

ABSTRACT
Chronic wasting disease (CWD) in cervids and bovine spongiform encephalopathy (BSE) in cattle are prion diseases that are caused by the same protein-misfolding mechanism, but they appear to pose different risks to humans. We are interested in understanding the differences between the species barriers of CWD and BSE. We used real-time, quaking-induced conversion (RT-QuIC) to model the central molecular event in prion disease, the templated misfolding of the normal prion protein, PrPc, to a pathogenic, amyloid isoform, scrapie prion protein, PrPSc. We examined the role of the PrPc amino-terminal domain (N-terminal domain [NTD], amino acids [aa] 23 to 90) in cross-species conversion by comparing the conversion efficiency of various prion seeds in either full-length (aa 23 to 231) or truncated (aa 90 to 231) PrPc. We demonstrate that the presence of white-tailed deer and bovine NTDs hindered seeded conversion of PrPc, but human and bank vole NTDs did the opposite. Additionally, full-length human and bank vole PrPcs were more likely to be converted to amyloid by CWD prions than were their truncated forms. A chimera with replacement of the human NTD by the bovine NTD resembled human PrPc. The requirement for an NTD, but not for the specific human sequence, suggests that the NTD interacts with other regions of the human PrPc to increase promiscuity. These data contribute to the evidence that, in addition to primary sequence, prion species barriers are controlled by interactions of the substrate NTD with the rest of the substrate PrPc molecule.
IMPORTANCE We demonstrate that the amino-terminal domain of the normal prion protein, PrPc, hinders seeded conversion of bovine and white-tailed deer PrPcs to the prion forms, but it facilitates conversion of the human and bank vole PrPcs to the prion forms. Additionally, we demonstrate that the amino-terminal domain of human and bank vole PrPcs requires interaction with the rest of the molecule to facilitate conversion by CWD prions. These data suggest that interactions of the amino-terminal domain with the rest of the PrPc molecule play an important role in the susceptibility of humans to CWD prions.
INTRODUCTION
Chronic wasting disease (CWD), bovine spongiform encephalopathy (BSE; mad cow disease) and Creutzfeldt-Jakob disease (CJD) are prion diseases of cervids, cattle, and humans, respectively. These neurodegenerative diseases are caused by the pathogenic misfolding of the normal prion protein (PrPc) to an amyloid conformation (scrapie prion protein, PrPSc), the accumulation of which causes neuronal death (1, 2). CWD was first identified in the western United States in the 1960s in captive mule deer (Odocoileus hemionus hemionus) (3) and has since spread into free-ranging and farmed cervid populations in 24 states and in Norway, the Republic of Korea, and two Canadian provinces (4). BSE was identified in the United Kingdom in the 1980s and is hypothesized to have been spread as a result of feeding animal by-products to cattle (5, 6). The BSE epidemic led to the culling of over 4.5 million cattle, and over 200 people died from variant CJD, a form of CJD thought to be acquired from consumption of BSE-tainted beef (7–12). So far, there is no evidence of transmission of CWD to humans, but the BSE epidemic suggests that prions have zoonotic potential.
Efforts to define the host range of both BSE and CWD have included experimental infections of nonhost species. CWD inoculations have resulted in prion propagation in cattle, sheep, hamsters, voles, felines, ferrets, and some nonhuman primates (13–20). BSE inoculations have resulted in prion propagation in deer, sheep, and some nonhuman primates (21–26). Moreover, natural BSE infections have occurred in felines and several species of zoo ungulates, in addition to humans (9, 11, 12). Transgenic mice expressing human PrPc (TgHu) have been used to model human susceptibility to animal prion diseases. Published reports of CWD inoculation of TgHu mice have reported no prion propagation although studies employing protein misfolding cyclic amplification (PMCA) have yielded more ambiguous results (27). Prion propagation has been reported in approximately 50% of TgHu mice inoculated with BSE (28–35). Despite these reports, prion species barriers remain mostly empirical. While the most important known factor in transspecies transmission is the compatibility of the PrPc amino acid sequences, there is evidence that the tertiary and quaternary structures of PrPSc also influence prion transmission (36–39).
Multiple studies have identified specific PrPc regions that may control susceptibility or resistance to conversion by nonhomologous prions, but the role of the amino-terminal domain (N-terminal domain [NTD], which we define as amino acids [aa] 23 to 90, according to mouse numbering) in species barrier maintenance has not been tested (40, 41). Transgenic mice with an amino-terminal PrPc truncation that resulted in expression of only aa 90 to 231 had no overt changes in phenotype compared to that of wild-type mice, while larger truncations caused spontaneous neurodegenerative disease (42). A transgenic mouse expressing chimeric mouse-hamster PrPc (aa 90 to 231) had reduced susceptibility to prion infection, which varied with the strain of mouse-adapted scrapie used for infection (43). Mice that expressed mouse aa 90 to 231 propagated prions upon inoculation but had delayed disease onset and lower PrPSc titers (44). Mice expressing mouse aa 88 to 231 had reduced susceptibility to infection compared with mice expressing full-length PrPc, but disease onset was accelerated when the inoculum consisted of PrPSc with the same truncation (45, 46). Overall, these results suggest that amino acids 23 to 90 are involved in prion propagation and disease progression.
Evidence from in vitro studies indicates that the amino-terminal fragment of the protein (amino acids 23 to 144) will misfold easily without addition of a prion seed and that the human fragment converted faster than either the hamster or mouse fragment (47). Other investigators determined that the aggregation mechanism and resulting aggregate morphology were heavily dependent on physical conditions (pH) and truncation of the protein (aa 90 to 231 versus the full-length protein) (48, 49). These in vitro data indicate that the absence of aa 23 to 90 has dramatic effects on the mechanism and on the likelihood of misfolding. We were interested in the region from aa 23 to 90 not only for its effect on amyloidogenicity but also for its role in preventing (or facilitating) templated misfolding between species.
We chose to compare full-length recombinant PrPc (rPrPc; aa 23 to 231) to truncated rPrPc comprised of aa 90 to 231 for several reasons: (i) the PrP fragment of aa 27 to 30, an important molecule identified early in prion disease research, is comprised of amino acids 90 to 231 (50); (ii) amino acids 23 to 90 comprise the N2 cleavage product in the disease state (51); and (iii) the fragment of aa 90 to 231 is commonly used as the substrate for prion detection in real-time, quaking-induced conversion (RT-QuIC) assays (52). Specifically, we hypothesized that the amino terminal domain (amino acids 23 to 90; NTD) decreases an rPrPc molecule's propensity to form amyloid and that it plays a role in defining in vitro species barriers. We have assessed species barriers using real-time, quaking-induced conversion (i) because it makes possible assessment of conversion efficiency (by detection of amyloid formation in real time) and (ii) because we can study the effects of changes to the PrPc primary structure without the complexity of a transgenic mouse or cell culture system.
MATERIALS AND METHODS
Recombinant PrP cloning and expression.
