

Abstract
Single prolonged stress (SPS) has been used to examine mechanisms via which stress exposure leads to post-traumatic stress disorder symptoms. SPS induces fear extinction retention deficits, but neural circuits critical for mediating these deficits are unknown. To address this gap, we examined the effect of SPS on neural activity in brain regions critical for extinction retention (i.e., fear extinction circuit). These were the ventral hippocampus (vHipp), dorsal hippocampus (dHipp), basolateral amygdala (BLA), prelimbic cortex (PL), and infralimbic cortex (IL). SPS or control rats were fear conditioned then subjected to extinction training and testing. Subsets of rats were euthanized after extinction training, extinction testing, or immediate removal from the housing colony (baseline condition) to assay c-Fos levels (measure of neural activity) in respective brain region. SPS induced extinction retention deficits. During extinction training SPS disrupted enhanced IL neural activity and inhibited BLA neural activity. SPS also disrupted inhibited BLA and vHipp neural activity during extinction testing. Statistical analyses suggested that SPS disrupted functional connectivity within the dHipp during extinction training and increased functional connectivity between the BLA and vHipp during extinction testing. Our findings suggest that SPS induces extinction retention deficits by disrupting both excitatory and inhibitory changes in neural activity within the fear extinction circuit and inducing changes in functional connectivity within the Hipp and BLA.
Previous studies have paired animal stress protocols with Pavlovian fear conditioning to examine stress-induced changes in neurobiological function that lead to persistent fear memory expression in post-traumatic stress disorder (PTSD) (Kohda et al. 2007; Armario et al. 2008; Baran et al. 2009; Wilber et al. 2011; Knox et al. 2012b,c; Hoffman et al. 2014; Bowers and Ressler 2015; Maren and Holmes 2015). One animal stress model that has been used to model persistent fear in PTSD is single prolonged stress (SPS). SPS refers to serial exposure to restraint, forced swim, and ether (Liberzon et al. 1997, 1999b; Armario et al. 2008; Yamamoto et al. 2009). SPS results in neuroendocrinological symptoms that mimic PTSD. These include enhanced negative feedback of the hypothalamic–pituitary–adrenal (HPA) axis and enhanced glucocorticoid receptor expression (Yehuda et al. 1996, 2006; Liberzon et al. 1997, 1999b; Ganon-Elazar and Akirav 2012; Knox et al. 2012c; George et al. 2013). SPS also mimics other features of PTSD such as deficits in excitatory tone in the medial prefrontal cortex (mPFC) (Bremner et al. 1999; Shin et al. 2004; Knox et al. 2010), enhanced locus coeruleus–norepinephrine reactivity (Southwick et al. 1993; Liberzon et al. 1999a; George et al. 2012), and deficits in the ability to retain fear extinction (Milad et al. 2008, 2009; Knox et al. 2012b,c; George et al. 2015; Keller et al. 2015a,b), which can be defined as maintaining inhibition of fear expression to a previously extinguished fear conditioned stimulus (CS). While SPS consistently results in extinction retention deficits (Knox et al. 2012b,c; George et al. 2015; Keller et al. 2015a,b), neural circuitry critical for mediating these deficits remain unknown.
Previous studies have shown that neural plasticity in the infralimbic cortex (IL), basolateral amygdala (BLA), and ventral hippocampus (vHipp) are critical for extinction retention (Milad and Quirk 2002; Santini et al. 2004; Sierra-Mercado et al. 2006, 2011; Vidal-Gonzalez et al. 2006; Burgos-Robles et al. 2007; Corcoran and Quirk 2007). The prelimbic cortex (PL) is critical for conditioned fear expression (Corcoran and Quirk 2007) and vHipp input to the PL is critical for regulating expression of conditioned fear (Orsini et al. 2011; Sotres-Bayon et al. 2012). The dorsal hippocampus (dHipp) is critical for contextual modulation of extinction retention (Westbrook et al. 2000; Corcoran and Maren 2001, 2004; Bouton et al. 2006; Orsini et al. 2011). Thus, a neural circuit (referred to here as the fear extinction circuit) comprised of the PL, IL, dHipp, BLA, and vHipp is critical for extinction retention.
Previous studies have examined neural circuitry through which stress disrupts extinction memory. Using chronic stress these studies have observed that mechanisms via which stress disrupts extinction memory involve enhancing BLA and dHipp neural activity during extinction testing, while disrupting increases in IL neural during extinction testing (Wilber et al. 2011; Hoffman et al. 2014). However, SPS is an acute stressor and the behavioral mechanisms via which of chronic stress and SPS lead to extinction retention deficits are different (Miracle et al. 2006; Wilber et al. 2011; Knox et al. 2012b,c), as well as neurobiological processes within the fear extinction circuit that are disrupted by both types of stressors (Moghaddam 1993; Mitra et al. 2005; Cui et al. 2008; Knox et al. 2010, 2012c). Thus, examining neural circuitry through which SPS exposure leads to extinction memory deficits has the potential to expand upon our understanding of how stress can disrupt the retention of extinction memory.
The goal of this study was to examine the effects of SPS on neural activity in the fear extinction circuit at baseline and during extinction training and testing. To accomplish this goal, a set of SPS and control rats were fear conditioned, then subjected to extinction training and testing (CS-fear condition). A second set of SPS and control rats were presented with CSs only during fear conditioning, then subjected to extinction training and testing (CS-only condition). This group was used to control for changes in neural activity due to repeated CS presentation. After extinction training or testing subsets of rats were euthanized to assay c-Fos levels in the fear extinction circuit. We used c-Fos as our measure of neural activity, because it is an excellent measure for mapping functional changes in neural activity across brain regions (Sagar et al. 1988) and is often used to map neural circuit in experiments that examine fear and extinction circuits (e.g. see Santini et al. 2004; Knapska and Maren 2009; Orsini et al. 2011; Hoffman et al. 2014; Lazenka et al. 2014) . We also assayed c-Fos levels in a third set of SPS and control rats after immediate removal from the housing colony to establish baseline c-Fos levels. By comparing c-Fos levels in SPS and control rats during extinction training and testing to baseline c-Fos levels in respective sets of rats, we were able to detect increases as well as decreases in c-Fos levels during extinction training and testing. The experimental design is illustrated in Figure 1. Finally, we subjected c-Fos levels to factor analysis to examine functional connectivity within the fear extinction circuit (see Materials and Methods for more details)
Figure 1.
