

Abstract
Signal transducer and activator of transcription 6 (STAT6) transmits signals from cytokines IL-4 and IL-13 and is activated in allergic airway disease. We are developing phosphopeptide mimetics targeting the SH2 domain of STAT6 to block recruitment to phosphotyrosine residues on IL-4 or IL-13 receptors and subsequent Tyr641 phosphorylation to inhibit the expression of genes contributing to asthma. Structure–affinity relationship studies showed that phosphopeptides based on Tyr631 from IL-4Rα bind with weak affinity to STAT6, whereas replacing the pY+3 residue with simple aryl and alkyl amides resulted in affinities in the mid to low nM range. A set of phosphatase-stable, cell-permeable prodrug analogues inhibited cytokine-stimulated STAT6 phosphorylation in both Beas-2B human airway cells and primary mouse T-lymphocytes at concentrations as low as 100 nM. IL-13-stimulated expression of CCL26 (eotaxin-3) was inhibited in a dose-dependent manner, demonstrating that targeting the SH2 domain blocks both phosphorylation and transcriptional activity of STAT6.
Graphical abstract
INTRODUCTION
Signal transducer and activator of transcription 6 (STAT6) transmits signals from cytokines interleukin 4 (IL-4) and IL-13.1–3 Under normal conditions, STAT6 is required for helper T cell type 2 (Th2) development in the context of allergic inflammatory responses and is hypothesized to contribute to the polarization of macrophages to the M2 phenotype.4 Upon receptor binding, both IL-4 and IL-13 induce phosphorylation of tyrosine residues located within the intracellular region of IL-4Rα (Figure 1). STAT6, via its SH2 domain, is recruited to these phosphotyrosine residues and is itself phosphorylated on Tyr641 by associated Janus kinases 1 and 3 (JAK1 and JAK3). Tyrosine phosphorylated STAT6 (pSTAT6) then dimerizes via reciprocal SH2 domain-pTyr interactions, allowing it to translocate to the nucleus, where it participates in the transcription of allergy-related genes. The IL-4/IL-13/JAK/STAT6 pathway plays a key role in asthma pathogenesis.1,2 Patients have elevated levels of IL-4 and IL-13 in their airways and elevated pSTAT6 levels have been found in bronchial epithelium.5 Stat6 knockout mice do not develop airway hyperresponsiveness (AHR) or lung pathology associated with asthma.6
Figure 1.
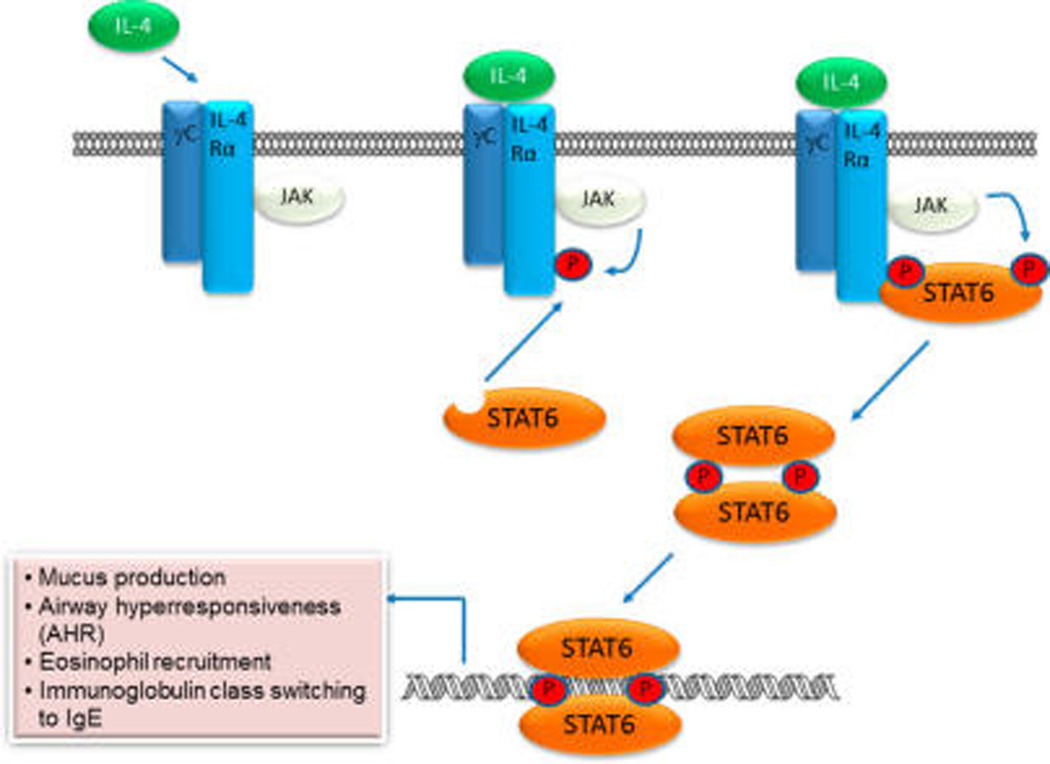
Schematic representation of the IL-4/JAK/STAT6 pathway. Note the IL-13 signals in an analogous manner using the IL-13R1/IL-4Rα coreceptor.
Intervention in the IL-4/IL-13/JAK/STAT6 pathway at several points for the treatment of asthma is being actively pursued by the pharmaceutical industry and academic laboratories.1,2 These include small molecules without described inhibitory mechanisms,7–12 non-SH2 domain directed dimerization inhibitors,13 JAK inhibitors,14,15 siRNA,6 decoy oligonucleotides,16 phosphopeptides targeting the SH2 domain of STAT6,17–19 antibodies targeting IL-13,20 and antibodies targeting IL-4Rα.20–22 Our approach is to target the SH2 domain of STAT6 with small molecule peptidomimetics to block recruitment to IL-4Rα, thereby preventing subsequent phosphorylation of Tyr641 by JAKs, dimerization, translocation to the nucleus, and transcription of genes, leading to airway inflammation. Proof of principle that steric blockade of the SH2 domain of STAT6 prevents recruitment to IL-4Rα, phosphorylation of Tyr641, and subsequent transcriptional activity leading to asthma symptoms has been demonstrated using phosphopeptides derived from Tyr631 of IL-4Rα, the docking site for STAT6,18 and Tyr641 of STAT6,17,19 with cell penetration peptides attached.
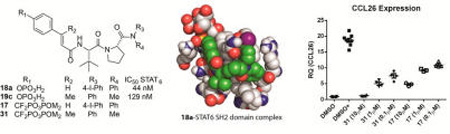
To the best of our knowledge, no systematic structure–affinity or structure–activity studies of phosphopeptide ligands of the SH2 domain of STAT6 have appeared in the literature. In this communication, we report truncation and amino acid substitutions on a phosphopeptide based on the sequence surrounding Tyr631 of IL-4Rα to gain an understanding of the determinants for binding affinity that could be used in the design of STAT6 antagonists. In parallel, a structure–affinity study of phosphate analogues of a phosphopeptide mimic reported in the patent literature23,24 was also conducted. A molecular docking study illustrated key intramolecular interactions that help explain the higher affinity of the mimics compared to the receptor-derived peptide ligand. Bis-POM prodrugs of phosphatase-stable phosphonodifluoromethyl analogues of a set of the new inhibitors provided a structure–activity relationship for inhibition of IL-4 and IL-13 stimulated phosphorylation of Tyr641in intact human airway epithelial cells. Several inhibitors induced significant inhibition at 100–500 nM in this cell line as well as in a human breast cancer cell line and primary murine T lymphocytes. Expression of the downstream target, CCL26 (eotaxin-3) was inhibited, which demonstrates that, in addition to inhibition of Tyr641 phosphorylation, this class of compounds blocks transcriptional activity of STAT6.
RESULTS
Affinity of Phosphopeptides for STAT6
To understand basic requirements for phosphopeptide binding to the SH2 domain and to possibly find a lead for peptide mimetic development, we screened a series of peptides based on the STAT6 docking site of IL-4Rα, Tyr 631. As SH2 domains typically recognize phosphotyrosine and two to four residues on its C-terminal side,25,26 we prepared a series of N-terminal pTyr phosphopeptides. The fluorescence polarization assay of Wu et al.27 was used to monitor affinity. A construct consisting of an N-terminal His6 tag, the coiled-coil domain, the DNA binding domain, the linker domain, and the SH2 domain of STAT6 was cloned, expressed in Escherichia coli, and purified by nickel IMAC chromatography for use in these studies. Overall, the peptides displayed rather weak avidity to STAT6 with IC50 values ranging from 2–6 µM (Table 1). Peptide length was important as affinities decreased from the octapeptide 1 (IC50 = 2 µM) to the tetrapeptide 5 (IC50 = 6 µM). Substitution of alanine for lysine at position pY+1 resulted in a significant reduction in affinity, while other amino acids such as tertiary leucine (Tle) and norleucine (Nle) were equipotent to lysine (7–9, Table 2). Thus, this position requires at the minimum β substitution and the ε-amino group of lysine does not provide interaction energy. A small screen of N-terminal hydrophobic appendages showed no significant effect on affinity (10–13, Table 2), and therefore further elaboration of this position was not pursued. Replacement of pTyr with constrained surrogates 4-phosphoryloxycinnamide (14) and 7-phosphoryloxyindole-2-caboxamide (15), which provided notable improvement in affinity for phosphopeptide inhibitors of STAT3,28 provided no significant increase in binding to STAT6 in this series of peptides (Table 3). Interestingly, incorporation of 7-phosphoryloxybenzofuran-2-caboxamide (16) led to a substantial decrease in affinity. This suggests that the NH group of the indole may be close to a hydrogen bond acceptor such as a carbonyl oxygen which would repel the oxygen atom of the benzofuran. In summary, phosphopeptides based on Tyr631 of the IL-4Rα displayed low affinity for STAT6 protein and were not useful as leads for inhibitor development.
Table 1.
Affinity of Phosphopeptides Based on Tyr631 of IL-4Rα for STAT6
| phosphopeptide | IC50 (µM) | |
|---|---|---|
| 1 | Ac-pTyr-Lys-Pro-Phe-Gln-Asp-Leu-Ile-NH2 | 1.8 ± 0.27 |
| 2 | Ac-pTyr-Lys-Pro-Phe-Gln-Asp-Leu-NH2 | 2.4 ± 0.66 |
| 3 | Ac-pTyr-Lys-Pro-Phe-Gln-Asp-NH2 | 4.5 ± 0.033 |
| 4 | Ac-pTyr-Lys-Pro-Phe-Gln-NH2 | 4.2 ± 0.25 |
| 5 | Ac-pTyr-Lys-Pro-Phe-NH2 | 6.1 ± 1.2 |
| 6 | Ac-pTyr-Lys-Pro-NH2 | 5.2 ± 0.096 |
Table 2.
N-Terminal Derivatization and Amino Acid Substitutions at Position pY+1 in Tetrapeptide Xaa-pTyr-Ybb-Pro-Phe-NH2
| Xaa | Ybb | IC50 (µM) | |
|---|---|---|---|
| 5 | CH3CO | Lys | 6.1 ± 1.2 |
| 7 | CH3CO | Tle | 4.0 ± 1.2 |
| 8 | CH3CO | Nle | 5.2 ± 1.2 |
| 9 | CH3CO | Ala | 14.9 ± 1.6 |
| 10 | PhCH2CH2CO | Lys | 8.1 ± 2.7 |
| 11 | PhCH2CO | Lys | 4.1 ± 2.3 |
| 12 | PhCO | Lys | 3.5 ± 0.78 |
| 13 | PhCH=CHCO | Lys | 6.6 ± 0.37 |
Table 3.
Effect of Constrained pTyr Surrogates on the Affinity of Xaa-Lys-Pro-Phe-NH2
| Xaa | IC50(µM) | ||
|---|---|---|---|
| 5 | Ac-pTyr | 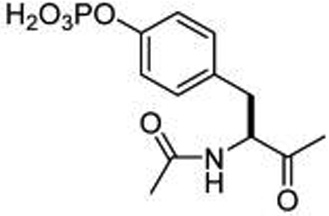 |
6.1 ± 1.2 |
| 14 | pCinn | 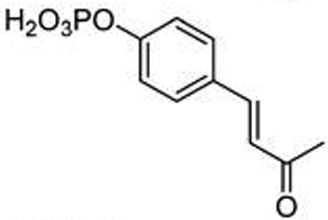 |
8.6 ± 2.8 |
| 15 | p-Indole | 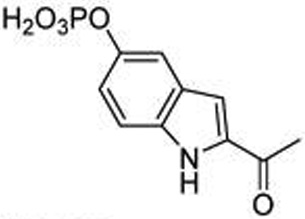 |
3.7 ± 0.052 |
| 16 | p-Benzofuran | 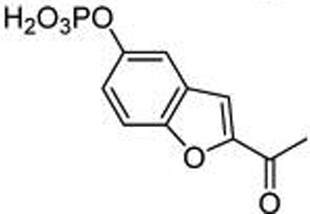 |
89.6 ± 1.5 |
In the late1990s, researchers from Tularik, Inc., recognized the potential of inhibition of STAT6 for the treatment of allergic airway disease and developed phosphopeptide mimics targeting the SH2 domain.23,24 Structure–affinity studies were reported, but only one compound with the potential to inhibit pSTAT6 in intact cells was described in the patents (17, Figure 2). Compound 17 is a phosphopeptide mimic in which phosphotyrosine is replaced by the conformationally constrained cinnamate unit. Stability to phosphatases was designed by replacing phosphate with the phosphonodifluoromethyl group.29 The negatively charged phosphonate oxygens were blocked with carboxyesterase-labile pivaloyloxymethyl (POM) groups as a means to permeate cells.30 Importantly, no biological evaluation was given, either in the patents or in peer-reviewed literature; therefore, it was not known whether this was a viable approach for the inhibition of STAT6 phosphorylation. Using our convergent methodology for the synthesis of phosphopeptide mimic prodrugs targeting SH2 domains,31,32 we improved the synthesis of 17 and found that it indeed inhibited both IL-4 and IL-13-stimulated phosphorylation of STAT6 in Beas-2B cells, an immortalized human bronchial epithelial cell line. Significant inhibition of pSTAT6 was observed at concentrations ≥1 µM.33
Figure 2.