The coding regions for the bank vole and bovine prion proteins (PRNP genes) were kindly provided by Glenn Telling and Jifeng Bian, and we obtained cDNA for the white-tailed deer (WTD), human, and chimeric PRNP genes from Genewiz, Inc. We cloned the sequences for the full-length PrP (amino acids 23 to 231) into the pet100D expression system (Life Technologies). We cloned the sequences for the truncated PrP (amino acids 90 to 231) into a vector kindly provided by Byron Caughey. We transformed and amplified the plasmids in Escherichia coli Top 10 cells (Life Technologies) and then stored the plasmids in E. coli BL21 Star cells (Life Technologies) in glycerol stocks at −80°C. To express PrP, we added BL21 cells from the glycerol stock to 5 ml of LB medium and antibiotics (5 μg/ml ampicillin for full-length constructs and 25 μg/ml kanamycin for truncated constructs) and grew the culture overnight, with shaking at 37o. We used the 5-ml culture to inoculate 1 liter of LB medium plus antibiotics as described above. We added auto-induction reagents for a final concentration of 0.5 M (NH4)2SO4, 1 M KH2PO4, 1 M Na2HPO4, 0.5% glycerol, 0.05% glucose, 0.2% α-lactose, and 0.001 M MgSO4. We harvested the bacteria when the optical density at 600 nm (OD600) reached approximately 3.0 for full-length constructs and 1.7 for truncated constructs. We either froze the cell pellets at −80°C for up to 5 days or immediately lysed the cells and harvested inclusion bodies, which we isolated according to the manufacturer's protocol with Bug Buster and Lysonase (EMD-Millipore).
Recombinant PrP purification.
We solubilized inclusion bodies in 8 M guanidine hydrochloride (GdnHCl) and 100 mM Na2HPO4 and rotated the solution overnight at room temperature. We bound the denatured rPrP to Superflow nickel resin (Qiagen) and refolded the rPrPc at a rate of 0.75 ml/min on the column in buffer with a gradient from 6.0 M GdnHCl, 100 mM Na2HPO4, and 10 mM Tris (pH 8.0) to the same buffer with no GdnHCl (wash buffer). We used a gradient from the wash buffer to 1.0 M imidazole, 100 mM NaH2PO4, and 10 mM Tris (pH 5.5) with a flow rate of 2.0 ml/min to elute full-length PrP. For truncated PrP, the final gradient ended with 0.5 M imidazole. We filtered and dialyzed the eluted rPrP at a concentration of ∼0.4 mg/ml in two changes of 4.0 liters of 20 mM NaH2PO4 (pH 5.5). After dialysis, we filtered and stored the rPrP at 4°C, its concentration having been determined by the A280 and Beer's law. We confirmed the purity of the rPrP samples by gel electrophoresis (12% bis-Tris gels with 1× morpholinepropanesulfonic acid [MOPS; Bio-Rad], 190 V, 55 min) and staining with Coomassie brilliant blue (Bio-Rad).
Preparation of prion seed material.
We homogenized brain tissue from deer, cattle, or humans at 10% weight/volume in 1× phosphate-buffered saline (PBS) using a FastPrep bead beater (MB Biomedicals). Single-use aliquots of 10% homogenate were stored at −80°C. Both prion-positive and prion-negative brain materials were prepared. Prion-positive brain material was used for the experiments, while prion-negative brain was used for the unseeded controls. The BSE sample was classical BSE (cBSE), and the sporadic CJD (sCJD) sample was MM1 (type 1).
Real-time, quaking-induced conversion (RT-QuIC) for full-length rPrP.
We combined 0.1 mg/ml rPrP with a premixed reaction solution that contained a final concentration of 400 mM NaCl, 1 mM EDTA, and 20 mM NaH2PO4. Then, we added freshly made thioflavin T (ThT; final concentration, 10 μM) for a final volume of 98 μl per well in a 96-well plate. We thawed aliquots of 10% brain homogenate and serially diluted the samples into 1× PBS plus 0.1% SDS. We added 2 μl of diluted brain seed to 98 μl of protein substrate mix in the well of an optical-bottom, 96-well plate. An RT-QuIC experiment consisted of 400 cycles (100 h) of shaking and incubation at 45°C; specifically, plates were shaken for 1 min (700 rpm, double orbital) followed by 1 min of rest. Fluorescence (450-nm excitation and 480-nm emission; 20 flashes/well) was recorded every 15 min using a gain setting of 1700 in a FLUOstar microplate reader (BMG Labtech). To compare full-length PrP directly to truncated PrP with the same molarity (0.6 nM), we increased the concentration of full-length rPrP to 0.14 mg/ml and that of the truncated rPrP to 0.10 mg/ml (see Fig. 2). These conditions were optimized for use with full-length rPrP. For the experiments shown in Fig. 4, conditions were as follows: 0.6 nM rPrP, 320 mM NaCl, and 0.1% SDS at 42°C.
FIG 2.
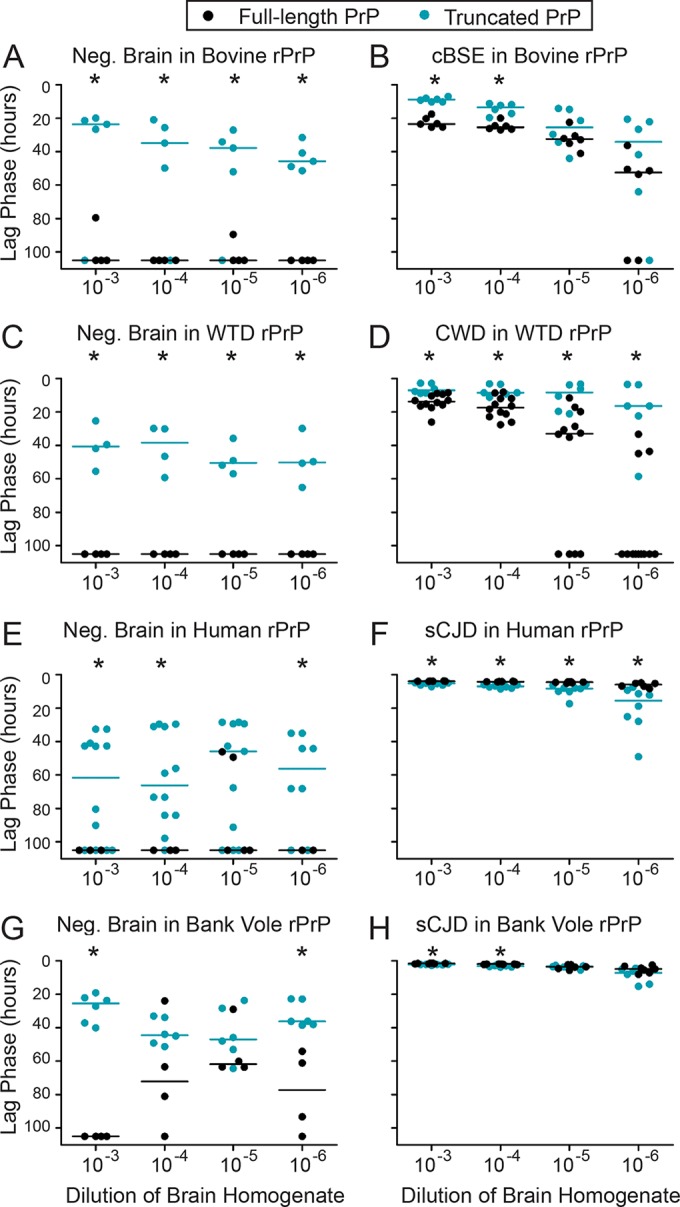
Removal of the amino-terminal domain affects rate of seeded conversion and increases spontaneous conversion of rPrP. Brain homogenate (prion positive or negative) was added to the homologous substrate, in either its full-length or truncated version. Reaction conditions were designed for increased amyloidogenicity and were the same for all experiments shown in this figure (RT-QuIC conditions for full-length PrP are as described in Materials and Methods). Scatterplots represent raw data, and lines indicate the medians. *, P < 0.05 (WMW test), for the difference between the lag phases for full-length and truncated substrates. (A) Truncated bovine rPrP spontaneously formed amyloid faster than full-length bovine rPrP (in the presence of normal brain homogenate). (B) cBSE seeded truncated bovine rPrP faster than full-length. (C) Truncated white-tailed deer rPrP spontaneously formed amyloid faster than full-length white-tailed deer rPrP (in the presence of normal brain homogenate). (D) CWD seeded truncated white-tailed deer rPrP faster than full-length. (E) Truncated human rPrP spontaneously formed amyloid faster than full-length human deer rPrP (in the presence of normal brain homogenate). (F) sCJD seeded full-length human rPrP faster than truncated. (G) Truncated bank vole deer rPrP spontaneously formed amyloid faster than full-length bank vole rPrP (in the presence of normal brain homogenate). (H) sCJD seeded full-length bank vole rPrP faster than truncated.