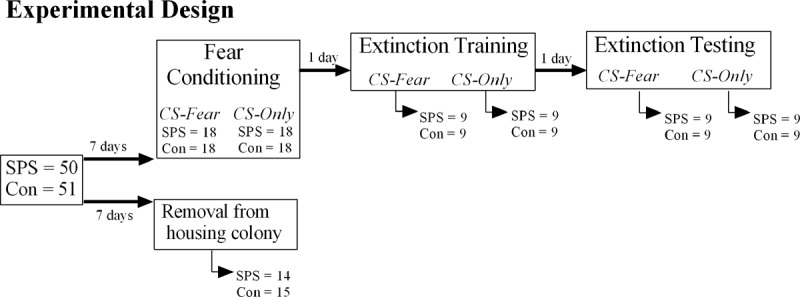
Experimental design used in this study. Animals in the CS-only condition were presented with tones during fear conditioning in the absence of any footshock. Arrows under the boxes indicate the number of animals that were euthanized to assay c-Fos in the fear extinction circuit.
Results suggest that during extinction training, SPS disrupted enhancements in IL neural activity and inhibition of BLA neural activity. SPS also disrupted inhibition of BLA and vHipp neural activity during extinction testing. SPS exposure disrupted functional connectivity within the dHipp during extinction training and increased functional connectivity between the vHipp and BLA during extinction testing. Finally, the results raise the possibility that changes in neural activity believed to be important for inhibiting conditioned fear expression is driven by repeated CS presentation as opposed to acquisition of an extinction rule (CS-no UCS).
Results
Behavioral results
Freezing during all behavioral testing was analyzed using stress × condition × trial factor designs with post hoc tests where appropriate. Animals in the CS-fear condition had higher levels of conditioned freezing during all behavioral sessions when compared with the CS-only condition. This was revealed by main effects of condition for fear conditioning [F(1,67) = 34.202, P < 0.001], extinction training [F(1,67) = 115.644, P < 0.001], and extinction testing [F(1,31) = 7.465, P = 0.01]. SPS resulted in enhanced freezing during the extinction test, which was revealed by a significant main effect of stress [F(1,31) = 5.347, P = 0.01]. However, this effect was only observed in the CS-fear condition [simple effect of stress: F(1,15) = 5.346, P = 0.035] and was most pronounced during baseline and the first block of trials of the extinction test. There were no stress effects observed in the CS-only condition [simple term for SPS: F(1,16) = 0.398, P = 0.537]. There were no stress effects during fear conditioning or extinction training (Ps > 0.05). These findings are illustrated in Figure 2.
Figure 2.
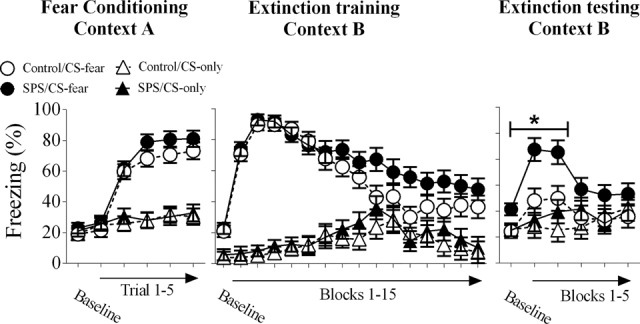
Behavioral results of this study. All animals in the CS-fear condition displayed enhanced freezing to the CS in comparison to animals in the CS-only condition in all of the behavioral tests. SPS enhanced conditioned freezing during the extinction test (i.e., extinction retention deficits). Asterisk indicates significant stress effect at the P < 0.05 level. One control rat was removed from analysis in this study, because freezing during extinction testing was >3 standard errors of the mean away from the group mean of the control/CS-fear group during extinction testing. (SPS) single prolonged stress.
c-Fos immunoreactivity
Representative c-Fos stained brain sections are illustrated in Figure 3. Signal activity illustrated in Figure 3 did not represent nonspecific secondary antibody binding to tissue or endogenous fluorescent activity of brain tissue (see Supplemental Material).
Figure 3.
Representative images in the vmPFC, dHipp, BLA, and vHipp obtained after performing c-Fos immunocytochemistry and scanning brain slides using the Li-cor Odyssey scanner. (vmPFC) ventromedial prefrontal cortex, (IL) infralimbic cortex, (PL) prelimbic cortex, (dHipp) dorsal hippocampus, (BLA) basolateral amygdala, (vHipp) ventral hippocampus.
vmPFC
Baseline PL c-Fos levels were equivalent between SPS and control rats [t(27) = 1.136, P = 0.266]. There was a significant increase in PL c-Fos levels during extinction training in all rats. This effect was revealed by a significant one-sample t-test for PL c-Fos levels collapsed across all groups [t(33) = 10.127, P < 0.001]. Neither SPS nor CS-fear treatment affected PL c-Fos levels during extinction training [P > 0.05]. During extinction testing, there was a significant increase in PL c-Fos levels [one-sample t-test collapsed across groups: t(33) = 5.399, P < 0.001]. However, there were no stress, condition, or stress × condition effects (P > 0.05). These results are illustrated in Figure 4A.
Figure 4.

Effects of stress and condition on c-Fos levels in the fear extinction circuit. White bars represent control rats, whereas gray bars represent SPS rats. Stripped bars represent rats in the CS-Fear (CS-F) condition, while nonstripped bars represent rats in the CS-Only (CS-O) condition. (A) During extinction training, there was enhanced vmPFC c-Fos levels and enhanced PL c-Fos levels during extinction testing. SPS attenuated enhanced IL c-Fos levels during extinction training. (B) There were no SPS effects on c-Fos levels in the dHipp, but levels were inhibited during extinction testing. (C) In the BLA, SPS attenuated inhibited c-Fos levels during extinction training and testing, while (D) SPS attenuated inhibited c-Fos levels in the vHipp during extinction testing. SPS had no effect on baseline c-Fos levels in any brain region. (IL) infralimbic cortex, (PL) prelimbic cortex. (+) indicates a significant one-sample t-test at the P < 0.05 level, and (*) indicates a significant stress effect at the P < 0.05 level.
Baseline IL c-Fos levels were equivalent between SPS and control rats [t(27) = 1.631, P = 0.115]. There was a significant increase in IL c-Fos levels during extinction training in control rats, and this effect was attenuated in SPS rats [main effect of stress: F(1,30) = 5.147, P = 0.031]. However, in both SPS and control rats, there was significant increases in IL c-Fos levels relative to baseline as evidenced by significant one-sample t-test for control [t(16) = 3.519, P = 0.006] and SPS [t(16) = 4.745, P < 0.001] rats. During extinction testing IL c-Fos levels were not affected by the stress or condition factors or the interaction of these factors (Ps > 0.05). These results are illustrated in Figure 4A.
dHipp
Baseline dHipp c-Fos levels were equivalent between SPS and control rats [dCA1: t(26) = 0.893, P = 0.38; dCA3: t(26) = 0.918, P = 0.367; dDG: t(26) = 0.848, P = 0.404]. Neither stress nor condition had any effects on c-Fos levels in the dHipp during extinction training (Ps > 0.05). Also, during extinction training, c-Fos levels in all regions of the dHipp were not significantly changed from baseline (Ps > 0.05).
