

Abstract
Metabolites are building blocks of cellular function. These species are involved in enzyme-catalyzed chemical reactions and are essential for cellular function. Upstream biological disruptions result in a series of metabolomic changes, and as such the metabolome holds a wealth of information that is thought to be most predictive of phenotype. Uncovering this knowledge is a work in progress. The field of metabolomics is still maturing; the community has leveraged proteomics experience when applicable and developed a range of sample preparation and instrument methodology along with myriad data processing and analysis approaches. Research focuses have now shifted toward a fundamental understanding of the biology responsible for metabolomic changes. There are several types of metabolomics experiments including both targeted and untargeted analyses. While untargeted, hypothesis generating, workflows exhibit many valuable attributes, challenges inherent to the approach remain. This Critical Insight comments on these challenges, focusing on the identification process of LC-MS based untargeted metabolomics studies – specifically in mammalian systems. Biological interpretation of metabolomics data hinges on the ability to accurately identify metabolites. The range of confidence associated with identifications that is often overlooked is reviewed, and opportunities for advancing the metabolomics field are described.
Graphical Abstract
The ultimate goal of metabolomics is the comprehensive study of the low molecular weight molecules within an organism. Metabolites are the result of both biological and environmental factors, and as such provide great potential to bridge knowledge of genotype and phenotype. Metabolomics is often likened to its proteomics sibling and has leveraged proteomics experience, but the field has evolved with inherently different challenges including the identification process. Peptides and proteins are typically a linear polymer and can be sequenced. Proteins are inferred by matching of identified experimental peptides against in-silico fragmentation spectra. Metabolites are more challenging to annotate. These small molecules often lack a common building block, although there is common use of the elements C, H, O, N, S, P, and potentially heteroatoms. The idea that untargeted mass spectrometry (MS)-based metabolomics analysis will result in a large list of ‘identified’ small molecules that can be mapped to networks and pathways is often assumed, yet high confidence analyte assignments/identifications may not be made owing to the fundamental challenges of the metabolomic identification processes. For example, features (i.e., mass-to-charge ratio and retention time pairs) can be assigned to a vast number of tentative or preliminary structures, or there may be no candidate matches in curated databases. Because metabolomics database content will likely always be considered incomplete – lacking a genetic template such as that for proteomics, in-silico metabolite databases can provide guidance and in some cases validation, but will not fit all metabolomic studies. Validation of retention times and MS/MS fragmentation data with a reference standard is nearly always required for confident metabolite identification.
Since its inception, the metabolomics field focus has shifted from detecting changes to understanding the biology leading to the changes [1], thus the accuracy of metabolite assignments is extremely important. In this Critical Insight, we will discuss various challenges inherent to LC-MS based metabolomics and describe the ranges of confidence for small molecule annotations when performing global metabolomic analyses, a concept essential for applying metabolomic data toward a better understanding of the mechanisms of human health and disease.
LC-MS based Metabolomics: Strengths and Challenges
Metabolomics experiments aim to characterize diverse classes of small molecules from a variety of sample types (e.g., cell extracts, culture media, urine, serum, etc.). The metabolomics community has leveraged numerous aspects of proteomics methodology such as separation technologies, state-of-the-art instrumentation, and data processing approaches. However, there are fundamental differences in MS-based metabolomics versus proteomics that are important to recognize. Table 1 outlines the strengths and challenges in contemporary metabolomics relative to proteomic analyses.
Table 1.
Current Scale, Strengths, and Challenges of MS-based Metabolomics and MS-based Proteomics.
| Metabolomics | Proteomics |
|---|---|
| Scale | |
|
|
| Strengths | |
|
|
| Challenges | |
|
|
The annotated human metabolome is considered to be less complex than the proteome [2,3], yet the diverse chemical structures exhibit a wide range of concentration, solubility, polarity, and volatility [4].Proteomics samples often require a multi-step preparation that may involve cell lysis, purification, enzymatic digestion, and solid phase extraction [5]. Sample preparation for metabolomics involves cell lysis and metabolite extraction [6], although purification and fractionation can also be performed. Metabolomics analyses are challenged by an analytes’ rapid temporal dynamics and sample composition reflecting endogenous and exogenous species (e.g., drugs, toxins, microorganisms, and nutrients) [1]. While proteomic analyses can often differentiate organism species based on protein sequence [6] (which is particularly useful in microbiome studies), species determination in metabolomics is challenging because often small molecules are common across different organisms [7]. This can, however, be advantageous for metabolomics animal model studies as knowledge of physical properties guiding identifications can be shared across species. Another major difference between proteomic and metabolomic technologies involves the interpretation of fragmentation data. Known protein sequences and enzyme cleavage patterns enable predictable peptide sequences and fragmentation spectra. Further, the large size of protein molecules often results in multiple peptides being observed thus increasing confidence of protein identification. This is in contrast to metabolomics studies, where the small size and wide array of molecular structures of metabolites results in a singular species with no consensus fragmentation pattern.