FIG 4.
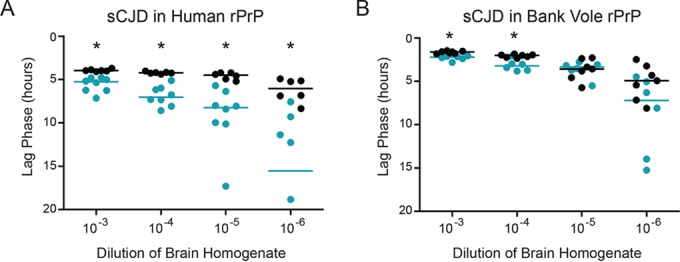
sCJD converts human and bank vole rPrPs very efficiently. These data are also displayed in Fig. 2, but they are shown here on a smaller y axis, which makes it easier to visualize the differences between the points. As described in the legend of Fig. 2, prion-positive human brain homogenate was added to human rPrPc substrate in either its full-length or truncated version (A) and to bank vole rPrPc, in either its full-length or truncated form (B). Reaction conditions were designed for increased amyloidogenicity and were the same for all experiments shown in this figure (RT-QuIC conditions for full-length PrP are as described in Materials and Methods). Scatterplots represent raw data, and lines indicate the medians. *, P < 0.05 (WMW test), for the difference between the lag phases for full-length and truncated substrates.
RT-QuIC for truncated PrP.
We combined 0.07 mg/ml rPrP with a premixed reaction solution that contained a final concentration of 320 mM NaCl, 1 mM EDTA, and 20 mM NaH2PO4. We added freshly made thioflavin T (ThT; final concentration, 10 μM) for a final volume of 98 μl per well in a 96-well plate. We thawed and serially diluted aliquots of 10% brain homogenate into 1× PBS plus 0.05% SDS. We added 2 μl of diluted brain seed to 98 μl of protein substrate mix in the well of an optical-bottom, 96-well plate. An RT-QuIC experiment consisted of at least 250 cycles (62.5 h) of shaking and incubation at 42°C; specifically, plates were shaken for 1 min (700 rpm, double orbital), followed by 1 min of rest. Fluorescence (450-nm excitation and 480-nm emission; 20 flashes/well) was recorded every 15 min using a gain setting of 1700 in a FLUOstar microplate reader (BMG Labtech). These conditions were optimized for use with truncated rPrP.
Data analysis and statistics.
The RT-QuIC readout is fluorescence, which was recorded at 15-min intervals (Fig. 1A). To assess the efficiency of reactions, we compared the lag phase for amyloid formation. First, we calculated a fluorescence threshold by averaging the baseline fluorescence for every well in the plate and then adding 5 standard deviations (SD). We used Omega software (BMG Labtech) to determine the lag phase, i.e., the time at which the fluorescence in a given well exceeded the threshold (Fig. 1B). If the fluorescence for a sample never crossed the threshold during the experiment, we estimated a value of 105 h (an example is marked by a gray X in Fig. 1B). Raw lag phase data are displayed as scatterplots with lines to indicate the medians. We analyzed the lag phase data using nonparametric, rank-based tests (Wilcoxon-Mann-Whitney [WMW] and Wilcoxon signed-rank tests). If all values for one sample were equal, WMW tests were invalid, so we used Wilcoxon signed-rank tests instead. We chose these tests for several reasons: (i) our data are often nonnormal (right skewed due to replicate wells that do not cross the threshold in the course of the experiment); (ii) our number of replicates is relatively small (8 to 12); and (iii) we must choose values to substitute for the replicate wells that do not cross the threshold. The choice of a substitute value would affect the mean (and the results of any statistical tests that relies on means, like t tests), but would not affect rank-based statistical analysis since those values will have the longest lag phase regardless of which value we choose. We chose to represent the median instead of mean for the same reason: the median is not affected by our choice of a value for wells that do not cross the threshold. We described differences as statistically significant at a P value of <0.05, which is indicated in the figures.
FIG 1.
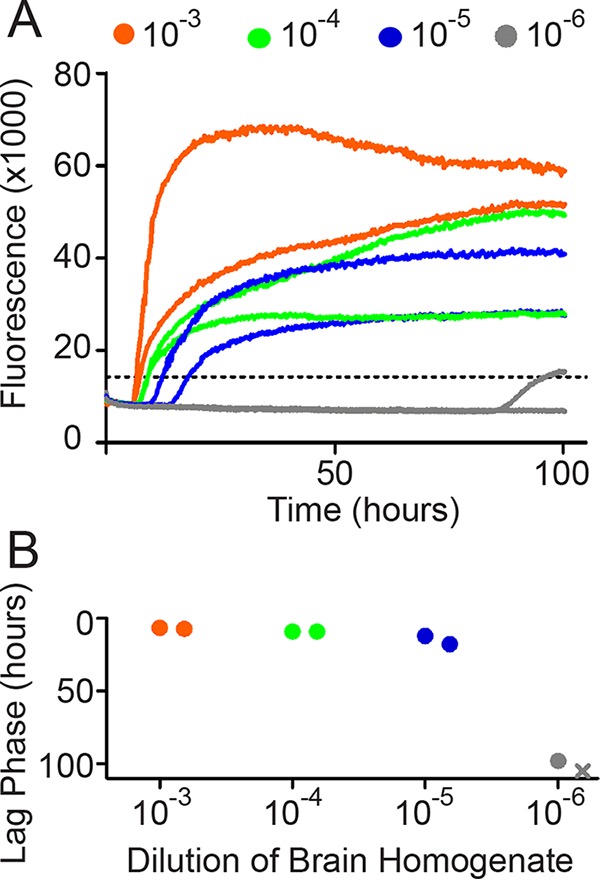
Data analysis workflow. (A) Raw data are recorded as fluorescence every 15 min. We show two technical replicates for this example. (B) We plot the time required to reach the threshold (+5 SD above the mean background) versus the dilution of 10% brain homogenate added to the reaction mixture. The X indicates an estimated value for a sample that did not cross the threshold during the experiment. We have reversed the y axis to make it easier to identify the fastest conversions as the highest points.
RESULTS
Removal of the amino-terminal domain affects efficiency of seeded conversion and increases spontaneous conversion of rPrPc.
To investigate the effect of the amino-terminal domain (NTD) on the propensity of rPrPc to form amyloid, we added the homologous prion seed (or negative brain material) to each species' rPrPc, either truncated or full-length (e.g., CWD to deer PrP). To ensure that any differences in the median amyloid formation lag phase would be due to differences in the rPrP/seed compatibility, not to other reaction conditions, we performed the RT-QuIC reactions with 0.6 nM rPrP, salt, SDS, and EDTA and at the same temperatures (full-length conditions are described in Materials and Methods).
