

Abstract
Anion exchanger 2 (AE2), the principal bicarbonate secretor in the human biliary tree, is down‐regulated in primary biliary cholangitis. AE2 creates a “bicarbonate umbrella” that protects cholangiocytes from the proapoptotic effects of bile salts by maintaining them deprotonated. We observed that knockdown of AE2 sensitized immortalized H69 human cholangiocytes to not only bile salt‐induced apoptosis (BSIA) but also etoposide‐induced apoptosis. Because the toxicity of etoposide is pH‐independent, there could be a more general mechanism for sensitization of AE2‐depleted cholangiocytes to apoptotic stimuli. We found that AE2 deficiency led to intracellular bicarbonate accumulation and increased expression and activity of soluble adenylyl cyclase (sAC), an evolutionarily conserved bicarbonate sensor. Thus, we hypothesized that sAC regulates BSIA. H69 cholangiocytes and primary mouse cholangiocytes were used as models. The sAC‐specific inhibitor KH7 not only reversed sensitization to BSIA in AE2‐depleted H69 cholangiocytes but even completely prevented BSIA. sAC knockdown by tetracycline‐inducible short hairpin RNA also prevented BSIA. In addition, sAC inhibition reversed BSIA membrane blebbing, nuclear condensation, and DNA fragmentation. Furthermore, sAC inhibition also prevented BSIA in primary mouse cholangiocytes. Mechanistically, sAC inhibition prevented Bax phosphorylation at Thr167 and mitochondrial translocation of Bax and cytochrome c release but not c‐Jun N‐terminal kinase activation during BSIA. Finally, BSIA in H69 cholangiocytes was inhibited by intracellular Ca2+ chelation, aggravated by thapsigargin, and unaffected by removal of extracellular calcium. Conclusions: BSIA is regulated by sAC, depends on intracellular Ca2+ stores, and is mediated by the intrinsic apoptotic pathway; down‐regulation of AE2 in primary biliary cholangitis sensitizes cholangiocytes to apoptotic insults by activating sAC, which may play a crucial role in disease pathogenesis. (Hepatology 2016;64:522‐534)
Abbreviations
- AE2
anion exchanger 2
- ANOVA
analysis of variance
- BSIA
bile salt‐induced apoptosis
- cAMP
cyclic adenosine 3′,5′‐monophosphate
- GCDC
glycochenodeoxycholate
- HBSS
Hanks' balanced salt solution
- JNK
c‐Jun N‐terminal kinase
- MCU
mitochondrial calcium uniporter
- MOMP
mitochondrial outer membrane permeabilization
- NaCDC
sodium chenodeoxycholate
- PARP
poly(adenosine diphosphate ribose) polymerase
- PBC
primary biliary cholangitis
- RT‐qPCR
reverse‐transcription quantitative polymerase chain reaction
- sAC
soluble adenylyl cyclase
- shRNA
short hairpin RNA
- tmAC
transmembrane adenylyl cyclase
Primary biliary cholangitis (PBC) is a chronic fibrosing cholangiopathy characterized by a cholestatic serum enzyme pattern, serum antimitochondria antibodies, and a striking female predominance.1 Female preponderance and the results of sibling studies in monozygotic and dizygotic twins2 and genome‐wide association studies3, 4 support an inherited genetic predisposition and are in line with a model autoimmune disease. However, the exact pathogenetic mechanisms and the etiology remain unclear.
In search of the disease mechanism, Prieto, Medina, and colleagues struck upon the frequent association with exocrine failure (salivary glands, lacrimal glands, pancreas, etc.) in PBC patients and hypothesized that impaired bicarbonate secretion might underlie these exocrine pathologies. They found that anion exchanger 2 (AE2, SLC4A2), a chloride/bicarbonate exchanger responsible for biliary bicarbonate secretion, is down‐regulated in the apical membrane of both hepatocytes and cholangiocytes among PBC patients5, 6 and in the salivary glands of patients with Sjögren syndrome.7 Furthermore, PBC patients show impaired secretin‐stimulated biliary bicarbonate secretion, which improved after ursodeoxycholate treatment.8
The improvement of the serum liver tests and the secretin responsiveness following ursodeoxycholate treatment in PBC patients underline the importance of bicarbonate secretion. On the basis of this, a defective “biliary bicarbonate umbrella” was proposed as a common pathology of fibrosing cholangiopathies.9 The biliary bicarbonate umbrella hypothesis states that biliary epithelia, by secreting bicarbonate, generate an alkaline barrier that renders bile salts in their polar, deprotonated, and membrane‐impermeant status. When the bicarbonate secretory machinery fails, as may be expected during down‐regulation of AE2 expression, cholangiocytes may become vulnerable to millimolar concentrations of bile salt monomers that normally pass along the bile ducts. In hepatocytes, bile salts have been demonstrated to activate the intrinsic apoptosis pathway by increasing cytosolic free Ca2+,10 enhancing reactive oxygen species production,11 and releasing cytochrome c as a result of Bax‐mediated permeabilization of the mitochondrial outer membrane.12 Using immortalized H69 human cholangiocytes as a model, Hohenester, Maillette de Buy Wenniger, and colleagues demonstrated that bile salt toxicity is pH‐dependent and that AE2 knockdown sensitized H69 cholangiocytes to bile salt‐induced apoptosis (BSIA).13