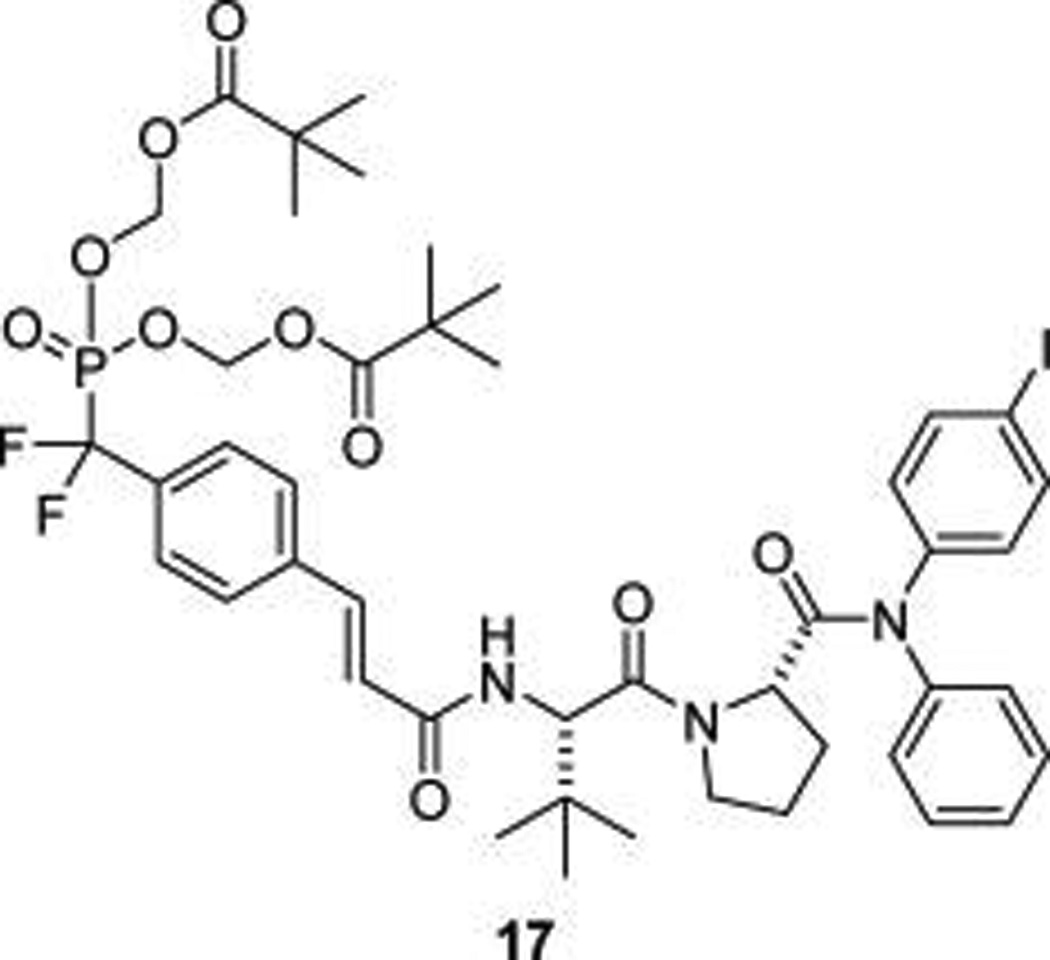
Lead STAT6 inhibitor (from ref 33).
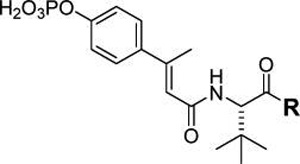
We embarked on new studies to test structural requirements of phosphate-containing analogues of prodrug 17 for affinity for the SH2 domain of STAT6. Interestingly, compound 18a (IC50 = 0.04 µM) was approximately 100-fold more potent than the phosphopeptides based on the IL-4Rα docking site (1–16), which exhibited IC50 values of 2–15 µM. Given that 18a has the cinnamate group of 14 and the Tle-Pro group of 7, we conclude that placing the aryl groups closer to the proline in 18a than that of Phe in the peptides is responsible for the increase in affinity. For STAT3 inhibitors, we found that substitution of a methyl group on the β position of the phosphocinnamate pTyr surrogate enhanced affinity 2–3-fold (pCinn vs βMpCinn).31 In keeping with these results, substitution of βMpCinn for pCinn produced a 2-fold increase in affinity 19a vs 18a (Table 4). 7-Phosphoryl-indole 2-carboxylate (20) reduced affinity compared to 4-phosphoryloxycinnamate, also consistent with our observations of inhibitors of STAT3.28
Table 4.
Effect of Constrained pTyr Surrogates on Affinity for Phosphodipeptide R-Tle-Pro-N(Ph)(4-I-Phe) for STAT6
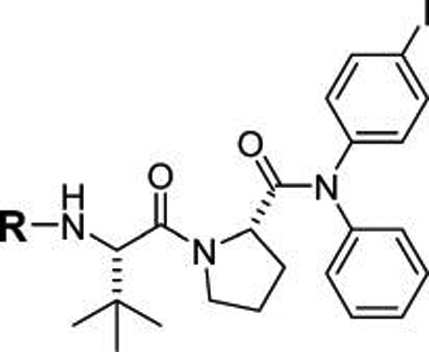 | ||
|---|---|---|
| R | IC50(µM) | |
| 18a | pCinn βMpCinn |
0.044 ± 0.016 |
| 19a | 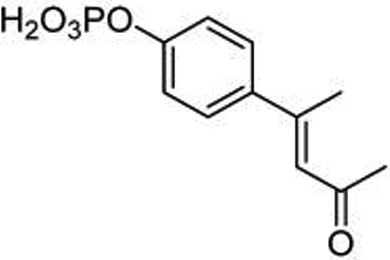 |
0.028 ± 0.006 |
| 20 | p-Indole | 0.14 ± 0.06 |
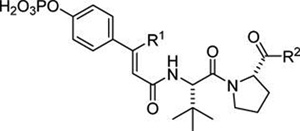
The literature describing 17 emphasized C-terminal bis-aryl and aryl,benzyl amides and all of the compounds in the reported structure–affinity relationship studies possessed these structural features.23,24 To test the requirements of aryl groups, we prepared a series of analogues of 18a and 19a with varying C-terminal amide substitution (Table 5). In the cinnamate series, the diphenylamide (18b) has a 3-fold higher IC50 than 18a, indicating that the iodine contributed to affinity. Methylanilide 18c is 4-fold lower in affinity than diphenylamide 18b, suggesting that aromatics are indeed important at the C-terminus. Removal of the methyl group (18d) resulted in further loss in affinity. Not surprisingly, in the β-methylcinamate series, the same trend in affinity loss was observed with reduced aryl and methyl substitution on the C-terminal amide nitrogen (19a–d), although the affinities in this series were higher than those of 18a–d. No gain in affinity was achieved by adding one or two methylene unit spacers to anilide 19d, see 19e and 19f. The cyclohexyl,methyl amide (19g) was 2-fold less avid than the phenyl,methyl amide, 19c. The dimethylamide, 19h, is significantly lower in affinity than the phenyl,-methyl amide. Affinity is improved with the larger diethylamide, 19i. These results show that while aryl groups are important at the C-terminus, reasonable affinity can be achieved with alkyl amides.
Table 5.
Effect of C-Terminal Substitution on Affinity for STAT6
 | |||
|---|---|---|---|
| compd | R1 | R2 | IC50 (µM) |
| 18a | H | NPh(4-I-Ph) | 0.044 ± 0.016 |
| 18b | H | NPh2 | 0.120 ± 0.04 |
| 18c | H | N(Me)Ph | 0.87 ± 0.03 |
| 18d | H | NHPh | 1.25 ± 0.34 |
| 19a | Me | NPh(4-I-Ph) | 0.028 ± 0.006 |
| 19b | Me | NPh2 | 0.089 ± 0.01 |
| 19c | Me | N(Me)Ph | 0.12 ± 0.03 |
| 19d | Me | NHPh | 0.27 ± 0.07 |
| 19e | Me | NHCH2Ph | 0.34 ± 0.07 |
| 19f | Me | NHCH2CH2Ph | 0.25 ± 0.06 |
| 19g | Me | N(Me)(C6H11) | 0.23 ± 0.14 |
| 19h | Me | N(Me)2 | 0.74 ± 0.34 |
| 19i | Me | N(Et)2 | 0.38 ± 0.09 |
Structural requirements of proline were probed (Table 6). For these experiments N-methylanilides were employed at the C-terminus. Replacing proline with the acyclic, N-methylated amino acid, sarcosine, 21, resulted in approximately 2-fold lower affinity than the proline-containing analogue, 19c, suggesting that relative to sarcosine, the conformational constraint of proline is important. Interestingly, alanine (22) was equipotent to proline, whereas N-methylation of the alanine (23) reduced affinity 4-fold. This suggests that the Tle–Pro peptide bond is likely in the trans conformation when bound to STAT6. Given that Xaa–Yaa peptide bonds sample significant cis conformations when Yaa is an imino acid, such as N-methyl alanine, the lower affinity of 23 relative to 22 may be due to the lower effective concentration of the trans conformer in the former.34
Table 6.
Effect of Proline Replacements on βMpCinn-Tle-Xaa-N(Me)Ph
| Xaa | IC50 (µM) | |
|---|---|---|
| 19c | proline | 0.12 ± 0.03 |
| 21 | sarcosine | 0.27 ± 0.05 |
| 22 | alanine | 0.12 ± 0.016 |
| 23 | N-methyl-alanine | 0.48 ± 0.094 |
Contributions of the atoms of the C-terminal amide bond were studied by replacement with conformationally constrained vinyl analogues (Table 7). Substituting the CONH group of 19d with a simple carbon–carbon double bond (24) reduced affinity over 10-fold. Extending the aliphatic chain by two methylene groups (25) increased affinity to 0.5 µM. Reduction of the vinyl group of 25 to an alkane (26) resulted in a slight decrease in affinity. Reduction of the carbonyl group of the methyl,phenyl amide of 19c resulted in a 4-fold loss in affinity (27). Thus, the carbonyl and nitrogen atoms of the C-terminal amide contribute to affinity for the SH2 domain of STAT6.
Table 7.
Effect of Modifications of the C-Terminal Amide Bond
 | ||
|---|---|---|
| # | R | IC50 (µM) |
| 24 |  |
3.43 ± 0.95 |
| 25 | 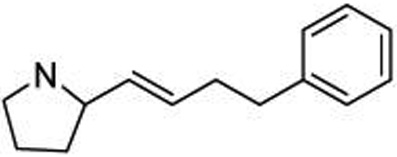 |
0.54 ± 0.11 |
| 26 | 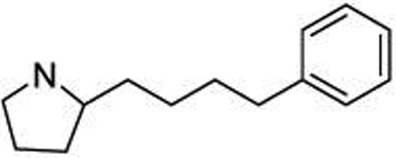 |
1.07 ± 0.17 |
| 27 | 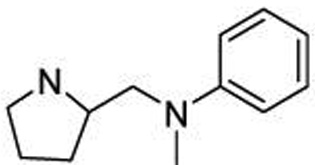 |
0.41 ± 0.05 |
Computed Structure of 18a Bound to the SH2 Domain of STAT6
To date, our attempts to crystallize STAT6 either alone or in the presence of a phosphopeptide have not been successful. To elucidate potential interactions between 18a (the phosphate analogue of 17) and STAT6, we employed a molecular modeling strategy that combines an AutoDock-based incremental protocol of flexible protein–ligand docking (DINC) and molecular dynamics simulations.35,36 A homology model of STAT6, based on the template structure of STAT1 (PDB ID 1YVL, chain A), was obtained from ModBase.37 Using DINC,35 18a was docked to the SH2 domain of STAT6. The complex was submitted to molecular dynamics simulation in explicit water. We obtained five clusters through k-mean clustering (k = 5). The sizes of the clusters were 173, 115, 101, 59, and 52 frames. Throughout the simulation, RMSD variations of 18a were largely between 1 and 2.5 Å, indicating that the peptide mimetic in the complex was stable. In the computed models, the inhibitor interacts with the surface of the protein and there do not appear to be deep pockets (Figure 3A,B). Hydrogen bonds are illustrated in the interaction diagram of Figure 3C and the stereo rendering in Figure 3D. The phosphate participates in ionic interactions with Lys544 and Arg562 and hydrogen bonding interactions with the main chains of Asp565 and Ser566, which are analogous to phosphate interactions with STAT138,39 and STAT340 observed in X-ray crystallographic structures and computational models of phosphopeptides bound to STAT3.28,36 These hydrogen bonds were very stable and were present in greater than 90% of the trajectory frames throughout the molecular dynamics simulation. The exception was the interaction between O2 of the phosphate and one of the quanidino NH2 groups of Arg562 (75% of trajectory frames). The hydrogen bond between the NH of Tle and the main chain C=O of Asn588 is typical of phosphopeptide interactions with SH2 domains and was present in 83% of the trajectory frames. Also noted was a very stable hydrogen bond between the C=O of Tle and the main chain NH of Gln590 (100% of trajectory frames). Interactions between the C=O of the pY+1 residue and the NH of Glu638 (analogous to Gln590) were also seen in computed models of phosphopeptides bound to STAT3.36,41 The aryl group of the cinnamate and the C-terminal iodophenyl group “sandwich” the side chain of Gln590, which extends away from the protein surface (Figures 3A,B,D). Not surprisingly, there were several hydrophobic interactions with the SH2 domain of STAT6, which are depicted as orange surfaces in Figure 3A and which are summarized in the interaction diagram in Figure 3C. The tert-butyl group of Tle interacts with the side chain methylene groups of Glu587 and the side chain of His 575. The C-terminal benzene rings interact with a hydrophobic surface comprised of the side chains of Ile589, Phe592, Leu609, Leu612, Ile606, and the side chain CH2 groups of Arg605. This hydrophobic surface does not appear to be able to accommodate the side chain benzene ring of Phe at position pY+3 in the peptides, which is extended from the proline by the α and β carbons. Thus, the peptides exhibit lower affinity than the mimetics.
Figure 3.