We found for all species that truncated rPrPc was more prone to spontaneous conversion (after addition of negative brain material) than the full-length substrate (Fig. 2A, C, E, and G). The addition of cBSE brain to truncated bovine rPrPc (aa 90 to 231) resulted in faster amyloid conversion than addition of the same seed to full-length bovine rPrPc (Fig. 2B). Likewise, the addition of CWD seed to truncated white-tailed deer rPrPc resulted in faster amyloid conversion than addition of the same CWD material to full-length white-tailed deer rPrPc (Fig. 2D).
The addition of sporadic CJD (sCJD) MM1 seed to truncated or full-length human rPrPc (methionine at position 129) resulted in very fast amyloid conversion. However, conversion of the truncated human rPrPc was significantly slower than that of the full-length human rPrPc (Fig. 2F). Because the bank vole and human PRNP sequences are very similar in the NTD (Fig. 3), we compared truncated to full-length bank vole rPrPc as well. As with the human rPrPc, both the truncated and full-length bank vole rPrPcs formed amyloid very quickly upon addition of sCJD prions (which have been demonstrated to convert bank vole PrP effectively [53, 54]). Also as with the human rPrPc, conversion of the full-length bank vole rPrPc was statistically faster than that of truncated rPrPc (53, 54) (Fig. 2H). Because the conversions of human and bank vole rPrPcs were so fast, the data are also shown on a smaller scale (Fig. 4A and B).
FIG 3.

Primary sequence alignment. The amino acid sequences of bovine, human, bank vole, and the N-Hu-Bo-C and N-Bo-Hu-C chimeras are aligned. Green letters indicate differences between human and bovine NTD sequences, and red letters indicate differences between bank vole and human PrPc NTD sequences. The red bars indicate the junctions where human and bovine NTDs meet in the chimeras.
The amino-terminal domain is not essential for preferential seeding of rPrP species by their native prions.
Once we completed the direct comparisons of truncated rPrP to full-length rPrP, we modified the reaction conditions used for the truncated rPrP to improve specificity. We decreased the salt, SDS, PrP concentration, and temperature to reduce the amyloidogenicity of the reaction mixture. Experiments shown in subsequent figures were performed under these more specific conditions (details are given in Materials and Methods).
We hypothesized that the NTD may play a role in defining an rPrPc molecule as bovine or human or white-tailed deer. For the RT-QuIC assay, we expected a prion seed to cause the most efficient conversion in its host substrate (38), but we hypothesized that this might no longer be the case when the NTD was removed. Therefore, we added each prion of interest (cBSE, CWD, and sCJD) to its homologous substrate and to a nonhomologous substrate. For cBSE, CWD, and sCJD, there was no indication that the NTD is necessary for the prion to preferentially seed its host rPrPc. cBSE prions caused amyloid conversion faster in bovine rPrPc than in nonhost rPrPc (white-tailed deer), whether the rPrPc was amino-terminally truncated or full-length (Fig. 5A and B). CWD prions converted white-tailed deer rPrPc faster than nonhost rPrPc (bovine), whether the rPrPc was truncated or full-length (Fig. 5C and D). sCJD prions caused amyloid conversion faster in human rPrPc than in the nonhost (bovine) protein, whether or not the rPrPc was truncated (Fig. 5E and F). Therefore, the NTD was not essential for prion seeds to most efficiently seed their host rPrPcs. The amyloid formation in unseeded controls is indicated by the dotted lines in Fig. 5, and the distribution of these data is displayed in Fig. 6. Additionally, each data set in Fig. 5 was compared to unseeded controls, and the results are shown in Table 1.
FIG 5.
The amino-terminal domain is not essential for preferential seeding of rPrP species by their native prions. Prion-positive or prion-negative brain homogenate was added to both a homologous rPrPc substrate and a nonhost substrate, either truncated or full-length. Full-length rPrP experiments were performed under RT-QuIC conditions for full-length PrP, as described in Materials and Methods. Truncated rPrP experiments were performed under RT-QuIC conditions for truncated PrP, as described in Materials and Methods and unlike the conditions used for the experiments shown in Fig. 2. Scatterplots represent raw data, and lines indicate the medians. Dotted lines indicate the median lag phases for unseeded misfolding for truncated substrates (species are identified by color). *, P < 0.05 (WMW test), for the difference between the lag phases for full-length and truncated substrates. (A) cBSE prions converted full-length bovine rPrPc faster than white-tailed deer rPrPc. (B) cBSE prions also converted bovine rPrPc faster than white-tailed deer rPrPc when the substrates were truncated. (C) CWD prions converted white-tailed deer rPrPc faster than bovine rPrPc when the substrates were full-length. (D) CWD prions converted white-tailed deer rPrPc faster than bovine rPrPc when the substrates were truncated. (E) sCJD prions converted human rPrPc faster than bovine rPrPc when the substrates were full-length. (F) sCJD prions converted human rPrPc faster than bovine rPrPc when the substrates were truncated.
FIG 6.

Distribution of unseeded controls. A frequency histogram of the negative controls (negative brain material) is displayed for full-length bovine substrate (A), truncated bovine substrate (B), full-length human substrate (C), truncated human substrate (D), full-length white-tailed deer (WTD) substrate (E), and truncated WTD substrate (F). Data represent the frequency of unseeded controls crossing the threshold for each lag phase.
TABLE 1.
Statistical analysis of data in Fig. 5
| Prion seed and substrate | Fig. no. | Dilution | Pa |
|---|---|---|---|
| BSE | |||
| FL bovine | 5A | 10−3 | <0.05 |
| 10−4 | <0.05 | ||
| 10−5 | <0.05 | ||
| 10−6 | <0.05 | ||
| FL WTD | 10−3 | <0.05 | |
| 10−4 | <0.05 | ||
| 10−5 | NS | ||
| 10−6 | NS | ||
| Truncated bovine | 5B | 10−3 | <0.05 |
| 10−4 | <0.05 | ||
| 10−5 | <0.05 | ||
| 10−6 | NS | ||
| Truncated WTD | 10−3 | <0.05 | |
| 10−4 | <0.05 | ||
| 10−5 | NS | ||
| 10−6 | <0.05 | ||
| CWD | |||
| FL WTD | 5C | 10−3 | <0.05 |
| 10−4 | <0.05 | ||
| 10−5 | <0.05 | ||
| 10−6 | NS | ||
| FL bovine | 10−3 | <0.05 | |
| 10−4 | <0.05 | ||
| 10−5 | <0.05 | ||
| 10−6 | NS | ||
| Truncated WTD | 5D | 10−3 | <0.05 |
| 10−4 | <0.05 | ||
| 10−5 | <0.05 | ||
| 10−6 | NS | ||
| Truncated bovine | 10−3 | NS | |
| 10−4 | NS | ||
| 10−5 | NS | ||
| 10−6 | NS | ||
| sCJD | |||
| FL human | 5E | 10−3 | <0.05 |
| 10−4 | <0.05 | ||
| 10−5 | <0.05 | ||
| 10−6 | <0.05 | ||
| FL bovine | 10−3 | <0.05 | |
| 10−4 | <0.05 | ||
| 10−5 | <0.05 | ||
| 10−6 | <0.05 | ||
| Truncated human | 5F | 10−3 | <0.05 |
| 10−4 | <0.05 | ||
| 10−5 | <0.05 | ||
| 10−6 | <0.05 | ||
| Truncated bovine | 10−3 | <0.05 | |
| 10−4 | NS | ||
| 10−5 | NS | ||
| 10−6 | NS |
Difference from the unseeded control value. NS, not significant.
The amino-terminal domain enables CWD conversion of human rPrPc in vitro.
