in annular fusion events required to fully seal the NE (Olmos et al, 2015; Vietri et al, 2015). Such a mechanism likely also acts to repair the dramatic NE ruptures observed as migrating cells move through constrictive channels (Denais et al, 2016; Raab et al, 2016). Thus, while nuclear pores may present a membrane topology ideal for recognition by ESCRT‐III, the canonical ESCRT‐mediated activity would be predicted to seal the very holes required for NPC assembly. A clue to solving this paradox might be found in decades‐old data, where double membrane seals were observed over defective NPCs in nup116Δ cells (Wente & Blobel, 1993). The sealing of NPCs lacking Nup116, a component of the central transport channel, was proposed to protect cells from defective NPCs; NPC sealing might proceed through the expansion of the nuclear pore membranes and their subsequent fusion through an ESCRT‐III compatible mechanism (Wente & Blobel, 1993). Therefore, it is plausible that ESCRT mechanisms that act at defective NPCs and NE ruptures are mechanistically similar.
These NE‐specific examples of ESCRT function build on a much larger body of work establishing that ESCRTs act in membrane scission steps throughout the cell (McCullough et al, 2013; Hurley, 2015; Olmos & Carlton, 2016). While ESCRTs function at multiple subcellular locales, the precise mechanism of ESCRT‐mediated membrane scission remains to be fully understood, although it is generally thought it will require the remarkable capacity of ESCRT‐III subunits to form polymers that generate and stabilize negative membrane curvature as membrane scaffolds (Ghazi‐Tabatabai et al, 2008; Hanson et al, 2008; Lata et al, 2008b; Saksena et al, 2009; Henne et al, 2012; Cashikar et al, 2014; Shen et al, 2014; Chiaruttini et al, 2015). There are multiple ESCRT‐III proteins in budding yeast and multicellular eukaryotes, with yeast Snf7 and its mammalian orthologue CHMP4B being the most abundant (Teis et al, 2008); members of the ESCRT‐III family are thought to share a similar structure, with a basic core domain of four alpha helices whose assembly into a homo or hetero‐polymer is autoinhibited by acidic helices that fold back onto this core (Muzioł et al, 2006; Zamborlini et al, 2006; Shim et al, 2007; Kieffer et al, 2008; Lata et al, 2008a; Bajorek et al, 2009; Xiao et al, 2009; Tang et al, 2015). A key question therefore is how ESCRT‐III subunits come together to form potentially unique filaments with context‐dependent biophysical properties (Cashikar et al, 2014). Such flexibility is exceptionally highlighted by the unexpected discovery that the ESCRT‐IIIs IST1 and CHMP1B form a heteropolymer capable of scaffolding tubules with positive membrane curvature (McCullough et al, 2015).
To fully understand how ESCRTs function during NPC biogenesis and NE repair will require a more extensive molecular understanding of the NE‐specific arm of the ESCRT pathway. For example, in vitro and in vivo analyses predominantly of the endocytic ESCRT arm have defined a stepwise activation of Snf7 polymerization by the sequential binding of the ESCRT‐II, Vps25, to the ESCRT‐III, Vps20, which in turn releases the autoinhibition of Snf7 to induce filament formation (Teis et al, 2008, 2010; Im et al, 2009; Saksena et al, 2009; Henne et al, 2012). Bro1‐domain‐containing proteins like ALIX are also capable of activating Snf7 polymerization, although they do so through a distinct Snf7‐binding interface (McCullough et al, 2008). The ESCRT‐IIIs Vps2 and Vps24 are thought to alter the Snf7 filament helicity (Saksena et al, 2009; Henne et al, 2012) and recruit the AAA‐ATPase Vps4, which stimulates ESCRT‐recycling in a manner that might directly contribute to membrane scission (Teis et al, 2008; Saksena et al, 2009; Adell et al, 2014). Interestingly, ESCRT‐III‐mediated NPC assembly quality control is independent of VPS25 and VPS20, raising the possibility that Snf7 activation at the NE might require specific factors distinct from those at endosomes (Webster et al, 2014).
As the recruitment of ESCRT‐III family members to different cellular compartments requires site‐specific adaptor molecules (McCullough et al, 2013; Hurley, 2015), it is possible that the adaptors themselves might contribute to the activation mechanism. In budding yeast, the conserved LAP2‐emerin‐MAN1 (LEM) integral INM proteins Heh1/Src1 and Heh2 were suggested to be NE‐specific Snf7 adaptors, but a direct interaction was not established (Webster et al, 2014). Moreover, the “orphan” ESCRT CHMP7 (Horii et al, 2006) might help to recruit CHMP4B to seal NE holes at the end of mitosis (Vietri et al, 2015), but how CHMP7 is itself recruited to the NE remains unknown.
Recent work in budding yeast suggests that the orthologue of CHMP7 (Chm7) is a chimera of ESCRT‐II and ESCRT‐III domains and was proposed to be an integral part of an ER‐specific arm of the ESCRT pathway (Bauer et al, 2015). Most interestingly, a genetic interaction between CHM7 and APQ12 (Bauer et al, 2015), which encodes a protein thought to modulate membrane fluidity in a way that is required for NPC assembly and NE morphology (Scarcelli et al, 2007), supports a model in which Chm7 acts at the NE. Here, we uncover a temporal and spatial network of functional interactions that comprise a NE‐specific, Chm7‐dependent quality control pathway that appears to seal nuclear pores to protect nuclear‐cytoplasmic compartmentalization. Using a combination of genetic and biochemical approaches, we define integral INM proteins as key factors for Chm7 recruitment and regulation at the NE. Perturbations that functionally ablate this pathway aggravate genetic backgrounds that are associated with defective NPC structures, while in an apparent gain‐of‐function scenario, this pathway is aberrantly activated and may compromise normal NPC assembly.
Results
Chm7 localizes to a focus on the NE
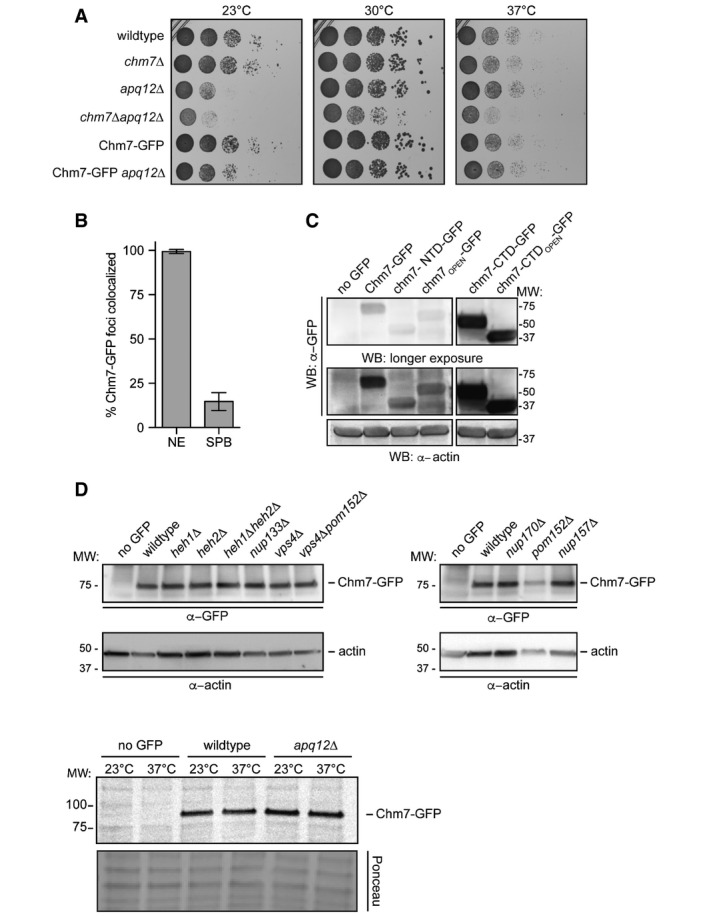
To begin to explore how Chm7 might contribute to NE function, we examined its steady‐state distribution by localizing a functional (Fig EV1A) Chm7‐GFP fusion protein expressed from the chromosomal CHM7 locus. Chm7‐GFP showed a diffuse cytosolic fluorescence with a discrete accumulation in a focus that coincides with a NE marker (Nup170‐mCherry) in ~26% of cells (Figs 1B and 2B). Timelapse analysis suggests that the focus (and sometimes two foci, see also Fig 7B for steady‐state quantification of foci number) dynamically samples the NE and can be inherited by daughter cells (Movie EV1). These properties are reminiscent of SPBs yet the Chm7‐GFP foci failed to colocalize with the SPB component Mps3‐mCherry (Figs 1D and EV1B), suggesting that Chm7 is associated with a distinct NE structure.
Figure EV1. Expression levels of Chm7‐GFP and truncations.
- Tenfold serial dilutions of the indicated yeast strains grown at 23, 30 or 37°C for 2–4 days.
- Plot of the percentage of Chm7‐GFP foci that colocalize with the NE or with Mps3‐mCherry‐labeled SPBs. Error bars are SD from the mean from three independent experiments where > 50 Chm7‐GFP foci were counted.
- Relative levels of GFP fusions of Chm7 and truncations assessed by Western blot with anti‐GFP antibody and ECL detection; anti‐actin is a loading control. Left and right panels are from same nitrocellulose membrane at same exposure. Note that constructs in left panel are expressed from the endogenous CHM7 promoter, whereas right panel constructs are expressed behind the GPD promoter. Numbers reflect positions of molecular weight (MW) standards.
- Western blots (anti‐GFP) detecting Chm7‐GFP levels in the indicated strains with anti‐actin loading control or Ponceau‐stained membrane to assess relative protein loads.
Source data are available online for this figure.
Figure 1. Chm7 localizes to a focus on the NE.
- Schematic of the predicted domain architecture of Chm7 and truncations. Green circle is a Chm7‐specific insertion (relative to Vps25; see Appendix Fig S1). MIM is microtubule interacting motif. Red stripes denote putative autoinhibitory helices. Numbers are amino acid residues.
- Deconvolved fluorescence micrographs of Chm7‐GFP and truncations with Nup170‐mCherry NE marker and merge of green and red channels. Scale bar is 5 μm.
- Scatter plot of the mean fluorescence intensity with SD of individual NE foci (horizontal line is mean). n > 50 foci were measured for each GFP fusion; P‐values from unpaired Student's t‐test. ***P ≤ 0.001; ****P ≤ 0.0001.
- Deconvolved fluorescence micrographs of Chm7‐GFP and Mps3‐mCherry with merge of green and red images (bottom). Scale bar is 5 μm.
- Tenfold serial dilutions of chm7Δapq12Δ strains grown at the indicated temperatures with either an empty pRS416 plasmid (−) or those expressing the indicated Chm7 constructs (pRS416‐HA‐CHM7, pRS416‐HA‐chm7‐NTD, pRS416‐HA‐chm7‐CTD). See Appendix Fig S2A for protein levels.
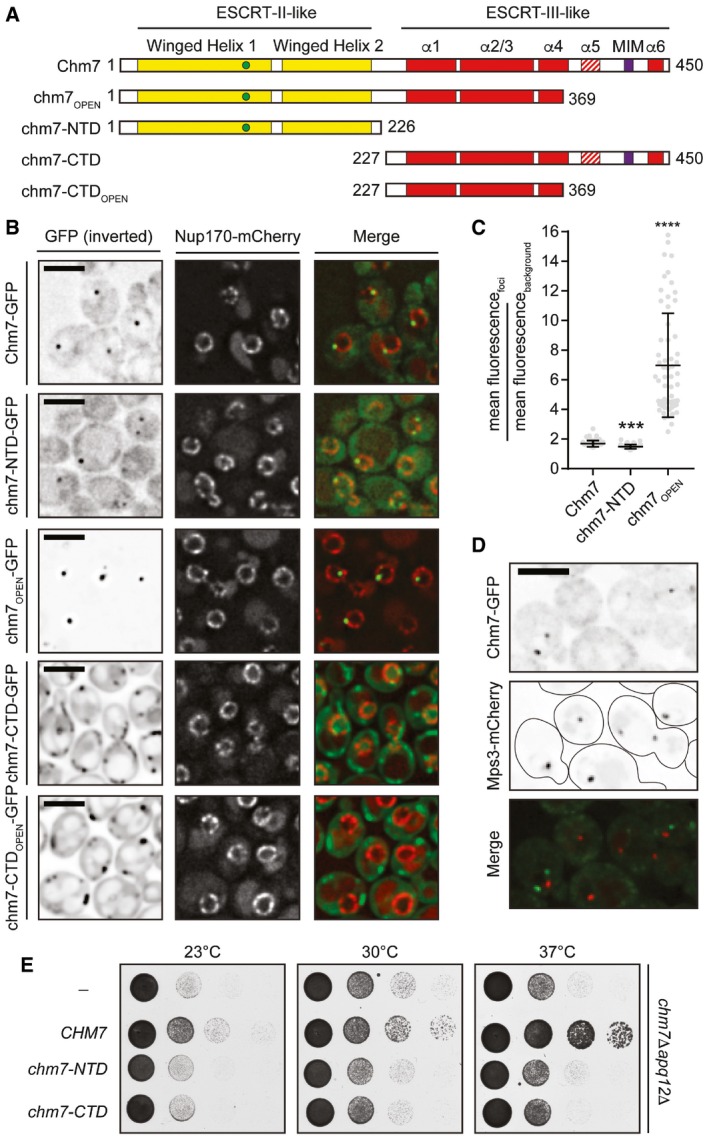
Figure 2. Heh1 is required for Chm7 recruitment to the NE subdomain.
- Deconvolved inverted fluorescence micrographs Chm7‐GFP and truncations in the indicated null strains. Scale bar is 5 μm.
- Plot of the percentage (± SD) of cells with Chm7‐GFP (and truncations) NE foci from (A). Data are from three independent replicates where > 200 cells were counted for each strain.
- Deconvolved inverted fluorescence micrographs of Chm7‐GFP in a heh1Δ strain containing either an empty pRS426 plasmid (−) or those expressing HEH1 (pRS426‐HEH1) or HEH2 (pRS426‐HEH2).
- Plots of the percentage of cells (± SD) with Chm7‐GFP NE foci from (C). Data are from three independent replicates where > 200 cells were counted for each strain. P‐values from unpaired Student's t‐test. **P ≤ 0.01.
Figure 7. Perturbation of NPC assembly leads to NE accumulation of Chm7.