There were no main effects of stress, condition, or stress × condition interaction (Ps > 0.05) for dHipp c-Fos levels during extinction testing. However, c-Fos levels in all regions of the dHipp were significantly decreased relative to baseline [dCA1: t(34) = 4.268, P < 0.001; dCA3: t(34) = 5.141, P < 0.001; dDG: t(34) = 3.05, P = 0.012]. These results are illustrated in Figure 4B.
BLA
Baseline c-Fos levels in the LA [t(26) = 0.973, P = 0.34] and in the BA [t(26) = 1.061, P = 0.298] were equivalent between SPS and control rats. In the LA and in the BA, there was a decrease in c-Fos levels during extinction training in control rats, and this effect was disrupted in SPS rats. These results were revealed by significant main effects of stress for c-Fos levels in the LA [F(1,31) = 11.101, P = 0.002] and in the BA [F(1,31) = 9.162, P = 0.005] as well as one-sample t-test that was statistically significant for control rats [LA: t(16) = −4.768, P < 0.001; BA: t(16) = −3.737, P = 0.004], but not SPS rats [LA: t(17) = 0.47, P = 0.644; BA: t(16) = 0.787, P = 0.442].
LA and BA c-Fos levels were decreased in control rats during extinction testing, and this effect was disrupted in SPS rats. This was revealed by significant main effect of stress for c-Fos levels in the LA [F(1,31) = 4.766, P = 0.037], and a main effect of stress that approached significance in the BA [F(1,31) = 3.149, P = 0.086]. Also, there were significant one-sample t-test for control rats in the LA [t(16) = −4.14, P = 0.002] and in the BA [t(16) = −3.465, P = 0.006], whereas there were no significant one-sample t-test for SPS rats [LA: t(16) = −1.137, P = 0.271; BA: t(16) = −1.14, P = 0.27]. These results are illustrated in Figure 4C.
vHipp
Baseline vHipp c-Fos levels were equivalent between SPS and control rats [vCA: t(27) = 0.493, P = 0.626; vDG: t(27) = 0.315, P = 0.755]. Neither stress nor condition had any effects on c-Fos levels in the vHipp during extinction training (P > 0.05). Also, during extinction training, c-Fos levels in all regions of the vHipp were not significantly changed from baseline (P > 0.05).
In the vCA and in the vDG regions of the vHipp, there was a decrease in c-Fos levels in control rats during extinction testing and this effect was attenuated in SPS rats. This was revealed by significant main effect of stress for c-Fos levels in the vCA [F(1,29) = 5.128, P = 0.031] and vDG [F(1,29) = 6.694, P = 0.015]. Also, there were significant one-sample t-tests for control rats in the vCA [t(14) = −5.094, P < 0.001] and vDG [t(14) = −5.031, P < 0.001]. This effect was absent in SPS rats [vCA: t(17) = −0.817, P = 0.425; vDG: t(16) = −0.725, P = 0.478]. These results are illustrated in Figure 4D.
Factor analysis
Factor analyses were used to assay functional connectivity within the fear extinction circuit (see Materials and Methods). The results of these analyses are illustrated in Figure 5. In each factor analysis, the rotated component matrix, variance explained by the factor analysis, cartoon of factors in brain sections, and schematic of factors are illustrated. All factor analyses accounted for >80% of the variance in c-Fos levels in brain regions analyzed and each brain region in a particular factor had a correlation value >0.5.
Figure 5.

Representation of factor analyses performed in this study. In each box, the rotated component matrix, variance explained by each factor analysis, cartoon representation of the factor analysis, and schematic representation of the factor analysis is illustrated. Assignment of color (e.g., blue) and number (e.g., 1) to factor is arbitrary. Factor analyses revealed that functional connectivity in SPS rats was different to control rats under all conditions. Notably, the vHipp of SPS rats functionally connected to at least one other component of the fear extinction circuit under all conditions, whereas the vHipp of control rats was only functionally connected to other components of the fear extinction circuit in the CS-only condition during extinction testing. Furthermore, disrupted dHipp functional connectivity during extinction training was only observed in the SPS/CS-fear treatment, while selective BLA↔vHipp functional connectivity was only observed in the SPS/CS-fear treatment during extinction testing.
In all conditions analyzed, functional connectivity within the fear extinction circuit of SPS and control rats was different. At baseline, SPS exposure resulted in functional connectivity between vHipp and vmPFC (vHipp↔vmPFC). Functional connectivity between the vHipp and the dCA1 (vHipp↔dCA1) and disrupted functional connectivity within the dHipp was observed in SPS rats in the CS-fear condition during extinction training. Functional connectivity between the vHipp and the BLA (vHipp↔BLA) was also observed in SPS rats in the CS-fear condition during extinction testing. None of these patterns of functional connectivity were observed in the SPS/CS-only, control/CS-fear, and control/CS-only treatments, during extinction training and testing. During extinction testing, the vHipp became functionally disconnected in control rats in the CS-only condition. The only condition in which the vHipp was functionally connected to other components of the fear extinction circuit in control rats was during control/CS-only treatment during extinction testing (BLA↔dHipp↔vHipp).
Discussion
SPS effects on neural activity in individual components of the fear extinction circuit
In replication of previous studies (Knox et al. 2012b,c; George et al. 2015; Keller et al. 2015a,b), we observed that SPS disrupted extinction retention. Enhanced neural activity in the vmPFC was observed during extinction training, which is consistent with previous studies that have used electrophysiological and metabolic measures (Milad and Quirk 2002; Barrett et al. 2003). Enhanced PL neural activity was also observed in all rats during extinction testing. This is somewhat surprising, since the PL is critical for conditioned fear expression (Corcoran and Quirk 2007), and levels of conditioned fear were low in all rats except rats in the SPS/CS-fear condition. Even though studies have observed low PL neural activity during extinction testing (Knapska and Maren 2009; Wilber et al. 2011), at least one previous report has observed enhanced PL neural activity (Holmes et al. 2012). SPS only attenuated increases in IL neural activity during extinction training. There was also a failure to inhibit BLA neural activity in SPS rats during extinction training. vmPFC, and specifically IL, inhibition of BLA neural activity is believed to be critical for extinction retention (Sierra-Mercado et al. 2006, 2011; Herry et al. 2008; Knapska and Maren 2009; Do-Monte et al. 2015). This inhibition may occur via IL input to the intercalated region of the amygdala, which then results in inhibition of neural activity in the BLA (Royer and Pare 2002; Quirk et al. 2003; Likhtik et al. 2008; Cho et al. 2013). Thus, SPS may disrupt extinction retention by disrupting IL inhibition of BLA neural activity during extinction training, which disrupts neural plasticity critical for extinction retention.
