

Abstract
Proteins undergo structural fluctuations between nearly isoenergetic substates. Such fluctuations are often intimately linked with the functional properties of proteins. However, in some cases, such as in transmembrane ion transporters, the control of the ion transport requires that the protein is designed to restrict the motions in specific regions. In this study, we have investigated the dynamics of a membrane-bound respiratory oxidase, which acts both as an enzyme catalyzing reduction of O2 to H2O and as a transmembrane proton pump. The segment of the protein where proton translocation is controlled (“gating” region) overlaps with a channel through which O2 is delivered to the catalytic site. We show that the replacement of an amino acid residue with a small side chain (Gly) by one with a larger side chain (Val), in a narrow part of this channel, completely blocks the O2 access to the catalytic site and results in formation of a compartment around the site that is impermeable to small gas molecules. Thus, the protein motions cannot counter the blockage introduced by the mutation. These results indicate that the protein motions are restricted in the proton-gating region and that rapid O2 delivery to the catalytic site requires a gas channel, which is confined within a rigid protein body.
Keywords: membrane protein, cytochrome c oxidase, proton transfer, dynamics, respiratory oxidases
Membrane-bound transport proteins such as ion pumps and channels are highly selective and control (“gate”) the ion flow across the membrane. These properties require the protein matrix to be highly rigid in the selectivity and “gating” segments, where the gating may be accomplished by means of transitions between structurally well defined states (see, e.g., refs. 1 and 2). Because proteins are highly flexible and dynamic structures in general, the high degree of rigidity requires a unique design. In this study, we have investigated the structural dynamics of a membrane-bound proton-translocating enzyme, cytochrome c oxidase (CcO). This enzyme catalyzes the one-electron oxidation of four molecules of reduced cytochrome c (cyt.c2+) and the four-electron reduction of O2 to H2O:
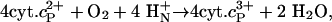 |
[1] |
in which the subscripts N and P denote the (relatively) negatively and positively charged sides of the membrane, respectively. The catalytic site where the O2 reduction takes place is located in the membrane-spanning part of the protein (see Fig. 1A). The enzyme uses the free energy provided by the exergonic electron-transfer reactions to pump protons, from the negative to the positive side, across the membrane (with a stoichiometry of one proton per electron transferred to O2), maintaining a proton electrochemical gradient that is used, for example, for synthesis of ATP. To accomplish proton pumping, the CcO must embody proton-transfer pathways spanning through the protein across the membrane. However, to prevent short-circuiting of the proton electrochemical gradient, it is crucial that there never be direct protonic contact between the two membrane sides (i.e., the proton conduction through the protein must be regulated). Such control is generally thought to be accomplished by locally modifying the proton-transfer rate between a proton donor and acceptor within the protein (for e.g., through a change in the distance or the medium between the donor and acceptor groups; see refs. 3–7). Because large-scale structural changes associated with proton pumping in CcO have not been observed, the control of proton transfer is likely to be accomplished by means of subtle changes in the position of protonatable residues and/or water molecules in the proton-transfer pathways. To prevent unspecific back leaks of protons, such a mechanism requires that the local surrounding protein structure be robust.
Fig. 1.

