

In this study, Saccilotto et al. investigated the molecular connection between proangiogenic signals and downstream gene expression during angiogenesis. By characterizing a Dll4 enhancer directing expression to endothelial cells at the angiogenic front, the authors identified MEF2 transcription factors as crucial regulators of sprouting angiogenesis directly downstream from VEGFA.
Keywords: sprouting angiogenesis, MEF2 transcription factors, HLX regulation, Dll4 regulation
Abstract
Angiogenesis, the fundamental process by which new blood vessels form from existing ones, depends on precise spatial and temporal gene expression within specific compartments of the endothelium. However, the molecular links between proangiogenic signals and downstream gene expression remain unclear. During sprouting angiogenesis, the specification of endothelial cells into the tip cells that lead new blood vessel sprouts is coordinated by vascular endothelial growth factor A (VEGFA) and Delta-like ligand 4 (Dll4)/Notch signaling and requires high levels of Notch ligand DLL4. Here, we identify MEF2 transcription factors as crucial regulators of sprouting angiogenesis directly downstream from VEGFA. Through the characterization of a Dll4 enhancer directing expression to endothelial cells at the angiogenic front, we found that MEF2 factors directly transcriptionally activate the expression of Dll4 and many other key genes up-regulated during sprouting angiogenesis in both physiological and tumor vascularization. Unlike ETS-mediated regulation, MEF2-binding motifs are not ubiquitous to all endothelial gene enhancers and promoters but are instead overrepresented around genes associated with sprouting angiogenesis. MEF2 target gene activation is directly linked to VEGFA-induced release of repressive histone deacetylases and concurrent recruitment of the histone acetyltransferase EP300 to MEF2 target gene regulatory elements, thus establishing MEF2 factors as the transcriptional effectors of VEGFA signaling during angiogenesis.
Sprouting angiogenesis, in which new blood vessels form from existing ones, is driven in response to insufficient supplies of nutrients and oxygen. This response involves an exquisitely regulated pattern of gene expression directly downstream from vascular endothelial growth factor A (VEGFA) stimulation (Hellström et al. 2007; Benedito et al. 2009). As a consequence of these stimuli, a subset of endothelial cells known as tip cells forms polarized filopodia protrusions and assumes the lead positions at the tip of each blood vessel sprout (Gerhardt et al. 2003). Neighboring angiogenic endothelial cells with greater proliferative activity, known as stalk cells, contribute to elongation, stability, and lumenization of the new sprout, while other endothelial cells maintain a quiescent state, thus ensuring the stability of the existing vasculature (Gerhardt et al. 2003).
While many factors can elicit an angiogenic response, and numerous pathways influence sprouting angiogenesis, the Notch signaling pathway is crucially required to coordinate endothelial cell behavior during vessel patterning downstream from VEGFA signaling. In particular, the specification of endothelial cells into tip and stalk cells is regulated by Delta-like ligand 4 (Dll4)/Notch signaling (Hellström et al. 2007). Higher levels of the Notch ligand DLL4 in tip cells results in increased Notch signaling in the neighboring stalk cell, which in turn actively suppresses the tip cell phenotype (Hellström et al. 2007; Jakobsson et al. 2010).
The mRNA expression patterns of many vascular genes, including Dll4, are precisely controlled at the angiogenic front (Claxton and Fruttiger 2004; del Toro et al. 2010; Strasser et al. 2010), supporting a role for transcriptional regulation in this process. However, the transcriptional networks that elicit this specific response to VEGFA gradients are very poorly understood. While a number of transcription factors influence angiogenesis (De Val and Black 2009), gene ablation studies often result in similar vascular phenotypes, making it challenging to ascribe specific roles to different transcription factors. Some implicated transcription factors, such as members of the ETS factor family, including ERG (Randi et al. 2009; Birdsey et al. 2015), play a role in the regulation of most endothelial-expressed genes (De Val and Black 2009), including those not expressed at the angiogenic front (e.g., Tie2), suggesting that they are unable to provide the required specificity individually. Furthermore, the lack of any characterized cis-regulatory elements (enhancers) that directly regulate differential expression at the angiogenic front in vivo has presented a major challenge when attempting to identify direct gene targets of transcriptional and signaling angiogenic networks. Many studies have focused instead on putative regulatory sequences identified by proximity to the core promoter. For example, multiple studies have focused on the Dll4 promoter region (Hayashi and Kume 2008; Roukens et al. 2010; Corada et al. 2013; Lizama et al. 2015), although recent transgenic mouse and zebrafish analyses have demonstrated that this region is neither sufficient nor required for Dll4 expression in vivo (Sacilotto et al. 2013; Wythe et al. 2013). A large body of work has now clearly determined that complex patterns of gene expression require multiple interactions between promoter and enhancer elements, the latter usually located away from the core promoter (for example, Kieffer-Kwon et al. 2013), stressing the need for a greater focus on distal enhancer regions when studying transcriptional pathways. Here we describe the first enhancer capable of directing precise, differential gene activity during angiogenic sprouting. By investigating the regulation of this enhancer, we uncovered an unexpected and essential role for MEF2 transcription factors in the regulation of Dll4 gene expression in tip cells and in the activation of gene expression during sprouting angiogenesis more generally in both physiological and pathological vascular growth.
Results
The Dll4 enhancer Dll4in3 directs expression to endothelial cells during sprouting angiogenesis
Previously, two arterial enhancers for the Notch ligand Dll4 have been described: one located within the third intron (referred to here as the Dll4in3 enhancer) (Sacilotto et al. 2013; Wythe et al. 2013) and the other located 12 kb upstream of the transcriptional start site (Dll4-12 enhancer) (Sacilotto et al. 2013). Similar to endogenous Dll4, both enhancers were active in arterial but not venous endothelial cells, an expression pattern precisely regulated by ETS, RBPJ, and SOXF transcription factors (Sacilotto et al. 2013; Wythe et al. 2013). However, endogenous Dll4 is also expressed in tip cells leading the formation of new vessel sprouts. Consequently, we investigated whether these Dll4 enhancers were able to direct reporter gene expression during sprouting angiogenesis. We detected Dll4in3-driven LacZ reporter gene activity in angiogenic vessels in a pattern closely mimicking that of endogenous Dll4, including expression in endothelial cells undergoing sprouting angiogenesis within the hindbrain at embryonic day 11 (E11) and in the postnatal retina (Fig. 1 A; Supplemental Fig. 1A–C). The reporter gene expression at the angiogenic front in the retina was specific and persisted throughout sprouting angiogenesis (Supplemental Fig. 1C).
Figure 1.
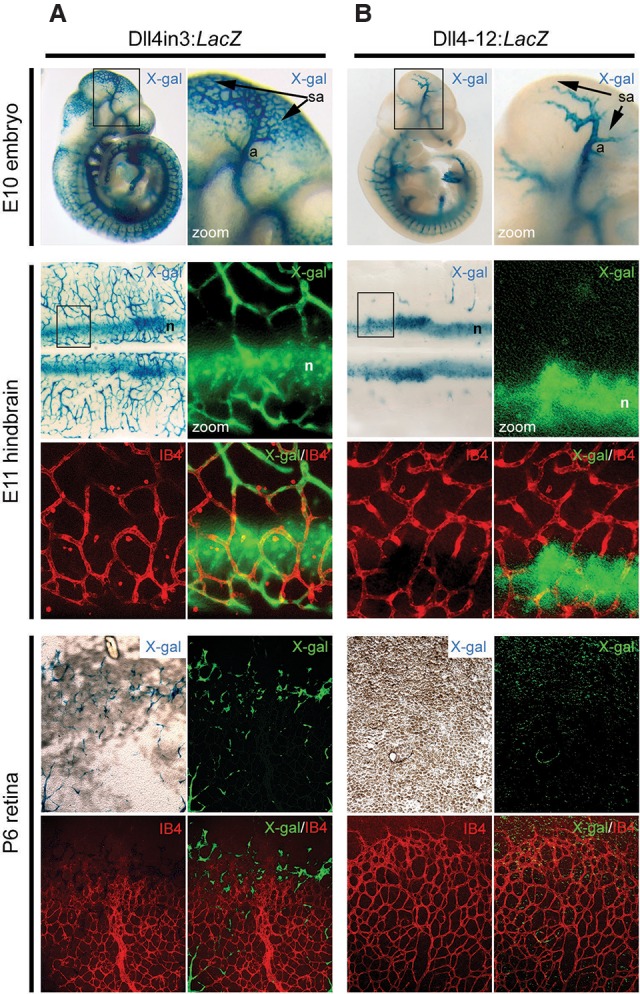
The Dll4in3 enhancer directs gene expression to endothelial cells during sprouting angiogenesis. (A) Representative images from Dll4in3:LacZ transgenic mice demonstrate enhancer activity in endothelial cells undergoing sprouting angiogenesis in the E10 embryo, E11 hindbrain, and postnatal day 6 (P6) retina. (B) Representative images from Dll4-12:LacZ transgenic mice demonstrate enhancer activity in arterial and neural tissues but no activity in endothelial cells during sprouting angiogenesis in E10 embryos, E11 hindbrains, or postnatal retinas. Enhancer activity was detected as X-gal activity (blue staining or green pseudocolor), and endothelial cells were detected by isolectin B4 (IB4) whole-mount immunostaining (red). (a) Artery; (sa) region of sprouting angiogenesis; (n) neuronal staining. See also Supplemental Figure 1.