During extinction testing, there was inhibited BLA and vHipp neural activity in control rats, and this effect was attenuated by SPS. This finding suggests that SPS may disrupt extinction retention by disrupting inhibition of neural activity in these two neural substrates. Even though pharmacological inhibition of the BLA and vHipp during extinction training disrupts extinction retention, this treatment may have no effect on extinction retention when applied immediately prior to extinction testing (Sierra-Mercado et al. 2011). However, pharmacological inhibition of neural activity in the BLA and vHipp may not mimic endogenous inhibition in the BLA and vHipp during extinction testing. Other studies have shown that BLA↔vHipp functional connectivity during extinction testing is critical for regulating levels of conditioned fear (Herry et al. 2008; Orsini et al. 2011). This finding raises the possibility that SPS could also disrupt extinction retention by enhancing BLA↔vHipp functional connectivity during extinction testing (see below).
SPS effects on functional connectivity within the fear extinction circuit
Using factor analysis to identify functional connectivity patterns among components of the fear extinction circuit consistently revealed increased vHipp functional connectivity in SPS rats. While BLA↔Hipp functional connectivity was observed in the control/CS-only treatment during extinction testing, selective BLA↔vHipp functional connectivity during extinction testing was only observed in the SPS/CS-fear treatment. BLA↔vHipp functional connectivity is critical for contextual modulation of conditioned fear expression during extinction testing (Orsini et al. 2011) and enhancing the activity of, “fear,” neurons in the BLA (Herry et al. 2008). Given that selective BLA↔vHipp functional connectivity during extinction testing was only observed in SPS rats, it is possible that selectively enhanced BLA↔vHipp functional connectivity during extinction testing contributes to extinction retention deficits in the SPS model by biasing conditioned fear expression. Further research identifying mechanisms of how SPS exposure selectively enhances BLA↔vHipp functional connectivity during extinction testing is needed.
Functional disconnection of the trisynaptic circuit of the dHipp (i.e., DG↔CA3↔CA1) was observed during extinction training in the SPS/CS-fear treatment. Disrupting neural activity, and thus functional connectivity, within the dHipp during extinction training disrupts extinction retention (Corcoran et al. 2005), and this could be due to improper representation of the extinction context (Corcoran et al. 2005). Enhanced baseline freezing during extinction testing was observed in SPS rats, which is consistent with a disruption in discrimination of the fear and extinction contexts. Thus, disruption of functional connectivity within the dHipp during extinction training could disrupt representation of the extinction context and lead to extinction retention deficits in the SPS model. The finding that SPS disrupts measures of neural plasticity in the dHipp believed to be critical for contextual memory is consistent with this hypothesis (Kohda et al. 2007; Yamamoto et al. 2008).
Neurobiological mechanisms by which SPS disrupts neural function within the fear extinction circuit
Through what mechanisms might SPS disrupt neural function within the fear extinction circuit? SPS decreases excitatory tone in the ventral medial prefrontal cortex (vmPFC) (Knox et al. 2010) and enhances apoptosis in the vmPFC (Zhao et al. 2014). Enhanced vmPFC apoptosis induced by SPS may be due to SPS-induced changes in signaling pathways in the endoplasmic reticulum that are critical for inhibiting apoptosis (Zhao et al. 2014; Wen et al. 2016). These SPS-induced changes in vmPFC function could underlie the decrease in IL neural activity during extinction training observed in this study. However, SPS-induced changes in excitatory tone and apoptosis are observed in the entire vmPFC and not just the IL (Knox et al. 2010; Zhao et al. 2014; Wen et al. 2016). Thus, it is unclear how these neurobiological changes could result in decreased neural activity in the IL, but not PL. Interestingly, SPS also enhances apoptosis in the BLA (Ding et al. 2010; Xiao et al. 2011). If SPS-induced BLA apoptosis resulted in selective loss of inhibitory GABAergic neurons in the BLA, then SPS-induced BLA apoptosis could contribute to the failure to inhibit neural activity during extinction training and testing in SPS rats. Finally, cannabinoid signaling at the CB1 receptor in the BLA and vHipp after SPS are critical for mediating the effects SPS has on contextual extinction memory (Ganon-Elazar and Akirav 2013). Thus, a failure to inhibit BLA and vHipp neural activity during extinction training and testing could stem from CB1 receptor signaling in these brain regions during SPS.
Chronic stress has similar effects on IL neural activity and extinction retention when compared with SPS. However, there are notable differences in mechanisms via which both stress protocols induce extinction retention deficits. Chronic stress decreases IL single unit activity and enhances dHipp and BLA neural activity (measured using c-Fos) during extinction testing (Wilber et al. 2011; Hoffman et al. 2014), while SPS disrupts IL neural activity during extinction training, disinhibits BLA neural activity during extinction training and testing, and disinhibits vHipp neural activity during extinction testing (see Results). Chronic stress increases glutamate levels and excitatory metabolites in the vmPFC (Moghaddam 1993), which may lead to impaired neural function in the IL (McEwen 2001; Izquierdo et al. 2006; Liu and Aghajanian 2008; Wilber et al. 2011). SPS decreases excitatory metabolites and induces apoptosis in the IL (Knox et al. 2010; Zhao et al. 2014). Chronic stress increases spine density on principal neurons in the BLA and enhances excitation within the BLA (Mitra et al. 2005; Qin et al. 2011; Leuner and Shors 2013; Padival et al. 2013), while SPS decreases BLA spine arborization and induces apoptosis (Cui et al. 2008; Ding et al. 2010; Xiao et al. 2011). Both chronic stress and SPS alter expression of glucocorticoid receptors (GRs) in emotional circuits (McEwen 2001; Armario et al. 2008; Yamamoto et al. 2009), though SPS has a more consistent enhancement in GRs in the hippocampus and vmPFC (Kohda et al. 2007; Knox et al. 2012c; Eagle et al. 2013; Ganon-Elazar and Akirav 2013; George et al. 2013, 2015). While chronic stress-induced changes in GR function may lead to extinction memory deficits (Gourley et al. 2009; Bingham et al. 2013; Segev et al. 2014), SPS-enhanced GR expression in emotional circuits may inhibit the development of extinction retention deficits in the SPS model (Keller et al. 2015b). Thus, it appears that chronic stress and SPS lead to extinction retention deficits via different neurobiological mechanisms.