Above, we showed that homologous prions are better seeds for their native rPrPcs, despite truncation (Fig. 5). However, when we assessed the behavior of nonhomologous prions in human rPrPc, we observed that the truncated human rPrPc is also less permissive to CWD prions than the full-length human rPrPc (Fig. 7A).
FIG 7.
The amino-terminal domain enables CWD conversion of human rPrPc. (A) Prion-positive or prion-negative brain homogenate from white-tailed deer was added to truncated or full-length human rPrPc. Experiments for both were performed under the same conditions, which are described in Materials and Methods. (B) Prion-positive or prion-negative brain homogenate from white-tailed deer was added to truncated or full-length bank vole rPrPc. Scatterplots represent raw data. *, P < 0.05 (WMW test), for the difference between the CWD susceptibilities of full-length and truncated substrates.
The amino-terminal domain improves CWD conversion of bank vole rPrPc in vitro.
Because bank vole and human PrPcs have only one amino acid difference in their NTDs (Fig. 3), we hypothesized that the full-length bank vole rPrPc might also be more susceptible to conversion by CWD prions than the truncated bank vole rPrPc.
We tested the effect of amino-terminal truncation on the promiscuity of the bank vole rPrPc (Fig. 7B). Again, CWD caused conversion slightly more efficiently in full-length bank vole PrPc than in truncated bank vole PrPc.
The human amino-terminal domain does not increase susceptibility when substituted into another rPrPc.
Since the human NTD behaved differently than that of bovine or white-tailed deer in its effect on amyloidogenicity, we hypothesized that the human NTD may confer increased promiscuity to the molecule and that the substitution of the human NTD into bovine rPrPc would increase the susceptibility of the chimeric rPrPc to CWD prions. To test this, we developed a chimera containing the human NTD and bovine carboxy-terminal domain (N-Hu-Bo-C). cBSE caused PrPc conversion with indistinguishable lag phases in the N-Hu-Bo-C chimera and bovine rPrPc, which suggests that the NTD substitution does not reduce the homologous seeding of bovine rPrPc with cBSE (Fig. 8A). sCJD prions converted the human rPrPc faster than the N-Hu-Bo-C chimera, indicating that the human NTD alone was not sufficient to define the rPrPc substrate as human (Fig. 8C). Finally, seeding efficiencies with CWD were equivalent in the N-Hu-Bo-C chimera and in bovine rPrPc, suggesting that the substitution of the human amino-terminal domain did not increase the propensity of bovine rPrPc to be converted by CWD prions (Fig. 8E).
FIG 8.

The human rPrPc is susceptible to conversion by CWD prions regardless of its amino-terminal domain sequence. However, the human amino-terminal domain does not confer increased susceptibility to other species' rPrPcs. Prion-positive or prion-negative brain homogenate from white-tailed deer (CWD), cattle (cBSE), or humans (sCJD) was added to full-length human, bovine, or chimeric rPrPc. These experiments were performed under RT-QuIC conditions for full-length PrP, as described in Materials and Methods. Scatterplots represent raw data, and lines indicate the medians. Dotted lines indicate the median lag phases for unseeded misfolding for truncated substrates (species are identified by color). Brackets indicate a significant difference between either full-length bovine or full-length human and the chimera (P < 0.05, WMW). (A) cBSE prions convert bovine and N-Hu-Bo-C chimeric rPrPcs with the same efficiencies, but human rPrPc is slower. (B) cBSE prions convert human and N-Bo-Hu-C chimeric rPrPcs with the same efficiencies, while the conversion of bovine rPrPc is faster. (C) sCJD prions convert bovine and N-Hu-Bo-C chimeric rPrPcs with the same efficiencies, but human rPrPc is faster. (D) sCJD prions convert human and N-Bo-Hu-C chimeric rPrPcs with the same efficiencies, while the conversion of bovine rPrPc is slower. (E) CWD prions convert bovine and N-Hu-Bo-C chimeric rPrPcs with the same efficiencies, but conversion of human rPrPc is faster. (F) CWD prions convert human and N-Bo-Hu-C chimeric rPrPcs with the same efficiencies, while the conversion of bovine rPrPc is slower.
CWD prions convert human rPrPc efficiently, regardless of amino-terminal domain sequence.
Given the above result, we alternatively hypothesized that human rPrPc is susceptible to conversion by CWD prions independent of NTD sequence, as long as the NTD is present. We created the N-Bo-Hu-C chimera, which substitutes the bovine NTD for the human NTD, to investigate the importance of the NTD in the CWD species barrier. When we added cBSE prions to the N-Bo-Hu-C chimera, the conversion of the chimera was statistically indistinguishable from that with the full-length human rPrPc (Fig. 8B). When we added sCJD prions, the N-Bo-Hu-C chimera likewise was statistically indistinguishable from full-length human rPrPc (Fig. 8D). Finally, when we added CWD prions, the chimera was statistically indistinguishable from full-length human rPrPc (Fig. 8F). These results suggest that the presence of an amino-terminal domain preserves the susceptibility of full-length human rPrPc to CWD prions even if it does not have exactly the same sequence as the human NTD.
DISCUSSION
Many researchers have investigated the role of the amino-terminal domain (NTD; aa 23 to 90) of PrPc in normal phenotype, disease progression, and amyloidogenicity (43–45, 47–49, 55–57). However, the role of the NTD in transspecies transmission of prions has not been reported. Here, we used real-time, quaking-induced conversion (RT-QuIC) to compare the conversion efficiency of white-tailed deer (WTD), bovine, bank vole, or human PrPc, with or without the NTD, in the presence of CWD, cBSE, and sCJD or without prions.
We demonstrated that truncated (aa 90 to 231) WTD, bovine, bank vole, and human PrPcs spontaneously form amyloid faster than the full-length proteins. This suggests that the NTD reduces the amyloidogenicity of PrPc. However, the efficiency of conversion in the presence of a prion seed relies upon the compatibility of the prion seed with the substrate and upon the amyloidogenicity of the PrPc substrate. Truncated bovine and WTD rPrPcs formed amyloid faster than full-length rPrPcs upon addition of a seed, whereas truncated human and bank vole rPrPcs converted more slowly than full-length rPrPcs upon addition of a seed. This suggested that the NTDs of human and bank vole may increase the compatibility of the seed and substrate in some way. Next, we tested the effect of the NTD on the permissiveness of rPrPc to conversion by homologous and heterologous prions. cBSE, CWD, and sCJD converted their native substrates more efficiently than nonnative substrates, regardless of truncation. This suggested that a prion seed is able to recognize its host PrPc without the NTD and that the NTD does not play an essential role in defining a PrPc molecule as one species or another. We observed previously that human rPrPc is susceptible to conversion by CWD prions (38). Here, we demonstrated that CWD converted truncated human rPrPc less efficiently than it converted full-length human rPrPc, suggesting that the susceptibility of human rPrPc to CWD prions may involve the NTD. The results are summarized in Table 2.
TABLE 2.
Summary of results for NTD of various species' rPrPc proteinsa
| Species PrPc | NTD role in permissiveness (Fig. 5) | NTD role in rate of misfolding (Fig. 2) | NTD required to recognize host (Fig. 3) | Chimera behaves like (Fig. 8) |
|---|---|---|---|---|
| Bank vole | ↑ | ↑ | No | |
| Bovine | ↔ | ↓ | No | |
| Human | ↑ | ↑ | No | |
| White-tailed deer | ↔ | ↓ | No | |
| N-Hu-Bo-C | Bovine | |||
| N-Bo-Hu-C | Human |
Additional details are provided in the indicated figures. Increased permissiveness or misfolding is indicated by an upward arrow. Decreased permissiveness or misfolding is indicated by a downward arrow. No effect is indicated by a double-headed arrow.