Targeted and Untargeted Metabolomic Studies
Figure 1 outlines the goals and the types of data sets that are generated in targeted and untargeted/global metabolomic studies. In general, targeted approaches are aimed at identifying and quantifying a limited number (tens to hundreds) of known metabolites, such as those commonly encountered in clinical analyses. Many untargeted, or hypothesis generating, approaches focus on acquiring data for as many species as possible, annotating metabolites, and reviewing both known and unknown metabolic changes. Data can be used for relative quantification across sample groups and to provide hypotheses that can be further studied with targeted approaches. There are two broad approaches for data acquisition in untargeted metabolomics studies. The first method uses full scan MS1 to generate accurate mass measurements for individual molecules (i.e., features) to permit statistical calculations followed by data dependent acquisition (DDA) of a subset of samples to guide identifications. Similar to conventional proteomics techniques, metabolomics DDA methods generate fragmentation patterns for metabolites exhibiting the highest signal intensity. A second untargeted metabolomics approach is based on data independent acquisition (DIA), where workflows integrate full MS1 with MS/MS fragmentation for all precursor ions either simultaneously (MSE [8]) or in finite mass ranges (SWATH [9]). DIA methods produce complicated fragmentation spectra and the link between precursor and product can be difficult to decipher. In downstream data analysis steps, fragment ions are matched with precursor ions based on retention time, mass, and drift time (when applicable). DIA allows fragmentation data to be acquired regardless of metabolite signal intensity. Both DDA and DIA approaches ultimately define features with a mass-to-charge ratio (m/z), retention time (RT), and drift time (DT) descriptors, among others. In the identification step, precursor ions and corresponding fragment ions are searched against databases for metabolite assignments.
Figure 1.

Untargeted versus Targeted Metabolomics Studies. Untargeted, or discovery-based, metabolomics focuses on global detection and relative quantitation of small molecules in a sample. In contrast, targeted, or validation-based, metabolomics focuses on measuring well-defined groups of metabolites, with opportunities for absolute quantitation.
One major advantage of untargeted metabolomics is the collection of data without pre-existing knowledge; however, this is accompanied with the caveat that certainly sample preparation and analytical methods have a direct impact on the qualitative results that are obtained. Owing to the diverse composition of the metabolome [10], sample preparation steps , separation methods, and instrument platform and parameters will influence the subset of metabolites detected.
Analytical Platforms
A variety of separation [liquid chromatography (LC), gas chromatography (GC) and capillary electrophoresis (CE)] and detection [MS and nuclear magnetic resonance (NMR)] methods are used for metabolomics experiments. We focus on LC-MS-based metabolomics as it has become a leading technology for both polar and nonpolar small molecule analyses and draws many parallels with LC-MS-based proteomics analyses referenced herein. LC methods are time-consuming (minutes to hours) compared to direct infusion or flow injection analyses (seconds to minutes) [11]. However, the ability of LC to increase both selectivity and data content makes it invaluable [12], particularly for complex metabolomics samples such as human blood where an average of three isomers or isobars per nominal mass are estimated [13].
The coupling of ion mobility (IM) separations with LC-MS based analyses represents an emerging technology (LC-IM-MS) for metabolomics research. Ion mobility resolves gas phase ions based on their size-to-charge ratio or gas phase packing efficiency, complementing polarity and mass separations. The addition of ion mobility separation offers increased peak capacity [2], the ability to decrease chromatography time without sacrificing resolution, and opportunities to separate co-eluting precursors [13]. Rapid (milliseconds) IM separations are well integrated into time scales of most MS platforms; multiple IM spectra are acquired for each LC peak, and multiple mass spectra (microsecond time scale) are acquired for each IM spectrum [14].In addition to improved mass spectra quality and increased selectivity, IM measurements can be used to determine collision cross sections (CCS) for individual metabolites. Unlike RT measurements, which vary based on column chemistry, mobile phase, and elution gradient, CCS values are physical properties and not influenced by MS or LC settings where inter-laboratory precision is reported to be at least <5% for over a broad range of molecules assayed [15]. Improvements to this precision are rapidly evolving with the development of more standardized protocols for CCS measurements.