Dll4in3 and Dll4-12 enhancers both contain the ETS-, RBPJ-, and SOXF-binding motifs essential for arterial expression (Sacilotto et al. 2013). Consequently, we investigated whether the Dll4-12 enhancer could also drive expression in the angiogenic sprout. However, although Dll4-12:LacZ activity appeared equally robust in arterial endothelial cells at both embryonic and postnatal stages, no transgene activity could be detected in endothelial cells undergoing active angiogenesis (Fig. 1B; Supplemental Fig. 1D,E). E11 hindbrains from multiple independent transgenic founders were examined to exclude the possibility of influences downstream from transgene insertion (Supplemental Fig. 1D). These results demonstrate that the Dll4in3 enhancer contains DNA sequences that convey unique transcriptional information and suggest that gene expression in sprouting angiogenesis is regulated by factors other than, or in addition to, ETS, RBPJ, and SOXF.
MEF2 transcription factors regulate Dll4 enhancer expression during sprouting angiogenesis in both physiological and pathological vessel growth
A comparison of the Dll4in3 and Dll4-12 enhancer sequences demonstrated that the angiogenic Dll4in3 enhancer contained consensus MEF2- and Forkhead C (FOXC)-binding motifs that were not present in the Dll4-12 sequence (Fig. 2A; Supplemental Fig. 2A,B). The Dll4in3 MEF2-binding motif bound MEF2A, MEF2C, and MEF2D proteins in electophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP) analysis (Supplemental Fig. 2C–E; Sacilotto et al. 2013; Wythe et al. 2013), whereas the putative FOX motif did not bind FOXC proteins in EMSA and had no detectable effect on Dll4in3:LacZ expression at E11 (Sacilotto et al. 2013). We therefore examined the role of the Dll4in3 MEF2-binding motif in enhancer activity in transgenic mice. Although mutation of the MEF2-binding motif present in Dll4in3 (Fig. 2B) had no effect on reporter gene expression in arterial endothelial cells (Fig. 2C; Sacilotto et al. 2013; Wythe et al. 2013), loss of MEF2 binding led to an almost total loss of reporter gene expression in embryonic hindbrains and postnatal retinas during sprouting angiogenesis (Fig. 2C; Supplemental Fig. 2F,G). These results therefore indicate that the MEF2-binding motif within the Dll4in3 enhancer is essential for directing gene expression in endothelial cells undergoing active sprouting angiogenesis.
Figure 2.
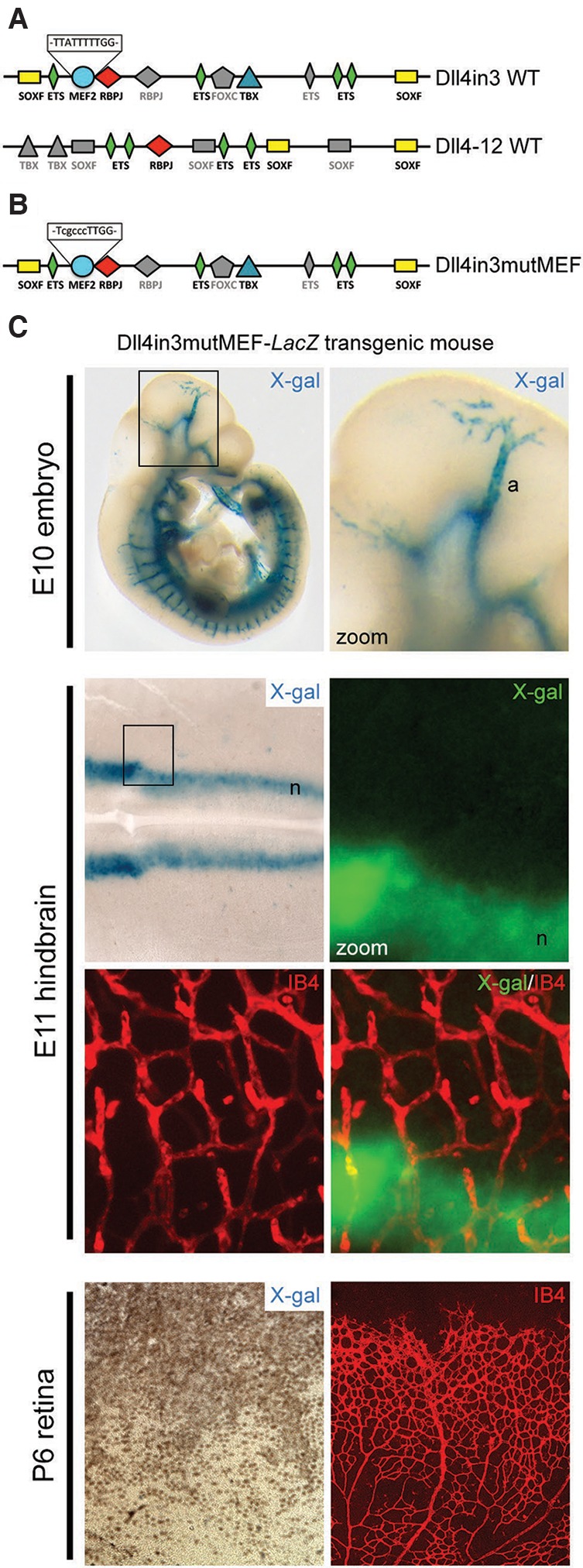
The Dll4in3 enhancer contains a MEF2-binding motif that is required for expression during sprouting angiogenesis. (A) Schematic representation of the binding motifs found within the mouse Dll4in3 and Dll4-12 enhancer sequences. Colored shapes represent sequences directly verified by EMSA analysis (Supplemental Fig. 2; Sacilotto et al. 2013), and gray shapes indicate consensus or near-consensus sequences that did not bind the cognate transcription factors in EMSA. (B) Schematic representation of the Dll4in3mutMEF enhancer sequence in which the MEF2-binding motif within the Dll4in3 enhancer has been mutated. (C) Representative images from Dll4in3mutMEF transgenic mice demonstrate that this enhancer does not direct endothelial expression during sprouting angiogenesis in E10 embryos, E11 hindbrains, or P6 retinas, although robust expression is detected in the arterial (a) and neuronal (n) compartments. See also Supplemental Figure 2.
Sprouting angiogenesis also occurs in the adult in response to physiological cues that support cycles of remodeling and repair and in pathological conditions, including cancer, where establishment of a vascular system is essential for tumour growth and metastasis (Kerbel 2008). Therefore, we examined the expression of the Dll4in3 enhancer during neovascular growth in adult mice. Similar to developmental angiogenesis, the Dll4in3:LacZ enhancer was able to direct reporter gene expression to new vessels during Matrigel plug and B16F10 melanoma-stimulated neovascularization (Fig. 3A; Supplemental Fig. 3A,B). As with the postnatal day 6 (P6) retina, expression was not detected in every endothelial cell (Supplemental Fig. 3A); instead, the pattern closely resembled that reported in Unc5bLacZ/+ mice, in which LacZ expression correlated with sprouting angiogenesis (Larrivee et al. 2007). Strikingly, mutation of the MEF2-binding motif within the Dll4in3 enhancer resulted in a total loss of reporter gene expression during neoangiogenesis (Fig. 3B; Supplemental Fig. 3C,D) despite the maintenance of transgene expression in the vasculature of some adult organs (Supplemental Fig. 3E). These results clearly demonstrate the importance of the MEF2-binding motif for Dll4 enhancer expression during both physiological and pathological angiogenesis.
Figure 3.

The Dll4in3 enhancer requires MEF2 binding to direct expression during adult neovascularization. (A) Representative images of Dll4in3:LacZ transgene expression during neovascularization into Matrigel plugs and B16F10 melanoma tumors demonstrate robust vascular X-gal staining. (B) Representative images of the mutant Dll4in3mutMEF:LacZ transgene demonstrate that the mutated enhancer was unable to direct any detectable reporter gene expression during adult neovascularization. See also Supplemental Figure 3.