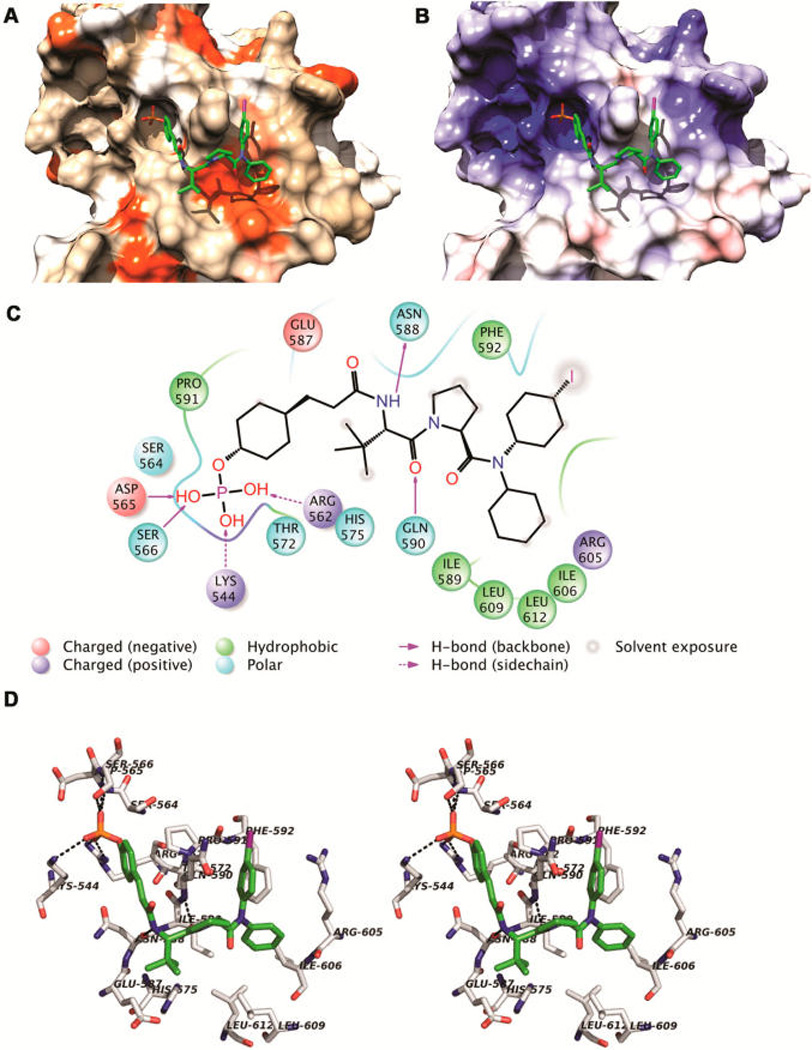
Computed structure of 18a in complex with a homology model of STAT6. A representative complex from the largest cluster from the final 10 ns of a molecular dynamics simulation is shown (see Methods). (A) STAT6 (comprised of the linker and SH2 domains) is shown in surface representation with coloring according to amino acid hydrophobicity on Kyte–Doolittle scale. (B) STAT6 is shown in surface representation with coloring according to APBS-based electrostatic potentials. (C) Intermolecular interaction diagram showing interactions between 18a and amino acids from the SH2 domain of STAT6. Structures in (A) and (B) were drawn using Chimera software, (C) was drawn using Maestro/Schrödinger software, and (D) was rendered in PyMOL.
Inhibition of Stat6 Phosphorylation in Intact Cells with Prodrugs Derived from Phosphopeptide Mimetics
A set of analogues of 17 (Chart 1) was synthesized to systematically evaluate the effect of structural modifications on the ability to inhibit STAT6 phosphosphorylation in intact cells (Figure 4). Beas-2B immortalized human airway epithelial cells were incubated with prodrugs for 2 h and stimulated with IL-4 for 1 h. Levels of pSTAT6 and total STAT6 proteins were evaluated by Western blotting of the cell lysates. The first series was a set of analogues of phosphates 18a–d. Starting with 17, possessing a phenyl, 4-iodophenyl amide, we removed the iodine (28) and then replaced one of the phenyl rings with a methyl group (29) then a hydrogen (30). As depicted in Figure 4A, removing the iodine from 1 resulted in a dramatic increase in potency (28). Significant inhibition was observed at 100 nM, and complete inhibition occurred at 500 nM. Substitution of one of the phenyl groups with a methyl group (29) produced a further increase in potency with significant inhibition at 50 nM and nearly complete inhibition at 100 nM. Replacing the methyl group with a hydrogen (30) resulted in approximately 25-fold less potency. Cellular viability (MTS assay, 72 h exposure, Figure 4C) did not parallel potency for STAT6 inhibition. The trend NPh,PhI < NPh2 < NMePh < NHPh indicates that smaller amides result in greater cytotoxicity. At concentrations ≤1 µM, diphenyl amide 28 and methylanilide 29 did not affect viability although pSTAT6 was completely inhibited. Thus, for these compounds, the reason for the observed cytotoxicity is not STAT6 phosphorylation inhibition. In contrast, for 30, the fraction of viable cells paralleled pSTAT6 inhibition. These results suggest that the C-terminal anilide is engaged in an as yet undefined off-target interaction that produces a cytotoxic effect.
Chart 1.
Structures of Prodrug Analogues of 17
Figure 4.
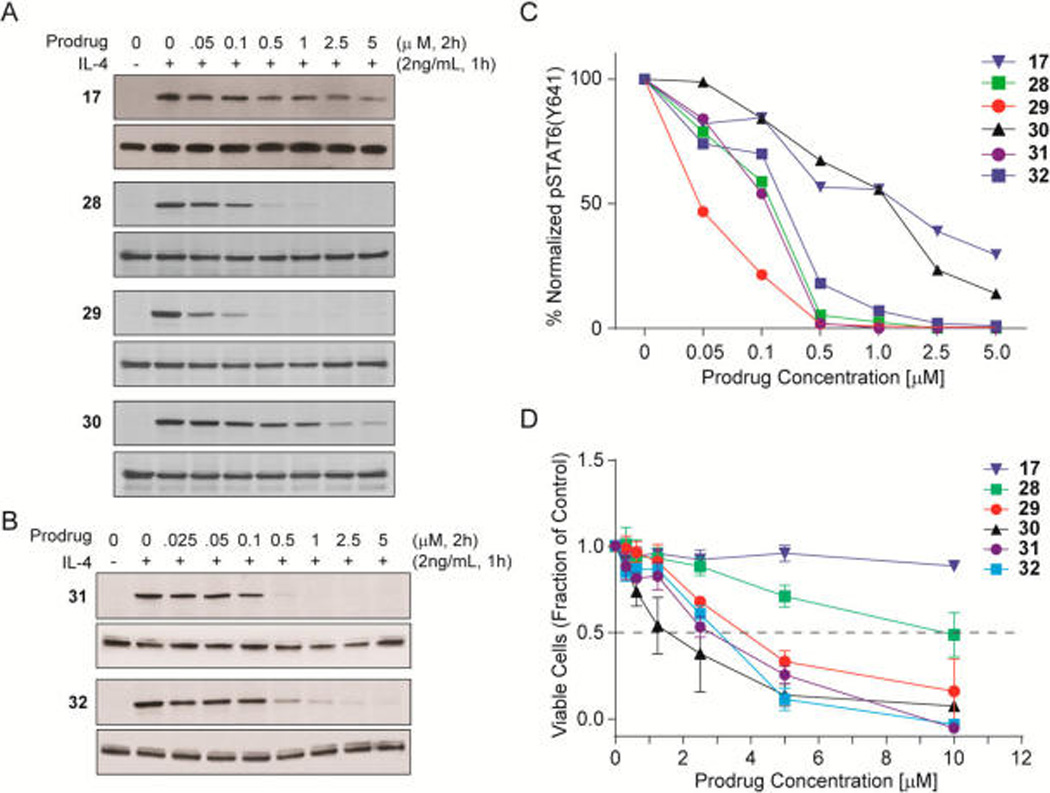
Effect of structure on inhibition of pSTAT6 and cytotoxicity. Bease-2B immortalized human epithelial airway cells were treated with the prodrugs for 2 h and were then stimulated with IL-4 for 1 h. Cells were lysed, and total and phosphorylated STAT6 were estimated by Western blots. For each pair of gels, the top is pSTAT6 and the bottom is total STAT6. (A) Prodrugs with cinnamoyl pTyr mimcs. (B) Prodrugs with β-methyl pTyr mimics. (C) Graphic representation the integrals of the bands in (A) and (B). (D) Effect of prodrugs on viability of Beas-2B cells. Cells were treated with the prodrugs for 72 h and viability was measured with MTT assays.
A second set of prodrugs was prepared to test the effect of β-methyl substitution on the cinnamate on cellular activity, as this substitution produced a small but significant increase in affinity of the phosphate analogues. Figure 4B shows that 31, the analogue of 29, retains potency and the β-methyl group has no noticeable effect on inhibition of pSTAT6. In 32, a cyclohexyl, methyl amide group replaced the phenyl,methyl amide of 31. As can be seen, this substitution also resulted in high potency. Cell viabilities of 31 and 32 were nearly identical to that of 29. Thus, the β-methyl group has no effect on cytotoxicity in this cell line.
Interestingly, cellular potency did not parallel affinity for STAT6 of the corresponding phosphates, 18a–d and 19c and 19g. Whereas affinity of the phosphate series decreased with decreasing aromatic character or decreasing methyl substitution, this was not the defining feature for the prodrug potency. Similar observations were made in the case of our prodrugs targeted to the SH2 domain of STAT3. For example, changing the C-terminal CONHBn to a simple methyl group in one series reduced affinity by a factor of 2 but increased cellular potency of the prodrug by 50-fold.31 In another series, substitution on the ring of a proline had no effect on affinity but increased cellular potencies from 20 to 100×.42 Thus, for this class of phosphonate prodrugs, biochemical affinity of the ligand for the target SH2 domain is not linearly predictive of cellular activity.
Other Cell Lines and Other SH2 Domains
The panel of prodrugs was tested for pSTAT6 inhibition in the triple negative breast cancer cell line, MDA-MB-468. After serum starvation, cells were treated with prodrugs followed by IL-4 stimulation which resulted in pSTAT6 inhibition. Overall, the pattern of inhibition paralleled that of the Beas-2B epithelial airway cells (Supporting Information Figure S1). Treatment of serum starved MDA-MB-468 cells with epidermal growth factor (EGF) activates the PI3K pathway leading to AKT phosphorylation,43 a process that is initiated by binding of the SH2 domain of p85, the regulatory subunit of PI3K, to pTyr residues on the EGF receptor.44 There was no clear trend for inhibition of pAkt as a function of structure. Whereas most compounds had no effect, 29 and 31 exhibited mild inhibition at the relatively high concentration of 5 µM. The exception was anilide 30, which potently inhibited pAkt at concentrations as low as 100 nM. MTS assays showed that for the panel of prodrugs, IC50 values for this cell line ranged from 5 to 10 µM. It is unclear if the decrease in pAkt is due to cross reactivity with the SH2 domains of p85 or due to other nonrelated offtarget effects that might affect this pathway.
The panel of prodrugs was assayed for the ability to inhibit the phosphorylation of STAT1, STAT3, STAT5, and STAT6 in primary mouse CD4+ T lymphocytes (Supporting Information Figure S2). Similarly to Beas-2B cells, inhibition of IL-4 stimulated pSTAT6 with 17 was relatively weak and the others stronger. We further found robust inhibition of IL-2 mediated STAT5 phosphorylation after treatment with 29, 28, 31, and 32. This is not surprising given the high homology in the SH2 domains of these two proteins. The hydrophobic surface mentioned above for STAT6 (Ile589, Phe592, Are605, Ile606, Leu609, and Leu612) is highly homologous in STAT5 (L643, Phe646, Are661, Leu662, Leu665, and Leu668). The side chain CH2 groups of Lys644 in STAT5 can also contribute to the hydrophobic binding site in an analogous manner to that of Gln590 in STAT6. Interestingly, anilide 30 showed selectivity for STAT6 as it was not an inhibitor of pSTAT5. None of the prodrugs affected the phosphorylation status of STAT1 or STAT3.
Inhibition of the Expression of CCL26 (Eotaxin-3)
Starting prodrug 17 and analogue 31 were tested for their ability to inhibit the expression of the downstream gene, CCL26 (eotaxin-3), a chemokine involved in the recruitment of eosinophils to sites of inflammation.45,46 Beas-2B cells were treated with the inhibitors, and after 24 h of exposure to IL-13, expression of CCL26 mRNA was assessed by quantitative PCR. As can be seen in Figure 5, addition of IL-13 led to robust expression of CCL26 mRNA (DMSO+). The expression of CCL26 mRNA was inhibited in a dose-dependent manner by both prodrugs. In both cases, >50% inhibition was achieved at 100 nM. The greater potency of 31 reflects its greater ability to inhibit STAT6 phosphorylation than 17. Thus, steric block of the STAT6 SH2 domain with these prodrugs not only inhibits activation (phosphorylation of Tyr641), it inhibits subsequent transcriptional activity.
Figure 5.
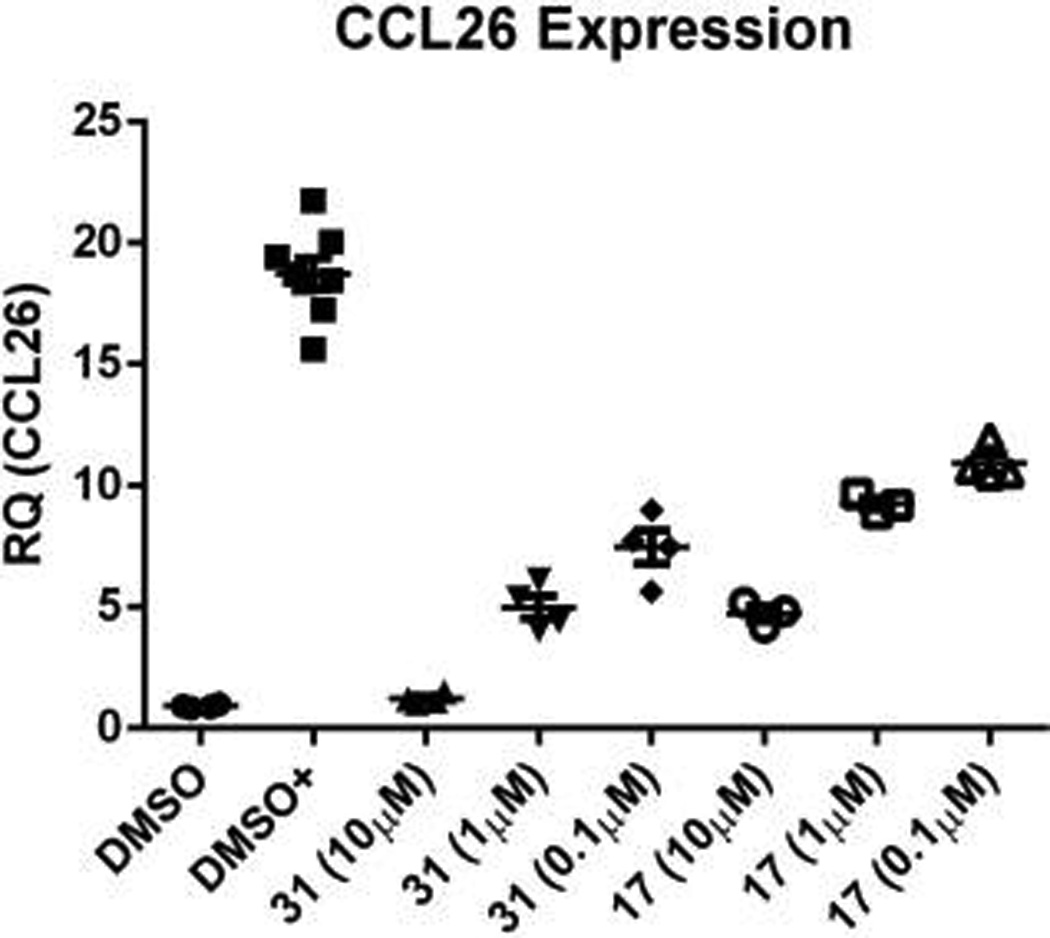
Prodrugs targeting the SH2 domain of STAT6 inhibit the expression of CCL26 mRNA. Beas-2B cells plated at 4 × 105 cells/well and incubated overnight in complete media, serum starved for 2 h, pretreated with drug for 2 h, stimulated with IL-13 overnight in serum free media, and total CCL26 mRNA was quantitated on cell lysates.