Analysis and Identification
Untargeted metabolomics data processing workflows incorporate several defined steps including noise filtering, peak detection, peak deconvolution, retention time alignment, and finally feature annotation. Importantly, features are not always metabolites; related species (e.g., isotopes, neutral losses, adducts) of a single metabolite may be present with different m/z values. Metabolite identification is necessary to draw biological conclusions from untargeted metabolomics data. Analyte identification can be performed by searching the experimental MS1 or MS/MS data through databases available to the public for free (e.g., ChemSpider (http://www.chemspider.com), METLIN [16], Human Metabolome DataBase (HMDB) [17], MassBank [18], mzCloud (https://www.mzcloud.org), GNPS (http://gnps.ucsd.edu/), and LipidBlast [19]) or for a nominal fee (e.g., NIST Mass Spectral Library (http://chemdata.nist.gov)). Batch searching MS/MS fragmentation spectra within these databases, however, is often not possible without commercial software. Given that numerous libraries are generally queried to maximize metabolome coverage, bioinformatics efforts are necessary to remove or reduce match redundancy. This process can be complicated since metabolite nomenclature is not entirely standardized and varies greatly by database.
Feature annotation is performed by comparing an experimental mass measurement to a database of known metabolites within a mass tolerance window to generate potential candidates. Thus, the development of high-resolution high-mass accuracy mass instruments has proven invaluable for discovery (MS1) and heuristic validation (MS2) metabolomics efforts. As illustrated in Figure 2, it is difficult for MS mass measurement alone to provide metabolite information beyond molecular formula, at best. Kind and Fiehn demonstrated that high mass accuracy measurements (<1 ppm error) were inadequate for determining the elemental composition of numerous metabolites [20]; notably, the authors later showed that isotope ratio measurements were more important than mass accuracy for determining the most probable elemental composition for small molecules [21]. Additional information, such as fragmentation data, is essential for structure elucidation of a mass measurement. Putative identifications require matching an experimental MS/MS spectrum with a reference fragmentation spectrum [24, 26]. Metabolomics spectral libraries have been created with experimental data from commercially available or synthesized standards. Significant efforts are being made to routinely update content as new compounds are analyzed, as such, these libraries are considered incomplete [22]. MS/MS data is often insufficient to differentiate structural and stereo-isomers. Orthogonal evidence is needed in these cases and when experimental MS/MS data is non-discriminating. LC and IM can be used to generate retention time and collision cross section information, respectively. Both of these separation methods are capable of resolving some isomeric/isobaric species. IM has even shown utility for differentiating lipids based on position of double bond, which is often unable to be accomplished by LC [23]. MS-based metabolomic studies are performed on numerous different instrument platforms; ion intensities and fragmentation patterns vary based on analytical conditions including instrument, ionization source, and collision energy [18]. For small molecules, a collision energy that depletes some precursors may have little effect on others. The selection of isobaric co-eluting precursor ions for fragmentation may further complicate experimental MS/MS data. MS/MS matching can be subjective. Scores are generated to represent similarities between experimental data from unknown and experimental data from the standard and assignments are often made using the best match. False positives and false negatives may be the result of low quality spectra and incomplete databases, respectively. Many opportunities exist for the development of methods to calculate these unknowns as well as a confidence metric for scoring MS/MS matches [12].
Figure 2.

An illustration of the amount of information density present at different levels of mass measurement accuracy, using the validated entries in the PubChem compound database. (a) The distribution of molecules in the PubChem compound database between 0 and 1000 Da, as surveyed in 2007, 2011, and 2015. As new compounds are discovered and archived, the distribution has shifted to lower mass, with most entries currently centered between 100 and 600 Da. Theoretical molecular formulas determined from chemical stability rules are illustrated by the dotted line, indicating that most of these entries are isomers. The inset zooms in on a 10 Da window where over half a million compounds are represented. (b) At increasing levels of mass accuracy, the number of possible molecular formulas can be reduced to a few thousand, but in one extreme case shown at 1 ppm, one formula is represented by over 10,000 isomers in the database. Mass spectrometry can significantly reduce complexity, but it cannot fully address molecular characterization without other dimensions of information. Reproduced with permission of Annual Review of Analytical Chemistry, Volume 9 © by Annual Reviews, http://www.annualreviews.org from reference [2].