MEF2 factors regulate endogenous DLL4 expression
We demonstrated previously that RBPJ and SOXF factors, both of which are essential for Dll4in3 enhancer activity in arterial endothelium, were also required for expression of the endogenous Dll4 gene and the acquisition of arterial fate more generally (Sacilotto et al. 2013). We therefore investigated whether MEF2 transcription factors regulate endogenous Dll4 during sprouting angiogenesis. Although MEF2 transcription factors have been implicated in vascular integrity and endothelial cell survival (Hayashi et al. 2004; Olson 2004; Chang et al. 2006), endothelial-specific ablation of MEF2C resulted in no detected embryonic defects (Xu et al. 2012) and did not alter Dll4in3 or endogenous Dll4 expression in early developing arteries (Wythe et al. 2013). However, Mef2Cfl/null;Tie2Cre/+ mice did exhibit increased vascular sprouting after oxygen-induced retinopathy, resembling Dll4LacZ/+ mice after similar insult (Lobov et al. 2007; Xu et al. 2012). All MEF2 factors bind a similar DNA motif and can be functionally redundant (Potthoff and Olson 2007; Liu et al. 2014). Since MEF2A, MEF2C, and MEF2D were each able to directly bind the Dll4in3 MEF2 motif (Supplemental Fig. 2D,E), we hypothesized that multiple MEF2 factors may regulate Dll4 expression. Supporting this, we detected expression of MEF2A, MEF2C, and MEF2D in endothelial cells from multiple tissues and with expression dynamics correlating to that of Dll4 after VEGFA (Fig. 4A,B; Supplemental Fig. 4A–D). Combined siRNA-mediated knockdown of MEF2A,C and MEF2D in human umbilical vein endothelial cells (HUVECs) resulted in significantly reduced Dll4 expression after VEGFA stimulation (Fig. 4C,D; Supplemental Fig. 4E,F). Furthermore, these MEF2 knockdown HUVEC cells demonstrated a reduction of tip cell competitiveness compared with control cells in VEGFA-induced mosaic spheroid sprouting assays (Supplemental Fig. 5A,B). The same phenotype was observed in VEGFA-induced sprouting of embryoid bodies after CRISPR/Cas9-mediated ablation of Mef2A and Mef2C in embryonic stem (ES) cells (Supplemental Fig. 5C–E). This reduced competitiveness was similar to previous reports demonstrating that Dll4LacZ/+ heterozygous ES cells with reduced DLL4 levels cannot compete with wild-type cells for the tip cell position during sprouting angiogenesis (Jakobsson et al. 2010).
Figure 4.

MEF2 factors regulate endogenous Dll4 expression and tip cell identity. (A) Whole-mount immunostaining demonstrates expression of MEF2A, MEF2C, and MEF2D transcription factors in endothelial cells from E11 hindbrains and in HUVECs. Bars, 100 μm. (B) Western blot analysis of MEF2A, MEF2C, MEF2D, and DLL4 expression in HUVECs 1 min to 8 h after VEGFA stimulation. Representative of four repeats. (C) RNAi-mediated knockdown of MEF2A, MEF2C, and MEF2D factors by pooled siRNAs in HUVECs results in loss of DLL4 expression, while VEGFR2 expression remains unaffected. (NS) Nontargeting control siRNA. Representative of two repeats. (D) Relative gene expression levels of DLL4, MEF2A, MEF2C, and MEF2D analyzed by quantitative RT–PCR (qRT–PCR) after RNAi-mediated knockdown and 1 h of VEGFA stimulation. Statistical analysis was performed on four biological replicates. Error bars indicate standard deviation. (E) Induced endothelial cell-specific (iEC) Mef2A/C double knockouts (Mef2A/C KO) show a significant reduction in DLL4 expression, as detected by Western blot of E12 hindbrain extracts. Cre recombination was induced by tamoxifen injection in pregnant mice at E9. (Ctrl) Cre-negative; (1–4) four different iEC Mef2A/C knockout littermates. Note the variability in DLL4 reduction among littermates, as expected from Cre recombination in response to tamoxifen. Comparative band intensity was normalized to ACTB levels and made relative to wild type. (F) Endogenous DLL4 expression (green) in representative control (Cre-negative) and iEC Mef2A/C knockout P5 retinas at the angiogenic front. All endothelial cells were detected by IB4 whole-mount immunostaining (red). (G) Relative gene expression levels of DLL4, MEF2A, MEFC, and MEF2D analyzed by qRT–PCR in five different P5 lungs taken from control (Cre-negative; CT) and knockout (iEC Mef2A/C KO) pups. Three technical replicates were used for each pup. Error bars indicate standard deviation.
To investigate whether depletion of MEF2 factors had a repressive effect on Dll4 expression in vivo, we next investigated the expression levels of DLL4 in mice after endothelial-specific induced deletion of Mef2A and Mef2C [Cdh5(PAC)Cre-ERT2;Mef2Afl/fl;Mef2Cfl/fl, denoted as induced endothelial cell-specific (iEC) Mef2A/C knockout]. Although Mef2D was not targeted in these mice, DLL4 levels were reduced in the highly angiogenic E11 hindbrains 3 d after induction of gene deletion (Fig. 4E), and substantially less DLL4 was detected at the angiogenic front in P5 retinas 4 d after the induction of gene deletion (Fig. 4F). Analysis of whole-lung extracts confirmed the significant decrease in Dll4 transcript levels concurrent with reduction in Mef2A and Mef2C despite the inclusion of nonvascular cells in this analysis (Fig. 4G). These results therefore support a key role for MEF2 factors in the regulation of endogenous Dll4 during sprouting angiogenesis and consequently on the differentiation of tip cells during this process.
MEF2 factors directly regulate many genes during sprouting angiogenesis
Although Dll4 expression was clearly reduced after induced endothelial deletion of Mef2A/C, it was notable that iEC Mef2A/C knockout P5 retinas did not exhibit the increased levels of sprouting angiogenesis and vessel branching reported as a consequence of Notch inhibition or Dll4 reduction alone (Hellström et al. 2007). Instead, iEC Mef2A/C knockout retinas displayed a vascular plexus with a significant reduction of vascular density and coverage accompanied by a reduced number of tip cells at the angiogenic front (Fig. 5). These results indicate that the loss of MEF2A/C results in an overall reduction of angiogenic sprouting and point to a potential role for MEF2 in regulating vascular growth more generally. To establish whether MEF2 factors directly regulate other angiogenesis-related genes and consequently the angiogenic process more generally, we analyzed genome-wide MEF2C-binding peaks associated with enhancer-specific histone marks in HUVECs from publicly available ChIP-seq (ChIP combined with high-throughput sequencing) data (Maejima et al. 2014). Supporting our earlier analysis, the Dll4in3 enhancer region contained a robust MEF2-binding peak, whereas no similar peak was found around the Dll4-12 nonangiogenic enhancer or elsewhere in the Dll4 locus (Supplemental Fig. 6A). Pan-genomic analysis of MEF2C-binding locations revealed a significant enrichment of enhancer-associated MEF2C binding near genes known to be up-regulated during sprouting angiogenesis (Fig. 6A,B; Supplemental Fig. 6B; Supplemental Tables 1, 2; del Toro et al. 2010; Strasser et al. 2010), suggesting a role for MEF2 factors in angiogenic gene activation beyond Dll4 and providing a potential explanation for the decreased vascular growth seen in the iEC Mef2A/C knockout retinas. No enhancer-associated MEF2-binding peaks were found within 200 kb of any other Notch pathway gene, including JAG1, JAG2, HES1, HEY1, and HEY2 (Supplemental Fig. 6C). Loci containing significant MEF2-binding peaks included the transcription factors ETS1, ELK3, and HLX, all of which are also implicated in the regulation of angiogenic behavior (Fig. 6C; De Val and Black 2009; Herbert et al. 2012). The human DNA sequences around these sites were tested in mosaic transgenic zebrafish at 26–30 h post-fertilization (hpf), confirming that they were bona fide angiogenic enhancers (Fig. 6D).
Figure 5.
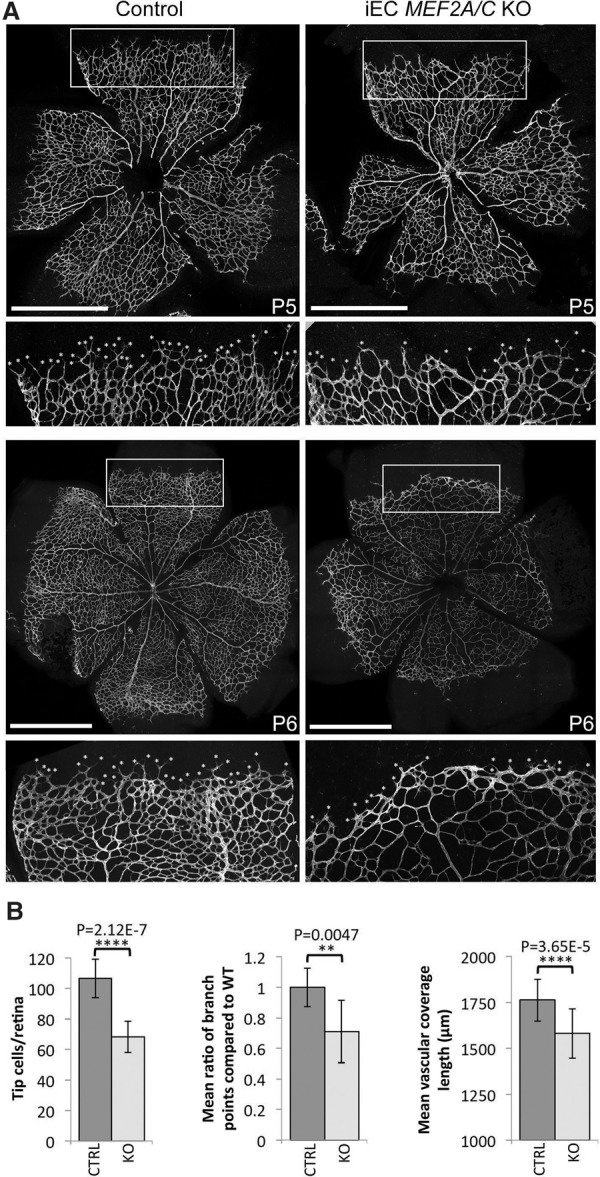
Induced endothelial deletion of Mef2A and Mef2C results in reduced retinal angiogenesis. (A) Representative P5 and P6 retinas taken from control (Cre-negative) and iEC Mef2A/C knockout (KO) pups and stained for IB4. Tip cells were detected by filopodia and are indicated by asterisks. The white box indicates region taken for zoom image. Bars, 1 mm. (B) Graphs demonstrating the mean number of tip cells, the ratio of the mean number of branch points, and the mean outgrowth length as measured by actual distance covered by the growing vessel from the center of the retina. Statistical analysis was performed on seven wild-type and nine knockout pups pooled from two different litters.