Chemistry
Phosphopeptide Synthesis
Phosphopeptides in Tables 1–3 were synthesized by manual solid phase techniques using the Fmoc protection scheme. C-termini were prepared as amides by the employment of Rink amide resin. N-Termini were capped with acetyl groups unless otherwise noted. Phosphotyrosine was introduced as the phosphoro-bis-dimethylamide.47–49 For the 5-phosphoryloxyindole (15) and 5-phosphoryloxybenzofurnan-2-carboxylates (16), the corresponding 5-hydroxy-2-carboxylates were coupled to resin-bound peptides and were phosphorylated using global phosphorylation with N,N-diisopropyl-di-tert-butylphosphoramidite in the presence of 1H-tetrazole. Peptides terminating in 4-phosphoryloxycinnamide and 4-phosphoryloxy-β-methylcinnamide were capped with the corresponding phosphorylated cinnamic acid-pentachlorophenyl esters as described previously.28 Peptides were cleaved from the resin using TFA:TIS:H2O.50 Peptides containing phosphoro-bis-dimethylamides were left overnight to deamidate the phosphate group.47–49 All phosphopeptides were purified by reverse phase HPLC and were the correct molecular weight as measured by electrospray mass spectrometry.
Synthesis of Phosphate Analogues of 17
We used convergent synthesis methodology to prepare phosphates for analysis of affinity for STAT6 protein in which modified dipeptides were synthesized and coupled to preformed constrained phosphotyrosine surrogates. For inhibitors 18–20 possessing constrained cinnamate and indole-based pTyr mimics (Table 4), diphenyl and phenyl,4-iodophenyl amines were not amenable to simple coupling to Boc-proline (33). In these cases, the first aryl group was installed as the anilide or 4-iodophenylamide of Boc-Pro-OH using water-soluble EDC as the coupling agent (34a,b) (Scheme 1). The second phenyl group was added by treatment with triphenylbismuth in the presence of cupric acetate and triethyl amine (35a,b).51,52 The Boc group was removed with TFA, and the resulting amine was coupled to Boc-tertiary-leucine with HBTU (36a,b). Again the Boc group was removed to give dipeptide amine salts 37a and 37b. Intermediate 37a was capped with pentachlorophenyl 4-phosphoryloxycinnamate (38a)28 and pentachlorophenyl β-methyl-4-phosphoryloxycinnamate (38b)31 to give inhibitors 18a and 19a (Table 4), respectively. Amine 37b was acylated with 38a and 38b to give 18b and 19b, respectively (Table 5). The alternative pTyr surrogate, 5-phosphoryloxyindole-2-carboxylate, was introduced by coupling 37a with 5-di-tertiary-butoxyphosphoryl-indoline-2-carboxylic acid (Scheme 2) (39) in the presence of HBTU and DIPEA. Treatment with TFA for an hour and purification by HPLC gave 20 (Table 4).
Scheme 1. Synthesis of Bis-arylamide Inhibitors of STAT6a.
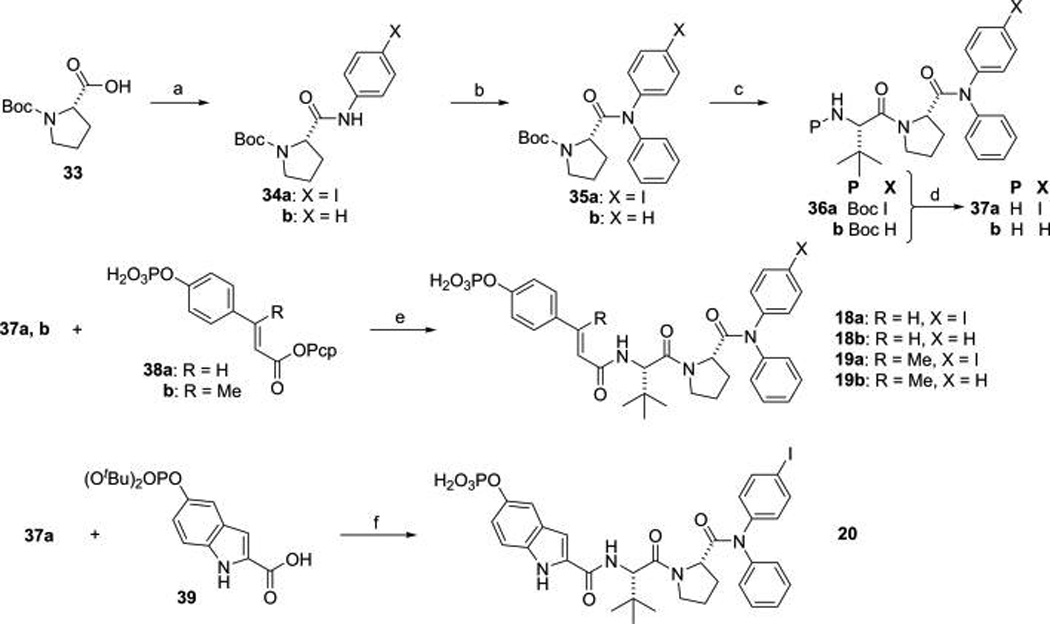
aReagents and conditions: (a) EDC, 4-iodoaniline (for 34a) or aniline (for 34b), DCM, rt 12 h; (b) Ph3Bi, Cu(OAc)2, TEA, DCM, 24 h; (c) (i) neat TFA, (ii) Boc-Tle-OH, HBTU, DIPEA, DCM rt 6 h; (d) neat TFA; (e) NMP, NMM, DMAP (cat), rt, 3 h; (f) (i) HBTU, DIPEA, DCM, 6 h, (ii) neat TFA, 1 h.
Scheme 2. Synthesis of the Phosphotyrosine Mimic, 2-(di-tert-Butylphosphoryloxy)indole-2-carboxylic Acida.
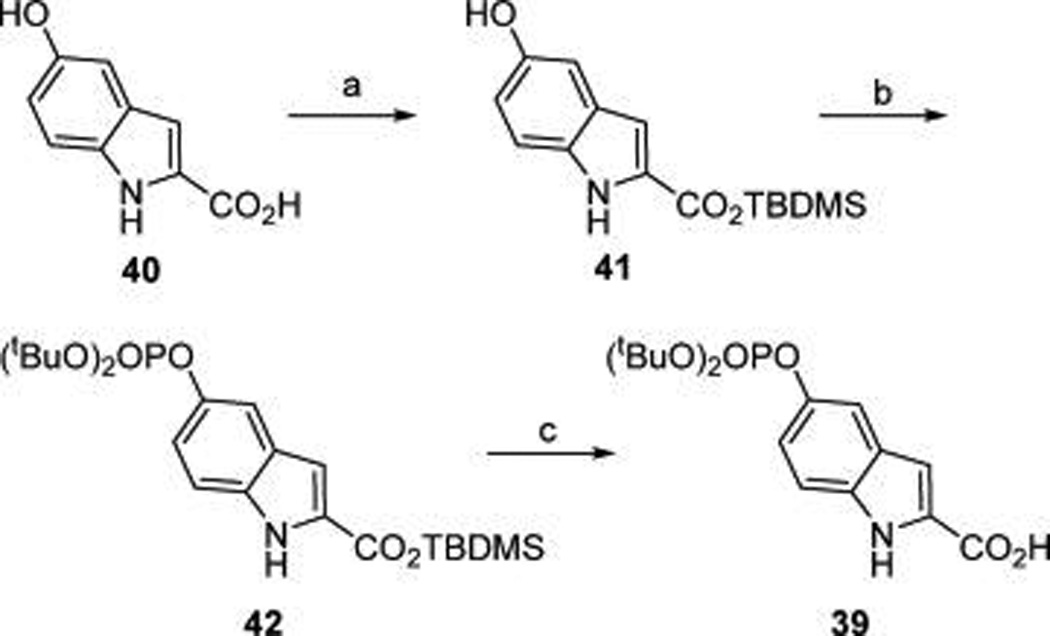
aReagents and conditions: (a) TBDMS-Cl, NMM; (b) (i) (tBuO2)-PNEt2, 1H-tetrazole, (ii) tBuOOH; (c) H3O+.
As shown in Scheme 2, 5-di-tertiary-butoxyphosphorylindoline-2-carboxylic acid (39) was prepared by protection of the carboxyl group of 5-hydroxyindole-2-carboxylate (40) as a tert-butyl dimethylsilyl (TBDMS) ester (41), followed by phosphitylation of the hydroxyl group with N,N-diethyl di-tert-butyl phosphoramidite and subsequent oxidation to the phosphate ester with tert-butyl hydrogen peroxide (42). Aqueous hydrolysis of the TBDMS ester gave the free carboxylate, 39 ready for capping 37a (Scheme 1).
Inhibitors in which the C-terminal amide was explored (18c,d, 19c–i, Table 5) were prepared by initial routine coupling of amines to Boc-proline using EDC (43c–i, Scheme 3). The Boc group was removed with TFA and Boc-tertiary-leucine was coupled with HBTU (44c–i). Again, the Boc group was removed and the dipeptides were capped with 38a or 38b to give inhibitors 18c–d and 19c–i.
Scheme 3. Synthesis of C-Terminal Amide Substitutions.
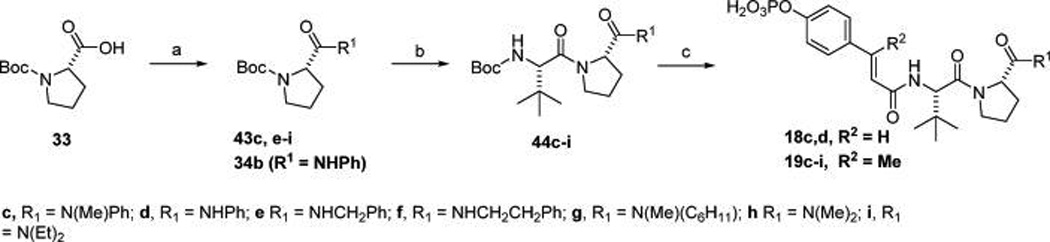
Reagents and conditions: (a) EDC, amine, DCM, rt, 12 h; (b) (i) neat TFA, (ii) HBTU, DIPEA, Boc-Tle-OH, (iii) neat TFA; (c) 38a or 38b, NMM, NMP, DMAP (cat).
Inhibitors 21–23 (Table 6), in which proline was replaced with nonconstrained sarcosine, alanine, and N-methylalanine, respectively, were prepared by simple coupling of N-methylaniline to the corresponding Boc-amino acid and elaborating as in Scheme 3.
Inhibitors 24–27 (Table 7) were synthesized to establish the importance of the C-terminal amide bond. As shown in Scheme 4, (S)-2-styryl-N-Boc-pyrrolidine (46) and (S)-2-(3-phenyl-propenyl)-N-Boc-pyrrolidine (48) were synthesized through a Wittig olefination of Boc-prolinal (45) and the corresponding triphenylphosphonium bromide derivative. Fully reduced (S)-2-(4-phenylbutyl)-N-Boc-pyrrolidine (50) was obtained by hydrogenation of 48 in the presence of 10%Pd-C. An analogue with a reduced amide bond was prepared by consecutive reductive-amination of Boc-prolinal with aniline (52) and then with formaldehyde (40% solution) (53). Intermediates 46, 48, 50, and 53 were elaborated to dipeptide amines 47, 49, 51, and 54 by TFA-mediated acidolysis of the Boc protecting group, HBTU-mediated coupling with Boc-Tle-OH, and then Boc cleavage with TFA as in Scheme 3. Treatment of the amine salts with 39b gave inhibitors 24–27.
Scheme 4. Synthesis of Prolyl Surrogates with Amide Bond Replacementsa.
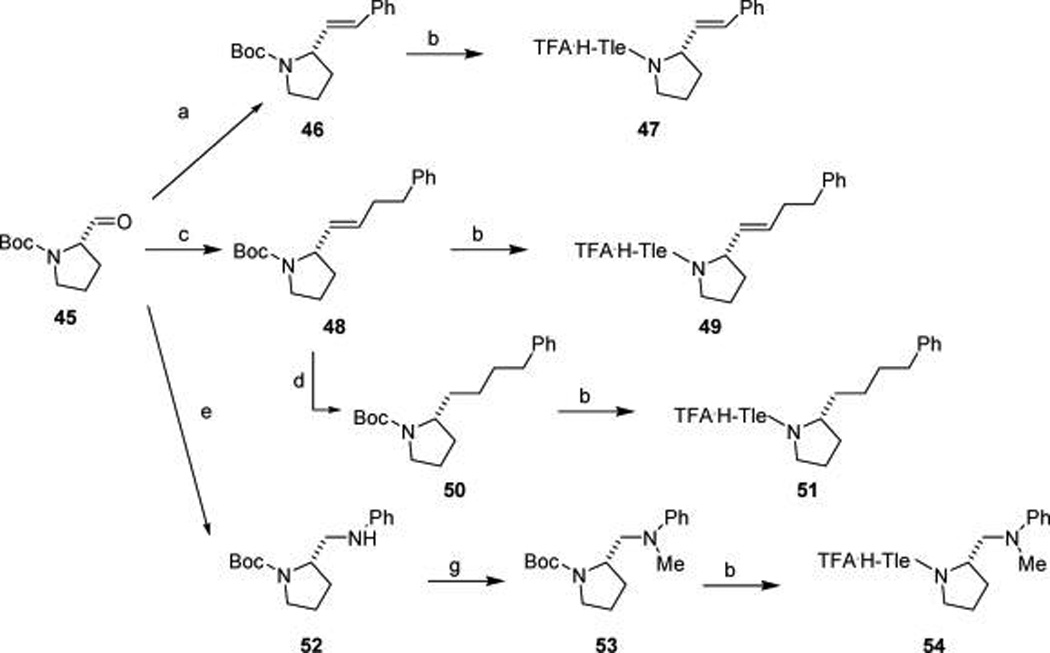
aReagents and conditions: (a) NaH, PhCH2PPh3Br, THF; (b) (i) TFA, (ii) Boc-Tle-OH, HBTU, DIPEA, (ii) TFA; (c) NaH, PhCH2CH2CH2PPh3Br, THF; (d) H2, 10%Pd-C; (e) aniline, NaCNBH3, MeOH-AcOH; (g) formaldehyde (40% solution), NaCNBH3, MeOH–AcOH.