A subset of experimental metabolomics data does not match any database entry. Null matches may represent truly new metabolites or simply known metabolites that are missing from or do not match the spectral database (e.g., in-source fragments, metabolites modified by enzyme activity, etc.) [24]. Characterization of these “unknown” unknowns requires significant effort – such as that often encountered in natural product discovery of secondary metabolites [25,45]. Algorithms geared toward predicting and comparing small molecule in silico and experimental MS/MS data are also currently available (e.g., MetFrag [26]), however significant opportunities exist for the refinement and further development of these tools. The addition of IM data is informative; mobility-mass correlations as well as CCS/mass ratios can guide unknown identifications by giving an idea of molecular class and by excluding unlikely candidates on the basis of structure. Established metabolomics labs and metabolomics centers have fixed chromatography methods that are robust, reliable and yield stable retention times. High quality RT and MS/MS fragmentation data of pure reference standards have been acquired for in-house libraries. These efforts certainly facilitate identification confidence, but are not feasible for most small research groups. Thus, leveraging methodologies and data with shared knowledge will benefit the entire metabolomics community.
Confidence levels
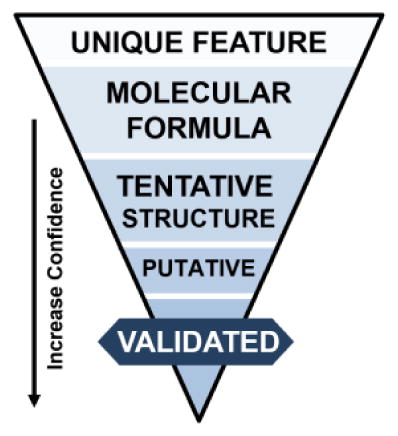
Metabolite annotation is the crucial link between acquired data and meaningful biological information. It is essential that the confidence of metabolite assignments is transparent. In 2007 the Chemical Analysis Working Group (CAWG) of the Metabolomics Standards Initiative (MSI) published a first stage of guidelines for reporting the minimum metadata relative to metabolite identification as a means to communicate the confidence of identifications [27]. Recently, revisions to these levels have been proposed to cover special cases where level determination may be unclear [28, 29]. We propose modest changes to include orthogonal IM-MS data as evidence for metabolite identification (Figure 3).
Figure 3.

Proposed workflow for metabolite identification confidence using multidimensional mass spectrometry. From top to bottom: Obtaining an exact mass measurement for a Unique Feature (Level 5) allows database searching, which here is illustrated by the over 61 million compounds indexed in PubChem at the time of this review. Subsequent levels of mass accuracy reduce the number of possible molecular formulas from over 200,000 (unit resolution), to ca. 10,000 at 1 ppm mass accuracy for the example mass of 354 Da. Using higher mass accuracy and/or a heuristic filtering approach obtains a unique Molecular Formula (Level 4), which still represents several thousand isomeric compounds. Tentative Structures (Level 3) match precursor m/z to a metabolite database and Putative Identifications (Level 2) match fragmentation data to metabolite MS/MS libraries. Obtaining a Validated Identification (Level 1) requires additional data evidence, such as tandem MS/MS, LC, IM, or measurements from other analytical techniques (optical spectroscopy or NMR) that match corresponding reference standard data under identical experimental conditions. Right portion of figure modified with permission of Annual Review of Analytical Chemistry, Volume 9 © by Annual Reviews, http://www.annualreviews.org from reference [2].
The highest confidence identification, a validated identification (Level 1), confirms a structure with a minimum of two independent and orthogonal data from a pure reference standard under identical analytical conditions. A lack of reference standard acquisition but predictive or externally acquired structure evidence, namely MS/MS data, exhibiting diagnostic fragments or neutral losses consistent with a specific structure would be considered a putative identification (Level 2). Preliminary identifications (Level 3) arise when accurate mass and isotopic distribution patterns produce tentative structures from database searches. Note, a single molecular formula typically renders multiple candidate structures. Our personal experience is that the majority of features detected by our methods result in preliminary identifications. Molecular formula candidates (Level 4) and a de-convoluted experimental m/z (Level 5) complete the less confident annotation classifications.