Figure 6.
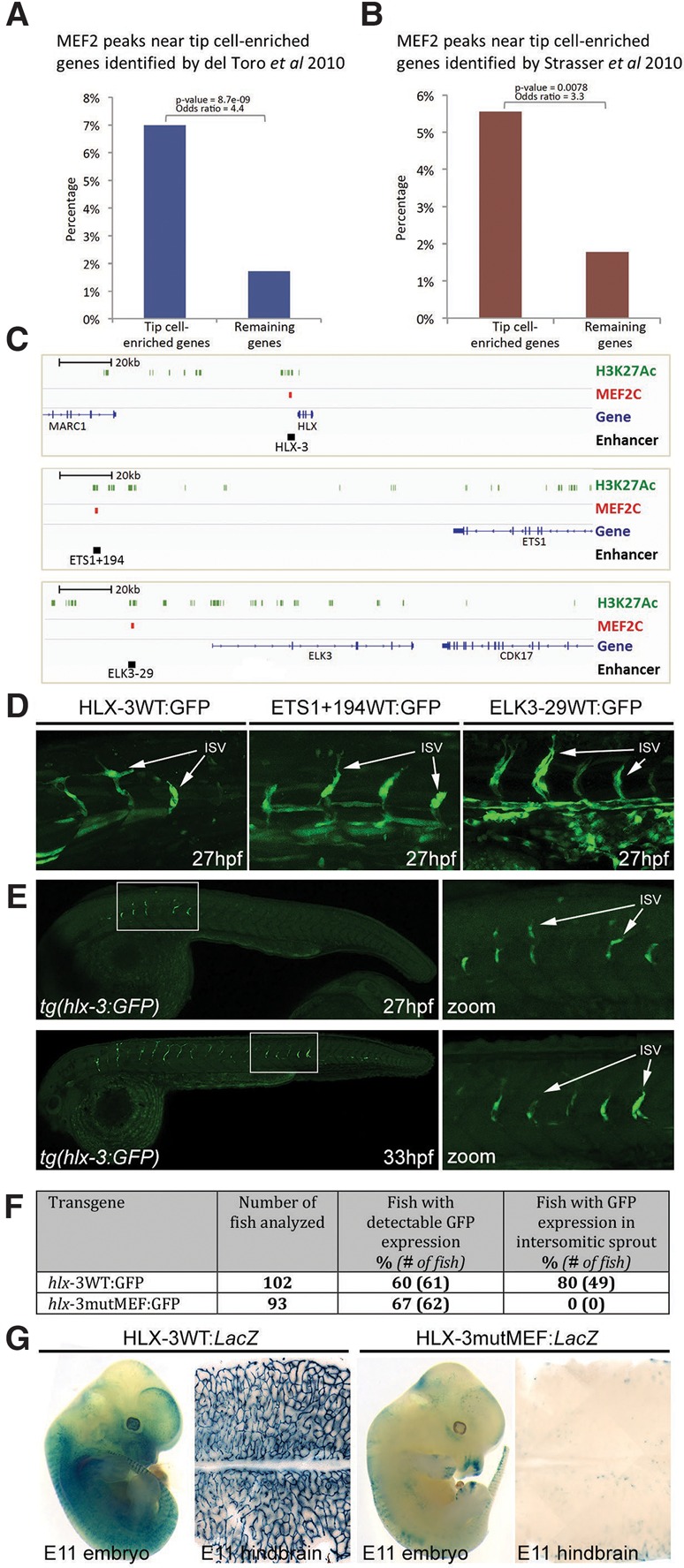
MEF2 binding is a shared feature of many angiogenesis enhancers. (A,B) MEF2C-binding peaks are enriched around 200 kb of genes associated with sprouting angiogenesis, as assessed by increased expression in the hypersprouting retina of Dll4+/− mice (P = 8.682 × 10−09) (del Toro et al. 2010) (A) or identified through laser capture microdissection of retinal tip cells (P = 0.007776) (Strasser et al. 2010) (B). (C) Genomic snapshots denoting MEF2C-binding sites within the loci for HLX, ETS1, and ELK3 transcription factors. H3K27ac peaks are indicated in green, MEF2C-binding peaks are indicated in red, and novel angiogenic enhancers are indicated in black. (D) tol2-mediated mosaic zebrafish transgenic for the human MEF2C-binding peak enhancers HLX-3, ETS1+194, and ELK3-29 demonstrate enhancer-driven GFP expression in sprouting endothelial cells (green). (ISV) Intersegmental vessels. (E) The stable transgenic zebrafish line tg(hlx-3:GFP) directs expression specifically to sprouting endothelial cells in the intersegmental vessels. (F) Summary of reporter gene expression detected in 32-hpf tol2-mediated mosaic transient transgenic zebrafish embryos. (G) Whole-mount embryos and hindbrains of representative E11 X-gal-stained transient transgenic embryos expressing HLX-3 wild-type:LacZ and HLX-3mutMEF:LacZ transgenes. See also Supplemental Figures 6–8.
The MEF2C-bound enhancer element 3 kb upstream of the HLX gene (HLX-3 enhancer) (Supplemental Fig. 7A) was particularly notable, as HLX/hlx expression is unusually specific to sprouting angiogenesis (Herbert et al. 2012; Prahst et al. 2014). Furthermore, HLX expression is also known to be downstream from VEGFA stimulation through EP300 binding at the same location where MEF2C was bound (Supplemental Fig. 7A; Zhang et al. 2013). We confirmed the sprout-specific expression pattern of this enhancer in a stable zebrafish transgenic line, tg(hlx-3:GFP) (Fig. 5E). This enhancer contained three MEF2 sites (Supplemental Fig. 7B), the strongest of which was able to bind MEF2A, MEF2C, and MEF2D in EMSA analysis at levels comparable with the Dll4in3 MEF2 motif (Supplemental Fig. 8). Mutation of these MEF2 sites resulted in total ablation of transgene expression in both transgenic zebrafish and mice (Fig. 6F,G; Supplemental Fig. 7C,D). Together, these results suggest that MEF2 factors play a crucial role in the transcriptional activation of multiple genes associated with sprouting angiogenesis.
MEF2 regulates tip cell genes downstream from VEGFA-mediated histone deacetylase (HDAC) derepression
Our data support a role for MEF2 factors in the activation of sprouting angiogenesis downstream from VEGFA signaling in endothelial cells. We therefore next investigated the mechanisms by which MEF2 factors are able to specifically activate gene transcription in sprouting, but not quiescent, endothelial cells. Although the transcriptional regulators of Mef2a and Mef2d in endothelial cells remain unknown, Mef2c transcription is known to be directly activated by ETS factors, including ETS1 (De Val et al. 2004). Therefore, MEF2 factors may lie at the top of a feed-forward loop during sprouting angiogenesis, activating the expression of Ets factors, which would then reinforce Mef2 expression and also collaborate with MEF2 factors in the activation of downstream sprouting angiogenic genes, including Dll4. This model is supported by our observations that mRNA and protein levels of MEF2A and MEF2C notably increase after VEGFA stimulation (Fig. 4B; Supplemental Fig. 4C).
However, this feed-forward loop is unlikely to be the only mechanism: The Mef2c enhancer itself is not specific to sprouting angiogenesis (De Val et al. 2004), ETS factors are essential for all endothelial gene expression (De Val and Black 2009; Randi et al. 2009), and MEF2 proteins can be detected in most endothelial cells (Fig. 4A; Supplemental Fig. 4A). Furthermore, ChIP analysis demonstrates that MEF2 factor binding to the DLL4in3 and HLX-3 enhancers occurs independently of VEGFA stimulation (Supplemental Fig. 9A,B), although both HLX expression (Testori et al. 2011) and DLL4 expression (Fig. 4B) are significantly up-regulated only after VEGFA stimulation. Therefore, we hypothesized that the ability of MEF2 factors to associate with transcriptional coactivators may be modified by VEGFA stimulation. The histone acetyltransferase EP300 has been shown to play a key role in transcriptional activation downstream from VEGFA in endothelial cells, where it is tightly associated with the enhancer-associated histone modification H3K27ac (Zhang et al. 2013). We therefore tested the dynamics of EP300 recruitment to the Dll4in3 enhancer and found a significant increase in binding after VEGFA stimulation (Fig. 7A). This result suggests that the ability of MEF2 factors to activate gene expression was affected directly by VEGFA-induced recruitment of EP300.