Chimeric PrPc molecules are commonly used in prion research to focus on the region of the protein responsible for resistance to prion conversion (58–62). We were particularly interested in whether the NTD of human PrPc could increase susceptibility to transspecies conversion by CWD prions, so we created a chimera with a human NTD substituted into bovine PrPc (N-Hu-Bo-C) and a chimera with the bovine NTD substituted into human PrPc (N-Bo-Hu-C). We hypothesized that the human NTD would increase promiscuity, so we tested the susceptibility of the N-Hu-Bo-C chimera to seeded conversion. However, N-Hu-Bo-C was not different from bovine rPrPc, whether homologous or nonhomologous prions were used as seeds. We concluded that the human NTD alone did not confer additional susceptibility to conversion. Alternatively, we hypothesized that the presence of any NTD to human substrate would have the effect of increasing the susceptibility of the chimera versus that of the truncated rPrPc. Indeed, we found that N-Bo-Hu-C had essentially the same permissiveness as full-length human rPrPc to conversion by human, cBSE, and CWD prions. The importance of the NTD for human rPrPc susceptibility recalls transgenic mouse studies wherein truncation around amino acid 90 results in reduced attack rate and PrPSc accumulation (43, 44). It is possible that the human and bank vole PrPcs have a site in their globular domains that interacts with the NTD to facilitate seeded conversion, but not spontaneous conversion.
Together, the above data suggest that the PrPc NTD alone does not control species barriers in vitro but that interaction of the amino-terminal domain with the rest of the protein may be involved in the susceptibility of human rPrPc to in vitro conversion by CWD prions. Multiple investigators have studied the role of specific regions of the PrPc core and carboxy-terminal domain in prion disease species barriers. Reports have identified the β2-α2 loop and amino acids 165 to 175 as regions controlling transspecies transmission of prions, particularly of CWD prions to humans (35, 40). A favored interpretation of these data describes a steric zipper, wherein very few amino acid changes can severely disrupt the tertiary structure. Several polymorphisms between human and WTD PrPcs exist in this region, which may explain the apparent resistance of humans to CWD (39, 40). However, our data indicate that the variation in the β2-α2 loop does not prevent in vitro conversion, which suggests that factors in addition to the PrPc-PrPSc interactions we modeled in vitro must influence in vivo conversion.
Our empirical observation that unseeded conversion of truncated rPrPc was more efficient than that of full-length rPrPc made us wonder whether the NTD impeded conversion universally or in a species-specific manner, serving to protect against spontaneous misfolding or against transspecies infection. We hypothesized that the NTD may play a protective role in transspecies prion conversion, perhaps in addition to the β2-α2 loop. However, our data suggest that the NTD does not control transspecies prion conversion but that it may contribute to the promiscuity of human and bank vole rPrPc. It is possible that the NTD, which is natively unfolded, has some interaction with other regions of the rPrPc or with the PrPSc seed. A recent report suggests a structural mechanism by which the NTD may interact with the globular domain (63). These observations suggest that full-length protein should be used in structural studies of human PrPc and of the mechanisms of its seeded or spontaneous conversion.
We found that human rPrPc can be readily converted to an amyloid state by CWD prions and that the NTD facilitates this conversion. As there is little evidence for the susceptibility of humans to CWD, the biologic significance of our observation remains to be determined. However, the role of the NTD in this in vitro phenomenon may be important to the in vivo mechanism as well. RT-QuIC, transgenic mouse bioassay, and PMCA measure different outcomes. This report compares the efficiency of initial amyloid formation, while bioassay and PMCA reflect total accumulation of protease-resistant PrPSc, which may explain the difference in the apparent susceptibilities of full-length human rPrPc in these models. The molecular underpinnings for species barriers and transspecies prion conversion remain a complex yet important problem in prion biology. We propose that an interaction between the amino-terminal domain and the globular domain facilitates in vitro susceptibility of human rPrPc to conversion by CWD prions. Such an interaction may be important in assessing the susceptibility of humans to animal prion diseases.
ACKNOWLEDGMENTS
We thank Maurice Bardsley and James Hope (Animal and Plant Health Agency, Surrey, United Kingdom) for the classical BSE samples and Wen-Quan Zou and Pierluigi Gambetti for providing the sporadic CJD samples. We thank Glenn Telling and Jifeng Bian for the bovine and bank vole PRNP sequence plasmids, and we thank Timothy Kurt (UC San Diego, San Diego, CA, USA) for the bank vole PrP sequence. We thank Julia Labadie for helpful discussions surrounding data display and statistical analysis of the data. We thank laboratory member Clare Hoover for her insight and suggestions. Finally, we thank Byron Caughey, whose assistance in establishing RT-QuIC in our laboratory was critical.
This work was supported by NIH grant R01-NS-061902 (E.A.H.). K.A.D. was supported by NIH fellowship F30-OD-021442.
REFERENCES
- 1.Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE. 1984. Purification and structural studies of a major scrapie prion protein. Cell 38:127–134. doi: 10.1016/0092-8674(84)90533-6. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner S. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 3.Williams ES, Young S. 1980. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis 16:89–98. doi: 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- 4.Haley NJ, Hoover EA. 2015. Chronic wasting disease of cervids: current knowledge and future perspectives. Annu Rev Anim Biosci 3:305–325. doi: 10.1146/annurev-animal-022114-111001. [DOI] [PubMed] [Google Scholar]
- 5.Wells GAH, McGill IS. 1992. Recently described scrapie-like encephalopathies of animals: case definitions. Res Vet Sci 53:1–10. doi: 10.1016/0034-5288(92)90076-E. [DOI] [PubMed] [Google Scholar]
- 6.Anderson RM, Donnelly CA, Ferguson NM, Woolhouse MEJ, Watt CJ, Udy HJ, MaWhinney S, Dunstan SP, Southwood TRE, Wilesmith JW, Ryan JBM, Hoinville LJ, Hillerton JE, Austin AR, Wells GAH. 1996. Transmission dynamics and epidemiology of BSE in British cattle. Nature 382:779–788. doi: 10.1038/382779a0. [DOI] [PubMed] [Google Scholar]
- 7.Scott MR, Will R, Ironside J, Nguyen H-OB, Tremblay P, DeArmond SJ, Prusiner SB. 1999. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci U S A 96:15137–15142. doi: 10.1073/pnas.96.26.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almond J, Pattison J. 1997. Human BSE. Nature 389:437–438. doi: 10.1038/38876. [DOI] [PubMed] [Google Scholar]