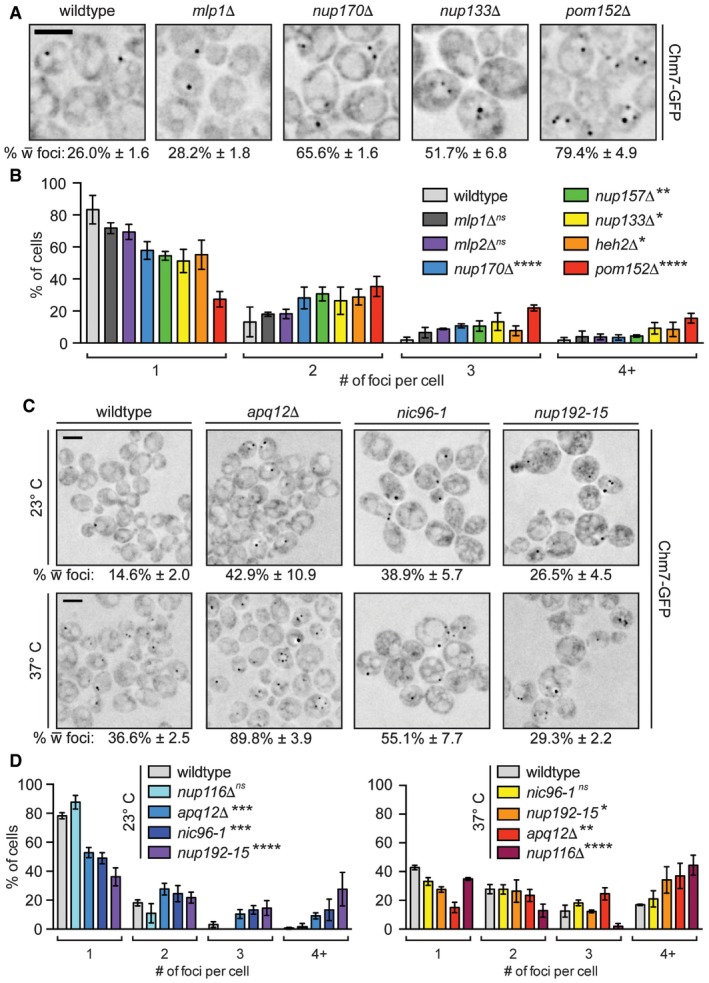
- Deconvolved inverted fluorescence micrographs of the indicated strains expressing Chm7‐GFP (percentages ± SD of the proportion of cells with Chm7‐GFP foci are shown at bottom of each panel, see Fig EV5B). Scale bar is 5 μm.
- Plot of the percentage of cells with the indicated number of Chm7‐GFP foci. Error bars are SD from the mean from three independent replicates of > 50 foci per strain. P‐values from two‐way ANOVA where ns represents P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001.
- Deconvolved inverted fluorescence micrographs of strains expressing Chm7‐GFP after 5 h at 23°C (top) or 37°C (bottom). Scale bar is 5 μm. Percentages ± SD of the proportion of cells with Chm7‐GFP NE foci are shown under each panel (see Fig EV5F).
- Plots of the percentage of cells from (C) with the indicated number of Chm7‐GFP foci at 23°C (left) and 37°C (right). Error bars are SD from the mean from three independent replicates of > 50 foci per strain. P‐values from two‐way ANOVA where ns is P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
As Chm7 is likely a chimera of ESCRT‐II and ESCRT‐III domains (Bauer et al, 2015), we next assessed how these domains contributed to the localization of the full‐length protein by generating GFP‐tagged chm7 truncations (Fig 1A). Interestingly, while the isolated ESCRT‐II domain (chm7‐NTD) was sufficient to localize to a focus on the NE in ~40% of cells (Figs 1B and 2B), the recruitment of a potentially “active” form of Chm7 lacking the putative auto inhibitory helices of the ESCRT‐III domain (chm7OPEN‐GFP; Fig 1A) was dramatically increased, being recruited to the NE subdomain in virtually 100% of cells (Fig 2B) with a ~fourfold increase in mean intensity (Fig 1B and C). This increase is likely relevant as the expression level of chm7OPEN‐GFP is equivalent (or lower than) Chm7‐GFP and chm7‐NTD‐GFP (Fig EV1C).
In order to visualize the distribution of the isolated ESCRT‐III domain of Chm7 (chm7‐CTD) and its predicted “activated” form chm7‐CTDOPEN, we expressed these constructs under control of the constitutive GPD promoter, which drove much higher protein levels than ESCRT‐II‐containing constructs (Fig EV1C). Both chm7‐CTD and chm7‐CTDOPEN accumulated in focal and tubular structures in the cytoplasm; chm7‐CTD‐GFP also resided in a diffusely cytosolic and nuclear pool that was absent from chm7‐CTDOPEN (Fig 1B). Taken together, these data support a model in which the ESCRT‐II domain of Chm7 is sufficient for NE targeting, which is amplified by the addition of the “activated” ESCRT‐III domain. However, only the full‐length Chm7 was capable of complementing the synthetic sickness of a chm7Δapq12Δ strain (Bauer et al, 2015), suggesting that individual ESCRT‐II and ESCRT‐III domains of Chm7 are unable to carry out its function in the NE‐ER system (Fig 1E).
Heh1 is required for Chm7 recruitment to the NE subdomain
In previous work, we proposed that the integral INM protein Heh2 contributed to the recruitment of Snf7 to the NE (Webster et al, 2014). We therefore assessed whether Heh2 and its paralog, Heh1, were necessary for the NE recruitment of Chm7. Strikingly, in heh1Δ cells, there was a complete loss of Chm7‐GFP foci on the NE also seen in heh1Δheh2Δ cells (Fig 2A and B). Surprisingly, Chm7‐GFP foci were more numerous and more prevalent in the cell population upon genetic deletion of HEH2 (Fig 2A and B); neither of these changes in distribution reflected alterations in Chm7‐GFP protein levels (Fig EV1D). However, consistent with the idea that both Heh1 and Heh2 are competent to physically interact with Chm7, Chm7‐GFP could be restored to the NE in heh1Δ cells by overexpressing either HEH1 or HEH2 (Fig 2C and D).
Similar results were obtained when we examined the distribution of the Chm7 truncations in heh1Δ and heh2Δ strains. Specifically, the NE foci formed by chm7‐NTD‐GFP (and even the more strongly recruited chm7OPEN) were completely abolished in the absence of HEH1 (Fig 2A and B). Cumulatively, these data support the conclusion that the ESCRT‐II chm7‐NTD domain is sufficient to target Chm7 to a NE focus in an Heh1‐dependent manner. The “activated” ESCRT‐III domain mediates the amplification of Chm7 accumulation in a way that is likely analogous to the “active”, polymerized form of other ESCRT‐III proteins.
Heh2 directly binds to Chm7 and Snf7
The Heh1 protein dependence of Chm7 NE recruitment (and the ability of HEH2 overexpression to recruit Chm7) raised the possibility that Heh1 and/or Heh2 might directly bind Chm7. We also reported previously that Heh2 can specifically interact with Snf7 in a complex cellular extract, but it was not clear whether this interaction is direct (Webster et al, 2014). Therefore, to test whether Chm7 and Snf7 directly bind the LEM domain integral INM proteins, we produced a recombinant heh2(1‐308) that encompasses the entire soluble extralumenal N‐terminal domain of Heh2 (Fig 3A). Unfortunately, despite exhaustive attempts, we were unable to generate a stable recombinant form of the soluble N‐terminal domain of Heh1. We therefore first assessed binding to recombinant bead‐bound GST‐Snf7 and GST‐snf7OPEN, which mimics the Snf7 active form (Fig 3B). As shown in Fig 3C, heh2(1‐308) bound to GST‐snf7OPEN preferentially to full‐length Snf7.
Figure 3. Heh2 directly binds Chm7 and Snf7.
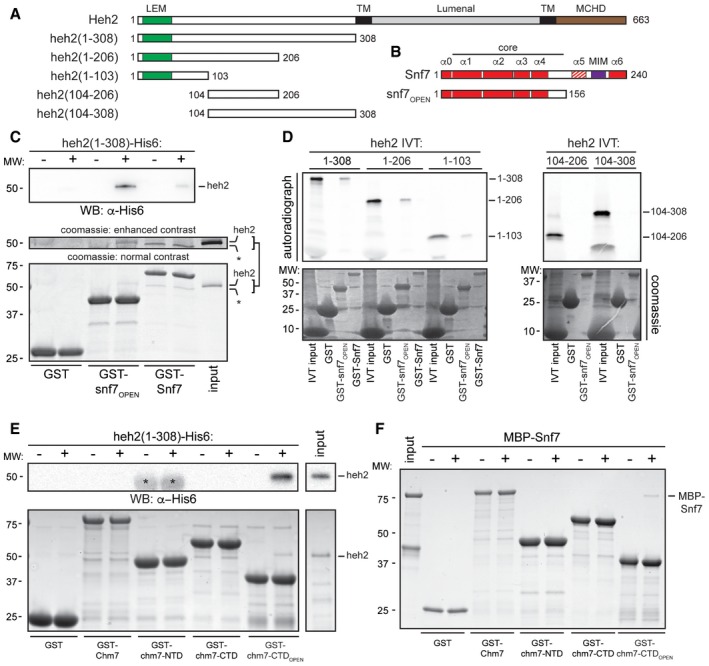
-
A, BSchematics of the domain organization and secondary structure of Heh2 and Snf7 and truncations with amino acid numbers. LEM is LAP2‐emerin‐MAN1 (green), TM is transmembrane (black), MCHD is MAN1 C‐terminal homology domain (brown), and MIM is microtubule interacting motif (purple). The striped α‐helix 5 represents the autoinhibitory helix.
-
CGST, GST‐Snf7, and GST‐snf7OPEN were immobilized on GT‐resin and incubated with buffer (−) or recombinant heh2(1‐308)‐His6. Bound proteins separated by SDS–PAGE were visualized by Coomassie stain and by Western blot with anti‐His6 antibody and ECL detection (WB; top panel). Middle panel shows indicated cropped region of the gel where contrast has been increased. Numbers on the side of the gel show position of molecular weight (MW) markers.
-
DIn vitro transcription translation (IVT) reactions generating radiolabeled (35S) truncations of Heh2 (inputs) were incubated with bead‐bound GST, GST‐snf7OPEN, or GST‐Snf7 before washing, elution, and detection of bound proteins by autoradiography (top panel) or Coomassie staining (bottom).
-
EAs in (C) except with GST‐Chm7 constructs. Upper panel shows Western blot (WB) with anti‐His6 antibody. Asterisk (*) indicates that chm7‐NTD is also expressed with a His6 tag. Bottom panel is Coomassie stained.
-
FAs in (E) except with MBP‐Snf7.
Despite the apparent specificity to the snf7OPEN construct over both GST and GST‐Snf7, we were concerned that the sub‐stoichiometric heh2(1‐308) binding might reflect a weak interaction that could be dislodged by non‐specific competition in vivo. However, we reproduced this specificity within an in vitro transcription translation mix in which non‐specific competitors are abundant, suggesting a biologically relevant interaction (Fig 3D). Further, by testing a series of truncations of the Heh2 N‐terminal domain, we narrowed the binding interface between Heh2 and Snf7 to the N‐terminal ~100 amino acids, which includes the LEM domain (Fig 3A and D); we were unable to test binding sufficiency with the LEM domain as we failed to produce a stable polypeptide. We nonetheless conclude that Heh2 directly binds to Snf7, thus providing a mechanistic basis for how this ESCRT‐III is recruited to the NE.
We next tested whether Heh2 could directly interact with Chm7 by producing recombinant Chm7 in addition to isolated chm7‐NTD, chm7‐CTD, and chm7‐CTDOPEN domains (Fig 1A). Remarkably, heh2(1‐308) directly and specifically interacted with the Chm7 ESCRT‐III domain in a manner analogous to its binding to Snf7, that is, it only bound to chm7‐CTDOPEN (Fig 3E). These data reinforce that the Chm7 C‐terminal domain might adopt an ESCRT‐III‐like structure recognized by Heh2.
As there are specific examples of ESCRT‐III proteins being capable of binding to ESCRT‐II and ESCRT‐III domains, we tested whether a MBP‐Snf7 fusion directly bound to Chm7 and the truncations. Interestingly, like Heh2, MBP‐Snf7 specifically interacted with the ESCRT‐III domain of Chm7 in its open form (Fig 3F). Surprisingly, however, these interactions were sub‐stoichiometric, suggesting that additional factors likely also contribute to binding, and perhaps activation, of Snf7 polymerization. As the Chm7 binding sites for Snf7 and Heh2 overlapped, an obvious hypothesis is that Snf7 might bind Chm7 cooperatively (or competitively) with Heh2. However, combining stoichiometric amounts of Snf7 and Heh2 did not alter their relative presence in GST‐chm7‐CTDOPEN‐bound fractions (Fig EV2). Thus, we suggest that other yet to be identified factors and/or physiological conditions are likely required for more robust binding between these proteins and Snf7 activation at the NE.
Figure EV2. Co‐incubation of Snf7 and Heh2 does not alter their binding to chm7‐CTDOPEN .
GST and GST fusions of Chm7 and truncations were immobilized on GT‐resin and incubated with buffer (−), recombinant heh2(1‐308)‐His6, or MBP‐Snf7. Bound proteins were separated by SDS–PAGE and visualized by Coomassie stain. Numbers on the side of the gel show position of molecular weight (MW) markers.