Figure 7.

VEGFA signaling leads to activation of MEF2 transcriptional activity. (A) Increased EP300 binding at the DLL4in3 enhancer after VEGFA stimulation in HUVECs analyzed by ChIP. Graph is representative of four biological replicates. (B) Decreased class II HDAC binding at the DLL4in3 enhancer after VEGFA stimulation in HUVECs analyzed by ChIP. The graph is representative of two biological replicates. (C) Relative DLL4 gene expression after VEGFA stimulation with and without trichostatin A (TSA), analyzed by qRT–PCR. Statistical analysis on four biological replicates. Error bars indicate standard deviation. (D) Representative Dll4in3:LacZ E13 hindbrains removed 24 h after in utero intracerebral injection of 10 μM HDAC inhibitor TSA or DMSO control. X-gal staining after DMSO control injection is weak, whereas more robust transgene expression was detected in hindbrains injected with TSA. (E) Relative gene expression levels of DLL4 and MMP10 after VEGFA stimulation with and without treatment with small molecule class II HDAC inhibitors BML-210 and MC-1568, analyzed by qRT–PCR. Statistical analysis was performed on three biological replicates. Error bars indicate standard deviation. (F) Proposed model of VEGFA-mediated activation of sprouting angiogenesis via the MEF2 transcription factor family. See also Supplemental Figure 9.
The activity of MEF2 factors in other cell types is known to be modulated by the mutually exclusive recruitment of EP300 and class II HDACs (Lu et al. 2000; Youn et al. 2000). Class II HDACs inhibit MEF2 factors by directly binding to them and consequently preventing the ability of MEF2 to recruit cofactors required for gene activation (Lu et al. 2000; Chan et al. 2003). Since VEGFA stimulation is known to directly induce class IIa HDAC phosphorylation and cytoplasmic accumulation (Ha et al. 2008a,b), we hypothesized that this may be the mechanism through which VEGFA signaling at the angiogenic front directly activates MEF2. ChIP analysis confirmed a significant decrease in binding of the class IIa HDACs HDAC4, HDAC7, and HDAC9 to the DLL4in3 enhancer after VEGFA stimulation (Fig. 7B). Furthermore, general inhibition of HDAC activity by trichostatin A (TSA) resulted in a significant increase in DLL4 transcription in HUVECs (Fig. 7C) and Dll4in3 enhancer activity both in vivo (Fig. 7D) and ex vivo (Supplemental Fig. 9C). To confirm that the repressive activity of HDACs on DLL4 expression occurred directly via MEF2 interaction, we tested the effects of two different small molecule inhibitors of class IIa HDACs. BML-210 works by specifically targeting and disrupting the MEF2:HDAC complex (Jayathilaka et al. 2012), whereas MC-1568 instead selectively stabilizes this complex (Nebbioso et al. 2009). Supporting the hypothesis that MEF2:HDAC binding directly inhibits DLL4 expression in nonangiogenic cells, treatment with BML-210 resulted in a significant increase in DLL4 transcription that is mirrored by that of MMP10, a known MEF2 target regulated by MEF2:HDAC interaction (Fig. 7E; Chang et al. 2006; Ha et al. 2008a). Conversely, treatment with MC-1568 did not increase DLL4 and MMP10 expression (Fig. 7E). Other genes known to be up-regulated or down-regulated by TSA in the presence of VEGF (Rafehi et al. 2014) but without a MEF2C-binding peak within 200 kb were not significantly affected by these compounds (Supplemental Fig. 9D).
In conclusion, these results support a model (Fig. 7F) in which VEGFA-triggered release of repressive HDACs and concurrent recruitment of the activating histone acetyltransferase EP300 to MEF2-bound enhancers results in MEF2-activated transcription of immediate downstream angiogenic target genes, including multiple ETS factors. This further reinforces the expression of Mef2 genes though a feed-forward loop, enhances the angiogenic transcriptional cascade, and drives the high levels of Dll4 expression required to form a leading tip endothelial cell. This activation of Notch signaling then triggers the lateral inhibition and guided vascular patterning essential for sprouting angiogenesis.
Discussion
It is well established that endothelial sprouting is controlled by VEGF signaling (Gerhardt et al. 2003). However, VEGF receptors use multiple signal transduction pathways and transcriptional effectors to obtain strikingly different vascular outcomes, and therefore the manner in which high levels of VEGFA establish the precise gene expression patterns required for sprouting angiogenesis has been a key question in vascular biology. In this study, we demonstrate that VEGFA-induced gene expression in endothelial cells at the angiogenic front is downstream from MEF2 factor-driven transcriptional activation. In addition to their roles in the endothelium, MEF2 factors are widely expressed in nonvascular tissues and play crucial roles in the development of cardiac and skeletal muscle, bone, neural crest, and T cells (Potthoff and Olson 2007). Consequently, it is likely that they collaborate with other transcription factors to achieve endothelial-specific responses. One potential partner is the ETS family of transcription factors. All angiogenic enhancers characterized in this study contained multiple ETS motifs in addition to MEF2 sites. Furthermore, loss of ETS motifs ablated Dll4in3 expression in both arteries and sprouting angiogenesis (Sacilotto et al. 2013; Wythe et al. 2013), suggesting that ETS factors cooperate with MEF2 factors in the vasculature. This is supported by the observation that ETS factors are both transcriptional activators of and activated by MEF2C (Fig. 6; De Val et al. 2004). Of note, the highly endothelial-expressed ETS transcription factor ERG has been implicated in the regulation of Dll4 in arteries downstream from VEGFA (Wythe et al. 2013) and in vascular growth, including in the angiogenic retina (Birdsey et al. 2015). However, it is unlikely that ERG or other ETS factors regulate angiogenic gene expression alone: Unlike MEF2 motifs, ETS-binding motifs are found in all known endothelial enhancers and promoters regardless of expression pattern within the vasculature (De Val and Black 2009), and ERG itself is expressed widely throughout the endothelium (Randi et al. 2009). Furthermore, we demonstrate here that Dll4in3 and HLX-3 enhancers with intact ETS motifs were unable to drive angiogenic expression after MEF2 motif ablation, although Dll4in3 was still able to drive arterial expression. Similarly, the wild-type Dll4-12 enhancer containing multiple functional sites for ETS but no MEF2 sites was unable to drive anything other than arterial expression. These results therefore instead support a model in which MEF2 factors cooperate with ETS factors, with ETS providing essential endothelial expression information, and MEF2 contributing to angiogenic sprout specificity.
The SOXF family of transcription factors may also play a role in MEF2-driven angiogenic gene activity. Unlike the widely expressed ETS and MEF2 factors, the SOXF factors (SOX7, SOX17, and SOX18) are primarily restricted to arterial and angiogenic endothelial cells in mammals (Corada et al. 2013; Lee et al. 2014). SOXF factors have already been implicated in the regulation of Dll4/Notch signaling (Corada et al. 2013; Sacilotto et al. 2013), but their precise role at the angiogenic front is not yet established. SOXF binding in combination with ETS factors is likely not sufficient for angiogenic expression, as multiple enhancers, including Dll4-12 and Ece1, robustly bind SOXF and ETS factors but do not drive expression during sprouting angiogenesis (Robinson et al. 2014). However, loss of SOX17 affected sprouting angiogenesis in the retina (Corada et al. 2013; Lee et al. 2014), and a role for SOXF factors in combination with MEF2 cannot be ruled out: The Dll4in3 MEF2 motif is adjacent to a functional SOXF-binding motif, and MEF2C is known to directly interact with SOX18 (Hosking et al. 2001). It would therefore be fascinating to directly compare the global binding patterns of SOX7, SOX17, and SOX18 with MEF2 factors in endothelial cells and establish the role of SOXF-binding motifs in our newly identified set of MEF2-dependent angiogenic enhancers.
In addition to collaborative binding with other transcription factors, our data also indicate that the specificity of MEF2 activation to sprouting angiogenesis is modulated by the removal of repressive class IIa HDAC binding and recruitment of EP300 downstream from VEGFA. However, MEF2 factors are able to confer signal responsiveness downstream from multiple signaling pathways. For example, multiple calcium-regulated protein kinases are able to modulate HDAC binding to MEF2 factors, and phosphorylation by MAP kinases can activate MEF2 (Potthoff and Olson 2007; Ge et al. 2009). The latter is particularly notable given that recent work implicates ERK as a specific effector of Vegfa signaling in the induction of angiogenic genes during sprouting (Shin et al. 2016). Furthermore, research in skeletal muscle also demonstrated that the MEF2 proteins can compete with NICD for binding to the MAML coactivator (Shen et al. 2006), suggesting that high levels of NICD in stalk cells may also have a repressive effect on MEF2 factors in the endothelium, reinforcing the lateral inhibition. Consequently, it is very likely that MEF2 transcriptional activity is modulated by a complex combination of transcriptional, translational, and post-translational modifications, all of which contribute to the gene expression pattern downstream from VEGF receptor signaling.