In the course of examining the consequence of AE2 down‐regulation in H69 cholangiocytes, we found that knockdown of AE2 sensitized H69 cholangiocytes not only to BSIA but also to etoposide‐induced apoptosis,13 although etoposide is not a weak acid. This suggests that a more general intracellular mechanism, affecting apoptosis in general, may be at play. Therefore, we wondered if AE2 down‐regulation, in addition to allowing more bile salts to enter the cells, sensitizes cholangiocytes to apoptotic stimuli as a result of intracellular bicarbonate retention, i.e., mediated by a bicarbonate‐responsive factor. Among the various intracellular acid‐base sensors, soluble adenylyl cyclase (sAC) seems to be the most attractive candidate14 because it can be directly stimulated by intracellular bicarbonate.15 Indeed, we have reported that in fibroblasts derived from Ae2a,b‐/‐ mice, sAC expression is increased, which leads to a constitutively increased production of cyclic adenosine 3′,5′‐monophosphate (cAMP).16sAC is an evolutionarily conserved member of the mammalian adenylyl cyclase family that was relatively recently discovered.17 sAC was originally identified with Mn2+‐dependent adenylyl cyclase activity in the cytosolic fraction of testes,18 where it is most abundant. Later, sAC expression was found in a wide range of tissues.14 The presence of sAC in human liver19 and mouse primary cholangiocytes20 has been demonstrated. Although both belong to class III nucleotidyl cyclases, sAC differs in several aspects from the transmembrane adenylyl cyclases (tmACs, ACDY1‐ACDY9).21 Firstly, sAC is not regulated by G proteins, nor is it activated by forskolin, a pan‐tmAC activator.17 Rather, sAC is activated by bicarbonate15 and fine‐tuned by Ca2+ and adenosine triphosphate.22, 23 Secondly, while tmACs are localized to the plasma membrane, sAC isoforms are found in several subcellular compartments, including the cytosol,24 mitochondria,25, 26 cytoskeleton,27 and nuclei.28 Thirdly, sAC produces cAMP in response to intracellular fluctuations of bicarbonate and free Ca2+, while tmACs relay the extracellular signals that activate G protein‐coupled receptors. In addition to its various roles in pH homeostasis and metabolic regulation,29, 30 sAC has been shown to mediate apoptosis in several models, including stimulated ischemia/acidosis in coronary endothelial cells,24 stimulated ischemia/reperfusion injury in cardiomyocytes,31 and oxysterol‐induced and oxidative stress‐induced apoptosis in vascular smooth muscle cells.32, 33 We therefore hypothesized that sAC is involved in BSIA in H69 cholangiocytes and is responsible for the sensitization to apoptotic triggers in the AE2 down‐regulated state.
Materials and Methods
Materials were purchased from Sigma‐Aldrich unless otherwise indicated.
Cell Culture
H69 cholangiocytes was cultured as described.34 The medium consisted of Dulbecco's modified Eagle's medium (Life Technologies)/Ham's nutrient mixture F‐12 (Life Technologies) (3:1) with 10% fetal bovine serum (Gibco) and the following supplements: 180 μM adenine, 865 nM insulin, 62.5 nM transferrin, 2 nM triiodothyronine, 1.1 μM hydrocortisone, 5.5 μM epinephrine, 1.67 nM epidermal growth factor, 37.5 U/mL penicillin, and 37.5 μg/mL streptomycin. The medium was supplemented with 1.75 g/L sodium bicarbonate and 20 mM 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid‐sodium hydroxide (pH 7.4) for use in a 5% CO2 incubator. Cells were passaged twice per week. For cell culture and experiments with tetracycline‐inducible sAC knockdown lines, Tet System Approved fetal bovine serum (Clontech) was used.
Statistics
All results are given as mean ± standard deviation. Statistical significance was determined by two‐tailed Student t test or one‐way analysis of variance (ANOVA) with Bonferroni's multiple comparison test using GraphPad Prism 5 (GraphPad Software, La Jolla, CA) with an α error of 0.05.
(For complete information, please refer to the Supporting Information.)
Results
H69 Cholangiocytes Express sAC and Respond to Increased Intracellular Bicarbonate
A significant sAC‐derived cAMP pool was demonstrated in H69 cholangiocytes in normal Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Supporting Fig. S1). To demonstrate bicarbonate responsiveness of sAC, we performed a cAMP accumulation assay in normal (chloride‐containing) and chloride‐free Hanks' balanced salt solution (HBSS). Switching extracellular medium from normal chloride‐containing HBSS to chloride‐free HBSS reversed the chloride gradient across the plasma membrane and forced AE2 to pump bicarbonate into cells, which led to intracellular alkalization (Supporting Fig. S2) and sAC activation (Fig. 1A). The sAC‐derived cAMP, defined as the difference in cAMP concentration in the presence and absence of the sAC‐specific inhibitor KH7, was elevated by 4.34 ± 0.84‐fold in chloride‐free HBSS compared to normal HBSS (Fig. 1B,C).
Figure 1.
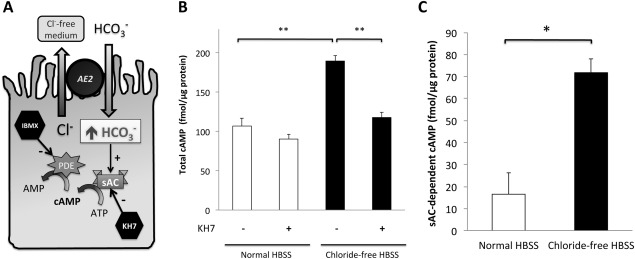
Presence of functional sAC in H69 cholangiocytes was demonstrated by bicarbonate‐stimulated cAMP accumulation assay. (A) Confluent H69 cholangiocyte monolayers were incubated in either normal HBSS or chloride‐free HBSS for 30 minutes. The latter reversed the chloride gradient across the plasma membrane and forced AE2 to take up bicarbonate. (B) Total cAMP was measured by an enzyme‐linked immunosorbent assay in the absence or presence of KH7, a specific inhibitor of sAC. Shown here is the average of two quadruplicate experiments. One‐way ANOVA, P = 0.0007. (C) From each independent quadruplicate experiment in (B), sAC‐derived cAMP was calculated (defined as the difference in total cAMP measured in the absence and presence of KH7). Two‐tailed Student t test, P = 0.03. * P < 0.05, **P ≤ 0.01. Abbreviations: AMP, adenosine monophosphate; ATP, adenosine triphosphate; IBMX, 3‐isobutyl‐1‐methylxanthine; PDE, phosphodiesterase.