Suggestions have been made to clarify the set of reporting standards with the inclusion of an evidence-based quantitative score [30]. With either a score or level-based system, the future of annotation is likely to be influenced by multiplexed technologies. Recently, Pacini et al. obtained five levels of small molecule data in a single DIA acquisition (LC,UV, IM, MS, MS/MS) [31]. Advances of multidimensional analytical approaches are inherently the most promising for the broadest metabolome coverage. Orthogonal in-line data can provide the needed evidence to meet minimum data requirements for confident identifications. At the present time LC and IM are successfully multiplexed with MS providing RT and CCS data, respectively, as supporting evidence. As IM-MS becomes more widespread and CCS data is populated in searchable metabolomic databases and libraries, identifications using this knowledge will increase metabolite assignment confidence. The class-specific relationship in IM is also valuable evidence to support both annotations of metabolites and exclusion of unlikely candidates. For example, only correlated molecular classes based on IM trend lines or retention times based on polarity may be considered for identification purposes. For metabolite candidates that lack an available reference standard, a quantitative structure retention relationship (QSRR) model can predict retention times [32] and computational calculations can estimate CCS values [33] to be used as evidence.
False Discovery Rate
False positive identifications are a significant challenge for metabolomics. As described above, annotations arise by querying neutral mass against a database of candidate small molecule masses. Neutral masses are inferred from experimental m/z, thus the presence of related isotope and adduct features may complicate neutral mass determination and potentially lead to false positive identifications. False positives can also arise during MS/MS spectrum matching. Statistical tools for estimating the error of metabolite-spectrum matches are necessary for evaluating the confidence of annotation results. The inclusion of experimental orthogonal data such as RT and CCS data will decrease false positives, but there is currently no agreed upon metric to assess False Discovery Rate (FDR) of metabolite identifications. In MS-based proteomic studies, target-decoy search based FDR calculations are widely accepted [34, 35]. Briefly, predicted peptide MS/MS spectra are used to create a reverse decoy database, and experimental data matches are used to estimate FDR. In principle, a target-decoy strategy could be used for metabolomics using a small molecule set exclusive of the experimental species, but since metabolomics databases are incomplete this approach is currently challenging [22]. There are reports of novel FDR methods exclusive of decoy approaches, though none has yet gained widespread acceptance. For example a simulation model that uses the rate of a match for elemental composition search queries [38], the incorporation of a spectrum similarity score with a completion score for GC × GC/TOF-MS data [36], and a mixture modeling method coined GREAZY for phospholipids [37] have all been suggested. Querying predicted molecular formula of experimental data against a decoy set of theoretically possible candidates has been proposed, however the inflated search space increases the chance of a false positive identification and it is difficult to distinguish artificial compositions from legitimate candidate metabolites [38].Potential inclusion of the Seven Golden Rules [21] may offer an approach to classify the legitimate candidate list for exclusion from the decoy elemental composition list.
Biological Analysis of Metabolites
Biological interpretation of metabolomics data, and ultimately systems biology studies, hinges on the ability to accurately identify metabolites so they can be mapped to pathways and networks. Data from an untargeted metabolomics experiment is challenging to visualize and interpret on account of the amount of data generated. This challenge is amplified by the fact that numerous features are identified with varying levels of confidence. Table 2 outlines several open source options for analyzing metabolomics data depending on identification confidence level (unique feature to validated identification, described in Confidence Levels section above). Subsets of data may be analyzed using different tools depending on the data obtained (e.g., MS/MS fragmentation spectra is often only available for higher abundance ions from DDA analyses). Statistical and multivariate analyses are applied to prioritize data; multiple hypothesis testing, data dimension reduction (e.g., Principal Component Analysis (PCA) scores or loadings plots and Partial Least Squares (PLS) modeling), and data visualization (e.g., cloud plots [39]) and clustering (e.g, Self-organizing Map (SOM) [40]) can reveal altered ion abundances and patterns that may be characteristic of the phenotype.
Table 2.