Previous work has already implicated MEF2 transcription factors in the maintenance of vascular integrity via HDAC7-mediated MMP10 repression and downstream from BMK1 (Hayashi et al. 2004; Olson 2004; Chang et al. 2006). Although these reports principally focused on MEF2C at least in part due to the cardiovascular phenotypes reported in the global MEF2C-null mice (Lin et al. 1998; Bi et al. 1999), endothelial-specific ablation of MEF2C resulted in no clear embryonic vascular defects (Xu et al. 2012). This result indicates that the vascular defects seen in the global MEF2C-null mice were principally downstream from cardiac dysfunction and suggests that MEF2 factor redundancy in the vasculature extends beyond the regulation of Dll4. This may mirror the situation in skeletal muscle regeneration, where an absolute requirement for MEF2 was revealed only after compound deletion of Mef2A, Mef2C, and Mef2D (Liu et al. 2014). However, although loss of endothelial MEF2C had no detected effects in physiological conditions, Xu et al. (2012) reported a significant increase in vascular recovery and a cognate decrease in pathological neovascularization in the Mef2C-null retinal vasculature after oxygen-induced retinopathy, indicating that loss of MEF2C results in increased sprouting after injury. These results, which correlate with work demonstrating reduced in vitro VEGFA-induced vascular sprouting after MEF2C overexpression (Sturtzel et al. 2014), are in agreement with the consequences of Dll4 perturbations: Reduced DLL4 levels result in increased retinal vascular recovery and decreased neovascularizion after oxygen-induced retinopathy (Xu et al. 2012) and increased sprouting in response to VEGFA (Sainson et al. 2005; Jakobsson et al. 2010), while overexpression of DLL4 reduces the in vitro responses to VEGFA (Williams et al. 2006). Therefore, we hypothesize that even limited reductions of Dll4 levels downstream from MEF2C perturbation disrupt Notch-mediated lateral inhibition, resulting in hypersprouting in conditions of pathological stress.
Functional redundancy within the MEF2 family may not be the only contributor to the variable phenotypes seen after Mef2 gene deletion. The multiple targets of MEF2 factors in the vasculature, particularly those involved in angiogenic sprouting, may also contribute to the challenges in detecting and understanding phenotypic changes after MEF2 factor modulation. The reduced vascular density, coverage, and tip cell numbers seen in iEC MEF2A/C retina vasculature would initially appear opposite to the hypersprouting seen after Dll4 reduction, although reduced Dll4 expression was consistently seen in these mice. However, this phenotype must be considered in light of other roles for MEF2 factors in the vasculature. MEF2 has been implicated in the regulation of vascular integrity downstream from MMP10 activation and in the activation of the transcription factors KLF2 and KLF4 (Parmar et al. 2006; Maejima et al. 2014), important regulators of inflammation, vascular tone, and stabilization (Atkins and Jain 2007). Additionally, our work here demonstrates a direct link between MEF2 factor and many other genes involved in sprouting angiogenesis. The challenge in understanding the phenotypes after Mef2 ablation comes because individual loss of different genes required during sprouting angiogenesis can result in vastly different consequences. While depletion of Dll4 levels can have a proangiogenic effect, loss or depletion of direct MEF2 targets ETS1, ELK3, and HLX can have anti-angiogenic effects in vivo (Pham et al. 2007; Wei et al. 2009; Herbert et al. 2012; Weinl et al. 2014). Furthermore, the vascular Notch pathway genes disregulated by DLL4 reduction are themselves, like all other vascular genes, likely to be direct ETS factor targets, suggesting that the hypersprouting routinely seen after Dll4 disruption may also be indirectly repressed by the general suppression of MEF2–ETS–Notch pathway gene activation. Consequently, while the balance of pro- and anti-angiogenic signaling appears perturbed only in MEF2C-null retina when pathologically stressed, the compound loss of MEF2A/C appears to tip the balance into an anti-angiogenic response, albeit one potentially mitigated by the maintained MEF2D signaling and possibly the disruption of Dll4-mediated lateral inhibition. These results also serve to illustrate the challenges of interpreting knockout animal models when studying transcription factors with multiple targets, particularly when redundancy must also be considered.
In conclusion, our work demonstrates that the analysis of enhancers can be a powerful approach to study the heterogeneity and complexity of signaling networks operating in angiogenesis in a variety of different settings.
Materials and methods
Cloning
The Dll4in3, Dll4in3mutMEF2, and Dll4-12 enhancers were as described previously (Sacilotto et al. 2013). The ETS1+194, ELK3-29, HLX-3, HLX-3mutMEF2, hlx-3, and hlx-3mutMEF enhancers were generated as custom-made, double-stranded linear DNA fragments (GeneArt Strings, Life Technologies). Reporter vectors were generated using Gateway technology (Invitrogen).
Animals
All UK animal procedures were approved by local ethical review and licensed by the UK Home Office. US animal procedures complied with US federal and institutional guidelines. Transgenic mice were generated by oocyte microinjection. Mosaic transgenic zebrafish embryos were generated using the tol2 system (Kawakami 2005), and GFP reporter expression was scored at 32 hpf. The tg(hlx-3:GFP) stable line was created by outcross of adult F0 carriers generated using the tol2 system. X-gal analysis of embryos and postnatal organs was as described (De Val et al. 2004). X-gal analysis in retinas was as described (Jakobsson et al. 2010), and hindbrains were dissected as described (Fantin et al. 2013) and treated like the embryos. For Matrigel assays, transgenic mice were subcutaneously injected into the flanks with BD Matrigel basement membrane matrix (BD) supplemented with 2 µg/mL fibroblast growth factor (Peprotech) and harvested 14 d after injection. For tumors, transgenic mice were subcutaneously injected with 100 µL of BD Matrigel basement membrane matrix (BD) containing 1 × 105 B16F10 melanoma cells and harvested at 12-mm diameter. iEC Mef2A/C knockout embryos and pups were obtained by crossing Mef2Aflox/flox;Mef2Cflox/flox mice (Vong et al. 2005; Akhtar et al. 2012) with Cdh5(PAC)Cre-ERT2 mice (Wang et al. 2010). Recombination was induced by tamoxifen injection in pregnant mice 9 d after a plug was detected, and embryos were harvested 3 d later, genotyped, and fixed in 4% PFA for 2 h. For postnatal analysis, tamoxifen injection occurred at P1, P2, and P3, and retinas and lungs were harvested at P5 and P6. Eyes were removed from pups, fixed with 4% PFA for 90 min, and rinsed in PBS, and then the retinas were dissected as described previously (Pitulescu et al. 2010). Whole lungs were also dissected, rinsed in PBS, and then snap-frozen in liquid nitrogen for subsequent RNA extraction.
Western blot
HUVEC pellets were lysed in RIPA buffer (50 mM Tris-HCL at pH 7.4, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA) supplemented with protease and phosphatase inhibitor cocktail (Roche). Hindbrains were lysed in Tris buffer (20 mM Tris at pH 9, 2% SDS) supplemented with protease and phosphatase inhibitor cocktail (Roche), boiled at 100°C, and then incubated at 750 rpm at 80°C. Insoluble material was removed, protein concentration was determined with the BCA protein assay kit (Thermo Scientific), and 10 μg of protein per lane was separated by SDS-PAGE. DLL4 was detected using anti-DLL4 (1:1000; Abcam, 7280) and membranes were reprobed with anti-β-Actin antibody ( 1:100,000; Abcam, clone AC-15). Band intensities of DLL4 and β-Actin were quantified with ImageLab software (Bio-Rad).
Immunostaining
Hindbrains and retinas were processed as described (del Toro et al. 2010; Pitulescu et al. 2010; Fantin et al. 2013), and tumors and E10 embryos were fixed in 4% PFA for 1 h on ice. After incubation in blocking solution (10% normal donkey serum, 0.1% [v/v] Triton X-100 in PBS), samples were incubated overnight at 4°C with the designated primary antibodies (MEF2A [Abcam], MEF2B [Abcam], MEF2C [Cell Signaling], MEF2D [BD], EP300 [Active Motif], HDAC4 [GeneTex], DLL4 [R&D Systems] isolectin B4 [IB4] [Vector Laboratories], and Erg [Abcam]) in 0.1% PBS-T. Samples were washed in PBS-T and incubated for 3 h with suitable species-specific Alexa fluor- or biotin-conjugated secondary antibodies in 0.1% PBS-T. Total numbers of branch points and tip cells and retinal outgrowth length were measured after IB4 staining using ImageJ software from pooled images of retinas from at least two independent litters.