Crystal structure of CcO (cytochrome aa3) from R. sphaeroides (Protein Data Bank ID code 1M56; ref. 13). (A) The catalytic site (heme a3 and CuB) and heme a are located within the membrane-spanning part of the protein. Hemes a and a3 are shown in light blue, and the Cu ions CuA and CuB are shown in purple. The positions of residues E(I-286) and G(I-283) are indicated, and a proton-transfer pathway (D pathway) leading from the surface to E(I-286) is indicated by an open arrow. The dashed open arrow indicates a possible exit route for pumped protons. Subunits (SU) I–IV are shown in different colors, as indicated. The solid lines indicate the approximate position of the membrane. (B) Close-up view of the O2 channel near the catalytic site illustrates how O2 and protons are transferred through the same region of the enzyme. The dark-blue shape in the middle of the picture is subunit I of CcO. Hemes a and a3, and CuB are shown together with E(I-286) and V(I-283) in the GV(I-283) mutant CcO. The red spheres represent water molecules in the D proton-transfer pathway. In “top view,” the O2 channel is indicated by a yellow arrow. Residues facing the O2 channel are shown in brown. The position of the V(I-283) side chain (green) in the GV(I-283) mutant CcO was determined from an energy-minimization calculation by using the program hyperchem (Hypercube, Gainesville, FL). The residue G(I-283) in the wild-type (WT) CcO is shown in blue. The atoms shown in light blue and red originate from the heme groups. As shown in the “side view,” a narrow segment of the O2 channel is found between residues F(I-282) and W(I-172). The drawings were prepared by using vmd software (50).
One method that has been used to investigate the dynamics of proteins is to monitor the transport kinetics of small gas molecules through the protein matrix. For example, investigation of the kinetics of O2 or CO binding to myoglobin, combined with theoretical studies, have been instrumental to visualization of the functional role of the protein dynamics and transitions between conformational substates (8–10).
In CcO, the free energy that is necessary for ion translocation is provided by reduction of O2 to H2O at a catalytic site consisting of a heme group (heme a3) and a Cu ion (CuB) (Fig. 1). The electron donor to CcO, cytochrome c, initially transfers an electron to a dinuclear Cu site (CuA) from where electrons are transferred consecutively to another heme group, heme a, and to the catalytic site. The O2-reduction reaction is initiated by O2 binding to the reduced heme a3. This heme group also binds other small ligands, such as CO, which can be reversibly removed by means of illumination. Both O2 and CO have been shown to bind transiently to CuB before binding to the reduced heme a3 (11, 12), which indicates the presence of a specific trajectory for the O2/CO transfer into the catalytic site. Several specific channels through which O2 is transported into the site have been identified in CcOs from Rhodobacter sphaeroides (13) (Fig. 1B), bovine heart (14), Thermus thermophilus (15), and Paracoccus denitrificans (16, 17). All of the proposed channels, which are common to the mitochondrial and bacterial CcOs, start in the membrane phase and merge near the catalytic site in the same region of the protein where the proton-gating machinery is likely to be located. Therefore, CcO is an ideal system for investigation of the structural dynamics of a membrane-bound ion transporter as an examination of the CO binding kinetics and can be used as a tool to investigate the protein dynamics in segments of the protein that are involved in the control of proton transport. In this study, we focus on the area around a Glu residue [E(I-286)] that acts as an internal proton donor to the catalytic site (for review, see ref. 18) and where the proton flux is controlled by the CcO (cytochrome aa3) from R. sphaeroides (see Fig. 1). We have investigated the kinetics of ligand (CO and O2) binding to a mutant of this CcO in which the O2 channel was blocked by replacement of a small residue, Gly [G(I-283)], by a larger residue, [Val, GV(I-283)], near the entrance to a gaseous cavity surrounding the catalytic site.
Materials and Methods
Growth of Bacteria and CcO Purification. CcO was prepared from R. sphaeroides, which was grown aerobically in shake incubators. The His-tagged CcO was purified by using a Ni2+-NTA agarose column (Qiagen, Valencia, CA), as described in ref. 19, and dissolved in Hepes (100 mM, pH 7.4) supplemented with 0.1% n-dodecyl-β-d-maltoside (Glycon Biochemicals, Luckenwalde, Germany). The GV(I-283) mutant CcO was prepared as described in ref. 20. All chemicals were of the purest available grade.