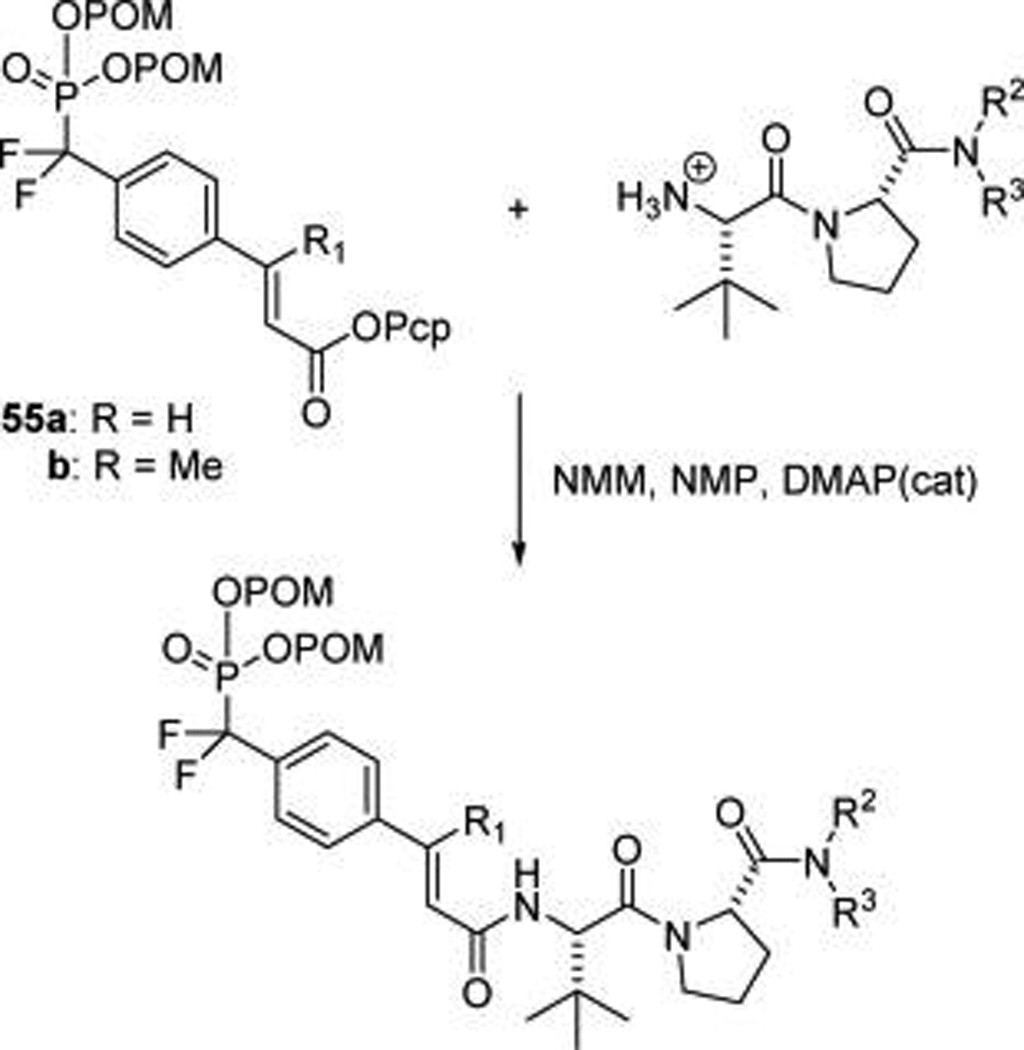
Cell permeable prodrugs (Chart 1) were synthesized (Scheme 5) by coupling dipeptide amines to pentachlorophenyl active esters of bis-POM protected phosphonodifloromethylcinnamates 55a or b that were synthesized as described previously.31,32 Couplings were carried out in NMP in the presence of N-methylmorpholine with DMAP catalysis. Products were purified by reverse phase HPLC with gradients of MeCN in H2O with no additives in the mobile phase.
Scheme 5. Synthesis of Prodrug Analogues of 17 (See Chart 1 for Structures).
CONCLUSIONS
The sequence surrounding Tyr631, the docking site of IL-4Rα, binds weakly to STAT6. This contrasts with STAT3 where a peptide derived from the docking site on gp130 (Tyr904) had low nM affinities.53,54 In the modified dipeptides reported by McKinney et al.,23,24 the aryl groups at pY+3 were closer to the main chain than the phenyl group of phenylalanine in the phosphopeptide series. Whereas C-terminal aryl amides produced the highest affinity, low nM binding constants could also be obtained with N-methyl anilides and dialkyl amides. The smaller hydrophobic groups of the latter likely do not penetrate as deeply into the proposed hydrophobic pocket as do benzene rings. Cellular potency of the bis-POM prodrugs was dependent on the substituents of the C-terminal amide but did not parallel affinity for STAT6 by the corresponding phosphates. Cellular activity of a peptidomimetic or any other drug is a function of many factors that may include (1) the intrinsic affinity of the drug for its target (e.g., enzyme, transcription factor, miRNA), (2) effectiveness of cell penetration, (3) efflux from the cell, (4) metabolic stability, (5) partitioning into membranes versus the cytosol, and other possible interactions. It is not clear which of these factors contributes to the deviation in cellular potency of the prodrugs from affinity for STAT6 of the corresponding phosphates. Inhibition of cell viability was inversely proportional to the size of the C-terminal amide with the smaller methylanilide and anilide analogues being most inhibitory. These apparent off-target effects can potentially induce more than one type of biological response in vivo, and it will be necessary to carefully monitor toxicity in animal studies. The prodrugs were quite potent in inhibiting expression of CCL26, suggesting that this class of compounds will have utility in modulating immune responses. In the case of asthma, cytotoxicity is detrimental, so care will have to be exercised to achieve the therapeutic window. The inhibitors were selective for STAT6 over STAT1 and STAT3. The basis of the selectivity can be explained as follows. At the base of the hydrophobic pocket that accommodates the C-terminal aromatic groups in STAT6 is Phe592. In the sequences of STAT1, STAT2, STAT3, and STAT4, the corresponding residue is a tyrosine. In the crystal structure of a phosphopeptide bound to STAT1,39 and in models calculated for phosphopeptide–STAT3 complexes,28,36,41 the side chain hydroxide of this tyrosine contacts a main chain peptide bond in a hydrogen bond which appears to be part of the recognition determinant for these SH2 domains. On the basis of these models, this hydroxyl group, and associated waters of hydration, in STAT1 and STAT3 would repel the C-terminal amide aromatic groups of the inhibitors, thus leading to selectivity for STAT6 (and STAT5). The observed cross reactivity to STAT5 in asthma is not likely to be a problem in clinical settings. As STAT5 was reported to be required for the asthma phenotype in a STAT6-independent manner,55 cross reactivity to STAT5 may actually contribute to in vivo efficacy. Evaluation of prodrugs in a fungal-induced murine model of asthma is in progress and will be reported under separate cover.
METHODS
Na-protected amino acids were purchased from, ChemImpex or NovaBiochem. HOBt was from ChemImpex. For the synthesis of phosphopeptides, Rink resin (Advanced Chemtech, Inc.) with a loading of 0.6 mmol/g was employed. Anhydrous DMF for amino acid solutions and aniline derivatives were from Aldrich. Other solvents were reagent grade and were used without further purification. NMR spectra were obtained on either a Bruker DPX 300 MHz spectrometer or a Bruker DRX 500 MHz spectrometer.
General Procedure for the Synthesis of Phosphopeptides
Solid phase syntheses were carried out manually using commercially available Rink resin. Resin, 0.2 g, was placed in a manual reactor and swollen and washed with 5 × 10 mL of DMF/CH2Cl2. Fmoc groups were removed with 3 × 6 mL of 20% piperidine/DMF for 5 min each. For coupling, 3-fold excesses of Fmoc-amino acids, DIC, and HOBt were used in 8–10 mL of DMF/CH2Cl2 and were allowed to proceed until resin samples tested negative with ninhydrin tests. After coupling and deprotection steps, resins were washed with 5 × 10 mL of DMF/CH2Cl2. On completion of the peptide chain, resins were washed with CH2Cl2 (3 × 10 mL) and were treated with TFA:TIS:H2O (95:2.5:2.5) (3 × 5 mL) for 15 min each. The combined filtrates volumes were reduced in vacuo. Peptides were precipitated in Et2O and collected by centrifugation. After drying, peptides were purified by reverse phase HPLC on a Varian Dynamax HPLC using a Phenomenex Luna C18 (2) 10 µM, 2.1 cm × 25 cm column. Gradients of 0.1% TFA–MeCN in 0.1% TFA–H2O with a flow rate of 20 mL/min were employed. For prodrugs, no TFA was used in the mobile phase. Peptides were tested for purity by reverse phase HPLC on a Waters Alliance HPLC using a Phenomenex Luna C18 (2) 5 µM 2.1 mm × 250 mm column at a flow rate of 0.4 mL/min. A gradient of 0–40% MeCN/30 min was used for phosphopeptides and peptide intermediates. For prodrugs the gradient was 10–80% MeCN/30 min. Phosphopeptides and prodrug intermediates were dried in vacuo over P2O5 at 37° for 24 h prior to use.53 All compounds were >95% pure (HPLC) before evaluation. Purities, yields, and mass spectral characteristics of phosphopeptides and prodrugs are provided in the Supporting Information.
Synthesis of 4-(Di-tert-butoxyphosphoryloxy)-Indole-2-carboxylic Acid, 39
To a suspension of 5-hydroxyindole-2-carboxylic acid (1.0 g, 5.64 mmol) and tert-butyldimethylsilyl chloride (0.85g, 5.64 mmol) in 10 mL of dry THF, N-methylmorpholine (0.62 mL, 5.64 mmol) was added at room temperature. After 15 min, 1H-tetrazole (1.6 g, 22.56 mmol) was added in one portion followed by the addition of di-tert-butyl N,N-diethylphosphoramidite (3.9 g, 11.3 mmol) in 15 mL of dry THF. After stirring for 2 h, 70% aq tert-butylhydroperoxide (3.0 mL, 16.9 mmol) was added at 0 °C and the mixture was stirred for 2 h more. The reaction was quenched with slow addition of an aqueous solution of 10% Na2S2O5 and stirred for 1.0 h. It was then diluted with ethyl acetate (30 mL) and extracted with an additional amount of ethyl acetate (2 × 20 mL). The combined organic parts were washed with water, followed by brine and dried (MgSO4). Solvent was removed, and the crude product was triturated with hexane–ether. The product was collected by filtration and dried to give 1.5 g (71%) of a white solid, which was used without further purification. 1H NMR (DMSO-d6, 300 MHz) δ 1.26 (s, 18H), 6.88–6.94 (m, 2H), 7.21–7.26 (m, 2H), 11.62 (s, 1H). 13C NMR (DMSO-d6, 75 MHz) δ 29.9, 83.1, 83.2, 107.8, 112.0, 113.6, 118.5, 127.3, 130.1, 134.8, 145.2, 145.3, 163.1. MS (ESI) m/z: (M + 1)+ calcd, 370.3; found, 370.4.
General Procedure for Boc-prolinamide Formation
A solution of Boc-proline (1.0 g, 4.64 mmol), the appropriate amine (1.0 equiv), and EDC (1.2 equiv) in 60 mL of dry dichloromethane was stirred for overnight. It was then transferred to a separatory funnel with an additional 20 mL of CH2Cl2 and washed with 5% HCl (2 × 30 mL) followed by 10% NaHCO3 (2 × 30 mL) and brine (1 × 20 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure. The residue was used as such for the next step or purified by flash chromatography with gradients of EtOAc in hexanes.
Boc-Pro-NHPhI, 34a
See ref 33.
Boc-Pro-NHPh, 34b
Yield 1.2 g, 90%, starting from 4.64 mmol of Boc-proline. 1H NMR (300 MHz, CDCl3) δ 1.5 (s, 9H), 1.91–1.95 (m, 3H), 2.52 (m, 1H) 3.44 (m, 2H), 4.44 (m, 1H), 7.1 (m, 1H), 7.3 (t, J = 7.6 Hz, 2H), 7.5 (d, J = 8.0 Hz, 2H), 9.5 (brs, 1H). MS (ESI) m/z: (M + 23)+ calcd, 313.2; found, 313.3
Boc-Pro-N(Me)Ph, 43c
Yield 1.1 g, 78%, starting from 4.64 mmol of Boc-proline. 1H NMR (300 MHz, CDCl3) δ 1.45 (s, 9H), 1.48 (isomer, s, 9H), 1.65–2.0 (m, 4H), 3.28 (s, 3H), 3.3–3.47 (m, 1H), 3.5–3.6 (m, 1H), 4.14 (m, 1H), 4.3 (isomer, m, 1H), 7.2–7.47 (m, 5H). MS (ESI) m/z: (M + 23)+ calcd, 327.2; found, 327.3.
Boc-Pro-NHCH2Ph, 43e
Yield 1.3 g, 92%, starting from 4.64 mmol of Boc-proline. 1H NMR (300 MHz, CDCl3) δ 1.4 (s, 9H), 1.82–1.98 (m, 2H), 2.1–2.43 (m, 2H), 3.4 (m, 2H), 4.2–4.65 (m, 3H), 6.4 (brs, 1H), 7.24–7.34 (m, 5H). MS (ESI) m/z: (M + 23)+ calcd, 327.2; found, 327.3.
Boc-Pro-NHCH2CH2Ph, 43f
Yield 1.2 g, 85%, starting from 4.64 mmol of Boc-proline. 1H NMR (300 MHz, CDCl3) δ 1.42 (s, 9H), 1.74–1.9 (m, 2H), 1.95–2.4 (m, 2H), 2.7–2.9 (m, 2H), 3.27–3.4 (m, 2H), 3.4–3.6 (m, 2H), 4.22 (m, 1H), 7.2–7.33 (m, 6H). MS (ESI) m/z: (M + 23)+ calcd, 341.2; found, 341.3.