Confidence Annotation, Statistical Evaluation, and Selected Bioinformatics Tools.
| Confidence | Statistical treatment | Utility | Bioinformatic tools | Ref |
|---|---|---|---|---|
|
Level 5 Unique m/z Feature & Level 4 Molecular Formula |
t-Test | Ranking significant differences | Most statistical software packages | |
| Principle Component Analysis (PCA) | ||||
| Partial Least Squares (PLS) Modeling | ||||
| Cloud Plot | Data visualization and prioritization | XCMS Online | [39] | |
| Volcano Plot | ||||
| Self-organizing Map (SOM) | Metabolite Expression Dynamics Inspection (MEDI) | [40] | ||
| Network activity prediction | Pathway/Network prediction without formal metabolite annotation | mummichog | [46] | |
|
Level 3 Tentative Structure |
MS1 database and MS2 spectral library | Matching parent ion exact mass and fragmentation patterns | ChemSpider, METLIN, HMDB, MassBank, mzCloud, LipidBlast, GNPS, NIST | [16,17, 18, 19] |
|
Level 2 Putative Identification & Level 1 Validation |
Pathway Analysis | Integration with known biology | MetaboAnalyst | [41] |
| Kyoto Encyclopedia of Genes and Genomes (KEGG) | [42] | |||
| Network Analysis | Mbrole (Metabolite Biological Role) | [43] | ||
| MetaCyc/BioCyc | [44] |
Most of the existing analysis tools require a list of identified metabolites to integrate biological knowledge [41–44]. New techniques for placing small molecules in a biological context are now being presented, relying on the integration of systems biology tools. For example, genomic and metabolomic data have been combined in a mining workflow to identify pharmaceutical candidates [45]. Another innovative approach utilizes the fact that single upstream biological disruptions result in a cascade of metabolomic changes. The creation of informatic strategies, such as mummichog [46], that predicts biological activity from MS1 data rather than formal MS2-dependent identifications is an attractive concept as it circumvents identification challenges. Importantly, a high level of agreement between identifications from mummichog results and conventional identification pipelines is found. This software uses the accurate mass of m/z features to map candidate metabolites to genome-scale metabolic networks and calculates local enrichment of metabolites to distinguish those networks from a stochastic distribution of metabolites [46]. Network modules are generated, as illustrated in Figure 4 which presents a comparison of metabolomic profiles of glucose 6-phosphate dehydrogenase deficient (G6PDd) and normal human erythrocytes, which reveal areas of network activity. These data are then used to focus additional efforts on validating the prioritized metabolites from the multitude of possibilities from database searching and isomeric species.
Figure 4.

Network module output from mummichog analysis of the qualitative and relative quantitative differences in metabolomic profiles of G6PDd deficient vs. normal human erythrocytes. Feature m/z values and significance measurements were used to predict metabolic activity networks without the use of conventional MS/MS identification workflows. Metabolites are colored blue (negative fold change) or red (positive fold change) and the size/color intensity represents the magnitude of fold change.
Summary
This is an exciting time for metabolomics research. Tremendous successes have been made to establish the necessary foundation for the field to mature. The metabolomics community now has the opportunity to address the high reward challenges associated with MS/MS data interpretation, database content, isomer resolution, identification confidence, and FDR estimation. Innovative research and development is essential, particularly at the interface of biomedical, cheminformatics, and bioinformatics fields. The metabolome is thought to be most predictive of phenotype thus novel ideas that address these challenges will allow the field to better understand mechanisms underlying health and disease.
Acknowledgments
The authors thank the many students, colleagues, and collaborators that have influenced their thinking in this area. Specific thanks are given to Jody C. May who conceptualized and constructed Figure 2 and elements of Figure 3, and to Jerry D. Holman for adaptation and presentation of algorithms in Figure 4. The authors are also grateful to our collaborators, Babu Tekwani - Research Institute of Pharmaceutical Sciences and the National Center for Natural Products Research & Department of Biomolecular Sciences at the University of Mississippi, Jagrati Jain - Department of Biomolecular Sciences at the University of Mississippi, and David W. Wright - Department of Chemistry at Vanderbilt University and Laboratories for Innovations in Global Health Technologies, for erythrocyte studies represented in Figure 4. Support for aspects of this work was provided by the National Institutes of Health/National Institute for General Medical Sciences (R01GM092218) and the National Institutes of Health/National Center for Advancing Translational Sciences (4UH3TR000491), the Vanderbilt University Center for Innovative Technology, Vanderbilt Institute of Chemical Biology; the Vanderbilt Institute for Integrative Biosystems Research and Education; and Vanderbilt University.