sAC is Up‐Regulated in AE2 Knockdown H69 Cholangiocytes
Next, we examined if AE2 down‐regulation has an effect on sAC. H69 cholangiocytes were transduced with lentivirus for short hairpin RNA (shRNA)‐mediated AE2 knockdown. Expression of sAC messenger RNA in H69 cholangiocytes was detected by reverse‐transcription quantitative polymerase chain reaction (RT‐qPCR) with a primer set targeting the second catalytic domain. sAC protein was detected by immunoblotting using the monoclonal mouse antibody R21 (kindly provided by Drs. L. Levin and J. Buck, Weill‐Cornell University. NY),27 which recognizes an epitope in the first catalytic domain. Knockdown of AE2 was confirmed by RT‐qPCR (Fig. 2A) and immunoblotting (Fig. 2B). sAC expression in AE2 knockdown H69 cholangiocytes was increased to 1.82 ± 0.44‐fold that of control H69 cholangiocytes (Fig. 2C), and the protein level also increased to 1.46 ± 0.22‐fold (Fig. 2D,E). In addition, AE2 knockdown in H69 cholangiocytes sensitized the cells to etoposide‐induced apoptosis (Fig. 2F), as reported.13
Figure 2.
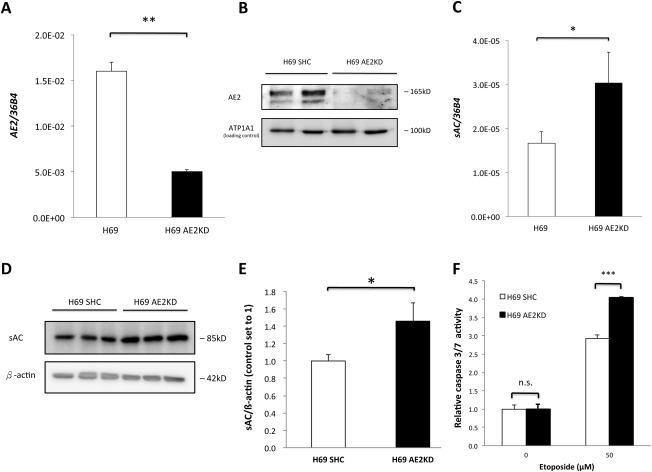
Knockdown of AE2 induced sAC expression. AE2 was knocked down by lentivirus‐mediated shRNA interference in H69 cholangiocytes and confirmed by RT‐qPCR (A) and immunoblot (B). Expression of sAC messenger RNA (C) and protein level (D) in both short hairpin control and AE2 knockdown H69 cholangiocytes was examined. Immunoblots were quantified by ImageJ (E). Two‐tailed Student t test, *P < 0.05, **P ≤ 0.01. (F) Control and AE2KD H69 cholangiocytes were incubated with 50 μM etoposide or 0.1% dimethyl sulfoxide (vehicle) for 16 hours. Caspase 3/7 activity was determined. One‐way ANOVA, P < 0.0001. The figure shows a representative experiment from a series with similar results. ***P ≤ .001. Abbreviations: ATP1A1, adenosine triphosphatase, sodium/potassium transporting, alpha 1 polypeptide; KD, knockdown; n.s., not significant; SHC, short hairpin control.
Inhibition of sAC Prevents Bsia
Because AE2 down‐regulation resulted in both increased sAC expression and enhanced BSIA,13 we hypothesized that increased sAC activity causes increased BSIA. Indeed, the sAC‐specific inhibitor KH7 dose‐dependently inhibited BSIA in both control and AE2 knockdown cholangiocytes (Fig. 3A). Strikingly, KH7 treatment not only redeemed the vulnerability to sodium chenodeoxycholate (NaCDC)‐induced apoptosis in AE2 knockdown cholangiocytes but at higher concentrations also prevented NaCDC‐induced apoptosis altogether. Total cAMP determination in the presence of 750 μM NaCDC showed that the protective effect of KH7 indeed paralleled a decrease in the total cAMP pool (Supporting Fig. S3).
Figure 3.