Neural activity driven by extinction learning versus repeated stimulus presentation
In neuroscientific models of extinction, it is assumed that changes in neural activity during extinction training are driven by acquisition of an extinction rule (i.e., CS-no UCS). In this study, changes in neural activity that are believed to be critical for extinction memory acquisition (i.e., increased IL neural activity, inhibited BLA, and vHipp neural activity) were observed in control rats that did not receive footshocks during fear conditioning (i.e., CS-only condition), and thus could not have acquired or expressed an extinction rule during extinction training and testing, respectively. This finding raises the possibility that some of the neural processes critical for reducing conditioned fear levels that occur during extinction training and testing are driven by repeated stimulus presentation and not acquisition of a CS-no UCS rule per se. Also, SPS effects on neural activity in the IL, BLA, and vHipp were observed in the CS-fear and CS-only conditions. This raises the possibility that subsets of neural function that are disrupted by SPS, and potentially critical for inhibiting conditioned fear expression, were driven by repeated stimulus presentation and not acquisition or expression of an extinction rule.
While the repeated CS presentation protocol adopted in this study resembles latent inhibition, there are a number of reasons why it is unlikely that a latent inhibition-like phenomena was invoked in the CS-only animals during extinction training. First, latent inhibition in the Pavlovian fear conditioning paradigm is typically driven by multiple CS presentation across days (Maren and Holt 2000; Marks et al. 2016), and not five CS presentation prior to fear conditioning. Second, latent inhibition is context specific. If the CS is presented in one context, but then fear conditioning is commenced in another context, then latent inhibition is not observed (Maren and Holt 2000; Westbrook et al. 2000; Miller et al. 2015). The repeated stimulus presentation protocol in this study involved a small number of CS presentations in one context, followed by an increase in repeated CS presentations in a novel context. This unique repeated stimulus presentation protocol may have selectively resulted in enhanced IL neural activity and inhibited BLA neural activity. Further research examining if this repeated CS presentation protocol can either reduce conditioned fear responding or enhance acquisition of extinction is needed.
Limitations
In this study, we used c-Fos to measure neural activity. This methodology allows for the measurement of neural activity in many brain regions at a common time point. This is advantageous, because changes in neural activity across many brain regions implicated in a behavioral phenomenon can be measured (Knapska and Maren 2009; Knox et al. 2012a; Hoffman et al. 2014). However, animals have to be euthanized in order to assay c-Fos and c-Fos cannot be used to measure neural activity on small time scales. These parameters make it difficult to directly link changes in c-Fos levels to changes in behavior within a session (e.g., during extinction training) and across sessions. For example, it is impossible to correlate inhibited BLA neural activity observed during extinction training with levels of conditioned freezing during extinction testing. Furthermore, certain types of GABAergic neurons do not express c-Fos (Staiger et al. 2002). As a result, further research is needed to better understand how changes in vHipp, IL, and BLA neural activity, as well as functional connectivity among these substrates, lead to extinction retention deficits in the SPS model.
Conclusion
The results of this study suggest that SPS may induce extinction retention deficits by disrupting IL inhibition of BLA neural activity and dHipp functional connectivity during extinction training, as well as disrupting inhibited BLA and vHipp neural activity and enhancing BLA↔vHipp functional connectivity during extinction testing. Deficits in PFC inhibition of BLA neural activity as well as dysfunction within the Hipp has been implicated in PTSD (Elzinga and Bremner 2002; Shin et al. 2006; Pitman et al. 2012). Given these findings, it would appear that the SPS model can be used to examine how trauma-induced changes in PFC, amygdala, and Hipp function in humans lead to persistent fear expression in PTSD.
Materials and Methods
Animals
One hundred and one adult male Sprague Dawley rats (150 g upon arrival), obtained from Charles River, were used in this study. Upon arrival all rats were pair housed until SPS exposure. All rats had ad libitum access to water and food initially, but then rats were fed 23 g of rat chow per day, which is the manufacturer's recommended diet (LabDiet). Throughout the study, rats had ad libitum access to water. Experimental manipulations commenced after rats had been in the housing colony for at least 5 d. Rats were on a 12-h light–dark cycle. All experimental procedures were performed in the animals’ light cycle and all behavioral tests were conducted between 9:00 a.m. and 2:00 p.m. All experiments were approved by the University of Delaware Institutional Animal Care and Use Committee following guidelines established by the NIH.
Experimental design, SPS, and behavioral procedures
All rats were initially assigned to the SPS or control stress group. SPS was conducted as previously described (Liberzon et al. 1997; Knox et al. 2010). SPS consisted of 2 h of restraint, 20 min of forced swim, and ether exposure until general anesthesia was induced. Rats assigned to the control group were placed in a novel room for the duration of SPS. After SPS, all rats were returned to the housing colony and a post-stress incubation period of 7 d elapsed prior to behavioral testing, because this period is needed to observe extinction retention deficits in the SPS model (Knox et al. 2012b).
All SPS and control rats were then divided into two groups. One group of SPS and control rats were removed from the housing colony and immediately euthanized. We used this group of rats to establish baseline c-Fos levels in the fear extinction circuit. The other group of SPS and control rats were further subdivided into two groups. One group was subjected to fear conditioning, then extinction training and testing (see below). We refer to this group as CS-fear. The other group of rats were presented with CSs in the absence of footshock during fear conditioning, then subjected to extinction training and testing. We refer to this group as CS-only. We used the CS-only treatment in order to control for changes in c-Fos levels that were driven by repeated CS presentation.
Fear conditioning and extinction sessions were conducted as previously described (Knox et al. 2012b,c; Keller et al. 2015b). A 10 sec auditory CS (2 kHz, 80 dB) coterminated with a 1 mA, 1 sec footshock unconditioned stimulus (UCS) in a distinct context (Context A). The CS-only group had CS presentations in the absence of footshocks. Extinction training commenced 1 d after fear conditioning and occurred in Context B with 30 CS presentations. Extinction testing commenced 1 d after extinction training in Context B and consisted of 10 CS presentations. Contexts A and B were created by manipulating multiple sensory cues (Knox et al. 2012b). All behavioral sessions consisted of a baseline period of 210 sec and interstimulus intervals (ISIs) of 60 sec. All behaviors were recorded and scored at a later time as described below.
c-Fos immunocytochemistry
Rats were euthanized via rapid decapitation either after immediate removal from the housing colony or 60 min after the start of extinction training or testing. Brains were then extracted and flash frozen in chilled isopentane and stored in a −80°C freezer until further processing. Brains were then thawed to −13°C in a cryostat (Leica CM1350) and 30 µm sections through the ventromedial prefrontal cortex (vmPFC), dHipp, BLA, and vHipp were mounted onto superfrost slides. Brain sections were then stored in a −80°C freezer until time of assay.
To perform c-Fos immunocytochemistry, sections were fixed in 4% paraformaldehyde in 0.2 M phosphate-buffered saline (PBS). Sections were then incubated in Triton X-100, rinsed in 0.1 M tris-buffered saline (TBS) and incubated in 3% goat serum. Sections were rinsed again in TBS and incubated with a rabbit polyclonal c-Fos antibody (Santa Cruz Biotechnology, sc-52) at a dilution of 1:500 in PBS overnight at 4°C. Sections were then rinsed in TBS containing 0.01% Tween-20 (TBS-T). After this, sections were incubated in a solution consisting of TBS, 1.5% goat serum, 0.1% Triton X-100, and goat anti-rabbit IgG antibody 800 CW (Li-cor Biotechnology 926-32211) in a dilution of 1:2000 for 2 h. Sections were rinsed in TBS-T, TBS, and then deionized water. Sections were then left to air dry overnight.