Preparation of the Fully Reduced Enzyme and CO-Binding Studies. Detergent-solubilized CcO was diluted to a final concentration of 4 μM in 100 mM Hepes and 0.1% n-dodecyl-β-d-maltoside (pH 7.4). The redox mediator phenazine methosulphate was added to a concentration of 5 μM. The sample was made anaerobic by repetitive evacuation on a vacuum line, followed by flushing with N2 and reduction by 2 mM ascorbate. To investigate binding of CO to the reduced CcO after reduction the N2, atmosphere in the cuvette was exchanged for CO (1 mM). Formation of the reduced CcO–CO complex was followed by measuring absorbance changes by using a Cary 400 Bio UV-Visible spectrophotometer (Varian). To study the binding of CO on shorter time scales, reduced CcO was mixed 1:1 with a CO-saturated buffer-detergent solution in a stopped-flow apparatus, connected to a diode-array detector (Applied Photophysics, Surrey, UK). In all experiments, the cuvette path length was 1.00 cm.
Measurements of CO Dissociation and Recombination. CO (1 mM) was added to the reduced enzyme, which results in formation of the CcO–CO complex in which CO is bound to heme a3. The CO ligand was photolyzed by a 6-ns laser flash at 532 nm (Brilliant B, Quantel, Santa Clara, CA), and absorbance changes were followed with a time resolution of ≈1 μs by using a flash-photolysis apparatus that was obtained from Applied Photophysics. For a detailed description of the equipment, see ref. 21.
Results
In CcO from R. sphaeroides, a Gly residue [G(I-283)] is found in a narrow region of a proposed O2 channel that connects the interior of the membrane with the catalytic site (Fig. 1). The G(I-283) residue, which does not have a side chain, was replaced by a Val [GV(I-283) mutant CcO] with a side-chain diameter of ≈5 Å. The GV(I-283) mutant CcO was reduced under N2 atmosphere. To determine the oxidation rate of the mutant CcO, O2 was added by opening the cuvette, followed by shaking. Absorbance changes at 445 nm, associated with oxidation of the heme groups, were followed as a function of time. As shown in Fig. 2, the mutation rendered CcO in which the reaction of the reduced enzyme with O2 was slowed from ≈1 ms (in the wild-type CcO in an air-equilibrated solution; data not shown; see, e.g., ref. 22) to minutes.
Fig. 2.

Oxidation of and CO binding to the reduced GV(I-283) CcO. Absorbance changes at 445 nm associated with reaction of O2 with the reduced GV(I-283) mutant CcO (i.e., oxidation of CcO, •). The O2-bidning rate is compared with that of CO binding to the reduced CcO (⋄, see also Fig. 3) observed at 430 nm under the same conditions. Here, the zero absorbance level is defined as the absorbance of the reduced CcO before addition of CO (see symbols at t < 0). In these experiments (• and ⋄) reduced CcO under N2 atmosphere was mixed (1:1) with an O2- or CO-saturated buffer (≈1 mM CO or ≈1.2 mM O2) by using a stopped-flow apparatus. Absorbance changes at 430 nm upon reduction of the mutant CcO after incubation of the oxidized CcO under CO atmosphere (0.5 mM) for 40 min are shown (▵). Here, the zero absorbance level is defined as the absorbance of the same sample in the reduced state (so that plots of ⋄ and ▵ can be compared directly). The lines have been added for ease of viewing. Conditions are as follows: 0.1 M Hepes, pH 7.4/0.1% n-dodecyl-β-d-maltoside; T = 22°C; ≈0.5 mM CO or ≈0.6 mM O2 after mixing (▵, obtained at 1 mM CO). Absorbance changes have been scaled to 1 μM reacting CcO. Negative absorbance changes are shown at 445 nm, for clarity.