Heh1‐Chm7 interactions can occur at the NE subdomain
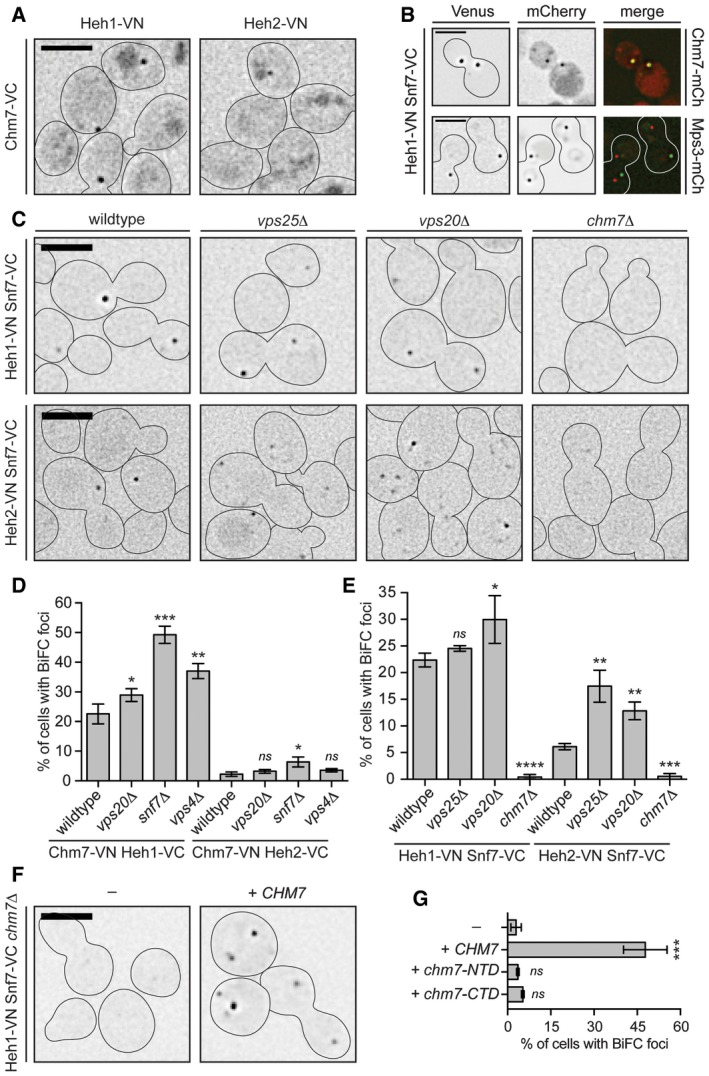
To assess whether direct biochemical interactions between Heh1, Heh2, Chm7 and Snf7 could occur at the NE, we turned to the Bifunctional Complementation strategy (BiFC)(Kerppola, 2008). Using this approach, bait and prey proteins are endogenously tagged with an N‐terminal (VN) and C‐terminal (VC) domain of the fluorescent protein Venus, respectively. When bait and prey proteins associate, Venus folds and fluoresces. As predicted by our direct binding studies, we observed BiFC signal between Heh2 and Chm7 as a solitary fluorescent focus (Fig 4A). Its low abundance (in only ~3% of cells; Fig 4D) contrasted with cells expressing the Heh1‐VN Chm7‐VC combination where we observed a focus in ~28% of cells (Fig 4D). This potential preference to Heh1 was anticipated, as Heh1 had a greater influence on the NE‐focal localization of Chm7 (Fig 2). A similar fluorescent focus was also observed in strains expressing Heh1‐VN and Heh2‐VN with Snf7‐VC (Fig 4C and E; Webster et al, 2014). Therefore, we hypothesized that the BiFC foci represented a functional organization of the ESCRT and Heh proteins at the same discrete subdomain of the NE recognized by Chm7. Consistent with this idea, the Heh1‐VN Snf7‐VC BiFC fluorescent foci colocalized with Chm7‐mCherry NE foci, but not with SPBs at steady state (Fig 4B).
Figure 4. Heh1‐Chm7 interactions occur at the NE subdomain.
-
ADeconvolved inverted fluorescence micrographs of BiFC signal of Chm7‐VC with either Heh1‐VN or Heh2‐VN. Cell borders are outlined. Scale bar is 5 μm.
-
BDeconvolved inverted fluorescence micrographs of yeast strains expressing Heh1‐VN and Snf7‐VC with either Chm7‐mCherry or Mps3‐mCherry and merge of both channels. Scale bar is 5 μm.
-
CDeconvolved inverted fluorescence micrographs of BiFC signal of the indicated VN and VC fusions in the indicated strains. Cell borders are outlined. Scale bar is 5 μm.
-
D, EPlots of the percentage of cells with BiFC signal of the indicated VN VC pairs in the indicated null backgrounds. Data are from three independent replicates where > 225 cells per strain per replicate were quantified. Error bars are SD from the mean of each replicate. P‐values from unpaired Student's t‐test where ns is P > 0.05. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
-
FDeconvolved inverted fluorescence micrographs of Heh1‐VN Snf7‐VC BiFC signal in a chm7Δ strain with either an empty plasmid (−; pRS416) or a plasmid expressing CHM7 (pRS416‐HA‐chM7). Cell borders are outlined. Scale bar is 5 μm.
-
GPlot of the percentage of cells with BiFC signal from (F). Error bars are SD from the mean of each replicate. P‐values from unpaired Student's t‐test where ns is P > 0.05. ***P ≤ 0.001.
Chm7 is required for Heh1/2‐Snf7 BiFC
The BiFC assay is also capable of reporting on dynamic interactions. For example, at endosomes, the functional interaction between Snf7 and Vps20 occurs after Vps25 action (Teis et al, 2010). As such, we failed to observe substantial BiFC signal at endosomes between Vps20‐VN and Snf7‐VC in the absence of VPS25 (Fig EV3A and B), despite the fusion proteins being produced at levels similar to wild‐type cells (Fig EV3C). This effect was specific, as Snf7‐VN Vps20‐VC BiFC signal was not reduced in vps24Δ, vps2Δ, bro1Δ, vps4Δ, and chm7Δ strains (Fig EV3A and B). Thus, BiFC is capable of providing a faithful readout of the pathway that leads to the activation of Snf7 at endosomes.
Figure EV3. Chm7 BiFC interactions.
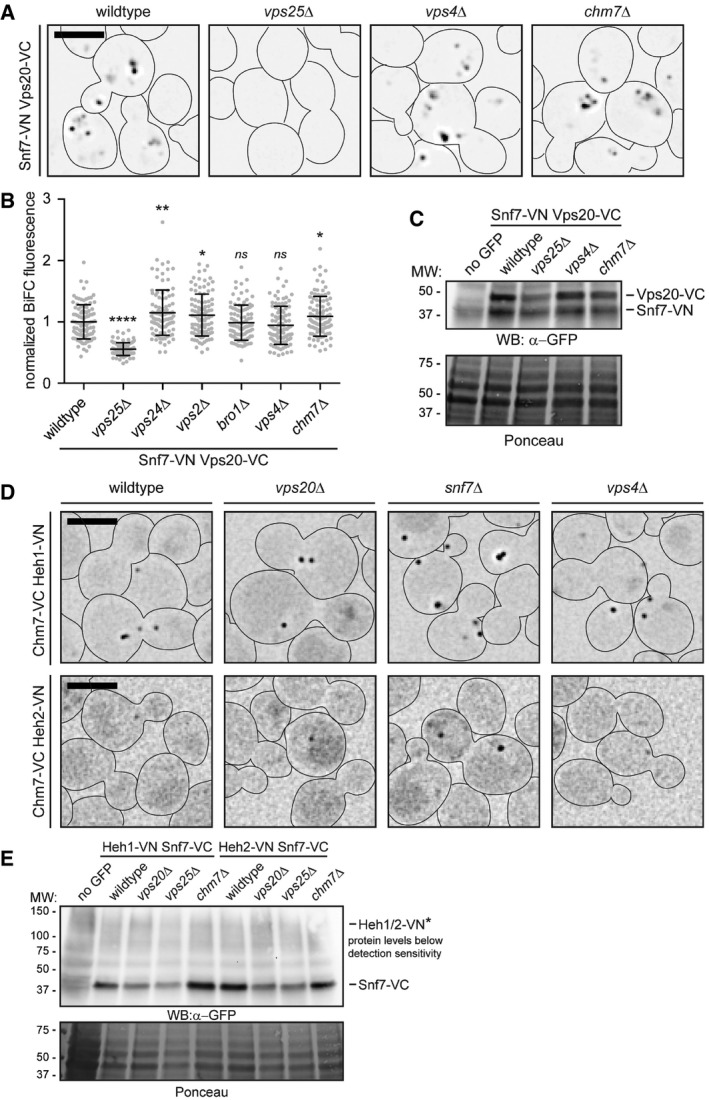
- Deconvolved inverted fluorescence micrographs of BiFC signal of Snf7‐VN and Vps20‐VC in the indicated genetic backgrounds. Cell borders are outlined. Scale bar is 5 μm.
- Plot of total cellular BiFC fluorescence of Snf7‐VN and Vps20‐VC in the indicated strains. Data are from three independent replicates where 100 cells per strain were quantified. Error bars are SD from the mean of each replicate. P‐values from unpaired Student's t‐test where ns is P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001.
- Western blot using polyclonal anti‐GFP antibodies to detect both VN and VC fusions of Snf7‐VN and Vps20‐VC in the indicated strains; detection by ECL. Bottom panel shows Ponceau‐stained nitrocellulose membrane as a total protein load reference. Numbers at the side show positions of molecular weight (MW) markers.
- Deconvolved inverted fluorescence micrographs of BiFC signal in the indicated yeast strains expressing Chm7‐VC and either Heh1‐VN or Heh2‐VN. Cell borders are outlined. Scale bar is 5 μm.
- Western blot of VN and VC fusions in the indicated strains detected using a polyclonal anti‐GFP antibody and ECL. To assess relative protein loads, bottom panel shows corresponding Ponceau‐stained nitrocellulose membrane.
Knowing that BiFC could provide insight into the functional state of ESCRT interactions, we tested whether deletion of ESCRTs altered NE‐specific interactions. Interestingly, the proportion of cells with Heh1/2‐VC Chm7‐VN BiFC signal was elevated in vps4Δ and snf7Δ cells but was minimally influenced by loss of VPS20, suggesting that Vps4 and Snf7 act downstream of the Heh1‐Chm7 interaction (Figs 4D and EV3D). In addition, and consistent with our prior genetic analysis (Webster et al, 2014), the deletion of the canonical Snf7 activation components (VPS25 and VPS20) did not diminish (and in fact slightly increased) BiFC signal of Heh1/2‐VN Snf7‐VC (Fig 4C and E). Most strikingly, the deletion of CHM7 virtually abolished BiFC between both Heh1/2‐VN and Snf7‐VC (Fig 4C and E) suggesting that the NE recruitment and/or activation of Chm7 is required for Heh1‐Snf7 BiFC, although we cannot formally rule out changes to Heh1‐VN levels as they were undetectable by immunoblot (Fig EV3E). However, consistent with the idea that these interactions require activation of Chm7, only the full‐length Chm7 (and not the isolated N or C‐terminal domains) was capable of restoring BiFC between Heh1‐VN and Snf7‐VC (Fig 4F and G). These data support a model in which Chm7 requires recruitment and activation to stimulate a synergistic set of interactions that link NE membrane proteins and ESCRTs. Interestingly, it is also likely that Chm7, Snf7, and Vps4 can interact at other subcellular locations, as BiFC signal between Chm7‐VN and Snf7‐VC (and Vps4‐VC) is observed throughout the cell and was not influenced by Heh1 and Heh2 (Fig EV4A–C).
Figure EV4. Chm7‐Snf7 interactions can occur outside of the NE.
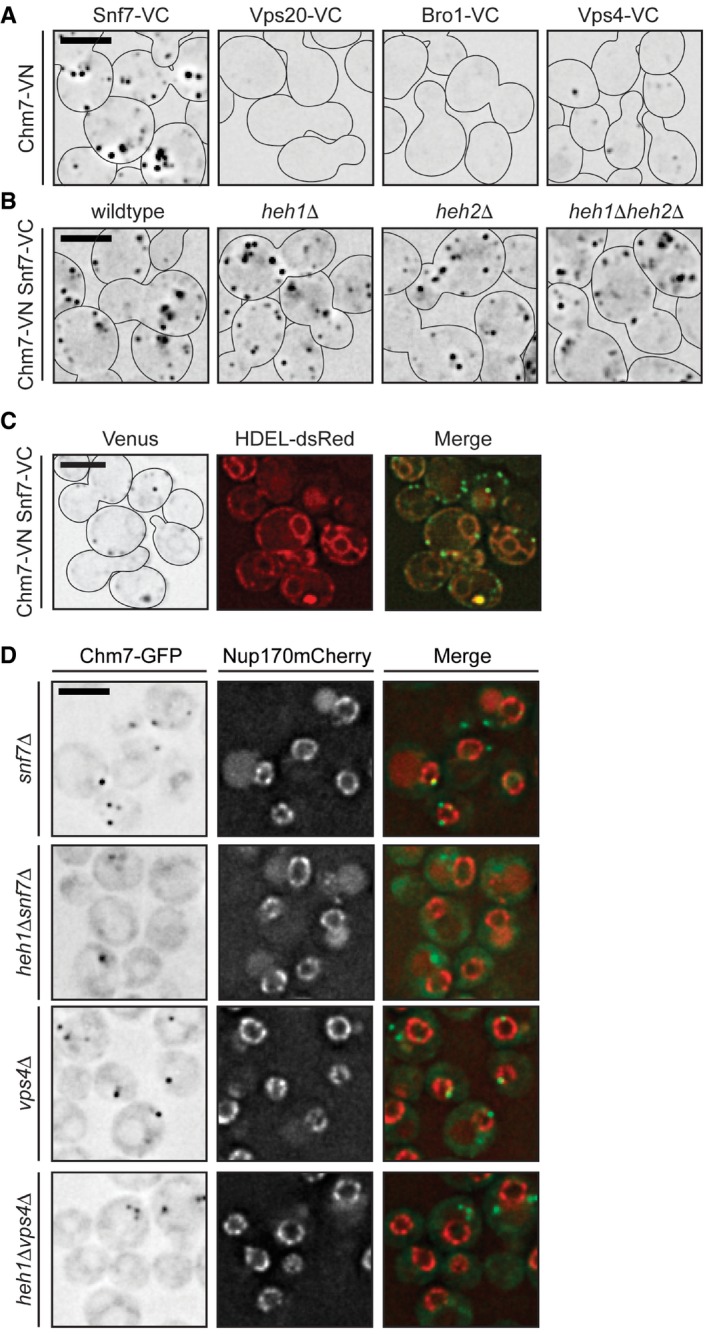
- Deconvolved inverted fluorescence micrographs of BiFC signal in wild‐type yeast strains expressing Chm7‐VN and the indicated VC fusions. Cell borders are outlined. Scale bar is 5 μm.
- Deconvolved inverted fluorescence micrographs of BiFC signal of Chm7‐VN and Snf7‐VC in the indicated null backgrounds. Cell borders are outlined. Scale bar is 5 μm.
- Deconvolved fluorescence micrographs of BiFC signal of Chm7‐VN and Snf7‐VC with NE/ER marker HDEL‐dsRED and merge. Note there is a small amount of bleed‐through of the HDEL‐dsRED into the Venus channel. Cell borders are outlined. Scale bar is 5 μm.
- Deconvolved fluorescence micrographs of Chm7‐GFP and Nup170‐mCherry (with merge) in the indicated strains. Scale bar is 5 μm.