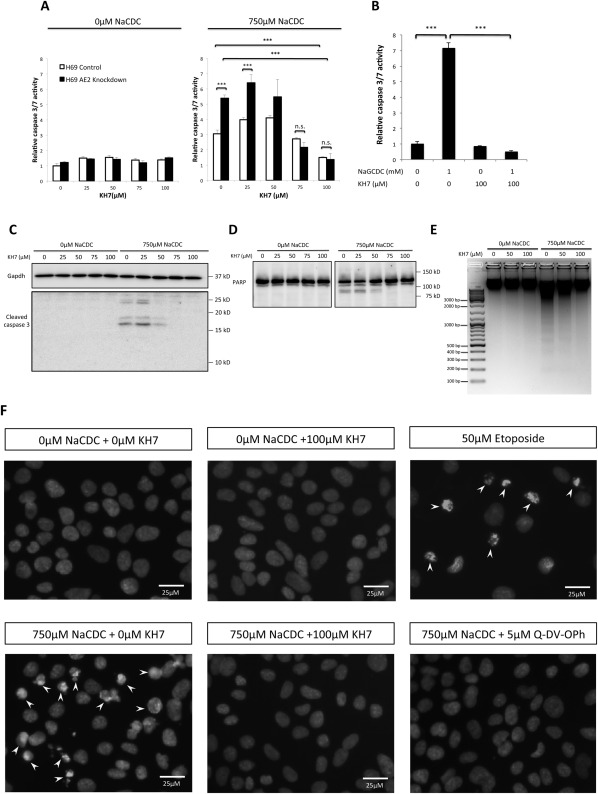
Inhibition of sAC prevented both unconjugated and conjugated BSIA. (A) Control and AE2 knockdown H69 cholangiocytes were incubated with increasing concentrations of sAC‐specific inhibitor KH7 in the absence or presence of 750 μM NaCDC at 37°C and 5% CO2 for 1 hour. Caspase 3/7 activity was determined. One‐way ANOVA, P < 0.0001. (B) Control and AE2 knockdown H69 cholangiocytes were incubated with 1 mM NaGCDC for 4 hours at pH 6.8 with or without KH7 in 37°C, 5% CO2 incubator. One‐way ANOVA, P < 0.0001. (C) H69 cholangiocytes were treated as in (A). Immunoblot for cleaved caspase 3 was performed. The antibody reacts with only cleaved caspase 3 (p17/p19), not procaspase 3. (D) Same treatment condition as (C), immunoblotted for PARP, an endogenous caspase 3/7 substrate. The full‐length PARP (∼113 kDa) was cleaved by caspase 3/7 into the 89‐kDa fragment. (E) DNA fragmentation assay. H69 cholangiocytes were incubated with 0, 50, and 100 μM KH7 in the absence or presence of 750 μM NaCDC for 1 hour. Genomic DNA was extracted and resolved on a 1.5% agarose gel. (F) H69 cholangiocytes were treated with or without 750 μM NaCDC in the presence or absence of 100 μM KH7 and 5 μM Q‐DV‐OPh (pan‐caspase inhibitor). Overnight treatment with 50 μM etoposide served as a positive control. Nuclei were visualized by DAPI staining. Arrowheads indicated apoptotic nuclear condensation. The figure shows a representative experiment from a series with similar results. ***P < 0.001. Abbreviations: Gapdh, glyceraldehyde 3‐phosphate dehydrogenase; KD, knockdown; n.s., not significant; SHC, short hairpin control.
To examine the relevance of this observation to PBC cholangiocytes, we repeated the same experiment with the predominant bile salt species in human bile, glycochenodeoxycholate (GCDC), the concentration of which can reach the millimolar range in hepatic bile. H69 cholangiocytes were incubated with 1 mM sodium GCDC (NaGCDC) at pH 6.8, a pH that can be expected in PBC bile due to impaired bicarbonate secretion.8 Consistent with the result from NaCDC‐induced apoptosis, KH7 treatment also prevented NaGCDC‐induced apoptosis (Fig. 3B). The reduced caspase 3/7 activity was not due to KH7 toxicity, as shown by WST‐1 viability assay (Supporting Fig. S4), or to reduced uptake of bile salts (Supporting Fig. S5).
To confirm the protective effect of KH7 on BSIA (Fig. 3A), immunoblots for cleaved caspase 3 and poly (adenosine diphosphate ribose) polymerase‐1 (PARP‐1), an established endogenous caspase 3/7 substrate, were performed. KH7 treatment dose‐dependently prevented cleavage of both caspase 3 (Fig. 3C) and PARP‐1 (Fig. 3D) during BSIA. In addition, KH7 treatment prevented NaCDC‐induced DNA fragmentation, an apoptotic signature that defines the irreversible fate to cell death (Fig. 3E). Consistently, analysis of apoptotic morphology revealed that inhibition of sAC reversed bile salt‐induced nuclear condensation (Fig. 3F) and plasma membrane blebbing (Supporting Fig. S6).
sAC‐Derived Camp Promotes Bsia, but Tmac‐Derived Camp Protects Against Bsia
To confirm that the antiapoptotic effect of KH7 treatment was specific, we analyzed BSIA in control and sAC knockdown H69 cholangiocytes. Interestingly, constitutive knockdown of sAC in H69 cholangiocytes led to growth arrest of the cell line. For this reason we used a tetracycline‐inducible sAC shRNA construct. Upon induction with doxycycline, the sAC protein level was reduced to 14% of control by 48 hours (Fig. 4A). Although less efficient compared to KH7 treatment (Fig. 3A), knockdown of sAC reduced caspase 3/7 activity by about 70% compared to control (Fig. 4B).
Figure 4.