The inert CO molecule has been used as an O2 analogue to investigate the kinetics of ligand binding to heme proteins. To investigate the binding kinetics of CO to heme a3 in the GV(I-283) mutant CcO, CO was added to the fully reduced CcO and the formation of the CcO–CO complex was monitored as an increase in absorbance at 430 nm (Figs. 2 and 3A). With the GV(I-283) CcO, an absorbance increase with a time constant of ≈30 ms (25% of the total amplitude) was followed by a biphasic slower increase with time constants of ≈5 s (25%) and 2,500 s (≈50%). Thus, in the major fraction of the CcO, population binding of CO to the GV(I-283) mutant CcO was much slower than to the wild-type CcO (τWT ≅ 10 ms at 1 mM CO; Fig. 3). The slower components in the CO-binding displayed approximately the same time constants as those of O2 binding to the reduced CcO (Fig. 2).
Fig. 3.

Binding of CO to GV(I-283) mutant and wild-type CcO. (A) Absorbance changes at 430 nm associated with binding of CO to the reduced wild-type (line) and GV(I-283) mutant (▴) CcO, as indicated. (B) When CO had bound to the reduced heme a3, it was photolyzed with a laser flash (decrease in absorbance at t = 0), and the rebinding of CO is shown as an increase in absorbance at 430 nm. Conditions were the same as in Fig. 2, except that the CO concentration was 1 mM in B.
To investigate whether CO could penetrate into the catalytic site pocket of the GV(I-283) mutant CcO without binding to heme a3, the oxidized CcO was incubated under CO atmosphere for 40 min (CO does not bind to the oxidized form of heme a3). No absorbance changes were observed during this incubation time, which shows that CO itself did not reduce the CcO. After 40 min, ascorbate and phenazine methosulphate were added and the formation of the reduced CcO–CO complex was followed at 430 nm (Fig. 2). The data show that CO binding was accelerated when the mutant CcO was incubated with CO before reduction, which indicates that there is space for at least one CO molecule in the cavity around the catalytic site.
After the enzyme–CO complex is formed, the ligand can be dissociated by means of illumination. Fig. 3B shows absorbance changes at 430 nm after pulsed illumination of the wild-type and GV(I-283) mutant CcO–CO complexes. The decrease in absorbance at t = 0, associated with dissociation of CO from heme a3, is followed by an increase, associated with recombination of the CcO–CO complex. As shown in Fig. 3, the CO recombination was significantly faster with the GV(I-283) [τ1 ≅ 40 μs (50%), τ2 ≅ 400 μs (25%), τ3 ≅ 7 ms (25%)] than with the wild-type CcO (τ ≅ 10 ms). Also, whereas the CO-recombination rate increases linearly with the CO concentration (in the range of 0–1 mM CO) (23) with the wild-type R. sphaeroides CcO, the rate was essentially insensitive to the bulk concentration of CO (data not shown) with the GV(I-283) enzyme. This result indicates that after flash-induced dissociation, the CO ligand does not leave the catalytic-site compartment of the mutant CcO.
In summary, the results shown in Figs. 2 and 3 indicate that the introduction of a restriction in the O2 channel results in a dramatically slowed binding of CO (and O2). As soon as the CO ligand is bound to heme a3 after flash-induced dissociation, it does not leave the heme pocket and it recombines faster than with the wild-type CcO.
Discussion
Analyses of the x-ray crystal structures of CcOs from different species, together with results from experimental and theoretical studies, indicate that there is a specific trajectory along which small gas molecules bind to heme a3. This trajectory passes through CuB, as illustrated by results showing that, upon flash photolysis of CO from heme a3, the ligand binds transiently to CuB before being released to the bulk solution (11, 12). Transport of O2 through a specific pathway was also supported by results from molecular-dynamics simulations (24). Wikström and colleagues (17, 25) investigated the O2-binding kinetics in a mutant of the P. denitrificans CcO in which a Val residue [V(I-287)], located in a putative O2 channel near the entrance into the O2-binding cavity at the catalytic site, was replaced by Ile. They found that, in this mutant CcO at 1 mM O2, the apparent first-order rate constant for O2 binding to heme a3 decreased by a factor of ≈30, indicating that a steric hindrance was introduced as a result of the mutation (17, 25). However, in contrast to the results from this study, when the mutation was introduced at V(I-287), CO could leave the catalytic site on the microsecond time scale upon pulsed illumination and the CO recombination rate was only slightly slower than with the wild-type CcO (17).