Chm7 accumulates in the SINC
To further assess the molecular determinants of Chm7 localization to the NE, we tested the distribution of Chm7‐GFP in knockout strains of different ESCRTs including vps20Δ, snf7Δ, and vps4Δ nulls. Consistent with the BiFC data, Chm7‐GFP accumulated in multiple puncta throughout the cell in both vps4Δ and snf7Δ cells, but was unaffected by deletion of VPS20 (Fig 5A). Moreover, NE foci could be specifically disrupted by deletion of HEH1 (Fig EV4D). In addition, as opposed to the single NE focus observed in wild‐type cells, Chm7‐GFP accumulated in multiple, fluorescently intense plaques reminiscent of the SINC in vps4Δ and snf7Δ cells, being most prevalent in vps4Δpom152Δ cells (Fig 5A; Webster et al, 2014). To assess whether these Chm7 localizations are equivalent to the previously described SINC, we simultaneously visualized Nup170‐mCherry. As shown in Fig 5B, there was a marked accumulation of Chm7‐GFP in SINCs such that virtually all SINCs contained Chm7 (Fig 5C). By contrast, there was little accumulation of Chm7‐GFP within morphologically similar clustered NPCs observed in nup133Δ cells (Fig 5C and D). To quantify the level of Chm7 SINC accumulation, we measured the fluorescence intensity of Chm7‐GFP in the SINC and compared these values to non‐SINC Chm7‐GFP foci that were similar to those seen in wild‐type cells. Clearly, there was substantially more Chm7‐GFP in SINCs, reaching a 10‐fold increase over non‐SINC compartments (Fig 5E). Moreover, this SINC accumulation likely occurred over several generations, as the levels of SINC‐enriched Chm7‐GFP correlated with the accumulation of Nup170‐mCherry (Fig 5F). Consistent with this idea, SINC‐associated Chm7‐GFP is retained in mother cells during mitosis (Movie EV2). Together, these data support that Chm7 can be recruited and further stabilized at a region of the NE enriched in (likely defective) NPC assembly intermediates. The enrichment of Chm7 in the SINC (over other ESCRTs that did not accumulate to this degree; Webster et al, 2014) also suggests that the accumulation of Chm7 in the absence of Vps4 and Pom152 might contribute to the underlying molecular pathology that leads to the SINC's expansion and toxicity.
Figure 5. Chm7 accumulates in the SINC.
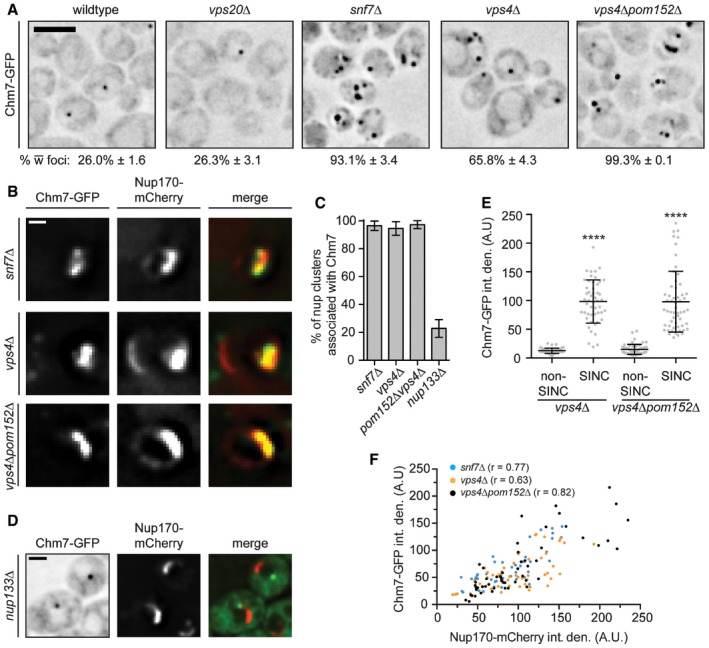
- Deconvolved inverted fluorescence micrographs of Chm7‐GFP in the indicated strains. Scale bar is 5 μm. The percentage of cells ± SD with Chm7‐GFP foci is indicated below each panel.
- Deconvolved fluorescence images of SINC‐containing nuclei in vps4Δ and vps4Δpom152Δ cells expressing Chm7‐GFP and Nup170‐mCherry (green, red, and merged images are shown). Scale bar is 1 μm.
- Plot of the proportion of nup clusters with Chm7‐GFP enrichment in the indicated genetic backgrounds. Error bars are SD of the mean from three independent replicates.
- Deconvolved fluorescence micrograph showing the lack of colocalization of Chm7‐GFP and clustered Nup170‐mCherry in a nup133Δ strain. Scale bar is 1 μm.
- Plot of the total fluorescence/integrated density (in arbitrary units, A.U.) of Chm7‐GFP within (and outside of) the SINC. Error bars represent SD deviation of the mean from > 50 SINC or non‐SINC accumulations pooled from three independent replicates. P‐values from unpaired Student's t‐tests with Welch's correction. ****P ≤ 0.0001.
- Correlation of the total fluorescence intensity/integrated density of SINC‐enriched Chm7‐GFP and Nup170‐mCherry within individual SINCs. Linear regression calculated from 150 SINCs pooled from three independent replicates; r is the linear correlation (Pearson's) coefficient.
Chm7 is required for SINC formation
To test the idea that Chm7 might be required to form the SINC in genetic backgrounds that promote its formation, we crossed vps4Δpom152Δ and chm7Δ cells and analyzed the growth of their progeny. Surprisingly, we observed a suppression of the impaired fitness of vps4Δpom152Δ strains after CHM7 deletion, as the vps4Δpom152Δchm7Δ strain grew indistinguishably from wild‐type cells (Fig 6A). We considered two possible models to explain how deletion of CHM7 might rescue the vps4Δpom152Δ growth defect. In one, endocytic sorting defects in vps4Δ strains could be rescued by deletion of CHM7. As shown in Fig 6B and C, this did not seem to be the case, as the distribution of the model endocytic cargo Sna3‐mCherry (Reggiori & Pelham, 2001; Russell et al, 2012) remained in class E‐compartments in both vps4Δ and vps4Δchm7Δ cells, and was not properly targeted to vacuoles as in wild‐type and chm7Δ cells (Fig 6C). Alternatively, in the second model, Chm7 might directly contribute to SINC formation, which would then be the underlying cause of toxicity. Consistent with such a model, GFP‐Nup49 no longer accumulated in the SINC in vps4Δchm7Δ or vps4Δpom152Δchm7Δ cells, despite its clear presence in CHM7‐containing backgrounds (Fig 6D and E). These surprising data suggest that the SINC is a product of a toxic gain‐of‐function of Chm7.
Figure 6. CHM7 is required for SINC formation.
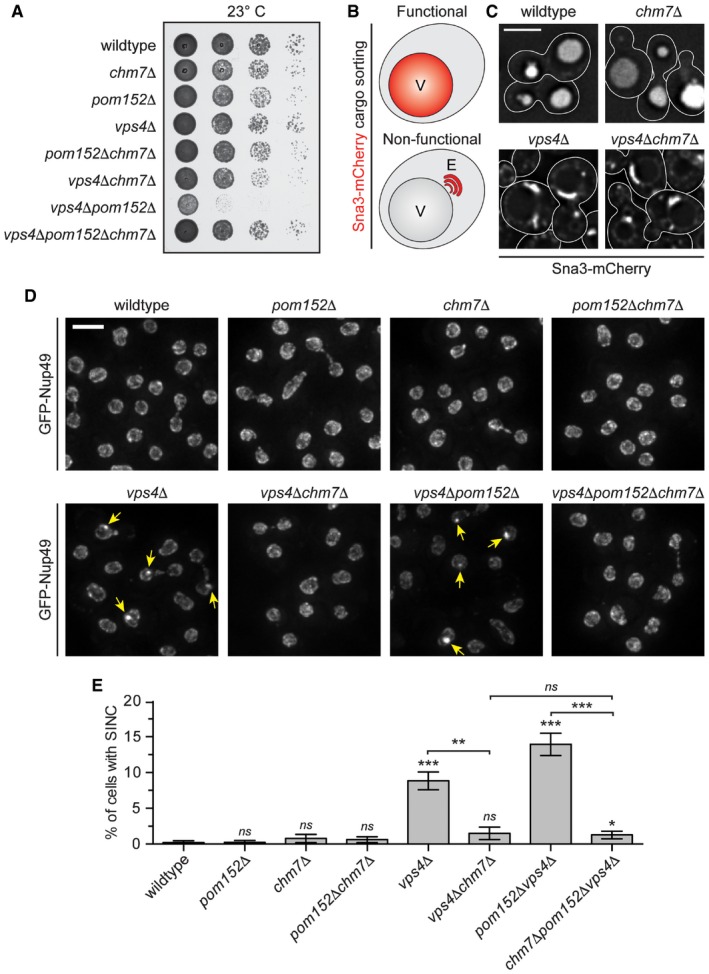
- Tenfold serial dilutions of the indicated yeast strains were grown on YPD at 23°C for 3 days before imaging.
- Schematic of Sna3‐mCherry localization. Sna3 accumulates in the vacuole (V) or class E compartment (E) in cells with, or lacking, ESCRT function, respectively.
- Deconvolved fluorescence micrographs of Sna3‐mCherry in the indicated genetic backgrounds. Scale bar is 5 μm.
- Deconvolved fluorescence micrographs (maximum intensity projections of a z‐series of images) of GFP‐Nup49 in the indicated yeast strains (arrows indicate SINCs). Scale bar is 5 ?m.
- Plot of proportion of cells in the indicated strains with SINCs. Error bars are the SD from the mean from three independent replicates quantifying > 300 cells for each strain. P‐values from unpaired Student's t‐test where ns is P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
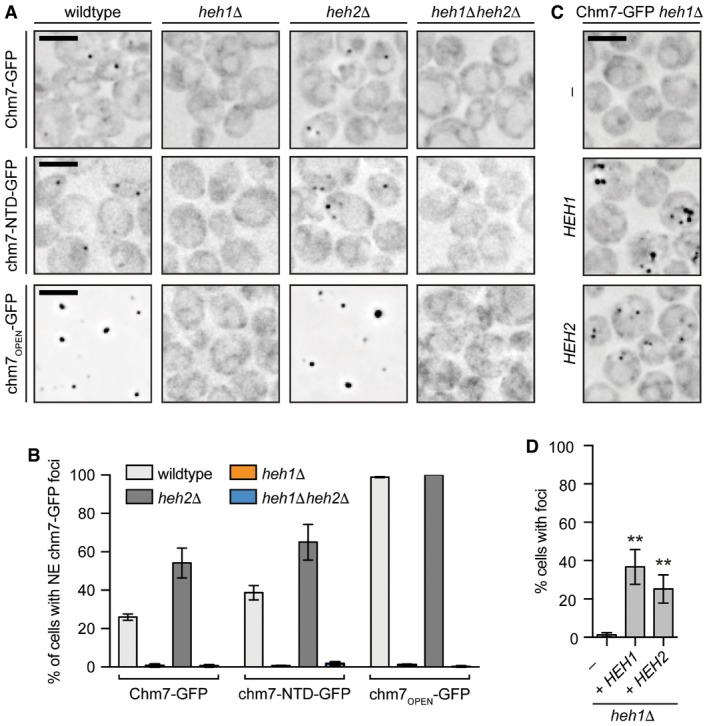
Perturbation to NPC assembly, not nuclear transport, affects Chm7 distribution
Because the SINC is filled with newly synthesized nups that lead to the formation of defective NPCs (Webster et al, 2014), we hypothesized that Chm7's enrichment in the SINC reflected an ability to recognize sites of assembling (or misassembling) NPCs. We therefore tested Chm7‐GFP localization in several yeast strains with deletions in non‐essential nup genes, including components of the membrane ring (Pom152), the inner ring (Nup170 and Nup157), the outer ring (Nup133), the central transport channel (Nup116), and nuclear basket (Mlp1 and Mlp2) (see Fig EV5A for diagram of NPC). We also tested established alleles in which NPC assembly is disrupted, including apq12Δ (Scarcelli et al, 2007), nic96‐1 (Zabel et al, 1996), and nup192‐15 (Kosova et al, 1999), although it is likely that NPC assembly is at least partially affected in all of the strains.
Figure EV5. Chm7‐GFP analysis in NPC‐defective strains.
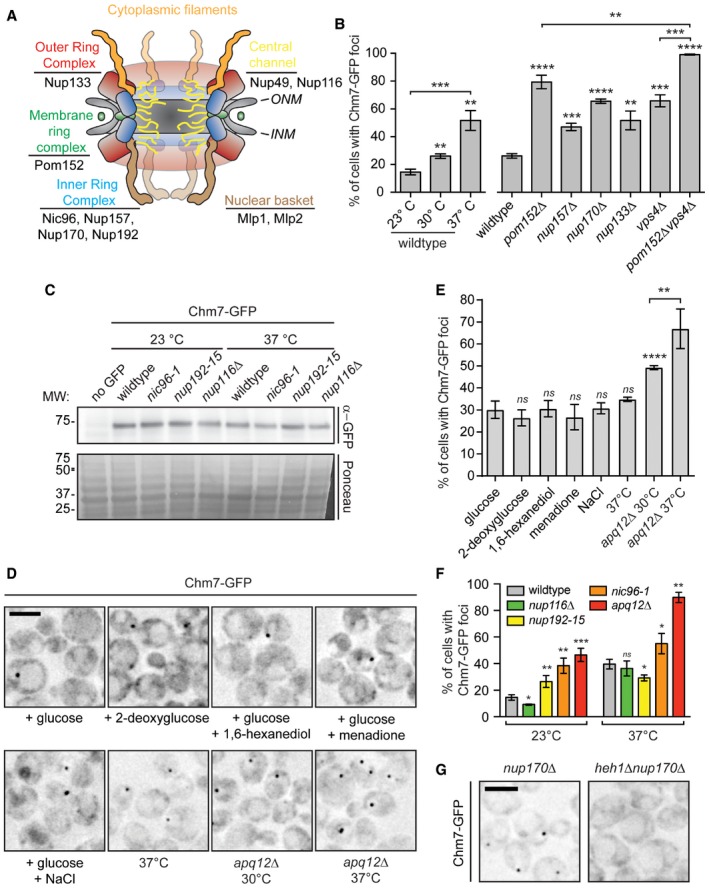
- Schematic of the yeast NPC depicting distinct nup subcomplexes and corresponding architectural units. Listed nups are referred to in the text.
- Plot of the percentage of cells with Chm7‐GFP foci in the indicated genetic backgrounds and temperatures (23 and 37°C temperature shifts were for 5 h; where not indicated, cells grown at 30°C). Error bars are SD from the mean from three independent experiments where > 175 cells per strain or condition were assessed. P‐values from unpaired Student's t‐test. **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
- Western blot using an anti‐GFP antibody and ECL to detect Chm7‐GFP levels in the indicated genetic backgrounds. Cells were grown at the indicated temperatures for 5 h prior to protein harvesting or 3 h for nup116Δ cells. Bottom panel is Ponceau‐stained nitrocellulose membrane to show relative protein loads.