Data analysis and statistical analysis
Freezing in all videos was analyzed using ANY-maze (Stoelting Inc.) as previously described (Knox et al. 2012c). Fear-conditioned freezing values were averaged in trials that consisted of a CS and respective ISI (e.g., CS1 and ISI1). Freezing during fear conditioning was analyzed using a stress (SPS vs. control) × condition (CS-only vs. CS-fear) × fear conditioning (baseline, trials 1–5) factor design. For extinction training, freezing across two extinction trials were averaged into a block and analyzed using a stress × condition × extinction training (baseline, blocks 1–15) factor design. Freezing during the extinction test was averaged into two-trial blocks and analyzed using a stress × condition × extinction test (baseline, blocks 1–5) factor design. If datum from a particular rat was >2 SEM away from the group mean, all data from this animal were removed from consideration.
The secondary antibody used in the c-Fos immunocytochemistry procedure fluoresces at 780 nm (i.e., near infrared fluorescence) (e.g. see Knox et al. 2012c). This activity was used to measure c-Fos levels in a specific brain region (see Supplemental Information). Dried brain sections, treated for c-Fos immunocytochemistry, were scanned at 21 µm resolution in the Odyssey scanner. Fluorescent activity in a particular brain region was then expressed as a percent change from activity in the corpus callosum. We referred to this as signal activity. Signal activity from all brain regions in the baseline condition was subjected to t-test (SPS versus control). Signal activity obtained from rats euthanized after extinction training and testing was normalized with respect to baseline signal activity. For example, IL signal activity from an SPS rat in the CS-only condition was normalized relative to averaged baseline IL signal activity of SPS rats. We refer to this as normalized activity. Normalized activity was constructed so that signal activity during extinction training and testing that was equal to baseline signal activity would yield a normalized score of 100% (i.e., (signal activity/averaged baseline activity) × 100). Normalized activity in the fear extinction circuit (i.e., IL, PL, dHipp (dCA1, dCA3, and dDG), BLA (lateral (LA) and basal (BA) regions separately), and vHipp (vCA, vDG)) during extinction training and testing was subjected to a stress × condition factor design.
All statistical tests were performed in IBM SPSS statistics 23. For all factor designs main and simple effects were analyzed using analysis of variance (ANOVA) while main and simple comparisons were analyzed using t-test with Bonferroni corrections applied where necessary. We also useed one sample t-tests to determine if a specific group mean was different from 100%. If multiple one-sample t-tests were performed for normalized activity, then Bonferroni corrections were also applied to these tests. All graphs plot means along with standard error of the means.
To examine functional connectivity within the fear extinction circuit, we applied factor analyses to the c-Fos data set. Factor analysis, when applied to a set of variables, identifies correlations among variables and groups these variables as a single factor, with the strength of the correlation determining whether variables are grouped into a factor (Cattell 1978). As a result, factor analysis applied to our c-Fos data set can be used to identify patterns of correlated neural activity (i.e., functional connectivity) among components of the fear extinction circuit. We applied factor analysis to signal activity in the IL, PL, dCA1, dCA3, dDG, LA, BA, vCA, and vDG of SPS and control rats. This was done separately in all conditions. Factors were extracted using principal component analysis. Only factors with eigenvalues >1 were considered (Kaiser rule), and the entire factor analysis was only considered valid if it accounted for >80% of the variance within a data set. For a variable to be considered as part of a factor, correlation of that variable with a factor had to be >0.5.
Competing interest statement
D.K., B.S., J.S., N.D., S.K., and T.D. have no conflict of interest concerning the findings presented in this manuscript.
Supplementary Material
Acknowledgments
The research in this report was funded by a pilot grant from the National Institutes of Health (1P20GM103653) awarded to D.K., a University of Delaware Research Foundation grant awarded to D.K., and the following grants, which supported confocal microscopy used in this study: P20 GM103446 (NIH), National Science Foundation EPSCoR grant IIA-1301765 (NSF), and the state of Delaware. We thank the following individuals for their help in conducting this study: Steve Albanese, Melanie Scicchitano, Elizabeth Schneider, Caroline Lawless, Marisa Chamness, Cristina Sosa, and Pauline V.C. David.
Footnotes
[Supplemental material is available for this article.]
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.043141.116.
References
- Armario A, Escorihuela RM, Nadal R. 2008. Long-term neuroendocrine and behavioural effects of a single exposure to stress in adult animals. Neurosci Biobehav Rev 32: 1121–1135. [DOI] [PubMed] [Google Scholar]
- Baran SE, Armstrong CE, Niren DC, Hanna JJ, Conrad CD. 2009. Chronic stress and sex differences on the recall of fear conditioning and extinction. Neurobiol Learn Mem 91: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett D, Shumake J, Jones D, Gonzalez-Lima F. 2003. Metabolic mapping of mouse brain activity after extinction of a conditioned emotional response. J Neurosci 23: 5740–5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham BC, Sheela Rani CS, Frazer A, Strong R, Morilak DA. 2013. Exogenous prenatal corticosterone exposure mimics the effects of prenatal stress on adult brain stress response systems and fear extinction behavior. Psychoneuroendocrinology 38: 2746–2757. [DOI] [PubMed] [Google Scholar]
- Bouton ME, Westbrook RF, Corcoran KA, Maren S. 2006. Contextual and temporal modulation of extinction: Behavioral and biological mechanisms. Biol Psychiatry 60: 352–360. [DOI] [PubMed] [Google Scholar]
- Bowers ME, Ressler KJ. 2015. An overview of translationally informed treatments for posttraumatic stress disorder: Animal models of Pavlovian fear conditioning to human clinical trials. Biol Psychiatry 78: E15–E27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Narayan M, Staib LH, Southwick SM, McGlashan T, Charney DS. 1999. Neural correlates of memories of childhood sexual abuse in women with and without posttraumatic stress disorder. Am J Psychiatry 156: 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ. 2007. Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron 53: 871–880. [DOI] [PubMed] [Google Scholar]
- Cattell RB. 1978. Use of factor analysis in behavioral and life sciences. Plenum, New York. [Google Scholar]