- 9.Bruce ME, Will R, Ironside J, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C. 1997. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature 389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 10.Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey LJ, Lantos P. 1997. The same prion strain causes vCJD and BSE. Nature 389:448–450. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 11.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. 1996. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 12.Will RG, Ironside JW, Zeidler M, Estibeiro K, Cousens SN, Smith PG, Alperovitch A, Poser S, Pocchiari M, Hofman A. 1996. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347:921–925. doi: 10.1016/S0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 13.Di Bari MA, Nonno R, Castilla J, D'Agostino C, Pirisinu L, Riccardi G, Conte M, Richt J, Kunkle R, Langeveld J, Vaccari G, Agrimi U. 2013. Chronic wasting disease in bank voles: characterisation of the shortest incubation time model for prion diseases. PLoS Pathog 9:e1003219. doi: 10.1371/journal.ppat.1003219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamir AN, Kunkle RA, Miller JM, Greenlee JJ, Richt JA. 2006. Experimental second passage of chronic wasting disease (CWDmuledeer) agent to cattle. J Comp Pathol 134:63–69. doi: 10.1016/j.jcpa.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 15.Hamir AN, Kunkle RA, Cutlip RC, Miller JM, Williams ES, Richt JA. 2006. Transmission of chronic wasting disease of mule deer to Suffolk sheep following intracerebral inoculation. J Vet Diagn Invest 18:558–565. doi: 10.1177/104063870601800606. [DOI] [PubMed] [Google Scholar]
- 16.Hamir AN, Kunkle RA, Cutlip RC, Miller JM, O'Rourke KI, Williams ES, Miller MW, Stack MJ, Chaplin MJ, Richt JA. 2005. Experimental transmission of chronic wasting disease agent from mule deer to cattle by the intracerebral route. J Vet Diagn Invest 17:276–281. doi: 10.1177/104063870501700313. [DOI] [PubMed] [Google Scholar]
- 17.Hamir AN, Cutlip RC, Miller JM, Williams ES, Stack MJ, Miller MW, O'Rourke KI, Chaplin MJ. 2001. Preliminary findings on the experimental transmission of chronic wasting disease agent of mule deer to cattle. J Vet Diagn Invest 13:91–96. doi: 10.1177/104063870101300121. [DOI] [PubMed] [Google Scholar]
- 18.Race B, Meade-White KD, Miller MW, Barbian KD, Rubenstein R, LaFauci G, Cervenakova L, Favara C, Gardner D, Long D, Parnell M, Striebel J, Priola SA, Ward A, Williams ES, Race R, Chesebro B. 2009. Susceptibilities of nonhuman primates to chronic wasting disease. Emerg Infect Dis 15:1366–1376. doi: 10.3201/eid1509.090253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartz JC, Marsh RF, McKenzie DI, Aiken JM. 1998. The host range of chronic wasting disease is altered on passage in ferrets. Virology 251:297–301. doi: 10.1006/viro.1998.9427. [DOI] [PubMed] [Google Scholar]
- 20.Mathiason CK, Nalls AV, Seelig DM, Kraft SL, Carnes K, Anderson KR, Hayes-Klug J, Hoover EA. 2013. Susceptibility of domestic cats to chronic wasting disease. J Virol 87:1947–1956. doi: 10.1128/JVI.02592-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dagleish MP, Martin S, Steele P, Finlayson J, Eaton SL, Sisó S, Stewart P, Fernández-Borges N, Hamilton S, Pang Y, Chianini F, Reid HW, Goldmann W, González L, Castilla J, Jeffrey M. 2015. Susceptibility of European red deer (Cervus elaphus elaphus) to alimentary challenge with bovine spongiform encephalopathy. PLoS One 10:e0116094. doi: 10.1371/journal.pone.0116094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dagleish MP, Martin S, Steele P, Finlayson J, Sisó S, Hamilton S, Chianini F, Reid HW, González L, Jeffrey M. 2008. Experimental transmission of bovine spongiform encephalopathy to European red deer (Cervus elaphus elaphus). BMC Vet Res 4:17. doi: 10.1186/1746-6148-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lasmézas CI, Comoy E, Hawkins S, Herzog C, Mouthon F, Konold T, Auvré F, Correia E, Lescoutra-Etchegaray N, Salès N. 2005. Risk of oral infection with bovine spongiform encephalopathy agent in primates. Lancet 365:781–783. doi: 10.1016/S0140-6736(05)17985-9. [DOI] [PubMed] [Google Scholar]
- 24.Lasmézas CI, Fournier J-G, Nouvel V, Boe H, Marcé D, Lamoury F, Kopp N, Hauw J-J, Ironside J, Bruce M, Dormont D, Deslys J-P. 2001. Adaptation of the bovine spongiform encephalopathy agent to primates and comparison with Creutzfeldt-Jakob disease: implications for human health. Proc Natl Acad Sci U S A 98:4142–4147. doi: 10.1073/pnas.041490898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lasmezas CI, Deslys JP, Demaimay R, Adjou KT, Lamoury F, Dormont D, Robain O, Ironside J, Hauw JJ. 1996. BSE transmission to macaques. Nature 381:743–744. doi: 10.1038/381743a0. [DOI] [PubMed] [Google Scholar]
- 26.Houston F, Foster JD, Chong A, Hunter N, Bostock CJ. 2000. Transmission of BSE by blood transfusion in sheep. Lancet 356:999–1000. doi: 10.1016/S0140-6736(00)02719-7. [DOI] [PubMed] [Google Scholar]
- 27.Barria MA, Telling GC, Gambetti P, Mastrianni JA, Soto C. 2011. Generation of a new form of human PrPSc in vitro by interspecies transmission from cervid prions. J Biol Chem 286:7490–7495. doi: 10.1074/jbc.M110.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sandberg MK, Al-Doujaily H, Sigurdson CJ, Glatzel M, O'Malley C, Powell C, Asante EA, Linehan JM, Brandner S, Wadsworth JD, Collinge J. 2010. Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J Gen Virol 91:2651–2657. doi: 10.1099/vir.0.024380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asante EA, Linehan JM, Gowland I, Joiner S, Fox K, Cooper S, Osiguwa O, Gorry M, Welch J, Houghton R, Desbruslais M, Brandner S, Wadsworth JDF, Collinge J. 2006. Dissociation of pathological and molecular phenotype of variant Creutzfeldt-Jakob disease in transgenic human prion protein 129 heterozygous mice. Proc Natl Acad Sci U S A 103:10759–10764. doi: 10.1073/pnas.0604292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, Welch J, Hill AF, Lloyd SE, Wadsworth JDF, Collinge J. 2002. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J 21:6358–6366. doi: 10.1093/emboj/cdf653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collinge J, Palmer MS, Sidle KC, Hill AF, Gowland I, Meads J, Asante E, Bradley R, Doey LJ, Lantos PL. 1995. Unaltered susceptibility to BSE in transgenic mice expressing human prion protein. Nature 378:779–783. doi: 10.1038/378779a0. [DOI] [PubMed] [Google Scholar]
- 32.Wadsworth JDF, Asante EA, Desbruslais M, Linehan JM, Joiner S, Gowland I, Welch J, Stone L, Lloyd SE, Hill AF, Brandner S, Collinge J. 2004. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science 306:1793–1796. doi: 10.1126/science.1103932. [DOI] [PubMed] [Google Scholar]
- 33.Wilson R, Plinston C, Hunter N, Casalone C, Corona C, Tagliavini F, Suardi S, Ruggerone M, Moda F, Graziano S, Sbriccoli M, Cardone F, Pocchiari M, Ingrosso L, Baron T, Richt J, Andreoletti O, Simmons M, Lockey R, Manson JC, Barron RM. 2012. Chronic wasting disease and atypical forms of bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild-type levels of human prion protein. J Gen Virol 93:1624–1629. doi: 10.1099/vir.0.042507-0. [DOI] [PubMed] [Google Scholar]