References
- 1.Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016 doi: 10.1038/nrm.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.May JC, McLean JA. Advanced Multidimensional Separations in Mass Spectrometry: Navigating the Big Data Deluge. Annu Rev of Anal Chem. 2015;9 doi: 10.1146/annurev-anchem-071015-041734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beisken S, Eiden M, Salek RM. Getting the right answers: understanding metabolomics challenges. Expert Rev Mol Diagn. 2015;15:97–109. doi: 10.1586/14737159.2015.974562. [DOI] [PubMed] [Google Scholar]
- 5.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 6.Fuhrer T, Zamboni N. High-throughput discovery metabolomics. Anal Biotechnol. 2015;31:73–78. doi: 10.1016/j.copbio.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB. Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol. 2004;22:245–252. doi: 10.1016/j.tibtech.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Plumb RS, Johnson KA, Rainville P, Smith BW, Wilson ID, Castro-Perez JM, et al. UPLC/MSE; a new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Comm Mass Spectrom. 2006;20:1989–1994. doi: 10.1002/rcm.2550. [DOI] [PubMed] [Google Scholar]
- 9.Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, et al. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol Cell Proteomics. 2012:11. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sana TR, Waddell K, Fischer SM. A sample extraction and chromatographic strategy for increasing LC/MS detection coverage of the erythrocyte metabolome. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;871:314–321. doi: 10.1016/j.jchromb.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 11.Fuhrer T, Zamboni N. High-throughput discovery metabolomics. Anal Biotechnol. 2015;31:73–78. doi: 10.1016/j.copbio.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Cajka T, Fiehn O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal Chem. 2016;88:524–545. doi: 10.1021/acs.analchem.5b04491. [DOI] [PubMed] [Google Scholar]
- 13.Kaplan KA, Hill HH. Metabolomics Using Ion Mobility Mass Spectrometry. In: Lutz NW, Sweedler JV, Wevers RA, editors. Methodologies for Metabolomics-Experimental Strategies and Techniques. Cambridge University Press; 2013. pp. 185–204. [Google Scholar]
- 14.May JC, Goodwin CR, McLean JA, Lyubimov AV. Gas-Phase Ion Mobility-Mass Spectrometry (IM-MS) and Tandem IM-MS/MS Strategies for Metabolism Studies and Metabolomics. In: Lyubimov A, editor. Encyclopedia of Drug Metabolism & Drug Interactions. John Wiley & Sons, Inc; 2012. [Google Scholar]
- 15.Paglia G, Williams JP, Menikarachchi L, Thompson JW, Tyldesley-Worster R, Halldórsson S, et al. Ion mobility derived collision cross sections to support metabolomics applications. Anal Chem. 2014;86:3985–3993. doi: 10.1021/ac500405x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith CA, Maille GO, Want EJ, Qin C, Trauger SA, Brandon TR, et al. METLIN: A Metabolite Mass Spectral Database. Ther Drug Monit. 2005;27:747–51. doi: 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- 17.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horai H, Arita M, Kanaya S, Nihei Y, Ikeda T, Suwa K, et al. MassBank: a public repository for sharing mass spectral data for life sciences. J Mass Spectrom. 2010;45:703–714. doi: 10.1002/jms.1777. [DOI] [PubMed] [Google Scholar]
- 19.Kind T, Liu KH, Yup Lee D, DeFelice B, Meissen JK, Fiehn O. LipidBlast - in-silico tandem mass spectrometry database for lipid identification. Nat Methods. 2013;10:755–758. doi: 10.1038/nmeth.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kind T, Fiehn O. Metabolomic database annotations via query of elemental compositions: mass accuracy is insufficient even at less than 1 ppm. BMC Bioinformatics. 2006;7:234. doi: 10.1186/1471-2105-7-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kind T, Fiehn O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics. 2007;8:105. doi: 10.1186/1471-2105-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuda F. Rethinking Mass Spectrometry-Based Small Molecule Identification Strategies in Metabolomics. Mass Spectrom (Tokyo) 2014;3:S0038. doi: 10.5702/massspectrometry.S0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groessl M, Graf S, Knochenmuss R. High resolution ion mobility-mass spectrometry for separation and identification of isomeric lipids. Analyst. 2015;140:6904–6911. doi: 10.1039/c5an00838g. [DOI] [PubMed] [Google Scholar]
- 24.Tachibana C. What's next in 'omics: The metabolome. Science. 2014;345:1519–1521. [Google Scholar]