- Deconvolved inverted fluorescence micrographs of Chm7‐GFP in wild‐type or apq12Δ strains treated as indicated for 45 min. Scale bar is 5 μm.
- Plot of the percentage of cells with Chm7‐GFP foci in wild‐type cells or apq12Δ cells treated as indicated. Data are from three independent replicates of > 150 cells per condition. Error bars represent SD from the mean. P‐values are from unpaired Student's t‐test where ns is P > 0.05; **P ≤ 0.01 ; ****P ≤ 0.0001.
- Plot of the percentage of cells with Chm7‐GFP foci in the indicated strains after 5 h at the indicated temperature or 3 h for nup116Δ. Data are from three independent replicates of > 100 cells. Error bars represent SD from the mean. P‐values from unpaired Student's t‐test where ns is P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
- Deconvolved inverted fluorescence micrographs of Chm7‐GFP in the indicated strains. Scale bar is 5 μm.
First, there was a general increase in both the percentage of cells (Figs 7A and EV5B) and number of foci per cell (Fig 7B) in many of the non‐essential nup knockout strains, including nup170Δ, nup133Δ, and nup157Δ nulls. The total levels of Chm7‐GFP were not altered (Fig EV1D) and Chm7‐GFP recruitment remained dependent on HEH1 (Fig EV5G). These effects were nonetheless specific, as the level of Chm7 accumulation at the NE was indistinguishable from wild‐type after disruption of two nuclear basket proteins, Mlp1 and Mlp2 (Fig 7A and B). In addition, pom152Δ cells showed particularly robust changes to Chm7 distribution, with an increase from ~26% to ~80% of cells with Chm7‐GFP NE foci, ~70% of which had more than one focus (Fig 7A and B). As Chm7 responded to deletion of components in several nup subcomplexes, we wondered whether a non‐specific disruption of nuclear transport might underlie the mechanism of Chm7 recruitment. We therefore tested whether the global inhibition of active nuclear transport or the NPC diffusion barrier affected Chm7 recruitment by treating cells with the energy poison 2‐deoxyglucose (Shulga et al, 1996, 2000; Timney et al, 2006) or 1,6 hexanediol (Ribbeck & Görlich, 2002; Shulga & Goldfarb, 2003), respectively. As shown in Fig EV5D and E, neither of these treatments influenced Chm7 distribution.
Interestingly, for temperature‐sensitive strains like nup116Δ (Wente & Blobel, 1993), apq12Δ, nic96‐1, and nup192‐15, there were clear differences in the properties of Chm7 NE recruitment. For example, while at both the permissive (23°C) and non‐permissive temperatures (37°C) of nic96‐1 and nup192‐15 cells there was a significant increase in the number of cells with Chm7‐GFP foci (Figs 7C and EV5F) and a larger distribution in the number of foci per cell (Fig 7D), the most significant changes were observed under conditions in which these alleles are only partially compromised (i.e., lower temperatures). In contrast, in both apq12Δ and nup116Δ cells, the most significant changes to foci number occurred at the higher temperature (Figs 7C and D, and EV5F). We also note that both the percentage of cells and number of foci/cell increased in wild‐type cells (albeit to a lesser extent) at 37°C (Figs 7D and EV5B), but this was not due to the triggering of a general stress response (Fig EV5D and E).
The most striking visual NE accumulation was observed in a subpopulation of nup116Δ cells at 37°C (Fig 8A) where Chm7‐GFP was no longer focal, but instead decorated virtually the entire nuclear periphery (Fig 8B). This result was particularly compelling, as the ultrastructure of the nup116Δ NEs at 37°C was previously shown to contain NE herniations and NPCs covered with double membrane seals (Wente & Blobel, 1993), observations that we reproduced (Fig 8C). Given the morphology of these structures, we considered that Chm7 might contribute to their formation. As a preliminary test to this hypothesis, we assessed whether CHM7 was required for viability of nup116Δ cells. As nup116Δ knockout cells were barely viable even at 23°C, these experiments were performed in the presence of a URA3‐expressing covering plasmid encoding NUP116 that is selected against in the presence of the drug 5‐fluoroorotic acid (5‐FOA). As shown in Fig 8D (left panel), without selective pressure all URA3‐containing strains had identical growth characteristics. In marked contrast, several colonies of nup116Δ cells grew on 5‐FOA plates but far fewer and smaller nup116Δchm7Δ colonies survived the 5‐FOA treatment (Fig 8D, right panel). Similar results were observed with nup116Δheh1Δ strains, while nup116Δheh2Δ cells failed to survive. Thus, nup116Δ cells show synthetic genetic aggravating interactions with CHM7, HEH1, and HEH2, supporting the conclusion that the recruitment of Chm7 to the NE protects nup116Δ cell viability.
Figure 8. nup116Δ cells require CHM7 for growth and accumulate Chm7 and NE herniations.
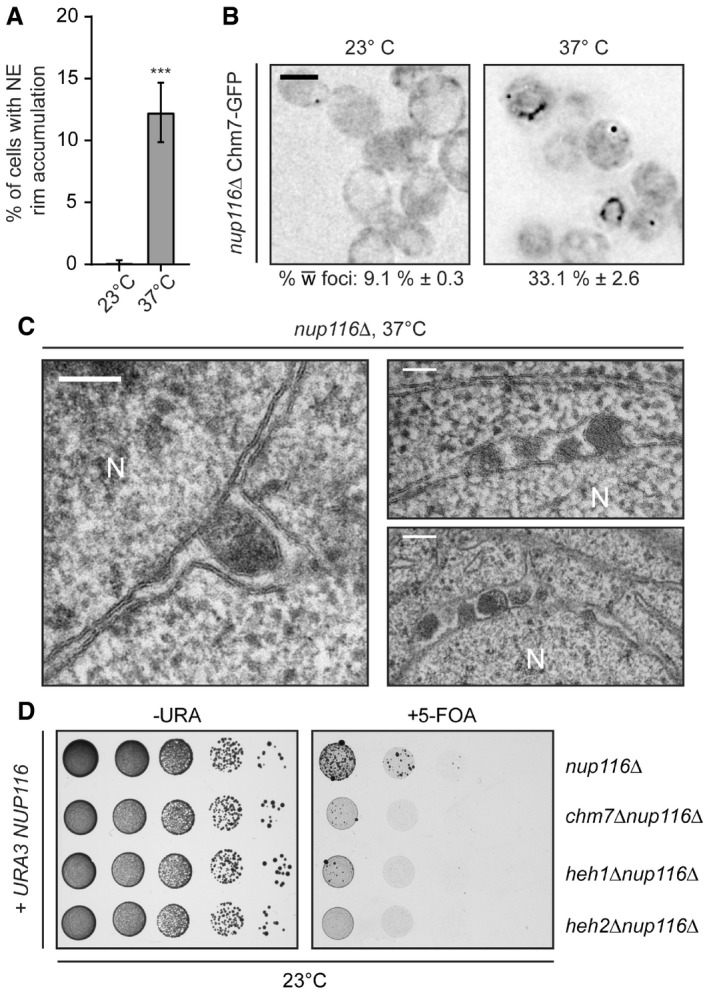
- Plot of the percentage of nup116Δ cells incubated for 3 h at 37°C with nuclear rim‐like accumulations of Chm7‐GFP. Error bars are SD from the mean from three independent replicates of > 100 cells. P‐values from unpaired Student's t‐test; ***P ≤ 0.001.
- Deconvolved inverted fluorescence micrographs of Chm7‐GFP in nup116Δ cells at either 23°C or grown for 3 h at 37°C. Percentages ± SD of the proportion of cells with Chm7‐GFP NE foci are shown under each panel (see Fig EV5F).
- Electron micrographs showing herniations of the NE in nup116Δ cells after incubation for 3 h at 37°C. N denotes nuclear interior. Scale bars are 100 nm.
- Tenfold serial dilutions of the indicated strains grown in medium permissive (‐URA) or restrictive (5‐FOA) of the retention of a URA3/NUP116 plasmid. Images acquired after incubation for 3 days (left) or 6 days (right) at 23°C.
Chm7 protects nuclear compartmentalization
In addition to influencing Chm7 NE recruitment in an analogous manner, nup116Δ and apq12Δ cells share a similar subset of genetic interactions with CHM7 (Bauer et al, 2015), HEH1, and HEH2 (Yewdell et al, 2011). Consistent with this, both strains also have similar NE herniations at 37°C (Scarcelli et al, 2007), although these phenotypes and growth rates are less severe in apq12Δ cells, making this strain more amenable to experimental manipulation. We therefore explored the function of Chm7 by testing the robustness of nuclear compartmentalization through the localization of a NLS‐GFP reporter in chm7Δ strains. In wild‐type cells, this reporter accumulates in the nucleus of all cells with a mean nuclear to cytosolic (N:C) ratio of ~1.6, which is slightly diminished at 37°C (Fig 9A and B). NLS‐GFP exhibited more elevated N:C ratios in apq12Δ cells as it was produced at higher levels in these strains (Appendix Fig S2B). Strikingly, however, at 37°C, ~35% of chm7Δapq12Δ cells showed a complete loss of nuclear compartmentalization with the NLS‐GFP reporter being evenly distributed throughout the nucleus and cytosol of these cells (Fig 9B and C). Thus, these results are consistent with the interpretation that the loss of viability of chm7Δapq12Δ cells is due to severe disruptions of the NE barrier either through leakage of defective NPCs or NE ruptures. Our data thus support a model in which Chm7 and the ESCRTs, which are recruited to the NE through interactions with LEM domain proteins, protect the nuclear compartment.
Figure 9. Chm7 protects nuclear compartmentalization.
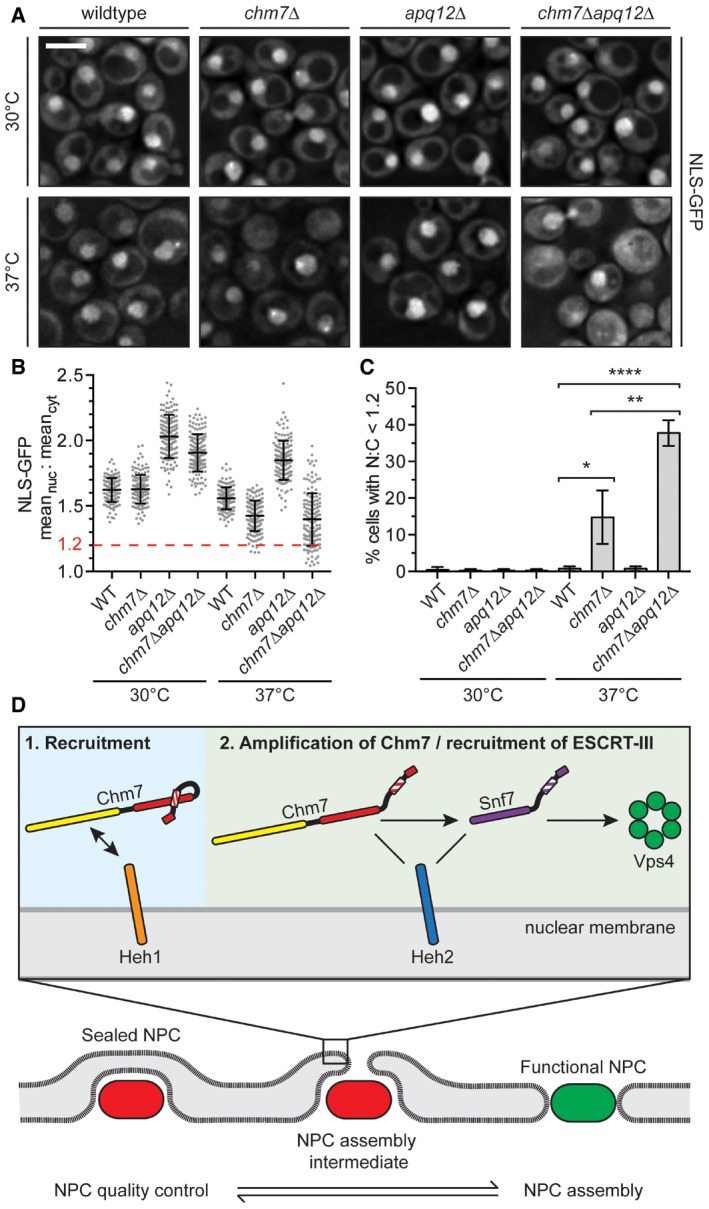
- Deconvolved fluorescence micrographs of NLS‐GFP in the indicated yeast strains at either 30°C or after 2 h at 37°C. Scale bar is 5 μm.
- Plot of the mean nuclear to cytosolic fluorescence intensity ratio of NLS‐GFP from cells represented in (A). Error bars are SD from the mean from three independent replicates of 150 cells from each strain. Only chm7Δ and chm7Δapq12Δ cells have N:C ratios below 1.2 (dotted red line).
- Re‐plotting of data from (B) showing the percentage of cells with a N:C NLS‐GFP fluorescence ratio < 1.2. Error bars are SD from the mean from three independent replicates of 150 cells/strain. P‐values from Student's t‐test; *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001.
- Model of a proposed stepwise recruitment and activation of Chm7 and other ESCRTs at defective NPCs (bottom, red), or NE holes.
Discussion
Nuclear compartmentalization is established by the enclosure of the genomic material by the double membrane NE and the selective transport and size‐selective diffusion barrier imposed by NPCs. Our work, and that of others, supports that the ESCRTs play integral roles in ensuring that both of these barrier mechanisms remain functional through the lifetime of a cell (Webster et al, 2014; Olmos et al, 2015; Vietri et al, 2015; Denais et al, 2016; Raab et al, 2016). In prior work, we argued that the complexity of NPC assembly, which requires the recruitment of hundreds of proteins to a NPC assembly site, as well as the fusion of the inner and outer nuclear membranes, is likely error‐prone and might pose a threat to NE integrity (Webster et al, 2014). We further proposed that Vps4 and Snf7 play a role in surveilling NPC assembly, but the mechanism of ESCRT function remained ill defined. This was due, in part, to challenges with visualizing a defined pool of ESCRT components at the NE. Our discovery that Chm7 localizes to NE foci has provided us with a unique window into these events, and we suggest that Chm7 (along with Heh1) defines a previously undiscovered NE structure that represents sites of ESCRT‐III function at the budding yeast NE.