A hydrophobic channel starting at a V-shaped cleft formed by subunit III has been identified in the P. denitrificans (16) and T. thermophilus (15) x-ray structures. This channel overlaps with one of the channels proposed for the bovine CcO (14). A putative O2 channel was also identified in the structure of the R. sphaeroides CcO on the basis of an analysis of the location of xenon atoms in the x-ray crystal structure (13). Even though the entrance of this channel is located in subunit I (between helices II and III), it converges with the channels starting in subunit III close to the catalytic site. The G(I-283) residue is located near this merging point, ≈8 Å from the heme a3 iron and 6 Å (toward the catalytic site) from a narrow segment of the channel confined between residues F(I-282) and W(I-172) (see “side view” in Fig. 1B). The V(I-287) residue, discussed above, is located near G(I-283) but is slightly closer to the heme a3–CuB binuclear center.
Specific substrate channels have been identified in other systems (for review, see refs. 26 and 27). For example, solvent-inaccessible intramolecular ammonia channels have been found in several enzymes (28, 29), where in the carbamoyl phosphate synthetase, the channel was suggested to be used for tunneling of an ammonia intermediate between two active sites separated by a distance of ≈96 Å (29, 30). The transfer of ammonia between the sites could be obstructed by introduction of a constriction within the tunnel (30). In the CO dehydrogenase/acetylcoenzyme A synthase, the active sites were found to be connected by a narrow CO-conducting channel extending over a distance of 138 Å through which the CO transfer is controlled by minor structural changes (see ref. 31). Also, in the Ni–Fe hydrogenases, specific hydrophobic channels were found to serve as pathways for H2 delivery to the buried catalytic site (32).
Early studies of fluorescence quenching of Trp residues in proteins showed that O2 could diffuse almost freely through the protein matrix (33, 34). However, results from later NMR studies indicated that small organic gas molecules reside for >1 ns in internal protein cavities, pointing to the importance of the internal cavities to allow for specific gas transport through proteins (35). The appearance of such cavities and dynamics are intimately linked and central for ligand binding, where the fluctuations in the atomic positions are crucial for permitting ligands to diffuse (8–10, 36). Consequently, a rigid region of a protein structure would require a specific channel to allow gas transport into a specific site.
The results from this study show that the GV(I-283) mutant CcO binds CO and O2 several orders of magnitude more slowly than the wild-type CcO. When CO is bound to heme a3 at the catalytic site, it is not released to the bulk solution after photolysis of the heme a3–CO bond. Also, the results indicate that there is room for at least one CO molecule in a cavity surrounding the heme a3–CuB center and that the connectivity between this cavity and the O2 channel is blocked in the GV(I-283) mutant CcO (see Fig. 2). Together, the results suggest that the protein matrix in the region around the catalytic site maintains a high degree of structural rigidity and that small gas molecules, such as CO and O2, cannot diffuse unspecifically through the protein matrix.
In addition to the O2 channels leading to the catalytic site of CcO, channels for water exit have also been proposed (14, 15, 37). The results from a recent study indicate that, in the R. sphaeroides CcO, water exits exclusively through a channel leading toward an Mg2+/Mn2+ site located “above” the hemes (37). Even though the water chains in CcO are highly dynamic structures (38), the results from the present study indicate that small gas molecules cannot diffuse through the water channel when the O2 channel is blocked, which presumably is due to a hydrophilic character of the water channel or to the water channel being blocked to O2 passage by the H2O molecules.