- 25.Sherrod SD, McLean JA. Systems-wide high-dimensional data acquisition and informatics using structural mass spectrometry strategies. Clinical Chemistry. 2016;62:77. doi: 10.1373/clinchem.2015.238261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf S, Schmidt S, Müller-Hannemann M, Neumann S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinformatics. 2010;11:148. doi: 10.1186/1471-2105-11-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI) Metabolomics. 2007;3:211–221. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeon J, Kurth D, Hollender J. Biotransformation pathways of biocides and pharmaceuticals in freshwater crustaceans based on structure elucidation of metabolites using high resolution mass spectrometry. Chem Res Toxicol. 2013;26:313–324. doi: 10.1021/tx300457f. [DOI] [PubMed] [Google Scholar]
- 29.Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, et al. Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ Sci Technol. 2014;48:2097–2098. doi: 10.1021/es5002105. [DOI] [PubMed] [Google Scholar]
- 30.Creek DJ, Dunn WB, Fiehn O, Griffin JL, Hall RD, Lei Z, et al. Metabolite identification: are you sure? And how do your peers gauge your confidence? Metabolomics. 2014;10:350–353. [Google Scholar]
- 31.Pacini T, Fu W, Gudmundsson S, Chiaravalle AE, Brynjolfson S, Palsson BO, et al. Multidimensional analytical approach based on UHPLC-UV-ion mobility-MS for the screening of natural pigments. Anal Chem. 2015;87:2593–2599. doi: 10.1021/ac504707n. [DOI] [PubMed] [Google Scholar]
- 32.Creek DJ, Jankevics A, Breitling R, Watson DG, Barrett MP, Burgess KE. Toward global metabolomics analysis with hydrophilic interaction liquid chromatography-mass spectrometry: improved metabolite identification by retention time prediction. Anal Chem. 2011;83:8703–8710. doi: 10.1021/ac2021823. [DOI] [PubMed] [Google Scholar]
- 33.Lanucara F, Holman SW, Gray CJ, Eyers CE. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat Chem. 2014;6:281–294. doi: 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- 34.Käll L, Storey JD, MacCoss MJ, Noble WS. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J Proteome Res. 2008;7:29–34. doi: 10.1021/pr700600n. [DOI] [PubMed] [Google Scholar]
- 35.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 36.Jeong J, Shi X, Zhang X, Kim S, Shen C. An empirical Bayes model using a competition score for metabolite identification in gas chromatography mass spectrometry. BMC Bioinformatics. 2011;12:392. doi: 10.1186/1471-2105-12-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kochen MA, Chambers MC, Holman JD, Nesvizhskii AI, Weintraub ST, Belisle JT, et al. Greazy: Open-Source Software for Automated Phospholipid Tandem Mass Spectrometry Identification. Anal Chem. 2016 doi: 10.1021/acs.analchem.6b00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsuda F, Shinbo Y, Oikawa A, Hirai MY, Fiehn O, Kanaya S, et al. Assessment of metabolome annotation quality: a method for evaluating the false discovery rate of elemental composition searches. PLoS One. 2009;4:e7490. doi: 10.1371/journal.pone.0007490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gowda H, Ivanisevic J, Johnson CH, Kurczy ME, Benton HP, Rinehart D, et al. Interactive XCMS Online: Simplifying Advanced Metabolomic Data Processing and Subsequent Statistical Analyses. Analytical Chemistry. 2014;86:6931–6939. doi: 10.1021/ac500734c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodwin CR, Sherrod SD, Marasco CC, Bachmann BO, Schramm-Sapyta N, Wikswo JP, et al. Phenotypic mapping of metabolic profiles using self-organizing maps of high-dimensional mass spectrometry data. Anal Chem. 2014;86:6563–6571. doi: 10.1021/ac5010794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Research. 2015;43:W251–W257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.López-Ibáñez J, Pazos F, Chagoyen M. MBROLE 2.0—functional enrichment of chemical compounds. Nucleic Acids Research. 2016 doi: 10.1093/nar/gkw253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Research. 2016;44:D471–D480. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maansson M, Vynne NG, Klitgaard A, Nybo JL, Melchiorsen J, Nguyen DD, et al. An Integrated Metabolomic and Genomic Mining Workflow To Uncover the Biosynthetic Potential of Bacteria. mSystems. 2016:1. doi: 10.1128/mSystems.00038-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, et al. Predicting network activity from high throughput metabolomics. PLoS Comput Biol. 2013;9:e1003123. doi: 10.1371/journal.pcbi.1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]