A major conceptual challenge is to consider how the ESCRT‐III machinery might interface with the NPC assembly pathway, given that both could act on the same intermediate (i.e., a NE pore). While it remains formally plausible that ESCRT‐III could help to drive membrane invaginations to support pore formation (Webster & Lusk, 2015), its established activities suggest that ESCRT‐III is most likely to fuse membranes together by driving membrane scission (i.e., to close NE holes), making this scenario less attractive (Hurley, 2015; Campsteijn et al, 2016), a point emphasized by recent work supporting a scission model acting to seal the NE at mitotic exit (Olmos et al, 2015; Vietri et al, 2015). In light of this, we suggest two models that would be consistent with ESCRT‐III functions at the NE and elsewhere. In one, defective NPCs (or NPC assembly intermediates) are removed from the NE, ultimately leaving NE pores that are sealed by ESCRT‐III. Such a mechanism would be conceptually analogous to coupling the removal of spindle microtubules by spastin and the sealing of the remaining NE hole by ESCRT‐III (Vietri et al, 2015). However, a pathway capable of degrading or breaking down NPCs has not yet been described (although nup phosphorylation, which drives NPC disassembly in mitosis in many eukaryotes could underlie such a mechanism, Laurell et al, 2011). Indeed, the NPC scaffold is remarkably stable in post‐mitotic cells (D'Angelo et al, 2009; Savas et al, 2012; Toyama et al, 2013), and even in yeast, the primary determinant of nup turnover is its rapid cell division, which dilutes “old” pools of nups (Christiano et al, 2014). Nonetheless, there is evidence in mammalian tissue culture lines that NPCs might be disassembled during interphase, suggesting that future studies will be necessary to fully assess this possibility (Dultz & Ellenberg, 2010). We favor, however, an alternative quality control model in which defective NPCs might be sealed off en bloc through membrane expansion and sealing that would not require NPC removal (Fig 9D). Importantly, since in theory a single defective NPC could lead to a loss of nuclear‐cytoplasmic compartmentalization, such a mechanism would mitigate the impact of a small number of defective NPCs or NPC assembly intermediates, allowing the remaining functional NPCs to effectively mediate regulated transport.
Evidence that defective NPCs might be sealed by exposed, closely apposed membranes comes from decades‐old electron micrographs of nup116Δ cells (and see Fig 8C), in which NPCs are enclosed by a double membrane (Wente & Blobel, 1993). Similar structures have been observed in apq12Δ cells (Scarcelli et al, 2007), as well as those expressing alleles of gle2 (Murphy et al, 1996), and may reside in the SINC as well (Webster et al, 2014). As the seal is a double membrane, a hypothetical intermediate would be the exposure (or expansion) of a “naked” nuclear pore membrane (Wente & Blobel, 1993); the closure of such an expanded or exposed membrane annulus would be compatible with an ESCRT‐driven scission step (Fig 9D). While it is possible that such events would impair viability by further compromising nuclear transport, loss of CHM7 exacerbates the already impaired fitness of nup116Δ (Fig 8D) and apq12Δ (Bauer et al, 2015) cells, suggesting that Chm7 promotes, in a protective mechanism, the response to defective NPC structures, consistent with the quality control mechanism described above. This model predicts that defective NPCs in chm7Δnup116Δ and chm7Δapq12Δ genetic backgrounds would fail to be sealed; indeed, we observe a loss of nuclear compartmentalization in these cells at higher temperatures (Fig 9A–C), in support of this model. We cannot exclude the possibility that the equilibration of the NLS‐GFP between the cytosol and nucleus in chm7Δapq12Δ cells is caused by NE ruptures, either independently or in concert with defective NPC assembly. However, given the similarity of the “substrate” that would be subjected to ESCRT‐dependent membrane scission at both NE ruptures and defective NPCs, we suggest that both likely proceed through a similar series of ESCRT‐III‐dependent events (Fig 9D).
Moreover, consistent with the idea that Chm7 acts at nascent and/or defective NPCs, our data strongly support that Chm7 is recruited to the NE upon blocks to NPC assembly, suggesting that Chm7 is poised to act at these sites. The close coupling of NPC assembly and the sealing off of NPCs (or perhaps the sealing of NE pores generated during defective assembly) makes considerable sense, as prolonged losses of nuclear compartmentalization are clearly deleterious to the cell. For example, NE ruptures are associated with increased DNA damage (Hatch et al, 2013; Harada et al, 2014; Maciejowski et al, 2015; Zhang et al, 2015; Denais et al, 2016; Raab et al, 2016). A key goal for the future is defining how Chm7 recognizes a defective NPC or weakened NE that might be prone to (or has just) rupture(d). Interestingly, our data are inconsistent with a model in which a change in nuclear transport efficiency or a disruption of the NPC diffusion barrier triggers NE repair (Fig EV5D and E), suggesting that there might be a mechanism of directly monitoring membrane integrity. While this could occur through surveillance of membrane properties, an alternative possibility is that the local release of luminal calcium could occur if NE integrity is lost; such a mechanism would have parallels with established modes of membrane repair, as local calcium release has been shown to play a role in the recruitment of ALG‐2 followed by ESCRT‐III to plasma membrane ruptures (Scheffer et al, 2014).
Analogous to the role of ALG‐2, our data suggest that Heh1 is the most upstream component of the NPC assembly surveillance pathway, as it is essential for Chm7 NE recruitment (Fig 2). Consistent with the general importance of Heh1 in this pathway, studies performed concurrently with ours support that in both fission yeast and in mammalian cell lines, the HEH1 orthologue LEM2 is required for Chm7 recruitment to the NE (Gu et al, 2016). Moreover, discontinuities in the NE have been observed in lem2Δ fission yeast (Gonzalez et al, 2012) suggesting they might be unable to recruit the ESCRT machinery to repair NE ruptures. While the relationship between NPC assembly and surveillance is less established in fission yeast and mammalian cells, we note that analogous genetic interactions exist between nup genes and ESCRTs in both budding and fission yeast (Frost et al, 2012), supporting the general conservation of these pathways.
Our data support a temporal model of ESCRT recruitment to the NE that is analogous to the stepwise process observed at endosomes, although this pathway is independent of Vps25 and Vps20. As diagramed in Fig 9D, we hypothesize that a defective NPC or NE rupture is recognized by Heh1, which recruits Chm7 via the chm7‐NTD, ESCRT‐II‐like domain. It remains unclear whether this is the result of a direct biochemical link between these proteins or whether Heh1 helps to establish a NE structure competent for chm7‐NTD binding. It will be particularly interesting to test whether the recently identified extended loop in the N‐terminus of mammalian CHMP7, which is required for its ER targeting, contributes to this interaction as this feature is at least partially conserved in yeast (Olmos et al, 2016; Appendix Fig S1).
The initial Heh1‐dependent recruitment of the chm7‐NTD to the NE appears to be followed by an amplification phase, which we interpret to involve Chm7 polymerization (Fig 9D). Consistent with this idea, the levels of Chm7 constructs that accumulate at the NE focus are increased when they contain its C‐terminal, ESCRT‐III domain, most dramatically when accompanied by deletion of its likely auto inhibitory helices, loss of which would be predicted to drive polymerization (Fig 1). Of course, it remains to be formally established whether Chm7 is capable of forming a polymer and whether its direct binding to Snf7 (Fig 3F) reflects the formation of a heterotypic polymer, or instead or in addition, whether this binding acts to stimulate Snf7 polymerization in analogy to the stepwise activation of Vps25 and Vps20 at endosomes. Nonetheless, as we observe substantial focal accumulations of Chm7 in snf7Δ and vps4Δ cells (Fig 5A), we suggest that these factors influence events downstream of Chm7 NE recruitment (Fig 9D). This proposed stepwise order of ESCRT function at the NE is consistent with mammalian cell culture studies in which CHMP7 is required for recruitment of other ESCRT‐III subunits like CHMP2 (Olmos et al, 2016) and CHMP4B (Vietri et al, 2015) to NE holes at the end of mitosis. Interestingly, our data also support that Chm7 function is locally regulated by other factors, including NE‐specific Heh2, which can directly bind ESCRT‐III domains in their active forms (Figs 3 and 9D).
Consistent with the idea that Chm7 activation might need to be tightly controlled to prevent it from taking on gain‐of‐function roles at the NE, the deletion of the factor required for ESCRT‐III polymer disassembly, Vps4, particularly in the context of pom152Δ, leads to the formation of the SINC (Fig 5). Our observation that SINC formation (and the aggravating synthetic growth delays of vps4Δpom152Δ cells) is dependent on CHM7 (Fig 6A), supports such a model of a toxic gain‐of‐function of Chm7 when it is not properly regulated. Interestingly, in a sister study in fission yeast, deletion of the CHM7 orthologue CMP7 also suppressed the growth delays and NE morphological abnormalities observed in fission yeast vps4Δ cells (as does LEM2/HEH1 deletion) suggesting the broad conservation of these functional interaction networks (Gu et al, 2016). We speculate that the over activation of a Chm7‐dependent NE pore closure machinery could lead to the inappropriate sealing of nascent NE pores essential for normal NPC assembly, which might explain why the SINC is enriched for newly synthesized NPCs without their full complement of nups (Webster et al, 2014). Together, this work reveals a role for Chm7 and ESCRT‐III components, which are recruited and regulated by integral NE membrane proteins of the LEM domain family, in protecting the nuclear compartment from a myriad of insults that could lead to a loss of nuclear compartmentalization.
Materials and Methods
Yeast strain generation and growth
All strains used in this study are listed in Table EV1. Gene knockouts and fluorescent tagging of endogenous genes were performed by PCR‐based integration using the pFA6a plasmid series (see Table EV2) as templates (Longtine et al, 1998; Van Driessche et al, 2005; Sung & Huh, 2007). Standard yeast protocols for transformation, mating, sporulation, and tetrad dissection were performed as described in Amberg et al (2005). Unless otherwise indicated, cells were grown to mid‐log phase in YPAD (1% bacto yeast extract [BD] 2% bacto peptone [BD], 2% d‐glucose [Sigma] and 0.025% adenine [Sigma]) or in complete synthetic medium (CSM) containing 2% d‐glucose and lacking one or more amino acids at 30°C. For comparing relative growth rates of yeast strains, equivalent numbers of cells from overnight cultures were spotted in 10‐fold serial dilutions onto YPD plates and imaged after 48 h at RT.
For rescue of synthetic sickness of chm7Δapq12Δ, CPL1299 was transformed with pRS416, pRS416‐HA‐CHM7, pRS416‐HA‐chm7‐NTD, or pRS416‐HA‐chm7‐CTD. To assess relative growth properties, transformants were spotted in 10‐fold serial dilutions onto –URA plates.
To assess epistasis of nup116Δchm7Δ, nup116Δheh1Δ, and nup116Δheh2Δ strains, a nup116Δ (BWCPL1622) covered with pRS416‐NUP116 was crossed with chm7Δ and heh1Δheh2Δ strains (BWCPL1634, CPL120), sporulated, and dissected. Growth of double knockout spores was assessed on –URA plates, or, on 5‐FOA to force loss of the pRS416‐NUP116 plasmid.
Assessing localization of Chm7‐GFP in strains that are temperature sensitive was performed as follows: Strains were grown overnight at the permissive temperature, divided, and diluted into two cultures at an OD600 of 0.2. Individual cultures were then grown at 23°C (for cold sensitive strains) or 37°C for times indicated in figure legends before imaging.
Deoxyglucose, hexanediol, and stress treatments
Cells were collected by centrifugation and resuspended in YPAD containing either 2% 1,6‐hexanediol (Shulga & Goldfarb, 2003), 500 mM NaCl (Hohmann, 2002), 0.5 mM menadione (Flattery‐O'Brien et al, 1993), or CSM supplemented with 10 mM 2‐deoxy‐d‐glucose (Shulga et al, 1996). All chemical treatments were performed for 45 min at 30°C before imaging.
Plasmids
All plasmids are listed in Table EV2. All plasmids generated in this study were verified by sequencing. To generate pRS416‐NUP116, pRS426‐HEH1, and pRS426‐HEH2, the NUP116, HEH1, and HEH2 genes were PCR amplified from isolated chromosomal DNA with 5′ and 3′ regions encompassing the native promoters and terminators. The PCR products were inserted into either pRS416 or pRS426 (Christianson et al, 1992) using homologous recombination in yeast. Briefly, either pRS416 or pRS426 was linearized and co‐transformed with either NUP116, HEH1, or HEH2 PCR products into W303a. Transformants were selected on CSM‐URA plates and grown overnight in CSM‐URA before plasmid isolation using a modified protocol in Amberg et al (2005) that incorporates a plasmid purification step on Qiagen miniprep columns (Qiagen). Plasmids purified from yeast were subsequently transformed into E. coli to isolate individual plasmid clones, which were confirmed by sequencing.
To generate pRS416‐HA‐CHM7, the CHM7 ORF was PCR amplified using Q5 DNA polymerase (New England Biolabs) with sense primers that contain the sequence for the hemagglutinin (HA) epitope using a genomic DNA template. The HA‐CHM7 PCR product was assembled along with a PCR amplified CHM7 promoter into pRS416 using the Gibson Assembly MasterMix (New England Biolabs). To generate pRS416‐HA‐chm7‐NTD, a stop codon was introduced into the codon of amino acid 227 of the Chm7 sequence using the Q5 site‐directed mutagenesis kit (New England Biolabs). To generate HA‐chm7‐CTD (Fig 1A), mutagenic primers were designed to delete the chm7‐NTD sequence from pRS416‐HA‐CHM7. The mutagenic PCRs were carried out using the Q5 site‐directed mutagenesis kit (New England Biolabs).
pFA6a‐TRP1MX6‐GPD was generated by excising the GPD promoter from pRS425‐GPD and subcloning it into pFA6a‐TRP1MX6 (Longtine et al, 1998) using restriction enzymes PacI and BglII.
All other plasmids were generated using the Gibson Assembly MasterMix (New England Biolabs) according to the manufacturer's instructions. Briefly, coding regions of plasmid inserts were PCR amplified from W303 chromosomal DNA using Q5 DNA polymerase (New England Biolabs) and assembled into either pCS2 (Promega) linearized with BamHI/EcoRI (New England Biolabs), pGEX‐6P1 (GE Life Sciences) linearized with BamHI/XhoI, or pET28a linearized with XbaI/XhoI.
Recombinant protein production and purification
E. coli BL21 (DE3) cells expressing GST fusion proteins were grown overnight, diluted to an OD600 of 0.15 in 2xYT, and allowed to reach an OD600 of 0.5 before the addition of IPTG to a final concentration of 0.5 mM. Cultures were then shifted to 24°C for 6 h before collecting cells by centrifugation. Cell pellets derived from 50 ml of culture were flash frozen in liquid nitrogen and stored at −80°C before lysis and protein purification.