In CcO from R. sphaeroides, the protons to be pumped, as well as the protons used in the O2-reduction reaction (substrate protons), are transferred through a proton-transfer pathway (“D pathway”), which consists of structurally ordered water molecules and protonatable and hydrophilic amino acid residues (39–41). The pathway starts at the protein surface on the negative side of the membrane and continues to a Glu residue [E(I-286)] (Fig. 1) from where protons are transferred either toward the catalytic site or to an acceptor of the pumped protons. Because both the substrate and pumped protons are transferred through the same pathway, the proton-transfer reactions from the region around E(I-286) must be controlled by the protein and distributed to different sites at specific times during the catalytic reaction. Also, as discussed in the introduction section, proton pumping requires the presence of at least one protonatable group within the protein which binds a proton from one side of the membrane in the “input configuration” and releases the proton to the other side in an “output configuration,” a phenomenon termed gating (see refs. 42–44). The gating machinery must be structurally robust to provide the specificity and to prevent the back leak of protons. As suggested from experimental studies and an analysis of the protein structure, the gating site(s) in CcO is presumably located in the area around E(I-286), the heme group propionates, and the catalytic site (see Fig. 1) (16, 45–48). We note that residue G(I-283) is located at a distance of ≈4 Å from E(I-286) (i.e., in a segment of the protein in which the gating of the pumped protons presumably takes place). Thus, the structural rigidity around the G(I-283)/E(I-286) region of the CcO is likely to be a consequence of the function of this enzyme as a proton pump.
Réat et al. (1) proposed a similar scenario for the light-driven proton pump bacteriorhodopsin (BR). They found that the protein is dynamically heterogeneous, such that the segment around the retinal is more rigid than the rest of the BR. They concluded that these properties are essential for the specificity of proton transfer in controlling the proton pumping. An example of related system that must maintain a rigid structure defining fixed distances between amino acid residues and the transported ion is the K+ channel, which conducts K+ at rates of up to 108 ions per second while excluding the smaller Na+. The selectivity for K+ is maintained because the carbonyl O2 atoms within the selectivity filter are arranged geometrically to mimic the position of water O2 atoms coordinating K+ in water solution (49). The variation in these distances must be minimized to exclude the Na+ ions while maintaining a low energy barrier for the entrance and exit of K+.
In conclusion, the results from this study indicate that the CcO structure is highly rigid in a domain of the protein through which protons and O2 are transferred. This rigidity is most likely to be a consequence of the necessity to control the flux of the substrate and pumped protons. Because the protongating region is located in close proximity to the catalytic site (i.e., near the source of the free energy used for proton pumping), the rigidity of this region requires that the substrate O2 be transported into the catalytic site through a specific channel rather than through conformational fluctuations of intraprotein cavities.
Acknowledgments
We thank Pia Ädelroth, Roderick MacKinnon, and Mikael Oliveberg for valuable discussions, and Håkan Lepp for the energy-minimization calculations of the mutant CcO. This work was supported by grants from the Swedish Research Council (to P.B.), Human Frontier Science Program Grant RG0135 (to R.B.G. and P.B.), and National Institutes of Health Grant HL16101 (to R.B.G.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: CcO, cytochrome c oxidase.