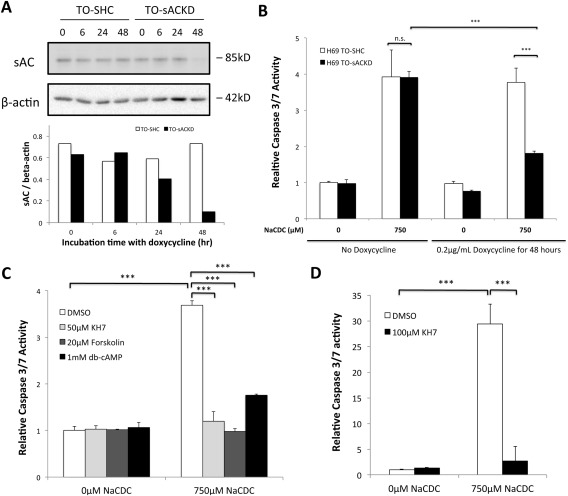
sAC‐derived cAMP promoted BSIA, while tmAC‐derived cAMP inhibited BSIA. (A) Control and inducible sAC knockdown H69 cholangiocytes were treated with 0.2 μg/mL doxycycline for the indicated periods of time. sAC knockdown was examined by immunoblotting. Immunoblot was quantified by ImageJ. Results are normalized to β‐actin. (B) Short hairpin control and sAC knockdown H69 cholangiocytes were induced with or without 0.2 μg/mL doxycycline for 48 hours and subsequently incubated with 750 μM NaCDC or vehicle for 1 hour. Caspase 3/7 activity was determined. One‐way ANOVA, P < 0.0001. The figure shows a representative experiment from a series of three independent experiments with similar results. (C) H69 cholangiocytes were incubated with or without 750 μM NaCDC in the presence of dimethyl sulfoxide (vehicle control), 50 μM KH7 (sAC‐specific inhibitor), 20 μM forskolin (tmAC‐specific activator), or 1 mM dibutyryl‐cAMP (membrane‐permeant cAMP analogue) for 4 hours in a 5% CO2, 37°C incubator. Caspase 3/7 activity was determined. One‐way ANOVA, P < 0.0001. (D) Primary mouse cholangiocytes were incubated with or without 750 μM NaCDC in the presence of dimethyl sulfoxide (vehicle control) or 100 μM KH7 for 3 hours in a 5% CO2, 37°C incubator. One‐way ANOVA, P < 0.0001. ***P < 0.001. Abbreviations: db‐cAMP, dibutyryl‐cAMP; DMSO, dimethyl sulfoxide; KD, knockdown; n.s., not significant; SHC, short hairpin control, TO, Tet‐operator.
We reasoned that sAC‐derived cAMP (cytosolic) could have a different effect on BSIA from tmAC‐derived cAMP (sub‐plasma membrane) due to different subcellular localizations. Indeed, both inhibition of sAC by KH7 and activation of tmAC by forskolin protected H69 cholangiocytes against BSIA (Fig. 4C), indicating that sAC‐derived cAMP was proapoptotic while tmAC‐dependent cAMP was antiapoptotic. Interestingly, treatment with a high concentration of dibutyryl‐cAMP (1 mM), which would overcome breakdown by phosphodiesterases and fully activate both sAC‐derived and tmAC‐derived signaling pathways, was protective. Subsequently, we investigated whether the observed role for sAC can also be demonstrated in primary cultured cholangiocytes. Cholangiocytes were isolated from wild‐type mouse liver and cultured on thick collagen gel layers. Importantly, in primary mouse cholangiocytes, BSIA was also inhibited by the sAC‐specific inhibitor KH7 (Fig. 4D).
Inhibition of sAC Prevents Mitochondrial Outer Membrane Permeabilization During Bsia
As bile salt has been reported to cause apoptosis through mitochondrial outer membrane permeabilization (MOMP) and subsequent cytochrome c release,12, 35 we examined if inhibition of sAC protected cholangiocytes from BSIA by preventing cytochrome c release. Indeed, NaCDC treatment released cytochrome c from mitochondria, which was dose‐dependently prevented by KH7 treatment (Fig. 5A). Because MOMP depends on the balance between antiapoptotic and proapoptotic actions of the BCL‐2 family proteins, we examined the subcellular localization of Bax, the effector BCL‐2 protein responsible for MOMP, during BSIA. Indeed, NaCDC induced Bax translocation from the cytosol to the mitochondria, which was dose‐dependently reduced by KH7‐mediated sAC inhibition (Fig. 5B).
Figure 5.
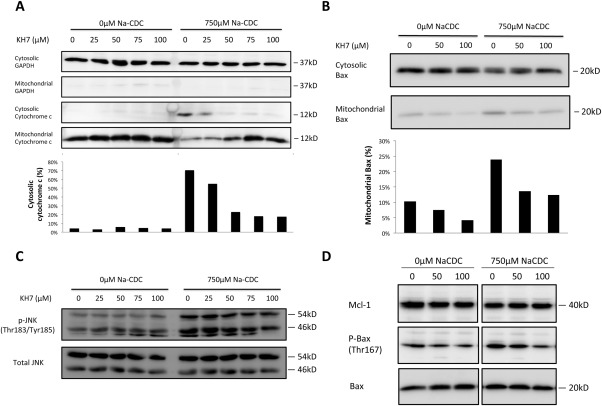
sAC inhibition prevented cytochrome c release and mitochondrial translocation of Bax during BSIA. (A) Cytochrome c release assay. H69 cholangiocytes were treated with increasing concentrations of KH7 in the absence or presence of 750 μM NaCDC for 1 hour in a 5% CO2, 37°C incubator. Cell fractionation was performed as described in Materials and Methods. Lysates of equal volume were subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and immunoblotted for glyceraldehyde 3‐phosphate dehydrogenase, a cytosolic marker, and cytochrome c. Cytochrome c immunoblots were quantified by ImageJ. Ratios of cytosolic signal to total signal (cytosolic plus mitochondrial) were calculated. (B) Bax translocation assay was performed and quantified as in (A). Percentage of mitochondrial Bax was calculated as mitochondrial signals divided by the sum of cytosolic and mitochondrial signals. (C) H69 cholangiocytes were treated as in (A). Whole‐cell lysates were used for immunoblot of total and phosphorylated JNK. (D) H69 cholangiocytes were treated as in (B). Whole‐cell lysates were used for immunoblot for myeloid cell leukemia 1, phospho‐Bax (Thr167), and total Bax. The figure shows a representative experiment from a series with similar results. Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; Mcl‐1, myeloid cell leukemia 1.