Boc-Pro-N(Me)C6H11, 43g
Yield 1.1 g, 76%, starting from 4.64 mmol of Boc-proline. 1H NMR (300 MHz, CDCl3) δ 1.05–1.2 (m, 1H), 1.25–1.48 (m, 14H), 1.5–1.73 (m, 4H), 1.74–2.25 (m, 6H), 2.8 (s, 1.5H), 2.9 (s, 1H), 2.92 (s, 1H), 3.4–3.7 (m, 2H), 4.4 (m, 0.5H), 4.5 (m, 0.5H), 4.65 (m, 0.5H). MS (ESI) m/z: (M + 23)+ calcd, 333.2; found, 333.3
Boc-Pro-NMe2, 43h
Yield 1.0 g, 79%, starting from 4.64 mmol of Boc-proline. 1H NMR (300 MHz, CDCl3) δ 1.4 (s, 9H), 1.46 (isomer, s, 9H), 1.78–1.92 (m, 4H), 1.96–2.23 (isomer, m, 4H), 2.96 (s, 3H), 2.97 (isomer, s, 3H), 3.06 (s, 3H), 3.1 (isomer, s, 3H), 3.4–3.5 (m, 2H), 3.53–3.66 (isomer, m, 2H). 4.55 (m, 1H), 4.67 (m, 1H). MS (ESI) m/z: (M + 23)+ calcd, 265.2; found, 265.3
Boc-Pro-NEt2, 43i
Yield 1.1 g, 87%, starting from 4.64 mmol of Boc-proline. 1H NMR (300 MHz, CDCl3) δ 1.08–1.18 (m, 6H), 1.2–1.28 (isomer, m, 6H), 1.4 (s, 9H), 1.45 (s, 9H), 1.76–1.92 (m, 4H), 1.97–2.23 (isomer, m, 4H), 3.2–3.7 (m, 12H, including isomer), 4.45 (m, 1H), 4.6 (m, 1H). MS (ESI) m/z: (M + 23)+ calcd, 293.2; found, 293.3
General Procedure for Arylation of Amides..51,52
To a stirred solution of amide 34a or b (5.0 mmol) in dry CH2Cl2 (50 mL) was added triphenylbismuth (2.0 equiv), Cu(OAc)2 (2.0 equiv), and dry triethylamine (3.0 equiv). After completion of the reaction (as monitored by HPLC), the solvent was evaporated and the residue was diluted with ether (150 mL) and filtered through Celite. The filtrate was then washed with 5% HCl (2 × 30 mL) followed by brine and was dried over MgSO4. Concentration under reduced pressure followed by purification by silica gel chromatography using 10% ethyl acetate–hexane mixture afforded the title compounds.
Boc-Pro-N(Ph)PhI, 35a
See ref 33.
Boc-Pro-NPh2, 35b
Yield 0.9 g, 71%, starting from 3.44 mmol of 35b. 1H NMR (600 MHz, CDCl3) δ 1.46 (s, 9H), 1.52 (s, 9H, isomer), 1.72–1.8 (m, 2H), 1.87–1.94 (m, 1H), 1.96–2.1 (m, 5H), 3.36–3.4 (m, 1H), 3.46–3.5 (m, 1H), 3.55–3.64 (m, 2H), 4.3 (m, 1H), 4.44 (m, 1H), 7.1–7.5 (m, 20H, including isomer). MS (ESI) m/z: (M + 23)+ calcd, 389.2; found, 389.3
General Procedure for Dipeptide Formation
A solution of the appropriate Boc-prolinamide (2.0 g) in 10 mL of 95%TFA/CH2Cl2 was stirred for 1 h. The solution was concentrated under vacuum. Residual TFA was stripped off with by the addition and evaporation of CCl4 (2 × 10 mL). The residue was treated with Boc-Tle-OH (1.2 equiv), HBTU (1.2 equiv), and DIEA (2.5 equiv) in 50 mL of dry CH2Cl2 for overnight. The organic layer was then diluted with an additional 50 mL of CH2Cl2 and washed with 5% HCl (3 × 30 mL) followed by 10% NaHCO3 (2 × 30 mL) and brine. After drying (MgSO4) and concentration under vacuum, crude products were sufficiently pure and were used as such for the next step.
Boc-Tle-Pro-N(Ph)PhI, 36a
See ref 33.
Boc-Tle-Pro-NPh2, 36b
Yield 0.7 g, 77%, starting from 1.9 mmol of H-Pro-NPh2. 1H NMR (600 MHz, CDCl3) δ 1.06 (s, 9H), 1.40 (s, 9H), 1.83 (m, 1H), 1.97–2.06 (m, 3H), 3.73 (m, 1H), 3.83 (m, 1H), 4.30 (d, J = 9.6 Hz, 1H), 4.55 (m, 1H), 5.38 (d, J = 9.6 Hz, 1H), 7.15–7.52 (m, 10H). MS (ESI) m/z: (M + 23)+ calcd, 502.3; found, 502.5
Boc-Tle-Pro-N(Me)Ph, 44c
Yield 0.69 g, 68%, starting from 2.44 mmol of H-Pro-N(Me)Ph. 1H NMR (600 MHz, CDCl3) δ 1.00 (s, 9H), 1.32 (s, 9H), 1.64–1.72 (m, 1H), 1.78 (m, 2H), 1.96 (m, 1H), 3.2 (s, 3H), 3.62 (m, 1H), 3.72 (m, 1H), 4.22 (d, J = 9.6 Hz, 1H), 4.34 (t, J = 7.4 Hz, 1H), 5.28 (d, J = 9.6 Hz, 1H), 7.26–7.38 (m, 5H). MS (ESI) m/z: (M + 23)+ calcd, 440.3; found, 440.5
Boc-Tle-Pro-NHPh, 44d
Yield 0.83 g, 79%, starting from 2.63 mmol of H-Pro-NHPh. 1H NMR (600 MHz, CDCl3) δ1.00 (s, 9H), 1.44 (s, 9H), 1.9 (m, 1H), 2.03 (m, 1H), 2.16 (m, 1H), 2.55 (m, 1H), 3.66 (m, 1H), 3.82 (m, 1H), 4.35 (d, J = 9.7 Hz, 1H), 4.83 (m, 1H), 5.26 (d, J = 9.6 Hz, 1H), 7.05 (m, 1H), 7.27 (m, 2H), 7.47 (m, 2H), 9.37 (brs, 1H). MS (ESI) m/z: (M + 23)+ calcd, 426.3; found, 426.4
Boc-Tle-Pro-NHCH2Ph, 44e
Yield 0.72 g, 71%, starting from 2.45 mmol of H-Pro-NHCH2Ph. 1H NMR (600 MHz, CDCl3) δ 0.88 (s, 9H), 1.42 (s, 9H), 1.85–1.9 (m, 1H), 1.92–2.0 (m, 1H), 2.14 (m, 1H), 2.41–2.45 (m, 1H), 3.61 (m, 1H), 3.75 (m, 1H), 4.26 (d, J = 9.6 Hz, 1H), 4.32 (dd, J = 5.4 and 15.0 Hz, 1H), 4.46 (dd, J = 6.6 and 15.0 Hz, 1H), 4.63 (dd, J = 2.4 and 7.8 Hz, 1H), 5.2 (d, J = 9.6 Hz, 1H), 7.23–7.30 (m, 5H). MS (ESI) m/z: (M + 23)+ calcd, 440.3; found, 440.4
Boc-Tle-Pro-NHCH2CH2Ph, 44f
Yield 0.53 g, 66%, starting from 1.83 mmol of H-Pro-NHCH2CH2Ph. 1H NMR (600 MHz, CDCl3) δ 0.96 (s, 9H), 1.42 (s, 9H), 1.88–1.96 (m, 2H), 2.06–2.09 (m, 1H), 2.28 (m, 1H), 2.74–2.81 (m, 2H), 3.47 (m, 2H), 3.61 (m, 1H), 3.77 (m, 1H), 4.3 (d, J = 9.6 Hz, 1H), 4.53 (m, 1H), 5.28 (d, J = 9.6 Hz, 1H), 6.9 (m, 1H), 7.16–7.21 (m, 3H), 7.27–7.3 (m, 2H). MS (ESI) m/z: (M + 23)+ calcd, 454.3; found, 454.5
Boc-Tle-Pro-N(Me)C6H11, 44g
Yield 0.62 g, 61%, starting from 2.38 mmol of H-Pro-N(Me)C6H11. 1H NMR (600 MHz, CDCl3) δ 1.04 (s, 6H), 1.05 (s, 3H), 1.3–1.4 (m, 4H), 1.4–1.44 (m, 11H), 1.53–1.61 (m, 1H), 1.62–1.7 (m, 2H), 1.74–1.8 (m, 1H), 1.81–1.9 (m, 2H), 1.9–2.00 (m, 1H), 2.06–2.24 (m, 2H), 2.8 (s, 1H), 2.95 (s, 2H), 3.6 (m, 0.5H), 3.73 (m, 1H), 3.86 (m, 1H), 4.3–4.4 (m, 1.5H), 4.83 (m, 0.5H), 4.9 (m, 0.5H), 5.35 (m, 1H). MS (ESI) m/z: (M + 23)+ calcd, 389.2; found, 389.3
Boc-Tle-Pro-NMe2, 44h
Yield 1.1 g, 60%, starting from 4.92 mmol of H-Pro-NMe2. 1H NMR (600 MHz, CDCl3) δ 1.04 (s, 9H), 1.42 (s, 9H), 1.86 (m, 1H), 1.95 (m, 1H), 2.1 (m, 1H), 2.18 (m, 1H), 2.94 (s, 3H), 3.11 (s, 3H), 3.74 (m, 1H), 3.86 (m, 1H), 4.33 (d, J = 9.6 Hz, 1H), 4.87 (m, 1H), 5.29 (d, J = 9.6 Hz, 1H). MS (ESI) m/z: (M + 23)+ calcd, 378.3; found, 378.4
Boc-Tle-Pro-NEt2, 44i
Yield 1.4 g, 64%, starting from 5.9 mmol of H-Pro-NEt2. 1H NMR (600 MHz, CDCl3) δ 1.03 (s, 9H), 1.1 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.42 (s, 9H), 1.86–1.96 (m, 2H), 2.12–2.18 (m, 2H), 3.28–3.42 (m, 3H), 3.53 (m, 1H), 3.73 (m, 1H), 3.87 (m, 1H), 4.32 (d, J = 10.2 Hz, 1H), 4.76 (m, 1H), 5.34 (d, J = 9.6 Hz, 1H). MS (ESI) m/z: (M + 23)+ calcd, 406.3; found, 406. 5
Synthesis of (S)-2-Styryl-N-Boc-pyrrolidine, 46.56
n-BuLi (1.0 mL, 2.5 mmol) was added dropwise via syringe over 10 min to a solution of benzyl-triphenylphosphonium bromide (1.0 g, 2.3 mmol) in 20 mL of dry THF under argon at −78 °C. The mixture was stirred for 1 h, then Boc-l-prolinal (0.46g, 2.3 mmol) in 20 mL of THF was added dropwise. The reaction was allowed to stir for 24 h at room temperature. The reaction mixture then quenched with 30 mL of saturated NH4Cl, diluted with ethyl acetate, washed with brine (2 × 25 mL), dried over sodium sulfate, and concentrated in vacuo. Silica gel chromatography (5:1 hexanes/EtOAc) yielded 0.480 g (76%). 1H NMR (CDCl3, 300 MHz) δ 1.42 (s, 9H), 1.8–2.2 (m, 4H), 3.45 (brs, 1H), 4.3–4.8 (m, 1H), 5.58–6.15 (m, 1H), 6.35–6.45 (m, 1H), 7.2–7.4 (m, 5H).
Synthesis of (S)-2-(4-Phenyl-but-1-enyl)-N-Boc-pyrrolidine, 48
The same procedure as described for the synthesis of 46 was followed starting with 1.0 g of (3-phenylpropyl)-triphenylphosphonium bromide (2.2 mmol) and 0.483 g of Boc-prolinal (2.2 mol). Yield: 0.452 g (1.5 mmol, 68%). 1H NMR (CDCl3, 300 MHz) δ 1.43 (s, 9H), 1.68– 1.92 (m, 4H), 2.37–2.52 (m, 2H), 2.56–2.81 (m, 2H), 3.3–3.42 (m, 2H), 4.45 (brs, 1H), 5.27–5.45 (m, 2H), 7.14–7.31 (m, 5H).
(S)-4-Phenbutyl-N-Boc-pyrrolidine, 50
A 50 mL round-bottom flask fitted with a three-way tap and balloon was charged with 400 mg of 10% Pd-C. Alkene 49 (405 mg) in 5 mL of EtOH was added via syringe and stirred for 2 h. Hydrogen gas was added as the balloon deflated. Upon complete reaction (TLC), the catalyst was removed by filtration. The product was concentrated in vacuo to yield a colorless oil, yield 376 mg (93%). 1H NMR (CDCl3, 300 MHz) δ 7.15–7.31 (m, 5H), 3.70–3.76 (br m, 1H), 3.26–3.41 (m, 2H), 2.58–2.64 (m, 2H), 1.59–1.93 (m, 7H), 1.49 (s, 9H), 1.30–1.35 (m, 3H).
Synthesis of N-Boc (S)-2-((Methyl(phenyl)amino)methyl)-pyrrolidine, 53
To a stirred solution of 45 (1.00 g, 5.02 mmol) and aniline (0.78 g, 6.02 mmol) in MeOH:acetic acid (9:1) (10 mL) was added NaCNBH3 (0.63 g, 10.04 mmol) at room temperature. The mixture was stirred for 1 h, evaporated to dryness, and the residue extracted with AcOEt (2 × 30 mL). The organic phase was washed with 5% NaHCO3, brine, dried (MgSO4), and evaporated. The product 52 (1.0 g, 3.2 mmol) was then dissolved in 10 mL of MeCN:acetic acid (9:1), and 37% formalin (10 mL) and sodium cyanoborohydride (0.6 g, 9.6 mmol) were added. The mixture was stirred for 4 h at room temperature. The solution was evaporated in vacuo, and the residue was dissolved in AcOEt (50 mL) and was washed with 1N KOH followed by brine. The organic phase was dried (MgSO4) and evaporated, and the crude product was purified by silica gel chromatography using 30% EtOAc–hexane mixture yielding 0.640 g (2.2 mmol) of 53 (61%). 1H NMR (CDCl3, 300 MHz) δ1.52 (s, 9H), 1.8–1.95 (m, 4H), 2.98 (s, 3H), 3.0–3.21 (m, 1H), 3.28–3.48 (m, 2H), 3.56–3.72 (m, 1H), 4.15 (m, 1H), 6.68 (m, 1H), 6.74–6.84 (m, 2H), 7.18–7.26 (m, 2H). MS (ESI) m/z: (M + H)+ calcd, 291.2; found, 291.3.