References
- 1.Réat, V., Patzelt, H., Ferrand, M., Pfister, C., Oesterhelt, D. & Zaccai, G. (1998) Proc. Natl. Acad. Sci. USA 95, 4970–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frauenfelder, H. & McMahon, B. (1998) Proc. Natl. Acad. Sci. USA 95, 4795–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gennis, R. B. (2004) Front. Biosci. 9, 581–591. [DOI] [PubMed] [Google Scholar]
- 4.Wikström, M. (2004) Biochim. Biophys. Acta 1655, 241–247. [DOI] [PubMed] [Google Scholar]
- 5.Mills, D. A. & Ferguson-Miller, S. (2003) FEBS Lett. 545, 47–51. [DOI] [PubMed] [Google Scholar]
- 6.Popovic, D. M. & Stuchebrukhov, A. A. (2004) J. Am. Chem. Soc. 126, 1858–1871. [DOI] [PubMed] [Google Scholar]
- 7.Tsukihara, T., Shimokata, K., Katayama, Y., Shimada, H., Muramoto, K., Aoyama, H., Mochizuki, M., Shinzawa-Itoh, K., Yamashita, E., Yao, M., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 15304–15309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frauenfelder, H., Sligar, S. G. & Wolynes, P. G. (1991) Science 254, 1598–1603. [DOI] [PubMed] [Google Scholar]
- 9.Brunori, M. & Gibson, Q. H. (2001) EMBO Rep. 2, 674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostermann, A., Waschipky, R., Parak, F. G. & Nienhaus, G. U. (2000) Nature 404, 205–208. [DOI] [PubMed] [Google Scholar]
- 11.Einarsdóttir, Ó., Dyer, R. B., Lemon, D. D., Killough, P. M., Hubig, S. M., Atherton, S. J., Lopez-Garriga, J. J., Palmer, G. & Woodruff, W. H. (1993) Biochemistry 32, 12013–12024. [DOI] [PubMed] [Google Scholar]
- 12.Lemon, D. D., Calhoun, M. W., Gennis, R. B. & Woodruff, W. H. (1993) Biochemistry 32, 11953–11956. [DOI] [PubMed] [Google Scholar]
- 13.Svensson-Ek, M., Abramson, J., Larsson, G., Törnroth, S., Brzezinski, P. & Iwata, S. (2002) J. Mol. Biol. 321, 329–339. [DOI] [PubMed] [Google Scholar]
- 14.Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono, R. & Yoshikawa, S. (1996) Science 272, 1136–1144. [DOI] [PubMed] [Google Scholar]
- 15.Soulimane, T., Buse, G., Bourenkov, G. P., Bartunik, H. D., Huber, R. & Than, M. E. (2000) EMBO. J. 19, 1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwata, S., Ostermeier, C., Ludwig, B. & Michel, H. (1995) Nature 376, 660–669. [DOI] [PubMed] [Google Scholar]
- 17.Riistama, S., Puustinen, A., Verkhovsky, M. I., Morgan, J. E. & Wikström, M. (2000) Biochemistry 39, 6365–6372. [DOI] [PubMed] [Google Scholar]
- 18.Namslauer, A. & Brzezinski, P. (2004) FEBS Lett. 567, 103–110. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell, D. M. & Gennis, R. B. (1995) FEBS Lett. 368, 148–150. [DOI] [PubMed] [Google Scholar]
- 20.Lee, A. (1998) Ph.D. thesis (University of Illinois at Urbana–Champaign, Urbana).
- 21.Brändén, M., Sigurdson, H., Namslauer, A., Gennis, R. B., Ädelroth, P. & Brzezinski, P. (2001) Proc. Natl. Acad. Sci. USA 98, 5013–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ädelroth, P., Ek, M. & Brzezinski, P. (1998) Biochim. Biophys. Acta 1367, 107–117. [DOI] [PubMed] [Google Scholar]
- 23.Aagaard, A., Gilderson, G., Gomes, C. M., Teixeira, M. & Brzezinski, P. (1999) Biochemistry 38, 10032–10041. [DOI] [PubMed] [Google Scholar]
- 24.Hofacker, I. & Schulten, K. (1998) Proteins 30, 100–107. [PubMed] [Google Scholar]