To further define the action of sAC, we examined the effect of sAC inhibition on c‐Jun N‐terminal kinase (JNK) and p38, two mitogen‐activated protein kinases known to activate Bax by Thr167 phosphorylation.36 Phosphorylation of p38 was affected by neither bile salts nor sAC inhibition (data not shown). JNK was activated by bile salt but was not affected by sAC inhibition (Fig. 5C). Bile salt treatment also resulted in increased Bax phosphorylation at Thr167, which was dose‐dependently prevented by sAC inhibition. Myeloid cell leukemia 1, an important antiapoptotic bcl‐2 family protein in cholangiocytes, was affected neither by bile salt nor by KH7 (Fig. 5D).
Bsia is Dependent on Intracellular Calcium Store
Bile salts are known to have calcium ionophore properties37, 38, 39 and could relay the apoptotic signal to sAC by increased cytosolic free Ca2+. To test this hypothesis, we pretreated H69 cholangiocytes with increasing concentrations of the acetoxymethyl ester of 1,2‐bis(2‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid to chelate intracellular free Ca2+. Indeed, intracellular Ca2+ chelation by 1,2‐bis(2‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid dose‐dependently inhibited BSIA (Fig. 6A).
Figure 6.
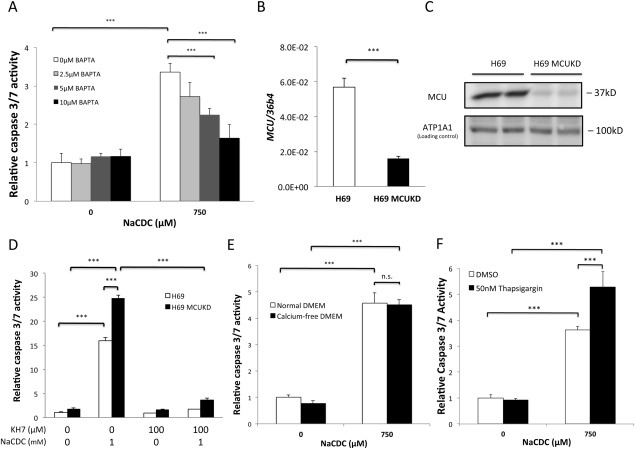
BSIA depends on intracellular calcium stores. (A) H69 cholangiocytes were loaded with increasing concentrations of 1,2‐bis(2‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid (BAPTA) and then treated with either vehicle or 750 μM NaCDC for 1 hour. Caspase 3/7 activities were determined. One‐way ANOVA, P < 0.0001. (B) MCU was knocked down by lentivirus‐mediated shRNA interference. Knockdown was confirmed by RT‐qPCR. Two‐tailed Student t test, P < 0.0001. (C) MCU knockdown was confirmed by immunoblotting. (D) Short hairpin control and MCU knockdown H69 cholangiocytes were treated with 1 mM NaCDC‐induced apoptosis in the absence or presence of sAC‐specific inhibitor KH7. One‐way ANOVA, P < 0.0001. (E) H69 cholangiocytes were incubated with 750 μM NaCDC in normal or Ca2+‐free medium. One‐way ANOVA, P < 0.0001. (F) H69 cholangiocytes were pretreated with or without 50 nM thapsigargin for 15 minutes and then with 0 μM or 750 μM NaCDC. The figure shows a representative experiment from a series with similar results. ***P < 0.001. Abbreviations: ATP1A1, adenosine triphosphatase, sodium/potassium transporting, alpha 1 polypeptide; BAPTA, 1,2‐bis(2‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid; DMEM, Dulbecco's modified Eagle's medium; DMSO, dimethyl sulfoxide; KD, knockdown; n.s., not significant.
sAC activity is synergistically stimulated by physiologic bicarbonate concentration (20‐25 mM) and free Ca2+ in a biphasic manner with a high‐affinity component (50% effective concentration = 300 nM) and a low‐affinity component (50% effective concentration = 300 μM).19, 22, 23 Therefore, the bile salt‐induced release of Ca2+ could theoretically activate sAC in both mitochondria and the cytosol, where free Ca2+ concentration could reach 100 μM and 1‐2 μM during calcium signaling, respectively.10, 40 To test which sAC isoform was involved, we examined BSIA in H69 cholangiocytes depleted of the mitochondrial calcium uniporter (MCU), the principal port of entry for cytosolic Ca2+. MCU knockdown reduces the mitochondrial calcium buffering capacity.40, 41 If mitochondrial sAC is involved in BSIA, MCU depletion is expected to be protective. On the other hand, if cytosolic sAC plays a role, MCU depletion is expected to aggravate apoptosis. MCU messenger RNA expression and protein level were reduced by 70% and 90%, respectively, in MCU‐depleted H69 cholangiocytes (Fig. 6B,C). However, MCU‐depleted H69 cholangiocytes were not more resistant to but, rather, were more susceptible to BSIA (Fig. 6D). Moreover, the enhanced apoptosis in MCU‐depleted H69 cholangiocytes could be inhibited by KH7 treatment, implying that cytosolic sAC, instead of mitochondrial sAC, was involved during BSIA.