General Procedure for Phosphopeptide/Phosphopeptidomimetic Formation
Boc-protected dipeptide (0.5 g) was treated with 5.0 mL of 95% TFA–CH2Cl2 for 1 h. After concentration under vacuum, the residue was diluted with 10 mL of water, neutralized with conc NH4OH, and lyophilized to obtain an off-white foam. The amine was then treated with the active ester (38a or 38b) (1.0 equiv) in 5 mL of dry NMP, N-methylmorpholine (NMM, 3.0 equiv), and a catalytic amount of DMAP (0.05 equiv). After completion of the coupling, as monitored by HPLC, 25 mL of ether was added to the reaction mixture and the resulting emulsion centrifuged. The clear supernatant was decanted off, and the crude precipitate was purified by HPLC using 0.1% TFA–MeCN–water. Fractions containing pure phosphopeptide were combined and lyophilized to obtain pure products as white powders. Each was further dried over P2O5 chamber under vacuum at 37 °C before affinity measurements.53 Yields, HRMS, and HPLC retention times are reported in the Supporting Information.
General Procedure for Prodrug Formation..31,32
The appropriate amine, derived from a Boc-protected dipeptide as described above, was coupled with the building block 55a or b (1.0 equiv) in the presence of 3 mL of dry NMP, dry NMM (3.0 equiv), and a catalytic amount of DMAP (0.05 equiv). After completion, checked by HPLC, the product was purified by RP-HPLC using acetonitrile–water (no TFA). The pure fractions were combined and lyophilized to obtain a white powder. Yields, HRMS, and HPLC retention times are reported in the Supporting Information.
Synthesis of βMpCinn-Tle-Sar-NMe(Ph), 21
To a stirred solution of Boc-Sar-OH (2.0 g, 10.6 mmol), N-methylaniline (1.2 g, 10.6 mmol), and HBTU (4.8 g, 12.7 mmol) in 50 mL of dry CH2Cl2, DIEA (3.7 mL, 21.2 mmol) was added and the reaction stirred overnight. The mixture was diluted with an additional 50 mL of CH2Cl2, and the solution then washed with 5% HCl (2 × 30 mL) followed by 10% NaHCO3 (2 × 30 mL) and brine (1 × 20 mL). The organic layer was dried (MgSO4) and evaporated to dryness under reduced pressure. The residue was treated with 10 mL of 95% TFA–CH2Cl2 for 1 h. The solvent was removed under vacuum and toluene (2 × 10 mL) was added and evaporated to get rid of residual TFA. The crude, foamlike material was treated with Boc-Tle-OH (1.2 equiv), HBTU (1.2 equiv), and DIEA (3.0 equiv) for 12 h in dry CH2Cl2 (50 mL). The solvent was evaporated in vacuo, and the residue was dissolved in EtOAc and washed with 5% HCl (3 × 30 mL), 10% NaHCO3 (2 × 30 mL), and brine. After drying (MgSO4), filtration, and evaporation, the residue was treated with 95%TFA–DCM for 1 h. The solvent was concentrated under vacuum and then diluted with water and neutralized the aqueous solution with NH4OH and lyophilized to give an off-white foam. The amine was then treated with 38b (1.0 equiv) in 5 mL of dry NMP, NMM (3.0 equiv), and a catalytic amount of DMAP (0.05 equiv). After completion of the coupling, as monitored by HPLC, 25 mL of ether was added to the reaction mixture and the resulting emulsion centrifuged. The clear supernatant was decanted off, and the crude precipitate was purified by HPLC using 0.1% TFA–MeCN/H2O. Fractions containing pure phosphopeptide were combined and lyophilized to obtain pure phosphopeptide/phosphopeptidomimetic as a white powder. It was further dried over P2O5 chamber under vacuum at 37 °C before being used for affinity measurement. The yield and HRMS, and HPLC retention times are provided in the Supporting Information.
Synthesis of βMpCinn-Tle-Ala-NMe(Ph) (22) and βMpCinn-Tle-(Me)Ala-NMe(Ph) (23)
These syntheses followed the same procedure as described for 21 starting with Boc-Ala-OH and Boc-N(Me)Ala-OH, respectively. Yields and HRMS and HPLC retention times are provided in the Supporting Information.
Fluorescence Polarization Assays
Peptides (200 µM) in “fluorescence polarization buffer” (FPB, 50 mM NaCl, 10 mM Hepes, 1 mM Na4EDTA, 2 mM DTT, and 1% NP-40) were placed in 96-well plates, and serial dilutions were carried out in the same buffer on a Biomek 2000 liquid handling robot. Aliquots (50 µL) of a solution of STAT6 (480 nM) and 20 nM of FAM-Ala-pTyr-Lys-Pro-Phe-Gln-Asp-Leu-Ile-NH2 in the same buffer were placed in wells of a second 96-well plate using the robot. To each well of the second plate was added 50 µL of the peptide solutions from the first plate. Fluorescence polarization was then read on a Tecan Infinite F200Pro plate reader. With use of Prizm 6, from GraphPad Software, Inc., the mP was plotted against the log of the peptide concentration and IC50 values were obtained from nonlinear regression analysis. Peptides were assayed in triplicate using three separate STAT6-probe preparations. IC50 values are reported as the mean of three IC50 values ± standard deviation.
Inhibition of Stat6 Tyrosine641 Phosphorylation in Intact Cells
Beas-2B cells were seeded on 6-well plates at a density of 3.0 × 105 cells/well the day before treatment. Powdered prodrugs were dissolved in DMSO to obtain 5–10 µM stock solutions. Aliquots of the stocks were added to the cell cultures to reach the desired final concentrations of the prodrugs. Cultures were incubated with prodrugs for 2 h and stimulated with IL-4 or IL-13 (2 ng/mL) for 1 h. Cells were harvested, washed with ice-cold phosphate buffer saline (PBS), and lysed with cell lysis buffer (Cell Signaling, no. 9803), supplemented with phenylmethanesulfonylfluoride (PMSF) serine protease inhibitor. After incubation on ice for 10 min, cells were collected by scraping the dishes, and the resulting suspensions were centrifuged at 13200 rpm for 15 min at 4 °C. The supernatants were transferred to clean tubes, and aliquots (5 µL) were used to measure the concentration of proteins using the modified Lowry protein assay (BioRad) on a Tecan Infinite Pro 200 plate reader. Levels of pSTAT6 and total STAT6 proteins were estimated by Western blotting. Quantification of Western blotting band density was performed using ImageJ sofware.
Western Blotting
Aliquots of the whole cell lysate (15–30 µg proteins) were separated on a 10% SDS-PAGE polyacrylamide gel and transferred to PVDF membranes. The membranes were blocked with a 3% BSA solution in TBST (1× Tris-buffered saline, 0.1% Tween 20) and probed with phospho-Stat6 (Tyr641) antibody (Cell Signaling, no. 9361), followed by incubation with horseradish peroxidase conjugated secondary antibody (BioRad). Levels of pStat6 protein were detected with ECL detection reagents (GE Healthcare Life Science), capturing the resulting chemiluminescence signal on X-ray films. PVDF filters were then stripped with stripping buffer at rt for 30 min, blocked with 3% BSA, and probed with Stat6 antibody (Cell Signaling, no. 9362 or Santa Cruz, sc-374021) whose signal was detected as described above.
Effect of Prodrugs on Beas-2B Cell Proliferation
Effects of prodrugs on cell proliferation were determined by MTS assay. Beas-2B cells were seeded on a 96-well plate (2000 cells/well). Compounds were dissolved in DMSO to make 10 mM stock solutions. Stocks were subjected to serial dilution in DMSO, and equal amounts of each dilution were added to the cultures. After incubation at 37 °C for 72 h, the culture medium was removed and a 1:5 MTS/PBS solution was added to each well. The dish was then incubated for an additional 1 h, and viable cells were estimated by measuring the optical density of the samples at 490 nm.
RT-PCR
RNA isolated from Beas-2B cells culture by Trizol (15596–026; Invitrogen, Grand Island, NY) was subjected to cDNA synthesis with TaqMan Reverse Transcription kit (N8080234; Applied Biosystems, Foster City, CA). Probes for CCL26 and β-actin were purchased from Applied Biosystems (Foster City, CA) to quantify relative expression using TaqMan Fast Universal PCR Master Mix (435189, Applied Biosystems, Foster City, CA) on a the 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA).
Mice
C57BL/6J mice were purchased from Jackson Laboratories. Mice were housed in the SPF animal facility at the M. D. Anderson Cancer Center, and all animal experiments were approved by Institutional Animal Care and Use Committee.
CD4+ T Cells Activation and Immunoblot Analysis
CD4+ T cells from C57BL/6J mice were sorted by AutoMACS (Miltenyi Biotec) and activated with plate-bound anti-CD3 and/or anti-CD28 (eBioscience) alone or together with IL2, IL-6, or IL-4 (Peprotech) for 15 min. Cells were lysed in kinase assay lysis buffer supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitors (10 mM NaF and 1 mM Na3VO4). Protein concentration was determined by Bio-Rad Bradford protein assay, and equal amounts of protein were loaded for immunoblot analysis with antibodies against phospho-STAT1 (Tyr701), phospho-STAT3 (Ser727), phospho-STAT5 (Tyr694), phosphor-STAT6 (Tyr641), STAT1, STAT3 (Cell Signaling), STAT5, and STAT6 (Santa Cruz Biotechnology). Antimouse IgG-HRP and antirabbit IgG-HRP secondary antibodies were from Thermo scientific.
Molecular Modeling
A homology model of STAT6 was obtained from ModBase, a database of comparative protein structure models.37 The homology model is based on the template structure of STAT1 (PDB ID 1YVL, chain A), and residues 445–631, comprising the SH2 domain and part of the linker domain, were used. Using DINC,35,36 18a was docked to the SH2 domain of STAT6. The docked structure was simulated in explicit water using a molecular dynamics program in AMBER (version 11). Potentials for STAT6 were assigned with AMBER’s ff99SB force field, and those of 18a were assigned with GAFF force field. Point charges for 18a were calculated using AM1-BCC charge model. The solvated complex was minimized in three rounds, with each round involving 1000 steps (10 steps of steepest descent minimization followed by 990 steps of conjugate gradient minimization). The solute (SH2 domain of STAT6) was restrained/fixed in each round. The restraint weight was set to 5.0, 2.0, and 0.05 (kcal/mol-Å2) in rounds 1, 2, and 3, respectively. The molecular dynamics protocol involved a temperature (300 K) equilibration step for 100 ps, a pressure equilibration step for 400 ps, followed by a production run of 20 ns at constant temperature and pressure. The time step used for the simulation was 2 fs, and the molecular dynamics trajectory was recorded every 1 ps. The structures of the complex derived from the last 10 ns of the trajectory were clustered using k-means (k = 5) clustering method, and the representative structure from the largest cluster was output as the model of 18a in complex with the STAT6 shown in Figure 3.
Supplementary Material
Acknowledgments
We are grateful to the American Asthma Foundation (award no. 11-0360), the Keck Center Computational Cancer Biology Training Program of the Gulf Coast Consortia (CPRIT RP101489), and the National Science Foundation (ABI 0960612 at Rice University) for support of this work. We are also grateful to the MDACC Institutional Research Grant and the CTT/TI-3D Chemistry & Molecularly Targeted Therapeutic Development Grant Program. We also acknowledge the Cancer Center Support Grant P30 CA016672 at MDACC for support of both the NMR facility and the Translational Chemistry Core Facility (mass spectrometry). Beas-2B cells were a kind gift from Walter Hittleman.
ABBREVIATIONS USED
- DIEA
diisopropylethylamine
- DIPCDI
diisopropylcarbodiimide
- Fmoc
9-fluorenylmethoxycarbonyl
- HBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HOBt
1-hydroxybenzotriazole
- IL-4
interleukin 4
- IL-13
interleukin 13
- JAK
Janus kinase
- pCinn
4-phosphoryloxycinnamide
- βMpCinn
β-methyl pCinn or [2E]3-(4-phosphoryloxyphenyl)-2-butenamide
- βMF2PmCinn
[2E]3-[4-[(phosphinyl)difluoromethyl]phenyl]-2-butenamide
- POM
pivaloyloxymethyl
- SAR
structure–activity relationship
- SH2 domain
Src homology 2 domain
- STAT3
signal transducer and activator of transcription 3
- STAT6
signal transducer and activator of transcription 6
- TES
triethylsilane
- Th2
helper T cell type 2
- TIS
triisopropylsilane
Footnotes
ASSOCIATED CONTENT
Supporting Information
- Yields and characterization of phosphopeptide inhibitors, phosphate analogues of 17, and prodrug analogues of 17; figures of the inhibition of pSTAT6 and pAkt in MDA-MB-468 breast cancer cells and STAT proteins in CD4+ T lymphocytes; NMR characterization of 19c and 31 (PDF) SMILES CSV file of compounds 1–55 (CSV)
The authors declare no competing financial interest.