- 34.Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D, Yuan J, Zheng M, Bai H, Deng H, Chen K, Jenny AL, O'Rourke K, Belay ED, Schonberger LB, Petersen RB, Sy MS, Chen SG, Gambetti P. 2005. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci 25:7944–7949. doi: 10.1523/JNEUROSCI.2467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamguney G, Giles K, Bouzamondo-Bernstein E, Bosque PJ, Miller MW, Safar J, DeArmond SJ, Prusiner SB. 2006. Transmission of elk and deer prions to transgenic mice. J Virol 80:9104–9114. doi: 10.1128/JVI.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sano K, Atarashi R, Ishibashi D, Nakagaki T, Satoh K, Nishida N. 2014. Conformational properties of prion strains can be transmitted to recombinant prion protein fibrils in real-time quaking-induced conversion. J Virol 88:11791–11801. doi: 10.1128/JVI.00585-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartz JC, Bessen RA, McKenzie D, Marsh RF, Aiken JM. 2000. Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J Virol 74:5542–5547. doi: 10.1128/JVI.74.12.5542-5547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davenport KA, Henderson DM, Mathiason CK, Hoover EA. 2015. Insights into chronic wasting disease and bovine spongiform encephalopathy species barriers using real-time conversion. J Virol 89:9524–9531. doi: 10.1128/JVI.01439-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurt TD, Sigurdson CJ. 2016. Cross-species transmission of CWD prions. Prion 10:83–91. doi: 10.1080/19336896.2015.1118603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurt TD, Jiang L, Fernández-Borges N, Bett C, Liu J, Yang T, Spraker TR, Castilla J, Eisenberg D, Kong Q, Sigurdson CJ. 2015. Human prion protein sequence elements impede cross-species chronic wasting disease transmission. J Clin Invest 125:1485–1496. doi: 10.1172/JCI79408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piening N, Nonno R, Di Bari M, Walter S, Windl O, Agrimi U, Kretzschmar HA, Bertsch U. 2006. Conversion efficiency of bank vole prion protein in vitro is determined by residues 155 and 170, but does not correlate with the high susceptibility of bank voles to sheep scrapie in vivo. J Biol Chem 281:9373–9384. doi: 10.1074/jbc.M512239200. [DOI] [PubMed] [Google Scholar]
- 42.Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. 1998. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93:203–214. doi: 10.1016/S0092-8674(00)81572-X. [DOI] [PubMed] [Google Scholar]
- 43.Uchiyama K, Miyata H, Yano M, Yamaguchi Y, Imamura M, Muramatsu N, Das NR, Chida J, Hara H, Sakaguchi S. 2014. Mouse-hamster chimeric prion protein (PrP) devoid of N-terminal residues 23–88 restores susceptibility to 22L prions, but not to RML prions in PrP-knockout mice. PLoS One 9:e109737. doi: 10.1371/journal.pone.0109737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flechsig E, Shmerling D, Hegyi I, Raeber AJ, Fischer M, Cozzio A, von Mering C, Aguzzi A, Weissmann C. 2000. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 27:399–408. doi: 10.1016/S0896-6273(00)00046-5. [DOI] [PubMed] [Google Scholar]
- 45.Supattapone S, Muramoto T, Legname G, Mehlhorn I, Cohen FE, DeArmond SJ, Prusiner SB, Scott MR. 2001. Identification of two prion protein regions that modify scrapie incubation time. J Virol 75:1408–1413. doi: 10.1128/JVI.75.3.1408-1413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Supattapone S, Bosque P, Muramoto T, Wille H, Aagaard C, Peretz D, Nguyen H-OB, Heinrich C, Torchia M, Safar J, Cohen FE, DeArmond SJ, Prusiner SB, Scott M. 1999. Prion protein of 106 residues creates an artificial transmission barrier for prion replication in transgenic mice. Cell 96:869–878. doi: 10.1016/S0092-8674(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 47.Surewicz WK, Jones EM, Apetri AC. 2006. The emerging principles of mammalian prion propagation and transmissibility barriers: insight from studies in vitro. Acc Chem Res 39:654–662. doi: 10.1021/ar050226c. [DOI] [PubMed] [Google Scholar]
- 48.Swietnicki W, Petersen R, Gambetti P, Surewicz WK. 1997. pH-dependent stability and conformation of the recombinant human prion protein PrP(90–231). J Biol Chem 272:27517–27520. doi: 10.1074/jbc.272.44.27517. [DOI] [PubMed] [Google Scholar]
- 49.Frankenfield KN, Powers ET, Kelly JW. 2005. Influence of the N-terminal domain on the aggregation properties of the prion protein. Protein Sci 14:2154–2166. doi: 10.1110/ps.051434005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weissmann C. 1996. Molecular biology of transmissible spongiform encephalopathies. FEBS Lett 389:3–11. doi: 10.1016/0014-5793(96)00610-2. [DOI] [PubMed] [Google Scholar]
- 51.Guillot-Sestier M-V, Sunyach C, Druon C, Scarzello S, Checler F. 2009. The α-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 284:35973–35986. doi: 10.1074/jbc.M109.051086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orru CD, Caughey B. 2011. Prion seeded conversion and amplification assays. Top Curr Chem 305:121–133. doi: 10.1007/128_2011_184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orrú CD, Groveman BR, Raymond LD, Hughson AG, Nonno R, Zou W, Ghetti B, Gambetti P, Caughey B. 2015. Bank vole prion protein as an apparently universal substrate for RT-QuIC-based detection and discrimination of prion strains. PLoS Pathog 11:e1004983. doi: 10.1371/journal.ppat.1004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nonno R, Bari MAD, Cardone F, Vaccari G, Fazzi P, Dell'Omo G, Cartoni C, Ingrosso L, Boyle A, Galeno R, Sbriccoli M, Lipp H-P, Bruce M, Pocchiari M, Agrimi U. 2006. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2:e12. doi: 10.1371/journal.ppat.0020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Radovanovic I, Braun N, Giger OT, Mertz K, Miele G, Prinz M, Navarro B, Aguzzi A. 2005. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J Neurosci 25:4879–4888. doi: 10.1523/JNEUROSCI.0328-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jones EM, Surewicz K, Surewicz WK. 2006. Role of N-terminal familial mutations in prion protein fibrillization and prion amyloid propagation in vitro. J Biol Chem 281:8190–8196. doi: 10.1074/jbc.M513417200. [DOI] [PubMed] [Google Scholar]
- 57.Jones EM, Surewicz WK. 2005. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell 121:63–72. doi: 10.1016/j.cell.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 58.Yokoyama T, Masujin K, Iwamaru Y, Imamura M, Mohri S. 2009. Alteration of the biological and biochemical characteristics of bovine spongiform encephalopathy prions during interspecies transmission in transgenic mice models. J Gen Virol 90:261–268. doi: 10.1099/vir.0.004754-0. [DOI] [PubMed] [Google Scholar]
- 59.Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. 1995. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 60.Telling GC, Scott M, Hsiao KK, Foster D, Yang SL, Torchia M, Sidle KC, Collinge J, DeArmond SJ, Prusiner SB. 1994. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci U S A 91:9936–9940. doi: 10.1073/pnas.91.21.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scott M, Groth D, Foster D, Torchia M, Yang S-L, DeArmond SJ, Prusiner SB. 1993. Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell 73:979–988. doi: 10.1016/0092-8674(93)90275-U. [DOI] [PubMed] [Google Scholar]
- 62.Scott MR, Safar J, Telling G, Nguyen O, Groth D, Torchia M, Koehler R, Tremblay P, Walther D, Cohen FE, DeArmond SJ, Prusiner SB. 1997. Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice. Proc Natl Acad Sci U S A 94:14279–14284. doi: 10.1073/pnas.94.26.14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Evans Eric GB, Pushie MJ, Markham Kate A, Lee H-W, Millhauser Glenn L. 2016. Interaction between prion protein's copper-bound octarepeat domain and a charged C-terminal pocket suggests a mechanism for N-terminal regulation. Structure 24:1057–1067. doi: 10.1016/j.str.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]