To identify the source of increased cytosolic free Ca2+, we performed BSIA in either normal or Ca2+‐free medium. Removal of extracellular calcium did not prevent BSIA, indicating that the increased cytosolic free Ca2+ from intracellular stores was sufficient (Fig. 6E). Consistently, thapsigargin pretreatment, which inhibited Ca2+ uptake by sarcoplasmic/endoplasmic reticulum Ca2+ adenosine triphosphatase, potentiated bile salt toxicity. Unexpectedly, elevation of cytosolic Ca2+ alone by calcium ionophore ionomycin was not sufficient to induce apoptosis (Supporting Fig. S7). Similarly, thapsigargin alone (i.e., in the absence of bile salt) caused no apoptosis within the time frame of the experiment (Fig. 6F).
Discussion
The present study sheds new light on the mechanism of BSIA in cholangiocytes and provides novel mechanistic insight into a potential role of AE2 down‐regulation in cholangiocytes of PBC patients.5, 6 We demonstrate that BSIA in both H69 human cholangiocytes and primary mouse cholangiocytes is regulated by sAC, that it involves Ca2+ release from intracellular stores, and that it is mediated by the intrinsic apoptotic signaling pathway.
AE2, an indispensable transporter of the biliary bicarbonate secretion machinery, is crucial for protecting cholangiocytes against bile salts.9 Recently, miR‐506, which is up‐regulated in PBC cholangiocytes, was demonstrated to down‐regulate AE2 posttranscriptionally in H69 cholangiocytes.42 Our observation that knockdown of AE2 sensitized H69 cholangiocytes to both pH‐dependent (bile salts) and pH‐independent (etoposide) stimuli suggests that AE2 down‐regulation has a general impact on cholangiocyte apoptosis. In the present study, we show that this generalized vulnerability in the AE2 down‐regulated state is caused by increased sAC expression and signaling. Knockdown of AE2 in H69 human cholangiocytes leads to increased sAC expression, which is consistent with our previous finding in Ae2a,b‐/‐ murine fibroblasts.16 Intracellular alkalinization secondary to the absence of AE2 further enhances sAC signaling. Our observation that inhibition of sAC not only reverses increased vulnerability of AE2‐depleted H69 cholangiocytes but also completely inhibits BSIA identifies sAC as a potentially important proapoptotic factor in the pathogenesis of PBC. Accordingly, other apoptotic insults (e.g., toxic compounds excreted into bile) in addition to bile salts may contribute to the exaggerated cholangiocyte apoptosis in PBC patients with reduced AE2 expression.
The mechanism of BSIA has been studied extensively in hepatocytes but much less so in cholangiocytes. We demonstrate here that in cholangiocytes bile salts activate the intrinsic apoptotic pathway, involving mitochondrial translocation of Bax, cytochrome c release, activation of caspase 3, cleavage of PARP, and DNA fragmentation, all of which can be reversed by sAC inhibition. Although activation of procaspase 8 was not investigated here, we did not observe cleavage of Bid, a caspase 8 substrate, during BSIA (not shown).
The signaling specificity of cAMP, a diffusible second messenger, is achieved by scaffolding of adenylyl cyclases, cAMP effectors, and selected substrates43 and by compartmentation of cAMP into microdomains bordered by phosphodiesterases.44 Therefore, cAMP generated in different microdomains can have different biological outcomes. This is exemplified by the observation that cytosolic cAMP disrupts endothelial cell barrier function while para‐plasma membrane cAMP is protective.45 Kumar et al. also demonstrated that sAC‐derived cAMP mediates acidosis‐induced apoptosis in coronary endothelial cells, while stimulating tmACs by forskolin is protective.24 As yet another example, we demonstrate here that sAC‐derived cAMP (in the cytosol) promotes BSIA, while tmAC‐derived cAMP (para‐plasma membrane) protects against BSIA. Furthermore, the effect of tmAC‐derived cAMP overruled that of sAC because activation of cAMP effectors of both tmACs and sAC by dibutyryl‐cAMP was antiapoptotic.
MOMP following apoptotic stimuli is the result of the interplay of the proapoptotic and antiapoptotic BCL‐2 family proteins,46 which are regulated by posttranslational modification such as phosphorylation. For example, the proapoptotic activity of Bax is enhanced when phosphorylated by JNK or p38 at Thr16736 but suppressed when phosphorylated at Ser184 by phosphatidylinositol 3‐kinase/Akt.47 In the present study, we show that chenodeoxycholate activates JNK but not p38. Although sAC inhibition did not prevent bile salt‐induced JNK phosphorylation, it did prevent Bax phosphorylation at Thr167, suggesting that sAC regulates bile salt‐induced apoptotic signaling downstream of JNK phosphorylation and upstream of MOMP.
We show that BSIA in H69 cholangiocytes depends on increased cytosolic Ca2+ released from intracellular stores. The bile salt‐triggered increase of cytosolic free Ca2+ most likely did not activate mitochondrial sAC because inhibition of mitochondrial calcium accumulation by MCU knockdown did not protect H69 cholangiocytes from BSIA but, rather, aggravated it. Importantly, this could be completely reversed by sAC inhibition, implying that Ca2+ released from intracellular stores activates cytosolic sAC. Interestingly, ionomycin treatment did not cause apoptosis, suggesting that elevated cytosolic free Ca2+ alone, albeit necessary, is not sufficient. Apparently, parallel signaling pathways, such as JNK activation, are required to induce apoptosis.