REFERENCES
- 1.Kuperman DA, Schleimer RP. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr. Mol. Med. 2008;8:384–392. doi: 10.2174/156652408785161032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oh CK, Geba GP, Molfino N. Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. Eur. Respir. Rev. 2010;19:46–54. doi: 10.1183/09059180.00007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walford HH, Doherty TA. STAT6 and lung inflammation. JAK-STAT. 2013;2:e25301. doi: 10.4161/jkst.25301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol. Res. 2012;53:11–24. doi: 10.1007/s12026-012-8291-9. [DOI] [PubMed] [Google Scholar]
- 5.Mullings RE, Wilson SJ, Puddicombe SM, Lordan JL, Bucchieri F, Djukanovic R, Howarth PH, Harper S, Holgate ST, Davies DE. Signal transducer and activator of transcription 6 (STAT-6) expression and function in asthmatic bronchial epithelium. J. Allergy Clin. Immunol. 2001;108:832–838. doi: 10.1067/mai.2001.119554. [DOI] [PubMed] [Google Scholar]
- 6.Darcan-Nicolaisen Y, Meinicke H, Fels G, Hegend O, Haberland A, Kuhl A, Loddenkemper C, Witzenrath M, Kube S, Henke W, Hamelmann E. Small interfering RNA against transcription factor STAT6 inhibits allergic airway inflammation and hyperreactivity in mice. J. Immunol. 2009;182:7501–7508. doi: 10.4049/jimmunol.0713433. [DOI] [PubMed] [Google Scholar]
- 7.Chiba Y, Todoroki M, Nishida Y, Tanabe M, Misawa M. A novel STAT6 inhibitor AS1517499 ameliorates antigen-induced bronchial hypercontractility in mice. Am. J. Respir. Cell Mol. Biol. 2009;41:516–524. doi: 10.1165/rcmb.2008-0163OC. [DOI] [PubMed] [Google Scholar]
- 8.Nagashima S, Hondo T, Nagata H, Ogiyama T, Maeda J, Hoshii H, Kontani T, Kuromitsu S, Ohga K, Orita M, Ohno K, Moritomo A, Shiozuka K, Furutani M, Takeuchi M, Ohta M, Tsukamoto S. Novel 7H-pyrrolo[2,3-d]pyrimidine derivatives as potent and orally active STAT6 inhibitors. Bioorg. Med. Chem. 2009;17:6926–6936. doi: 10.1016/j.bmc.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 9.Nagashima S, Nagata H, Iwata M, Yokota M, Moritomo H, Orita M, Kuromitsu S, Koakutsu A, Ohga K, Takeuchi M, Ohta M, Tsukamoto S. Identification of 4-benzylamino-2-[(4-morpholin-4-ylphenyl)amino]pyrimidine-5-carboxamide derivatives as potent and orally bioavailable STAT6 inhibitors. Bioorg. Med. Chem. 2008;16:6509–6521. doi: 10.1016/j.bmc.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 10.Nagashima S, Yokota M, Nakai E, Kuromitsu S, Ohga K, Takeuchi M, Tsukamoto S, Ohta M. Synthesis and evaluation of 2-{[2-(4-hydroxyphenyl)-ethyl]amino}pyrimidine-5-carboxamide derivatives as novel STAT6 inhibitors. Bioorg. Med. Chem. 2007;15:1044–1055. doi: 10.1016/j.bmc.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 11.Ohga K, Kuromitsu S, Takezawa R, Numazaki M, Ishikawa J, Nagashima S, Shimizu Y. YM-341619 suppresses the differentiation of spleen T cells into Th2 cells in vitro, eosinophilia, and airway hyperresponsiveness in rat allergic models. Eur. J. Pharmacol. 2008;590:409–416. doi: 10.1016/j.ejphar.2008.06.035. [DOI] [PubMed] [Google Scholar]
- 12.Tanabe T, Kanoh S, Tsushima K, Yamazaki Y, Kubo K, Rubin BK. Clarithromycin inhibits interleukin-13-induced goblet cell hyperplasia in human airway cells. Am. J. Respir. Cell Mol. Biol. 2011;45:1075–1083. doi: 10.1165/rcmb.2010-0327OC. [DOI] [PubMed] [Google Scholar]
- 13.Zhou L, Kawate T, Liu X, Kim YB, Zhao Y, Feng G, Banerji J, Nash H, Whitehurst C, Jindal S, Siddiqui A, Seed B, Wolfe JL. STAT6 phosphorylation inhibitors block eotaxin-3 secretion in bronchial epithelial cells. Bioorg. Med. Chem. 2012;20:750–758. doi: 10.1016/j.bmc.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 14.Kudlacz E, Conklyn M, Andresen C, Whitney-Pickett C, Changelian P. The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur. J. Pharmacol. 2008;582:154–161. doi: 10.1016/j.ejphar.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 15.Matsunaga Y, Inoue H, Fukuyama S, Yoshida H, Moriwaki A, Matsumoto T, Matsumoto K, Asai Y, Kubo M, Yoshimura A, Nakanishi Y. Effects of a Janus kinase inhibitor, pyridone 6, on airway responses in a murine model of asthma. Biochem. Biophys. Res. Commun. 2011;404:261–267. doi: 10.1016/j.bbrc.2010.11.104. [DOI] [PubMed] [Google Scholar]
- 16.Wang LH, Yang XY, Kirken RA, Resau JH, Farrar WL. Targeted disruption of stat6 DNA binding activity by an oligonucleotide decoy blocks IL-4-driven T(H)2 cell response. Blood. 2000;95:1249–1257. [PubMed] [Google Scholar]
- 17.McCusker CT, Wang Y, Shan J, Kinyanjui MW, Villeneuve A, Michael H, Fixman ED. Inhibition of experimental allergic airways disease by local application of a cell-penetrating dominant-negative STAT-6 peptide. J. Immunol. 2007;179:2556–2564. doi: 10.4049/jimmunol.179.4.2556. [DOI] [PubMed] [Google Scholar]
- 18.Stolzenberger S, Haake M, Duschl A. Specific inhibition of interleukin-4-dependent Stat6 activation by an intracellularly delivered peptide. Eur. J. Biochem. 2001;268:4809–4814. doi: 10.1046/j.1432-1327.2001.02417.x. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Li Y, Shan J, Fixman E, McCusker C. Effective treatment of experimental ragweed-induced asthma with STAT-6-IP, a topically delivered cell-penetrating peptide. Clin. Exp. Allergy. 2011;41:1622–1630. doi: 10.1111/j.1365-2222.2011.03853.x. [DOI] [PubMed] [Google Scholar]
- 20.Blease K. Therapeutics targeting IL-13 for the treatment of pulmonary inflammation and airway remodeling. Curr. Opin. Invest. Drugs. 2008;9:1180–1184. [PubMed] [Google Scholar]
- 21.Walsh GM. An update on emerging drugs for asthma. Expert Opin. Emerging Drugs. 2012;17:37–42. doi: 10.1517/14728214.2012.657625. [DOI] [PubMed] [Google Scholar]
- 22.Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, Wang L, Kirkesseli S, Rocklin R, Bock B, Hamilton J, Ming JE, Radin A, Stahl N, Yancopoulos GD, Graham N, Pirozzi G. Dupilumab in persistent asthma with elevated eosinophil levels. N. Engl. J. Med. 2013;368:2455–2466. doi: 10.1056/NEJMoa1304048. [DOI] [PubMed] [Google Scholar]
- 23.McKinney J, Raimundo BC, Cushing TD, Yoshimura H, Ohuchi Y, Hiratate A, Fukushima H. Preparation of peptides as inhibitors of STAT function. 6426331 B1. US. 2002
- 24.McKinney J, Raimundo BC, Cushing TD, Yoshimura H, Ohuchi Y, Hiratate A, Fukushima H, Xu F, Peto C. STAT4 and STAT6 binding dipeptide derivatives. 2001083517 20000503. WO. 2001
- 25.Zhou S, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Neel BG, Birge BR, Fajardo JE, Chou MM, Hanafusa H, Schaffhausen B, Cantley LC. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 26.Songyang Z, Shoelson SE, McGlade J, Olivier P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T, Ren R, Baltimore D, Ratnofsky S, Feldman RA, Cantley LC. Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol. Cell. Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu P, Brasseur M, Schindler U. A high-throughput STAT binding assay using fluorescence polarization. Anal. Biochem. 1997;249:29–36. doi: 10.1006/abio.1997.2158. [DOI] [PubMed] [Google Scholar]
- 28.Mandal PK, Limbrick D, Coleman DR, Dyer GA, Ren Z, Birtwistle JS, Xiong C, Chen X, Briggs JM, McMurray JS. Conformationally constrained peptidomimetic inhibitors of signal transducer and activator of transcription 3: evaluation and molecular modeling. J. Med. Chem. 2009;52:2429–2442. doi: 10.1021/jm801491w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burke TR, Jr, Smyth MS, Otaka A, Nomizu M, Roller PP, Wolf G, Case R, Shoelson SE. Nonhydrolyzable phosphotyrosyl mimetics for the preparation of phosphatase-resistant SH2 domain inhibitors. Biochemistry. 1994;33:6490–6494. doi: 10.1021/bi00187a015. [DOI] [PubMed] [Google Scholar]
- 30.Farquhar D, Khan S, Srivastva DN, Saunders PP. Synthesis and antitumor evaluation of bis[(pivaloyloxy)methyl] 2′-deoxy-5-fluorouridine 5′-monophosphate (FdUMP): a strategy to introduce nucleotides into cells. J. Med. Chem. 1994;37:3902–3909. doi: 10.1021/jm00049a009. [DOI] [PubMed] [Google Scholar]
- 31.Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, Kaluarachchi KK, Chen X, Bast RC, Liao WS, McMurray JS. Potent and selective phosphopeptide mimetic prodrugs targeted to the Src homology 2 (SH2) domain of signal transducer and activator of transcription 3. J. Med. Chem. 2011;54:3549–5463. doi: 10.1021/jm2000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mandal PK, Liao WS, McMurray JS. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org. Lett. 2009;11:3394–3397. doi: 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morlacchi P, Mandal PK, McMurray JS. Synthesis and in vitro evaluation of a peptidomimetic inhibitor targeting the Src homology 2 (SH2) domain of STAT6. ACS Med. Chem. Lett. 2014;5:69–72. doi: 10.1021/ml4003919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coleman DRIV, Kaluarachchi K, Ren Z, Chen X, McMurray JS. Solid phase synthesis of phosphopeptides incorporating 2,2-dimethyloxazolidine pseudoproline analogs: evidence for trans Leu-Pro peptide bonds in Stat3 inhibitors. Int. J. Pept. Res. Ther. 2008;14:1–9. [Google Scholar]
- 35.Dhanik A, McMurray JS, Kavraki LE. DINC: a new AutoDock-based protocol for docking large ligands. BMC Struct. Biol. 2013;13:S11. doi: 10.1186/1472-6807-13-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhanik A, McMurray JS, Kavraki LE. Binding modes of peptidomimetics designed to inhibit STAT3. PLoS One. 2012;7:e51603. doi: 10.1371/journal.pone.0051603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pieper U, Webb BM, Barkan DT, Schneidman-Duhovny D, Schlessinger A, Braberg H, Yang Z, Meng EC, Pettersen EF, Huang CC, Datta RS, Sampathkumar P, Madhusudhan MS, Sjolander K, Ferrin TE, Burley SK, Sali A. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2011;39:D465–D474. doi: 10.1093/nar/gkq1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Jr, Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- 39.Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, McMurray JS, Demeler B, Darnell JE, Jr, Chen X. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol. Cell. 2005;17:761–771. doi: 10.1016/j.molcel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 40.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 41.McMurray JS. Structural basis for the binding of high affinity phosphopeptides to Stat3. Biopolymers. 2008;90:69–79. doi: 10.1002/bip.20901. [DOI] [PubMed] [Google Scholar]
- 42.Mandal PK, Ren Z, Chen X, Kaluarachchi K, Liao WSL, McMurray JS. Structure-activity studies of phosphopeptidomimetic prodrugs targeting the Src homology 2 (SH2) domain of signal transducer and activator of transcription 3 (Stat3) Int. J. Pept. Res. Ther. 2013;19:3–12. doi: 10.1007/s10989-012-9313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M, Zinner R, Hung MC, Steck P, Siminovitch K, Mills GB. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999;18:7034–7045. doi: 10.1038/sj.onc.1203183. [DOI] [PubMed] [Google Scholar]
- 44.Workman P. Inhibiting the phosphoinositide 3-kinase pathway for cancer treatment. Biochem. Soc. Trans. 2004;32:393–396. doi: 10.1042/bst0320393. [DOI] [PubMed] [Google Scholar]
- 45.Hoeck J, Woisetschlager M. Activation of eotaxin-3/CCLl26 gene expression in human dermal fibroblasts is mediated by STAT6. J. Immunol. 2001;167:3216–3222. doi: 10.4049/jimmunol.167.6.3216. [DOI] [PubMed] [Google Scholar]
- 46.Hoshino A, Tsuji T, Matsuzaki J, Jinushi T, Ashino S, Teramura T, Chamoto K, Tanaka Y, Asakura Y, Sakurai T, Mita Y, Takaoka A, Nakaike S, Takeshima T, Ikeda H, Nishimura T. STAT6-mediated signaling in Th2-dependent allergic asthma: critical role for the development of eosinophilia, airway hyper-responsiveness and mucus hypersecretion, distinct from its role in Th2 differentiation. Int. Immunol. 2004;16:1497–1505. doi: 10.1093/intimm/dxh151. [DOI] [PubMed] [Google Scholar]
- 47.Chao H-G, Leiting B, Reiss PD, Burkhardt AL, Klimas CE, Bolen JB, Matsueda GR. Synthesis and application of Fmoc-O-[bis(dimethylamino)phosphono]tyrosine, a versatile protected phosphotyrosine equivalent. J. Org. Chem. 1995;60:7710–7711. [Google Scholar]
- 48.Ueki M, Goto M, Okumura J, Ishii Y. N,N′-dialkyldiamide-type phosphate protecting groups for Fmoc synthesis of phosphotyrosine-containing peptides: optimization of the alkyl group. Bull. Chem. Soc. Jpn. 1998;71:1887–1898. [Google Scholar]
- 49.Ueki M, Tachibana J, Ishii Y, Okumura J, Goto M. N,N′-Dialkyldiamide-type phosphate protecting groups for Fmoc synthesis of phosphotyrosine-containing peptides. Tetrahedron Lett. 1996;37:4953–4956. [Google Scholar]
- 50.Pearson DA, Blanchette M, Baker ML, Guindon CA. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989;30:2739–2742. [Google Scholar]
- 51.Barton DHR, Finet JP, Khamsi J. Copper salt catalysis of N-phenylation of amines by trivalent organobismuth compounds. Tetrahedron Lett. 1987;28:887–890. [Google Scholar]
- 52.Chan DMT. Promotion of reaction of N-H bonds with triarylbismuth and cupric acetate. Tetrahedron Lett. 1996;37:9013–9016. [Google Scholar]
- 53.Coleman DRt, Ren Z, Mandal PK, Cameron AG, Dyer GA, Muranjan S, Campbell M, Chen X, McMurray JS. Investigation of the binding determinants of phosphopeptides targeted to the SRC homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J. Med. Chem. 2005;48:6661–6670. doi: 10.1021/jm050513m. [DOI] [PubMed] [Google Scholar]
- 54.Ren Z, Cabell LA, Schaefer TS, McMurray JS. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg. Med. Chem. Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- 55.Takatori H, Nakajima H, Hirose K, Kagami S, Tamachi T, Suto A, Suzuki K, Saito Y, Iwamoto I. Indispensable role of Stat5a in Stat6-independent Th2 cell differentiation and allergic airway inflammation. J. Immunol. 2005;174:3734–3740. doi: 10.4049/jimmunol.174.6.3734. [DOI] [PubMed] [Google Scholar]
- 56.Kawabata T, Matsuda S, Kawakami S, Monguchi D, Moriyama K. Stereochemical diversity in asymmetric cyclization via memory of chirality. J. Am. Chem. Soc. 2006;128:15394–15395. doi: 10.1021/ja0670761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.