- Cho JH, Deisseroth K, Bolshakov VY. 2013. Synaptic encoding of fear extinction in mPFC-amygdala circuits. Neuron 80: 1491–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran KA, Maren S. 2001. Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J Neurosci 21: 1720–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran KA, Maren S. 2004. Factors regulating the effects of hippocampal inactivation on renewal of conditional fear after extinction. Learn Mem 11: 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran KA, Quirk GJ. 2007. Activity in prelimbic cortex is necessary for the expression of learned, but not innate, fears. J Neurosci 27: 840–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran KA, Desmond TJ, Frey KA, Maren S. 2005. Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J Neurosci 25: 8978–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Sakamoto H, Higashi S, Kawata M. 2008. Effects of single-prolonged stress on neurons and their afferent inputs in the amygdala. Neuroscience 152: 703–712. [DOI] [PubMed] [Google Scholar]
- Ding J, Han F, Shi Y. 2010. Single-prolonged stress induces apoptosis in the amygdala in a rat model of post-traumatic stress disorder. J Psychiatr Res 44: 48–55. [DOI] [PubMed] [Google Scholar]
- Do-Monte FH, Manzano-Nieves G, Quinones-Laracuente K, Ramos-Medina L, Quirk GJ. 2015. Revisiting the role of infralimbic cortex in fear extinction with optogenetics. J Neurosci 35: 3607–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle AL, Knox D, Roberts MM, Mulo K, Liberzon I, Galloway MP, Perrine SA. 2013. Single prolonged stress enhances hippocampal glucocorticoid receptor and phosphorylated protein kinase B levels. Neurosci Res 75: 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzinga BM, Bremner JD. 2002. Are the neural substrates of memory the final common pathway in posttraumatic stress disorder (PTSD)? J Affect Disord 70: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganon-Elazar E, Akirav I. 2012. Cannabinoids prevent the development of behavioral and endocrine alterations in a rat model of intense stress. Neuropsychopharmacology 37: 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganon-Elazar E, Akirav I. 2013. Cannabinoids and traumatic stress modulation of contextual fear extinction and GR expression in the amygdala-hippocampal-prefrontal circuit. Psychoneuroendocrinology 38: 1675–1687. [DOI] [PubMed] [Google Scholar]
- George SA, Knox D, Curtis A, Valentino R, Liberzon I. 2012. Altered locus coeruleus activity following single prolonged stress, a rodent model of PTSD. Eur J Neurosci 37: 901–909. [DOI] [PubMed] [Google Scholar]
- George SA, Stout SA, Tan M, Knox D, Liberzon I. 2013. Early handling attenuates enhancement of glucocorticoid receptors in the prefrontal cortex in an animal model of post-traumatic stress disorder. Biol Mood Anxiety Disord 3: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George SA, Rodriguez-Santiago M, Riley J, Rodriguez E, Liberzon I. 2015. The effect of chronic phenytoin administration on single prolonged stress induced extinction retention deficits and glucocorticoid upregulation in the rat medial prefrontal cortex. Psychopharmacology (Berl) 232: 47–56. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Kedves AT, Olausson P, Taylor JR. 2009. A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology 34: 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A. 2008. Switching on and off fear by distinct neuronal circuits. Nature 454: 600–606. [DOI] [PubMed] [Google Scholar]
- Hoffman AN, Lorson NG, Sanabria F, Foster Olive M, Conrad CD. 2014. Chronic stress disrupts fear extinction and enhances amygdala and hippocampal Fos expression in an animal model of post-traumatic stress disorder. Neurobiol Learn Mem 112: 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Fitzgerald PJ, MacPherson KP, DeBrouse L, Colacicco G, Flynn SM, Masneuf S, Pleil KE, Li C, Marcinkiewcz CA, et al. 2012. Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nat Neurosci 15: 1359–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo A, Wellman CL, Holmes A. 2006. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci 26: 5733–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SM, Schreiber WB, Staib JM, Knox D. 2015a. Sex differences in the single prolonged stress model. Behav Brain Res 286: 29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SM, Schreiber WB, Stanfield BR, Knox D. 2015b. Inhibiting corticosterone synthesis during fear memory formation exacerbates cued fear extinction memory deficits within the single prolonged stress model. Behav Brain Res 287: 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapska E, Maren S. 2009. Reciprocal patterns of c-Fos expression in the medial prefrontal cortex and amygdala after extinction and renewal of conditioned fear. Learn Mem 16: 486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox D, Perrine SA, George SA, Galloway MP, Liberzon I. 2010. Single prolonged stress decreases glutamate, glutamine, and creatine concentrations in the rat medial prefrontal cortex. Neurosci Lett 480: 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox D, Fitzpatrick CJ, George SA, Abelson JL, Liberzon I. 2012a. Unconditioned freezing is enhanced in an appetitive context: Implications for the contextual dependency of unconditioned fear. Neurobiol Learn Mem 97: 386–392. [DOI] [PubMed] [Google Scholar]
- Knox D, George SA, Fitzpatrick CJ, Rabinak CA, Maren S, Liberzon I. 2012b. Single prolonged stress disrupts retention of extinguished fear in rats. Learn Mem 19: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox D, Nault T, Henderson C, Liberzon I. 2012c. Glucocorticoid receptors and extinction retention deficits in the single prolonged stress model. Neuroscience 223: 163–173. [DOI] [PubMed] [Google Scholar]
- Kohda K, Harada K, Kato K, Hoshino A, Motohashi J, Yamaji T, Morinobu S, Matsuoka N, Kato N. 2007. Glucocorticoid receptor activation is involved in producing abnormal phenotypes of single-prolonged stress rats: A putative post-traumatic stress disorder model. Neuroscience 148: 22–33. [DOI] [PubMed] [Google Scholar]
- Lazenka MF, Selley DE, Sim-Selley LJ. 2014. DeltaFosB induction correlates inversely with CB(1) receptor desensitization in a brain region-dependent manner following repeated Delta(9)-THC administration. Neuropharmacology 77: 224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner B, Shors TJ. 2013. Stress, anxiety, and dendritic spines: What are the connections? Neuroscience 251: 108–119. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Krstov M, Young EA. 1997. Stress-restress: Effects on ACTH and fast feedback. Psychoneuroendocrinology 22: 443–453. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Abelson JL, Flagel SB, Raz J, Young EA. 1999a. Neuroendocrine and psychophysiologic responses in PTSD: A symptom provocation study. Neuropsychopharmacology 21: 40–50. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Lopez JF, Flagel SB, Vazquez DM, Young EA. 1999b. Differential regulation of hippocampal glucocorticoid receptors mRNA and fast feedback: Relevance to post-traumatic stress disorder. J Neuroendocrinol 11: 11–17. [DOI] [PubMed] [Google Scholar]
- Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Pare D. 2008. Amygdala intercalated neurons are required for expression of fear extinction. Nature 454: 642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RJ, Aghajanian GK. 2008. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: Role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci 105: 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Holmes A. 2015. Stress and fear extinction. Neuropsychopharmacology 41: 58–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Holt W. 2000. The hippocampus and contextual memory retrieval in Pavlovian conditioning. Behav Brain Res 110: 97–108. [DOI] [PubMed] [Google Scholar]