E. coli BL21 cells expressing His6‐MBP‐Snf7 construct were grown in 2xYT at 30°C. The cells were induced with 0.5 mM IPTG at OD600 0.8 and grown for 3 h. The His6‐MBP‐Snf7 protein was affinity purified in batch on Ni‐NTA resin (Qiagen) and eluted in 400 mM imidazole. To further increase purity, eluted fractions were affinity purified with an amylose resin (New England Biolabs) and eluted in 10 mM maltose.
Frozen cell pellets were resuspended in 15 ml lysis buffer (50 mM Tris pH 7.4, 300 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 10% glycerol, 0.5% NP‐40, 1 mM DTT, Roche complete protease inhibitors) and lysed by sonication (Branson Sonifier 450). Whole cell lysates were cleared by centrifugation for 15 min at 20,000 × g. Supernatants were incubated in batch with 100 μl of glutathione sepharose (GT) beads 4B (GE Healthcare) for 1 h, collected by centrifugation, and washed three times with lysis buffer before being used as bait in binding experiments (below) or released from beads (and GST) through incubation with HRV 3C protease (Thermo Scientific) overnight at 4°C.
Recombinant protein binding experiments
For Chm7 binding experiments, GST‐beads with bound GST or GST fusions of Chm7 were washed with a binding buffer of PBS containing 10% glycerol, 0.5% NP‐40, and 0.5 mM DTT. Beads were then incubated with purified His6‐MBP‐Snf7 or heh2(1‐308) generated from HRV 3C cleavage or in vitro transcription and translation (IVT; see below) for 1 h at 4°C, collected by centrifugation, and washed three times with binding buffer. Bound proteins were eluted with 30 μl of 2× SDS–PAGE sample buffer before separation by SDS–PAGE. Proteins were visualized by staining with SimplyBlue SafeStain (Invitrogen), Western blot, or autoradiography.
To assess competitive (or cooperative) binding of heh2(1‐308) and His6‐MBP‐Snf7 to fragments of Chm7, equimolar quantities of the purified proteins were combined with GT‐beads with bound GST, or GST fusions of Chm7 in binding buffer. GT‐bead‐bound proteins were quantified by Pierce Coomassie protein assay kit. The binding was carried out for 1 h at 4°C with the samples under gentle rotation. The beads were collected by centrifugation, washed three times with binding buffer, and eluted with 30 μl of 2× SDS–PAGE sample buffer to visualize on SDS–PAGE. In all binding experiments, the relative amount of input and bound fractions was loaded onto gels to allow a direct 1:1 comparison.
In vitro transcription and translation (IVT) and autoradiography
TNT Quick Coupled Transcription/Translation System (Promega) was used to generate radiolabeled fragments of Heh2. Specifically, individual 40 μl IVT reactions were performed for 90 min at 30°C and consisted of 600 ng of plasmid DNA encoding HEH2 fragments, 20 μCi [35S] methionine, and 10 μl of IVT mix. About 10 μl of the completed IVT reaction was used within individual binding experiments. SDS–PAGE gels with radiolabeled proteins were dried at 80°C for 1 h using a gel dryer (EC355, E‐C Apparatus Corporation) and exposed to autoradiography film (Bio‐Rad Molecular Imager FX Imaging Screen) for 3 days. [35S] Methionine‐labeled proteins were visualized using a Bio‐Rad Personal Molecular Imager (Bio‐Rad).
Western blotting
Proteins separated by SDS–PAGE were transferred to nitrocellulose for Western blotting with the following primary antibodies: anti‐GFP (gift of M. Rout), anti‐Snf7 (gift of D. Katzmann), and anti‐actin (mAbcam 8224). Primary antibodies were detected with anti‐rabbit HRP‐conjugated secondary antibodies followed by ECL. ECL visualized using a VersaDoc Imaging System (Bio‐Rad).
Fluorescence microscopy
All fluorescence images were acquired on a DeltaVision widefield deconvolution microscope (Applied Precision/GE Healthcare) equipped with a 100×, 1.40 numerical aperture objective (Olympus), solid state illumination, and an Evolve EMCCD camera (Photometrics). In all cases, Z‐stacks of images (0.2 μm sections) were acquired.
For timelapse experiments, cells were first immobilized on a 1.4% agarose pad containing CSM, 2% d‐glucose, 0.025% adenine, and sealed with VALAP (1:1:1, vaseline:lanolin:paraffin) before imaging.
Image processing and analysis
All fluorescence micrographs presented were deconvolved using the iterative algorithm in softWoRx (version 6.5.1; Applied Precision GE Healthcare) with subsequent processing and analyses performed in Fiji/ImageJ (Schindelin et al, 2015). Importantly, quantification of fluorescence intensities was performed on unprocessed images after background subtraction unless otherwise indicated.
To quantify the fluorescence intensity of Venus within BiFC experiments, the integrated density of Venus fluorescence within a central focal plane of an entire cell was measured.
Calculation of the mean intensity of individual Chm7‐GFP foci associated with the NE in the full length and truncated strains was measured and normalized to the mean fluorescence intensity of the extracellular background of the same image.
Similarly, the total fluorescence intensity of Chm7‐GFP associated (or not) with SINCs was measured using Nup170‐mCherry as a SINC landmark. To correlate the enrichment of Chm7‐GFP and Nup170‐mCherry in the SINC, the integrated density of Nup170‐mCherry and Chm7‐GFP fluorescence was measured and plotted on a correlation curve. The linear correlation coefficient (Pearson coefficient, r) was calculated using Prism (GraphPad).
To calculate the nuclear to cytosolic ratio of the NLS‐GFP reporter, the mean fluorescence of two identically sized regions of interest (one cytoplasmic, one within the nucleus) within a middle z‐section of individual cells was measured and related.
Plots and statistical analysis
All graphs and statistical analyses were generated and performed using Prism (GraphPad 6). P‐values for all graphs were generated with tests indicated within figure legends and are represented as follows: ns, P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. All error bars represent standard deviation from the mean.
Electron microscopy
To observe the ultrastructure of nup116Δ strains, unfixed cells were high‐pressure‐frozen using a Leica HMP100 at 2,000 psi and freeze‐substituted using a Leica Freeze AFS unit using 1% osmium tetroxide and 1% glutaraldehyde. Samples were infiltrated with Durcupan resin (Electron Microscopy Science) and cut using a Leica UltraCut UC7. Sections were collected on formvar/carbon coated nickel grids and stained with 2% uranyl acetate and lead citrate. Grids were viewed in a FEI Tecnai Biotwin TEM at 80 kv. Images were acquired using Morada CCD and iTEM (Olympus) software.
Author contributions
CPL, DJT, and BMW conceived of and designed all experiments and analyzed all data. DJT and BMW performed the majority of experiments with help from SEO with strain generation and image acquisition/analysis. Direct binding experiments were performed by JJ and SB. CPL supervised the project and wrote the manuscript with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Movie EV2
Source Data for Expanded View
Review Process File
Acknowledgements
We are grateful for discussions of unpublished data with Adam Frost and Jeremy Carlton; we thank Megan King for critical reading of the manuscript. We appreciate the generosity of Topher Carroll, Mike Rout, Rick Wozniak, and David Katzmann in sharing reagents and expertise and Morven Graham and Xinran Liu for EM. This work was supported by grants from the NIH GM105672 to C. Patrick Lusk. Brant Webster and David Thaller are also supported by NIH 5T32GM007223. Jens Jäger is supported by the Deutsche Forschungsgemeinschaft (Ja 2487/1‐1).
The EMBO Journal (2016) 35: 2447–2467
[The copyright line of this article was changed on 15 November 2016 after original publication.]
References
- Adell MAY, Vogel GF, Pakdel M, Müller M, Lindner H, Hess MW, Teis D (2014) Coordinated binding of Vps4 to ESCRT‐III drives membrane neck constriction during MVB vesicle formation. J Cell Biol 205: 33–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni‐Schmidt O, Williams R, Chait BT, Sali A, Rout MP (2007) The molecular architecture of the nuclear pore complex. Nature 450: 695–701 [DOI] [PubMed] [Google Scholar]
- Amberg DC, Burke D, Strathern JN (2005) Methods in yeast genetics. Cold Spring Harbor, NY: CSHL Press; [Google Scholar]
- von Appen A, Kosinski J, Sparks L, Ori A, DiGuilio AL, Vollmer B, Mackmull M‐T, Banterle N, Parca L, Kastritis P, Buczak K, Mosalaganti S, Hagen W, Andrés‐Pons A, Lemke EA, Bork P, Antonin W, Glavy JS, Bui KH, Beck M (2015) In situ structural analysis of the human nuclear pore complex. Nature 526: 140–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajorek M, Schubert HL, McCullough J, Langelier C, Eckert DM, Stubblefield W‐MB, Uter NT, Myszka DG, Hill CP, Sundquist WI (2009) Structural basis for ESCRT‐III protein autoinhibition. Nat Struct Mol Biol 16: 754–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer I, Brune T, Preiss R, Kölling R (2015) Evidence for a nonendosomal function of the Saccharomyces cerevisiae ESCRT‐III‐Like protein Chm7. Genetics 201: 1439–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui KH, von Appen A, DiGuilio AL, Ori A, Sparks L, Mackmull M‐T, Bock T, Hagen W, Andrés‐Pons A, Glavy JS, Beck M (2013) Integrated structural analysis of the human nuclear pore complex scaffold. Cell 155: 1233–1243 [DOI] [PubMed] [Google Scholar]
- Burke B, Stewart CL (2014) Functional architecture of the cell's nucleus in development, aging, and disease. Curr Top Dev Biol 109: 1–52 [DOI] [PubMed] [Google Scholar]
- Campsteijn C, Vietri M, Stenmark H (2016) Novel ESCRT functions in cell biology: spiraling out of control? Curr Opin Cell Biol 41: 1–8 [DOI] [PubMed] [Google Scholar]
- Casey AK, Chen S, Novick P, Ferro‐Novick S, Wente SR (2015) Nuclear pore complex integrity requires Lnp1, a regulator of cortical endoplasmic reticulum. Mol Biol Cell 26: 2833–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashikar AG, Shim S, Roth R, Maldazys MR, Heuser JE, Hanson PI (2014) Structure of cellular ESCRT‐III spirals and their relationship to HIV budding. Elife 3: e02184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadrin A, Hess B, San Roman M, Gatti X, Lombard B, Loew D, Barral Y, Palancade B, Doye V (2010) Pom33, a novel transmembrane nucleoporin required for proper nuclear pore complex distribution. J Cell Biol 189: 795–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Smoyer CJ, Slaughter BD, Unruh JR, Jaspersen SL (2014) The SUN protein Mps3 controls Ndc1 distribution and function on the nuclear membrane. J Cell Biol 204: 523–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaruttini N, Redondo‐Morata L, Colom A, Humbert F, Lenz M, Scheuring S, Roux A (2015) Relaxation of loaded ESCRT‐III spiral springs drives membrane deformation. Cell 163: 866–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiano R, Nagaraj N, Fröhlich F, Walther TC (2014) Global proteome turnover analyses of the yeasts S. cerevisiae and S. pombe . Cell Rep 9: 1959–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P (1992) Multifunctional yeast high‐copy‐number shuttle vectors. Gene 110: 119–122 [DOI] [PubMed] [Google Scholar]
- D'Angelo MA, Anderson DJ, Richard E, Hetzer MW (2006) Nuclear pores form de novo from both sides of the nuclear envelope. Science 312: 440–443 [DOI] [PubMed] [Google Scholar]
- D'Angelo MA, Raices M, Panowski SH, Hetzer MW (2009) Age‐dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 136: 284–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson RT, Lazarus MD, Hetzer MW, Wente SR (2009) ER membrane‐bending proteins are necessary for de novo nuclear pore formation. J Cell Biol 184: 659–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J (2016) Nuclear envelope rupture and repair during cancer cell migration. Science 352: 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan J‐A, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, Catanzaro JM, Daniel Ricketts M, Lamark T, Adam SA, Marmorstein R, Zong W‐X, Johansen T, Goldman RD, Adams PD, Berger SL (2015) Autophagy mediates degradation of nuclear lamina. Nature 527: 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet CM, Talamas JA, Hetzer MW (2010) Cell cycle‐dependent differences in nuclear pore complex assembly in metazoa. Cell 141: 1030–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drin G, Casella JF, Gautier R, Boehmer T, Schwartz TU, Antonny B (2007) A general amphipathic alpha‐helical motif for sensing membrane curvature. Nat Struct Mol Biol 14: 138–146 [DOI] [PubMed] [Google Scholar]
- Dultz E, Ellenberg J (2010) Live imaging of single nuclear pores reveals unique assembly kinetics and mechanism in interphase. J Cell Biol 191: 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flattery‐O'Brien J, Collinson LP, Dawes IW (1993) Saccharomyces cerevisiae has an inducible response to menadione which differs from that to hydrogen peroxide. J Gen Microbiol 139: 501–507 [DOI] [PubMed] [Google Scholar]
- Floch AG, Tareste D, Fuchs PFJ, Chadrin A, Naciri I, Léger T, Schlenstedt G, Palancade B, Doye V (2015) Nuclear pore targeting of the yeast Pom33 nucleoporin depends on karyopherin and lipid binding. J Cell Sci 128: 305–316 [DOI] [PubMed] [Google Scholar]
- Franz C, Walczak R, Yavuz S, Santarella R, Gentzel M, Askjaer P, Galy V, Hetzer M, Mattaj IW, Antonin W (2007) MEL‐28/ELYS is required for the recruitment of nucleoporins to chromatin and postmitotic nuclear pore complex assembly. EMBO Rep 8: 165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost A, Elgort MG, Brandman O, Ives C, Collins SR, Miller‐Vedam L, Weibezahn J, Hein MY, Poser I, Mann M, Hyman AA, Weissman JS (2012) Functional repurposing revealed by comparing S. pombe and S. cerevisiae genetic interactions. Cell 149: 1339–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazi‐Tabatabai S, Saksena S, Short JM, Pobbati AV, Veprintsev DB, Crowther RA, Emr SD, Egelman EH, Williams RL (2008) Structure and disassembly of filaments formed by the ESCRT‐III subunit Vps24. Structure 16: 1345–1356 [DOI] [PubMed] [Google Scholar]
- Gonzalez Y, Saito A, Sazer S (2012) Fission yeast Lem2 and Man1 perform fundamental functions of the animal cell nuclear lamina. Nucleus 3: 60–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Chen OS, Lajoie D, Ladinsky MS, Redd MJ, Nikolova L, Bjorkman PJ, Ullman KS, Sundquist WI, Frost A (2016) LEM2 and CHMP7 function in ESCRT‐dependent nuclear envelope closure in yeast and human cells. bioRxiv doi: 10.1101/049312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Roth R, Lin Y, Heuser JE (2008) Plasma membrane deformation by circular arrays of ESCRT‐III protein filaments. J Cell Biol 180: 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T, Swift J, Irianto J, Shin J‐W, Spinler KR, Athirasala A, Diegmiller R, Dingal PCDP, Ivanovska IL, Discher DE (2014) Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J Cell Biol 204: 669–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW (2013) Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154: 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch E, Hetzer M (2014) Breaching the nuclear envelope in development and disease. J Cell Biol 205: 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne WM, Buchkovich NJ, Zhao Y, Emr SD (2012) The endosomal sorting complex ESCRT‐II mediates the assembly and architecture of ESCRT‐III helices. Cell 151: 356–371 [DOI] [PubMed] [Google Scholar]