- 25.Riistama, S., Puustinen, A., Garcia-Horsman, A., Iwata, S., Michel, H. & Wikström, M. (1996) Biochim. Biophys. Acta 1275, 1–4. [DOI] [PubMed] [Google Scholar]
- 26.Huang, X., Holden, H. M. & Raushel, F. M. (2001) Annu. Rev. Biochem. 70, 149–180. [DOI] [PubMed] [Google Scholar]
- 27.Wilson Miles, E., Rhee, S. & Davies, D. R. (1999) J. Biol. Chem. 274, 12193–12196. [DOI] [PubMed] [Google Scholar]
- 28.Krahn, J. M., Kim, J. H., Burns, M. R., Parry, R. J., Zalkin, H. & Smith, J. L. (1997) Biochemistry 36, 11061–11068. [DOI] [PubMed] [Google Scholar]
- 29.Thoden, J. B., Holden, H. M., Wesenberg, G., Raushel, F. M. & Rayment, I. (1997) Biochemistry 36, 6305–6316. [DOI] [PubMed] [Google Scholar]
- 30.Huang, X. & Raushel, F. M. (2000) J. Biol. Chem. 275, 26233–26240. [DOI] [PubMed] [Google Scholar]
- 31.Grahame, D. A. (2003) Trends Biochem. Sci. 28, 221–224. [DOI] [PubMed] [Google Scholar]
- 32.Montet, Y., Amara, P., Volbeda, A., Vernede, X., Hatchikian, E. C., Field, M. J., Frey, M. & Fontecilla-Camps, J. C. (1997) Nat. Struct. Biol. 4, 523–526. [DOI] [PubMed] [Google Scholar]
- 33.Lakowicz, J. R. & Weber, G. (1973) Biochemistry 12, 4171–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calhoun, D. B., Vanderkooi, J. M., Woodrow, G. V., 3rd, & Englander, S. W. (1983) Biochemistry 22, 1526–1532. [DOI] [PubMed] [Google Scholar]
- 35.Otting, G., Liepinsh, E., Halle, B. & Frey, U. (1997) Nat. Struct. Biol. 4, 396–404. [DOI] [PubMed] [Google Scholar]
- 36.Tilton, R. F., Jr., Singh, U. C., Kuntz, I. D., Jr. & Kollman, P. A. (1988) J. Mol. Biol. 199, 195–211. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt, B., McCracken, J. & Ferguson-Miller, S. (2003) Proc. Natl. Acad. Sci. USA 100, 15539–15542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olkhova, E., Hutter, M. C., Lill, M. A., Helms, V. & Michel, H. (2004) Biophys. J. 86, 1873–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brzezinski, P. & Ädelroth, P. (1998) J. Bioenerg. Biomembr. 30, 99–107. [DOI] [PubMed] [Google Scholar]
- 40.Konstantinov, A. A., Siletsky, S., Mitchell, D., Kaulen, A. & Gennis, R. B. (1997) Proc. Natl. Acad. Sci. USA 94, 9085–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verkhovskaya, M. L., García-Horsman, A., Puustinen, A., Rigaud, J. L., Morgan, J. E., Verkhovsky, M. I. & Wikström, M. (1997) Proc. Natl. Acad. Sci. USA 94, 10128–10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wikström, M. (1998) Biochim. Biophys. Acta 1365, 185–192. [DOI] [PubMed] [Google Scholar]
- 43.Rich, P. R. (1995) Aust. J. Plant Physiol. 22, 479–486. [Google Scholar]
- 44.Brzezinski, P. & Larsson, G. (2003) Biochim. Biophys. Acta 1605, 1–13. [DOI] [PubMed] [Google Scholar]
- 45.Puustinen, A. & Wikström, M. (1999) Proc. Natl. Acad. Sci. USA 96, 35–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mills, D. A. & Ferguson-Miller, S. (2002) Biochim. Biophys. Acta 1555, 96–100. [DOI] [PubMed] [Google Scholar]
- 47.Michel, H. (1998) Proc. Natl. Acad. Sci. USA 95, 12819–12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rich, P. R., Junemann, S. & Meunier, B. (1998) J. Bioenerg. Biomembr. 30, 131–138. [DOI] [PubMed] [Google Scholar]
- 49.MacKinnon, R. (2003) FEBS Lett. 555, 62–65. [DOI] [PubMed] [Google Scholar]
- 50.Humphrey, W., Dalke, A. & Schulten, K. (1996) J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]