Cell culture and ChIP
HUVECS (pooled) (Lonza) were grown as described previously (Strasser et al. 2010; Sacilotto et al. 2013). For VEGF stimulation, cells were starved for 18 h in EBM-2 medium (Lonza) and then incubated for 1 h with EBM-2 supplemented with 25 ng/mL VEGFA165 (Peprotech, Lonza). ChIP assays were performed as described (Sacilotto et al. 2013) using antibodies against EP300 (Active Motif, 61401), HDAC4 (Proteintech, 60105-1), HDAC7 (Epigentek, A4007-050), HDAC9 (BioOrbyt, orb214926), Mef2A (Abcam, ab109420), Mef2C (Cell Signaling, 5030S), and Mef2D (BD, 610774). RNA isolation, cDNA synthesis, RT–PCR, and quantitative RT–PCR (qRT–PCR) were performed as described previously (Nikitenko et al. 2013) using primer/probes from Applied Biosystems. Dharmacon siRNA targeting human MEF2A, MEF2C, MEF2D, or the negative control sequence were purchased from GE Healthcare. Stealth siRNA targeting mouse Mef2A, Mef2C, and Mef2D were purchased from Life Technologies. See the Supplemental Material for sequences and catalog numbers. siRNAs were transfected into primary HUVECs or bEND cells expressing GFP (pGIPz-GFP-Puro HUVECs) at a final concentration of 100 nM using Oligofectamine (Life Technologies). MEF2A, MEF2C, or MEF2D stable knockdown was performed using lentiviral particles containing shRNAmir for MEF2A, MEF2C, MEF2D, or nonsilencing target (Open Biosystems).
Bioinformatic analysis of MEF2-enriched binding sites
MEF2C and corresponding H3K27ac ChIP-seq data were obtained using publicly available ChIP-seq data (Maejima et al. 2014) accessible at NCBI Gene Expression Omnibus database (Edgar et al. 2002), accession numbers GSE32547, GSE32644, GSE32693, and GSE41553. Raw reads were trimmed with Sickle, and duplicate PCR reads were removed with rmdup. Reads were then aligned to human genome build hg19 using Bowtie2, and peaks were called with MACS2. For the enrichment analysis, RefSeq genes were obtained from University of California at Santa Cruz, and antisense genes were removed. MEF2C peaks were required to reside <500 base pairs from a H3K27ac peak. Fisher's exact was used to determine enrichment of MEF2C peaks in or around genes up-regulated in tip cells.
HDAC inhibitor assays
HUVECS were starved for 16 h in EBM-2; treated with 400 nM TSA, 10 µM BML-210 (Abcam), 10 µM MC-1568 (Sigma), or DMSO (incubation times are in the figure legends); and stimulated with EBM-2 supplemented with 25 ng/mL VEGF-A for 1 h before harvesting for RNA extraction. Embryo culture in TSA was as described previously (Rojas et al. 2005) with modifications: Whole embryos (with their heads) were incubated for 17 h in either 100 µM TSA or control DMSO. In utero intracerebral injections were as described previously (Garcia-Moreno et al. 2014): Each E12 embryo was injected with ∼1 μL of 10 µM TSA or DMSO control, dyed with fast green into the fourth ventricle, left in utero for 24 h, and then harvested.
Gene expression analysis
RNA extraction was carried out using the RNAspin minikit (GE Healthcare) following the manufacturer's instructions from either HUVECs (directly lysed on the cell culture plate in R1 buffer) or whole-lung extracts (homogenized in liquid nitrogen using a pestle and mortar and subsequently lysed in R1 buffer). Total RNA (0.5–1 µg) was then retrotranscribed using random primers and SuperScript III (Life Technologies) as per the manufacturer's instructions, and the cDNA was analyzed by qRT–PCR (StepOne Plus, Life Technologies) using gene-specific TaqMan probes (Applied Biosystems).
Supplementary Material
Acknowledgments
We thank the Ludwig Institute for Cancer Research for support, N. Ahituv for providing E1b-GFP-GW and hsp-LacZ-GW vectors, H. Gerhardt for providing ES cells and sprouting assay protocol, E. Olson for providing the Mef2Aflox/flox mice, and M. Shipman for help with imaging. This work was supported by Ludwig Cancer Research Ltd., the Medical Research Council (MR/J007765/1 to N.S., K.L., G.B.-G., and S.D.V.), the Wellcome Trust (090532/Z/09/Z to B.D. and D.B., and 095718/Z/11/Z to S.P.H.), the British Heart Foundation (PG/10/83/28610 to N.S. and S.D.V., and PG/16/34/32135 to S.P.), the Breast Cancer Research Foundation (ANR00161 to A.H.), and Cancer Research UK (ANR0030 to E.B.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.290619.116.
Freely available online through the Genes & Development Open Access option.
References
- Akhtar MW, Kim M-S, Adachi M, Morris MJ, Qi X, Richardson JA, Bassel-Duby R, Olson EN, Kavalali ET, Monteggia LM. 2012. In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS One 7: e34863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins GB, Jain MK. 2007. Role of Krüppel-like transcription factors in endothelial biology. Circ Res 100: 1686–1695. [DOI] [PubMed] [Google Scholar]
- Benedito R, Roca C, Sörensen I, Adams S, Gossler A, Fruttiger M, Adams RH. 2009. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137: 1124–1135. [DOI] [PubMed] [Google Scholar]
- Bi W, Drake CJ, Schwarz JJ. 1999. The transcription factor MEF2C-null mouse exhibits complex vascular malformations and reduced cardiac expression of angiopoietin 1 and VEGF. Dev Biol 211: 255–267. [DOI] [PubMed] [Google Scholar]
- Birdsey GM, Shah AV, Dufton N, Reynolds LE, Osuna Almagro L, Yang Y, Aspalter IM, Khan ST, Mason JC, Dejana E, et al. 2015. The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/β-catenin signaling. Dev Cell 32: 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JKL, Sun L, Yang X-J, Zhu G, Wu Z. 2003. Functional characterization of an amino-terminal region of HDAC4 that possesses MEF2 binding and transcriptional repressive activity. J Biol Chem 278: 23515–23521. [DOI] [PubMed] [Google Scholar]
- Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN. 2006. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 126: 321–334. [DOI] [PubMed] [Google Scholar]
- Claxton S, Fruttiger M. 2004. Periodic Delta-like 4 expression in developing retinal arteries. Gene Expr Patterns 5: 123–127. [DOI] [PubMed] [Google Scholar]
- Corada M, Orsenigo F, Morini MF, Pitulescu ME, Bhat G, Nyqvist D, Breviario F, Conti V, Briot A, Iruela-Arispe ML, et al. 2013. Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat Commun 4: 2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Toro R, Prahst C, Mathivet T, Siegfried G, Kaminker JS, Larrivee B, Bréant C, Duarte A, Takakura N, Fukamizu A, et al. 2010. Identification and functional analysis of endothelial tip cell-enriched genes. Blood 116: 4025–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Val S, Black BL. 2009. Transcriptional control of endothelial cell development. Dev Cell 16: 180–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Val S, Anderson JP, Heidt AB, Khiem D, Xu S-M, Black BL. 2004. Mef2c is activated directly by Ets transcription factors through an evolutionarily conserved endothelial cell-specific enhancer. Dev Biol 275: 424–434. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin A, Vieira JM, Plein A, Maden CH, Ruhrberg C. 2013. The embryonic mouse hindbrain as a qualitative and quantitative model for studying the molecular and cellular mechanisms of angiogenesis. Nat Protoc 8: 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Moreno F, Vasistha NA, Begbie J, Molnar Z. 2014. CLoNe is a new method to target single progenitors and study their progeny in mouse and chick. Development 141: 1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge R, Tai Y, Sun Y, Zhou K, Yang S, Cheng T, Zou Q, Shen F, Wang Y. 2009. Critical role of TRPC6 channels in VEGF-mediated angiogenesis. Cancer Lett 283: 43–51. [DOI] [PubMed] [Google Scholar]
- Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, et al. 2003. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161: 1163–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha CH, Jhun BS, Kao H-Y, Jin Z-G. 2008a. VEGF stimulates HDAC7 phosphorylation and cytoplasmic accumulation modulating matrix metalloproteinase expression and angiogenesis. Arterioscler Thromb Vasc Biol 28: 1782–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha CH, Wang W, Jhun BS, Wong C, Hausser A, Pfizenmaier K, McKinsey TA, olson EN, Jin ZG. 2008b. Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J Biol Chem 283: 14590–14599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Kume T. 2008. Forkhead transcription factors regulate expression of the chemokine receptor CXCR4 in endothelial cells and CXCL12-induced cell migration. Biochem Biophys Res Commun 367: 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Kim S-W, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee J-D. 2004. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest 113: 1138–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström M, Phng L-K, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson A-K, Karlsson L, Gaiano N, et al. 2007. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445: 776–780. [DOI] [PubMed] [Google Scholar]
- Herbert SP, Cheung JYM, Stainier DYR. 2012. Determination of endothelial stalk versus tip cell potential during angiogenesis by H2.0-like homeobox-1. Curr Biol 22: 1789–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosking BM, Wang SC, Chen SL, Penning S, Koopman P, Muscat GE. 2001. SOX18 directly interacts with MEF2C in endothelial cells. Biochem Biophys Res Commun 287: 493–500. [DOI] [PubMed] [Google Scholar]
- Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A, et al. 2010. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12: 943–953. [DOI] [PubMed] [Google Scholar]
- Jayathilaka N, Han A, Gaffney KJ, Dey R, Jarusiewicz JA, Noridomi K, Philips MA, Lei X, He J, Ye J, et al. 2012. Inhibition of the function of class IIa HDACs by blocking their interaction with MEF2. Nucleic Acids Res 40: 5378–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K. 2005. Transposon tools and methods in zebrafish. Dev Dyn 234: 244–254. [DOI] [PubMed] [Google Scholar]
- Kerbel RS. 2008. Tumor angiogenesis. N Engl J Med 358: 2039–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer-Kwon K-R, Tang Z, Mathe E, Qian J, Sung M-H, Li G, Resch W, Baek S, Pruett N, Grøntved L, et al. 2013. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell 155: 1507–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrivee B, Freitas C, Trombe M, Lv X, DeLafarge B, Yuan L, Bouvree K, Bréant C, del Toro R, Brechot N, et al. 2007. Activation of the UNC5B receptor by Netrin-1 inhibits sprouting angiogenesis. Genes Dev 21: 2433–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Lee S, Yang H, Song S, Kim K, Saunders TL, Yoon JK, Koh GY, Kim I. 2014. Notch pathway targets proangiogenic regulator Sox17 to restrict angiogenesis. Circ Res 115: 215–226. [DOI] [PubMed] [Google Scholar]
- Lin Q, Lu J, Yanagisawa H, Webb R, Lyons GE, Richardson JA, olson EN. 1998. Requirement of the MADS-box transcription factor MEF2C for vascular development. Development 125: 4565–4574. [DOI] [PubMed] [Google Scholar]
- Liu N, Nelson BR, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. 2014. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc Natl Acad Sci 111: 4109–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizama CO, Hawkins JS, Schmitt CE, Bos FL, Zape JP, Cautivo KM, Borges Pinto H, Rhyner AM, Yu H, Donohoe ME, et al. 2015. Repression of arterial genes in hemogenic endothelium is sufficient for haematopoietic fate acquisition. Nat Commun 6: 7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ. 2007. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci 104: 3219–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Zhang CL, olson EN. 2000. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell 6: 233–244. [DOI] [PubMed] [Google Scholar]
- Maejima T, Inoue T, Kanki Y, Kohro T, Li G, Ohta Y, Kimura H, Kobayashi M, Taguchi A, Tsutsumi S, et al. 2014. Direct evidence for pitavastatin induced chromatin structure change in the KLF4 gene in endothelial cells. PLoS One 9: e96005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebbioso A, Manzo F, Miceli M, Conte M, Manente L, Baldi A, De Luca A, Rotili D, Valente S, Mai A, et al. 2009. Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC–MEF2 complexes. EMBO Rep 10: 776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitenko LL, Leek R, Henderson S, Pillay N, Turley H, Generali D, Gunningham S, Morrin HR, Pellagatti A, Rees MCP, et al. 2013. The G-protein-coupled receptor clr is upregulated in an autocrine loop with adrenomedullin in clear cell renal cell carcinoma and associated with poor prognosis. Clin Cancer Res 19: 5740–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN. 2004. Undermining the endothelium by ablation of MAPK–MEF2 signaling. J Clin Invest 113: 1110–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, et al. 2006. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest 116: 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham VN, Lawson ND, Mugford JW, Dye L, Castranova D, Lo B, Weinstein BM. 2007. Combinatorial function of ETS transcription factors in the developing vasculature. Dev Biol 303: 772–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulescu ME, Schmidt I, Benedito R, Adams RH. 2010. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc 5: 1518–1534. [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Olson EN. 2007. MEF2: a central regulator of diverse developmental programs. Development 134: 4131–4140. [DOI] [PubMed] [Google Scholar]
- Prahst C, Kasaai B, Moraes F, Jahnsen ED, Larrivee B, Villegas D, Pardanaud L, Pibouin-Fragner L, Zhang F, Zaun HC, et al. 2014. The H2.0-like homeobox transcription factor modulates yolk sac vascular remodeling in mouse embryos. Arterioscler Thromb Vasc Biol 34: 1468–1476. [DOI] [PubMed] [Google Scholar]
- Rafehi H, Balcerczyk A, Lunke S, Kaspi A, Ziemann M, KN H, Okabe J, Khurana I, Ooi J, Khan AW, et al. 2014. Vascular histone deacetylation by pharmacological HDAC inhibition. Genome Res 24: 1271–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randi AM, Sperone A, Dryden NH, Birdsey GM. 2009. Regulation of angiogenesis by ETS transcription factors. Biochem Soc Trans 37: 1248–1253. [DOI] [PubMed] [Google Scholar]
- Robinson AS, Materna SC, Barnes RM, De Val S, Xu S-M, Black BL. 2014. An arterial-specific enhancer of the human endothelin converting enzyme 1 (ECE1) gene is synergistically activated by Sox17, FoxC2, and Etv2. Dev Biol 395: 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, Val SD, Heidt AB, Xu S-M, Bristow J, Black BL. 2005. Gata4 expression in lateral mesoderm is downstream of BMP4 and is activated directly by Forkhead and GATA transcription factors through a distal enhancer element. Development 132: 3405–3417. [DOI] [PubMed] [Google Scholar]
- Roukens MG, Alloul-Ramdhani M, Baan B, Kobayashi K, Peterson-Maduro J, van Dam H, Schulte-Merker S, Baker DA. 2010. Control of endothelial sprouting by a Tel–CtBP complex. Nat Cell Biol 12: 933–942. [DOI] [PubMed] [Google Scholar]
- Sacilotto N, Monteiro R, Fritzsche M, Becker PW, Sanchez-del-Campo L, Liu K, Pinheiro P, Ratnayaka I, Davies B, Goding CR, et al. 2013. Analysis of Dll4 regulation reveals a combinatorial role for Sox and Notch in arterial development. Proc Natl Acad Sci 110: 11893–11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainson RCA, Aoto J, Nakatsu MN, Holderfield M, Conn E, Koller E, Hughes CCW. 2005. Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J 19: 1027–1029. [DOI] [PubMed] [Google Scholar]
- Shen H, McElhinny AS, Cao Y, Gao P, Liu J, Bronson R, Griffin JD, Wu L. 2006. The Notch coactivator, MAML1, functions as a novel coactivator for MEF2C-mediated transcription and is required for normal myogenesis. Genes Dev 20: 675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Beane T, Quillien A, Male I, Zhu LJ, Lawson ND. 2016. Vegfa signals through ERK to promote angiogenesis but not artery differentiation. Development doi: 10.1242/dev.137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser G, Kaminker J, Tessier-Lavigne M. 2010. Microarray analysis of retinal endothelial tip cells identifies CXCR4 as a mediator of tip cell morphology and branching. Blood 115: 5102. [DOI] [PubMed] [Google Scholar]
- Sturtzel C, Testori J, Schweighofer B, Bilban M, Hofer E. 2014. The transcription factor MEF2C negatively controls angiogenic sprouting of endothelial cells depending on oxygen. PLoS One 9: e101521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testori J, Schweighofer B, Helfrich I, Sturtzel C, Lipnik K, Gesierich S, Nasarre P, Hofer-Warbinek R, Bilban M, Augustin HG, et al. 2011. The VEGF-regulated transcription factor HLX controls the expression of guidance cues and negatively regulates sprouting of endothelial cells. Blood 117: 2735–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong LH, Ragusa MJ, Schwarz JJ. 2005. Generation of conditionalMef2cloxP/loxP mice for temporal- and tissue-specific analyses. Genesis 43: 43–48. [DOI] [PubMed] [Google Scholar]
- Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, et al. 2010. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465: 483–486. [DOI] [PubMed] [Google Scholar]
- Wei G, Srinivasan R, Cantemir-Stone CZ, Sharma SM, Santhanam R, Weinstein M, Muthusamy N, Man AK, Oshima RG, Leone G, et al. 2009. Ets1 and Ets2 are required for endothelial cell survival during embryonic angiogenesis. Blood 114: 1123–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinl C, Wasylyk C, Garrido MG, Sothilingam V, Beck SC, Riehle H, Stritt C, Roux MJ, Seeliger MW, Wasylyk B, et al. 2014. Elk3 deficiency causes transient impairment in post-natal retinal vascular development and formation of tortuous arteries in adult murine retinae. PLoS One 9: e107048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CK, Li J-L, Murga M, Harris AL, Tosato G. 2006. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 107: 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wythe JD, Dang LTH, Devine WP, Boudreau E, Artap ST, He D, Schachterle W, Stainier DYR, Oettgen P, Black BL, et al. 2013. ETS factors regulate Vegf-dependent arterial specification. Dev Cell 26: 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Gong J, Maiti D, Vong L, Wu L, Schwarz JJ, Duh EJ. 2012. MEF2C ablation in endothelial cells reduces retinal vessel loss and suppresses pathologic retinal neovascularization in oxygen-induced retinopathy. Am J Pathol 180: 2548–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn HD, Chatila TA, Liu JO. 2000. Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis. EMBO J 19: 4323–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Day DS, Ho JW, Song L, Cao J, Christodoulou D, Seidman JG, Crawford GE, Park PJ, Pu WT. 2013. A dynamic H3K27ac signature identifies VEGFA-stimulated endothelial enhancers and requires EP300 activity. Genome Res 23: 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.