- Hodge CA, Choudhary V, Wolyniak MJ, Scarcelli JJ, Schneiter R, Cole CN (2010) Integral membrane proteins Brr6 and Apq12 link assembly of the nuclear pore complex to lipid homeostasis in the endoplasmic reticulum. J Cell Sci 123: 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann S (2002) Osmotic stress signaling and osmoadaptation in yeasts. Microbiol Mol Biol Rev 66: 300–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horii M, Shibata H, Kobayashi R, Katoh K, Yorikawa C, Yasuda J, Maki M (2006) CHMP7, a novel ESCRT‐III‐related protein, associates with CHMP4b and functions in the endosomal sorting pathway. Biochem J 400: 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH (2015) ESCRTs are everywhere. EMBO J 34: 2398–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im YJ, Wollert T, Boura E, Hurley JH (2009) Structure and function of the ESCRT‐II‐III interface in multivesicular body biogenesis. Dev Cell 17: 234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokhi V, Ashley J, Nunnari J, Noma A, Ito N, Wakabayashi‐Ito N, Moore MJ, Budnik V (2013) Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep 3: 988–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK (2008) Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys 37: 465–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer C, Skalicky JJ, Morita E, De Domenico I, Ward DM, Kaplan J, Sundquist WI (2008) Two distinct modes of ESCRT‐III recognition are required for VPS4 functions in lysosomal protein targeting and HIV‐1 budding. Dev Cell 15: 62–73 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- King MC, Lusk CP (2016) A model for coordinating nuclear mechanics and membrane remodeling to support nuclear integrity. Curr Opin Cell Biol 41: 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosova B, Pante N, Rollenhagen C, Hurt E (1999) Nup192p is a conserved nucleoporin with a preferential location at the inner site of the nuclear membrane. J Biol Chem 274: 22646–22651 [DOI] [PubMed] [Google Scholar]
- Lata S, Roessle M, Solomons J, Jamin M, Göttlinger HG, Svergun DI, Weissenhorn W (2008a) Structural basis for autoinhibition of ESCRT‐III CHMP3. J Mol Biol 378: 818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S, Schoehn G, Jain A, Pires R, Piehler J, Göttlinger HG, Weissenhorn W (2008b) Helical structures of ESCRT‐III are disassembled by VPS4. Science 321: 1354–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurell E, Beck K, Krupina K, Theerthagiri G, Bodenmiller B, Horvath P, Aebersold R, Antonin W, Kutay U (2011) Phosphorylation of Nup98 by multiple kinases is crucial for NPC disassembly during mitotic entry. Cell 144: 539–550 [DOI] [PubMed] [Google Scholar]
- Lone MA, Atkinson AE, Hodge CA, Cottier S, Martínez‐Montañés F, Maithel S, Mène‐Saffrané L, Cole CN, Schneiter R (2015) Yeast integral membrane proteins Apq12, Brl1, and Brr6 form a complex important for regulation of membrane homeostasis and nuclear pore complex biogenesis. Eukaryot Cell 14: 1217–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T (2015) Chromothripsis and kataegis induced by telomere crisis. Cell 163: 1641–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marelli M, Lusk CP, Chan H, Aitchison JD, Wozniak RW (2001) A link between the synthesis of nucleoporins and the biogenesis of the nuclear envelope. J Cell Biol 153: 709–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Fisher RD, Whitby FG, Sundquist WI, Hill CP (2008) ALIX‐CHMP4 interactions in the human ESCRT pathway. Proc Natl Acad Sci USA 105: 7687–7691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Colf LA, Sundquist WI (2013) Membrane fission reactions of the mammalian ESCRT pathway. Annu Rev Biochem 82: 663–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Clippinger AK, Talledge N, Skowyra ML, Saunders MG, Naismith TV, Colf LA, Afonine P, Arthur C, Sundquist WI, Hanson PI, Frost A (2015) Structure and membrane remodeling activity of ESCRT‐III helical polymers. Science 18: 1548–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros N, Cibulka J, Mendiburo MJ, Romanauska A, Schneider M, Köhler A (2015) Nuclear pore basket proteins are tethered to the nuclear envelope and can regulate membrane curvature. Dev Cell 33: 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H (2015) Receptor‐mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 522: 359–362 [DOI] [PubMed] [Google Scholar]
- Murphy R, Watkins J, Wente SR (1996) GLE2, a Saccharomyces cerevisiae homologue of the Schizosaccharomyces pombe export factor RAE1, is required for nuclear pore complex structure and function. Mol Biol Cell 7: 1921–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzioł T, Pineda‐Molina E, Ravelli RB, Zamborlini A, Usami Y, Göttlinger H, Weissenhorn W (2006) Structural basis for budding by the ESCRT‐III factor CHMP3. Dev Cell 10: 821–830 [DOI] [PubMed] [Google Scholar]
- Olmos Y, Hodgson L, Mantell J, Verkade P, Carlton JG (2015) ESCRT‐III controls nuclear envelope reformation. Nature 522: 236–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos Y, Carlton JG (2016) The ESCRT machinery: new roles at new holes. Curr Opin Cell Biol 38: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos Y, Perdrix‐Rosell A, Carlton JG (2016) Membrane binding by CHMP7 coordinates ESCRT‐III dependent nuclear envelope reformation. Curr Biol 26: 2635–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon‐Duménil AM, Manel N, Piel M (2016) ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352: 359–362 [DOI] [PubMed] [Google Scholar]
- Rasala BA, Ramos C, Harel A, Forbes DJ (2008) Capture of AT‐rich chromatin by ELYS recruits POM121 and NDC1 to initiate nuclear pore assembly. Mol Biol Cell 19: 3982–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Pelham HR (2001) Sorting of proteins into multivesicular bodies: ubiquitin‐dependent and ‐independent targeting. EMBO J 20: 5176–5186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribbeck K, Görlich D (2002) The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J 21: 2664–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts P, Moshitch‐Moshkovitz S, Kvam E, O'Toole E, Winey M, Goldfarb DS (2003) Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae . Mol Biol Cell 14: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem A, Gruber R, Shorer H, Shaulov L, Klein E, Harel A (2009) Importin beta regulates the seeding of chromatin with initiation sites for nuclear pore assembly. Mol Biol Cell 20: 4031–4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothballer A, Kutay U (2013) Poring over pores: nuclear pore complex insertion into the nuclear envelope. Trends Biochem Sci 38: 292–301 [DOI] [PubMed] [Google Scholar]
- Russell MRG, Shideler T, Nickerson DP, West M, Odorizzi G (2012) Class E compartments form in response to ESCRT dysfunction in yeast due to hyperactivity of the Vps21 Rab GTPase. J Cell Sci 125: 5208–5220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KJ, McCaffery JM, Wente SR (2003) The Ran GTPase cycle is required for yeast nuclear pore complex assembly. J Cell Biol 160: 1041–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksena S, Wahlman J, Teis D, Johnson AE, Emr SD (2009) Functional reconstitution of ESCRT‐III assembly and disassembly. Cell 136: 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, Toyama BH, Xu T, Yates JR, Hetzer MW (2012) Extremely long‐lived nuclear pore proteins in the rat brain. Science 335: 942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarcelli JJ, Hodge CA, Cole CN (2007) The yeast integral membrane protein Apq12 potentially links membrane dynamics to assembly of nuclear pore complexes. J Cell Biol 178: 799–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, Jaiswal JK (2014) Mechanism of Ca2+‐triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun 5: 5646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Rueden CT, Hiner MC, Eliceiri KW (2015) The ImageJ ecosystem: an open platform for biomedical image analysis. Mol Reprod Dev 82: 518–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiter R, Hitomi M, Ivessa AS, Fasch E, Kohlwein SD, Tartakoff A (1996) A yeast acetyl coenzyme A carboxylase mutant links very‐long‐chain fatty acid synthesis to the structure and function of the nuclear membrane‐pore complex. Mol Biol Cell 16: 7161–7172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q‐T, Schuh AL, Zheng Y, Quinney K, Wang L, Hanna M, Mitchell JC, Otegui MS, Ahlquist P, Cui Q, Audhya A (2014) Structural analysis and modeling reveals new mechanisms governing ESCRT‐III spiral filament assembly. J Cell Biol 206: 763–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S, Kimpler LA, Hanson PI (2007) Structure/function analysis of four core ESCRT‐III proteins reveals common regulatory role for extreme C‐terminal domain. Traffic 8: 1068–1079 [DOI] [PubMed] [Google Scholar]
- Shulga N, Roberts P, Gu Z, Spitz L, Tabb MM, Nomura M, Goldfarb DS (1996) In vivo nuclear transport kinetics in Saccharomyces cerevisiae: a role for heat shock protein 70 during targeting and translocation. J Cell Biol 135: 329–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulga N, Mosammaparast N, Wozniak R, Goldfarb DS (2000) Yeast nucleoporins involved in passive nuclear envelope permeability. J Cell Biol 149: 1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulga N, Goldfarb DS (2003) Binding dynamics of structural nucleoporins govern nuclear pore complex permeability and may mediate channel gating. Mol Cell Biol 23: 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, Moore MJ, Budnik V (2012) Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 149: 832–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung M‐K, Huh W‐K (2007) Bimolecular fluorescence complementation analysis system for in vivo detection of protein‐protein interaction in Saccharomyces cerevisiae . Yeast 24: 767–775 [DOI] [PubMed] [Google Scholar]
- Talamas JA, Hetzer MW (2011) POM121 and Sun1 play a role in early steps of interphase NPC assembly. J Cell Biol 194: 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Henne WM, Borbat PP, Buchkovich NJ, Freed JH, Mao Y, Fromme JC, Emr SD (2015) Structural basis for activation, assembly and membrane binding of ESCRT‐III Snf7 filaments. Elife 4: e12548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teis D, Saksena S, Emr SD (2008) Ordered assembly of the ESCRT‐III complex on endosomes is required to sequester cargo during MVB formation. Dev Cell 15: 578–589 [DOI] [PubMed] [Google Scholar]
- Teis D, Saksena S, Judson BL, Emr SD (2010) ESCRT‐II coordinates the assembly of ESCRT‐III filaments for cargo sorting and multivesicular body vesicle formation. EMBO J 29: 871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timney BL, Tetenbaum‐Novatt J, Agate DS, Williams R, Zhang W, Chait BT, Rout MP (2006) Simple kinetic relationships and nonspecific competition govern nuclear import rates in vivo . J Cell Biol 175: 579–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR, Hetzer MW (2013) Identification of long‐lived proteins reveals exceptional stability of essential cellular structures. Cell 154: 971–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Driessche B, Tafforeau L, Hentges P, Carr AM, Vandenhaute J (2005) Additional vectors for PCR‐based gene tagging in Saccharomyces cerevisiae and Schizosaccharomyces pombe using nourseothricin resistance. Yeast 22: 1061–1068 [DOI] [PubMed] [Google Scholar]
- Vietri M, Schink KO, Campsteijn C, Wegner CS, Schultz SW, Christ L, Thoresen SB, Brech A, Raiborg C, Stenmark H (2015) Spastin and ESCRT‐III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 522: 231–235 [DOI] [PubMed] [Google Scholar]
- Vollmer B, Schooley A, Sachdev R, Eisenhardt N, Schneider AM, Sieverding C, Madlung J, Gerken U, Macek B, Antonin W (2012) Dimerization and direct membrane interaction of Nup53 contribute to nuclear pore complex assembly. EMBO J 31: 4072–4084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer B, Lorenz M, Moreno‐Andrés D, Bodenhöfer M, De Magistris P, Astrinidis SA, Schooley A, Flötenmeyer M, Leptihn S, Antonin W (2015) Nup153 recruits the Nup107‐160 complex to the inner nuclear membrane for interphasic nuclear pore complex assembly. Dev Cell 33: 717–728 [DOI] [PubMed] [Google Scholar]
- Walther TC, Askjaer P, Gentzel M, Habermann A, Griffiths G, Wilm M, Mattaj IW, Hetzer M (2003) RanGTP mediates nuclear pore complex assembly. Nature 424: 689–694 [DOI] [PubMed] [Google Scholar]
- Wandke C, Kutay U (2013) Enclosing chromatin: reassembly of the nucleus after open mitosis. Cell 152: 1222–1225 [DOI] [PubMed] [Google Scholar]
- Webster BM, Colombi P, Jäger J, Lusk CP (2014) Surveillance of nuclear pore complex assembly by ESCRT‐III/Vps4. Cell 159: 388–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster BM, Lusk CP (2015) ESCRTs breach the nuclear border. Nucleus 6: 197–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster BM, Lusk CP (2016) Border safety: quality control at the nuclear envelope. Trends Cell Biol 26: 29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente SR, Blobel G (1993) A temperature‐sensitive NUP116 null mutant forms a nuclear envelope seal over the yeast nuclear pore complex thereby blocking nucleocytoplasmic traffic. J Cell Biol 123: 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Chen X‐W, Davies BA, Saltiel AR, Katzmann DJ, Xu Z (2009) Structural basis of Ist1 function and Ist1‐Did2 interaction in the multivesicular body pathway and cytokinesis. Mol Biol Cell 20: 3514–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell WT, Colombi P, Makhnevych T, Lusk CP (2011) Lumenal interactions in nuclear pore complex assembly and stability. Mol Biol Cell 22: 1375–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel U, Doye V, Tekotte H, Wepf R, Grandi P, Hurt EC (1996) Nic96p is required for nuclear pore formation and functionally interacts with a novel nucleoporin, Nup188p. J Cell Biol 133: 1141–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamborlini A, Usami Y, Radoshitzky SR, Popova E, Palu G, Göttlinger H (2006) Release of autoinhibition converts ESCRT‐III components into potent inhibitors of HIV‐1 budding. Proc Natl Acad Sci USA 103: 19140–19145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C‐Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D (2015) Chromothripsis from DNA damage in micronuclei. Nature 522: 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Movie EV2
Source Data for Expanded View
Review Process File