- Marks WN, Cavanagh ME, Greba Q, Cain SM, Snutch TP, Howland JG. 2016. The Genetic Absence Epilepsy Rats from Strasbourg model of absence epilepsy exhibits alterations in fear conditioning and latent inhibition consistent with psychiatric comorbidities in humans. Eur J Neurosci 43: 25–40. [DOI] [PubMed] [Google Scholar]
- McEwen BS. 2001. Plasticity of the hippocampus: Adaptation to chronic stress and allostatic load. Ann N Y Acad Sci 933: 265–277. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. 2002. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70–74. [DOI] [PubMed] [Google Scholar]
- Milad MR, Orr SP, Lasko NB, Chang Y, Rauch SL, Pitman RK. 2008. Presence and acquired origin of reduced recall for fear extinction in PTSD: Results of a twin study. J Psychiatr Res 42: 515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, Rauch SL. 2009. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry 66: 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RR, Laborda MA, Polack CW, Miguez G. 2015. Comparing the context specificity of extinction and latent inhibition. Learn Behav 43: 384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miracle AD, Brace MF, Huyck KD, Singler SA, Wellman CL. 2006. Chronic stress impairs recall of extinction of conditioned fear. Neurobiol Learn Mem 85: 213–218. [DOI] [PubMed] [Google Scholar]
- Mitra R, Jadhav S, McEwen BS, Vyas A, Chattarji S. 2005. Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc Natl Acad Sci 102: 9371–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B. 1993. Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: Comparison to hippocampus and basal ganglia. J Neurochem 60: 1650–1657. [DOI] [PubMed] [Google Scholar]
- Orsini CA, Kim JH, Knapska E, Maren S. 2011. Hippocampal and prefrontal projections to the basal amygdala mediate contextual regulation of fear after extinction. J Neurosci 31: 17269–17277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padival M, Quinette D, Rosenkranz JA. 2013. Effects of repeated stress on excitatory drive of basal amygdala neurons in vivo. Neuropsychopharmacology 38: 1748–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, Milad MR, Liberzon I. 2012. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci 13: 769–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M, Xia Z, Huang T, Smith CB. 2011. Effects of chronic immobilization stress on anxiety-like behavior and basolateral amygdala morphology in Fmr1 knockout mice. Neuroscience 194: 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, Pare D. 2003. Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci 23: 8800–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Pare D. 2002. Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience 115: 455–462. [DOI] [PubMed] [Google Scholar]
- Sagar SM, Sharp FR, Curran T. 1988. Expression of c-Fos protein in brain: Metabolic mapping at the cellular level. Science 240: 1328–1331. [DOI] [PubMed] [Google Scholar]
- Santini E, Ge H, Ren K, Pena de Ortiz S, Quirk GJ. 2004. Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci 24: 5704–5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segev A, Rubin AS, Abush H, Richter-Levin G, Akirav I. 2014. Cannabinoid receptor activation prevents the effects of chronic mild stress on emotional learning and LTP in a rat model of depression. Neuropsychopharmacology 39: 919–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Orr SP, Carson MA, Rauch SL, Macklin ML, Lasko NB, Peters PM, Metzger LJ, Dougherty DD, Cannistraro PA, et al. 2004. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry 61: 168–176. [DOI] [PubMed] [Google Scholar]
- Shin LM, Rauch SL, Pitman RK. 2006. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci 1071: 67–79. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D Jr, Corcoran KA, Lebron-Milad K, Quirk GJ. 2006. Inactivation of the ventromedial prefrontal cortex reduces expression of conditioned fear and impairs subsequent recall of extinction. Eur J Neurosci 24: 1751–1758. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. 2011. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 36: 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Sierra-Mercado D, Pardilla-Delgado E, Quirk GJ. 2012. Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron 76: 804–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwick SM, Krystal JH, Morgan CA, Johnson D, Nagy LM, Nicolaou A, Heninger GR, Charney DS. 1993. Abnormal noradrenergic function in posttraumatic stress disorder. Arch Gen Psychiatry 50: 266–274. [DOI] [PubMed] [Google Scholar]
- Staiger JF, Masanneck C, Bisler S, Schleicher A, Zuschratter W, Zilles K. 2002. Excitatory and inhibitory neurons express c-Fos in barrel-related columns after exploration of a novel environment. Neuroscience 109: 687–699. [DOI] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, Quirk GJ. 2006. Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem 13: 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Han F, Shi Y, Li X. 2016. Role of the endoplasmic reticulum pathway in the medial prefrontal cortex in post-traumatic stress disorder model rats. J Mol Neurosci 59: 471–482. [DOI] [PubMed] [Google Scholar]
- Westbrook RF, Jones ML, Bailey GK, Harris JA. 2000. Contextual control over conditioned responding in a latent inhibition paradigm. J Exp Psychol Anim Behav Process 26: 157–173. [DOI] [PubMed] [Google Scholar]
- Wilber AA, Walker AG, Southwood CJ, Farrell MR, Lin GL, Rebec GV, Wellman CL. 2011. Chronic stress alters neural activity in medial prefrontal cortex during retrieval of extinction. Neuroscience 174: 115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Yu B, Wang HT, Han F, Shi YX. 2011. Single-prolonged stress induces apoptosis by activating cytochrome C/caspase-9 pathway in a rat model of post-traumatic stress disorder. Cell Mol Neurobiol 31: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Morinobu S, Fuchikami M, Kurata A, Kozuru T, Yamawaki S. 2008. Effects of single prolonged stress and D-cycloserine on contextual fear extinction and hippocampal NMDA receptor expression in a rat model of PTSD. Neuropsychopharmacology 33: 2108–2116. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Morinobu S, Takei S, Fuchikami M, Matsuki A, Yamawaki S, Liberzon I. 2009. Single prolonged stress: Toward an animal model of posttraumatic stress disorder. Depress Anxiety 26: 1110–1117. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Teicher MH, Trestman RL, Levengood RA, Siever LJ. 1996. Cortisol regulation in posttraumatic stress disorder and major depression: A chronobiological analysis. Biol Psychiatry 40: 79–88. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Yang RK, Buchsbaum MS, Golier JA. 2006. Alterations in cortisol negative feedback inhibition as examined using the ACTH response to cortisol administration in PTSD. Psychoneuroendocrinology 31: 447–451. [DOI] [PubMed] [Google Scholar]
- Zhao D, Han F, Shi Y. 2014. Effect of glucose-regulated protein 94 and endoplasmic reticulum modulator caspase-12 in medial prefrontal cortex in a rat model of posttraumatic stress disorder. J Mol Neurosci 54: 147–155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.