Because both Ca2+ and bicarbonate activate sAC synergistically and bile salts have calcium ionophore properties, BSIA is expected to be most prominent when intracellular bicarbonate concentration is elevated. In light of the pathogenesis of PBC, this is highly relevant as bicarbonate secretion of cholangiocytes is impaired as a result of AE2 down‐regulation,5, 6, 8 which renders cholangiocytes more sensitive to BSIA. Accordingly, we propose the following working hypothesis on how sAC is involved in the pathogenesis of PBC in the context of AE2 down‐regulation (Fig. 7). On the one hand, impaired bicarbonate secretion (due to reduced AE2 expression) weakens the protective “bicarbonate umbrella” and permits more bile salt to cross the plasma membrane. On the other hand, the retained bicarbonate induces sAC expression and activates sAC, thereby promoting BSIA. The impact of reduced AE2 expression in PBC cholangiocytes is thus 2‐fold.
Figure 7.
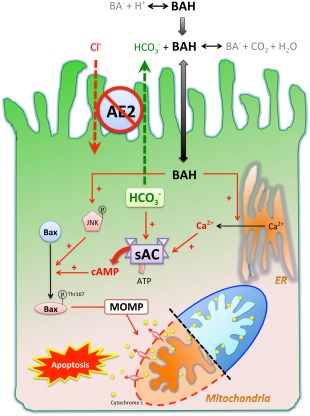
Working hypothesis of how sAC is involved in the pathogenesis of PBC in the context of AE2 down‐regulation. Protonated bile salts are nonpolar and enter cells at a higher rate than their deprotonated counterparts. AE2 on the apical membrane of cholangiocytes generates a bicarbonate umbrella that deprotonates the otherwise proapoptotic bile salts. Reduced AE2 expression in PBC cholangiocytes causes more bile acids to enter. Once inside the cells, bile salts cause JNK phosphorylation and activating phosphorylation of Bax at Thr167, which promote mitochondrial translocation of Bax, leading to MOMP, cytochrome c release, and apoptosis. Inhibition of sAC prevents bile salt‐induced Bax phosphorylation at Thr167 but not JNK phosphorylation. By acting as a calcium ionophore, bile salts cause Ca2+ release from endoplasmic reticulum, which is necessary but not sufficient for BSIA. On the other hand, the intracellular bicarbonate accumulation secondary to impaired secretion increases sAC activity synergistically with bile salt‐triggered Ca2+ release and enhances MOMP and apoptosis. Abbreviations: ATP, adenosine triphosphate; BA‐, deprotonated bile salt; BAH, protonated bile salt; ER, endoplasmic reticulum.
In conclusion, the present study demonstrates that sAC is a critical mediator of BSIA and an attractive therapeutic target. The bicarbonate anion, although indispensable in bile salt‐independent choleresis and reducing permeation of bile salts, is in fact a double‐edged sword in the hands of AE2 and sAC. In a state of reduced AE2 expression (PBC), intracellular bicarbonate accumulation as a result of reduced bicarbonate secretion activates sAC and makes cholangiocytes more vulnerable to bile salts and other apoptotic triggers.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28550/suppinfo.
Supporting Information
Potential conflict of interest: Nothing to report.
Supported by a ZonMW TOP grant (TOP 40‐00812‐98‐10054, to R.O.E.).
References
- 1. Selmi C, Bowlus CL, Gershwin ME, Coppel RL. Primary biliary cirrhosis. Lancet 2011;377:1600–1609. [DOI] [PubMed] [Google Scholar]
- 2. Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology 2004;127:485–492. [DOI] [PubMed] [Google Scholar]
- 3. Hirschfield GM, Liu X, Han Y, Gorlov IP, Lu Y, Xu C, et al. Variants at IRF5‐TNPO3, 17q12‐21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet 2010;42:655–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. Genome‐wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet 2011;43:329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Medina JF, Martinez A, Vazquez JJ, Prieto J. Decreased anion exchanger 2 immunoreactivity in the liver of patients with primary biliary cirrhosis. Hepatology 1997;25:12–17. [DOI] [PubMed] [Google Scholar]
- 6. Prieto J, Qian C, Garcia N, Diez J, Medina JF. Abnormal expression of anion exchanger genes in primary biliary cirrhosis. Gastroenterology 1993;105:572–578. [DOI] [PubMed] [Google Scholar]
- 7. Vazquez JJ, Vazquez M, Idoate MA, Montuenga L, Martinez‐Anso E, Castillo JE, et al. Anion exchanger immunoreactivity in human salivary glands in health and Sjogren's syndrome. Am J Pathol 1995;146:1422–1432. [PMC free article] [PubMed] [Google Scholar]
- 8. Prieto J, Garcia N, Marti‐Climent JM, Penuelas I, Richter JA, Medina JF. Assessment of biliary bicarbonate secretion in humans by positron emission tomography. Gastroenterology 1999;117:167–172. [DOI] [PubMed] [Google Scholar]
- 9. Beuers U, Hohenester S, de Buy Wenniger LJ, Kremer AE, Jansen PL, Elferink RP. The biliary HCO3 ‐ umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010;52:1489–1496. [DOI] [PubMed] [Google Scholar]
- 10. Spivey JR, Bronk SF, Gores GJ. Glycochenodeoxycholate‐induced lethal hepatocellular injury in rat hepatocytes. Role of ATP depletion and cytosolic free calcium. J Clin Invest 1993;92:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rodrigues CM, Fan G, Ma X, Kren BT, Steer CJ. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest 1998;101:2790–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodrigues CM, Ma X, Linehan‐Stieers C, Fan G, Kren BT, Steer CJ. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ 1999;6:842–854. [DOI] [PubMed] [Google Scholar]
- 13. Hohenester S, Wenniger LM, Paulusma CC, van Vliet SJ, Jefferson DM, Elferink RP, et al. A biliary HCO3 ‐ umbrella constitutes a protective mechanism against bile acid‐induced injury in human cholangiocytes. Hepatology 2012;55:173–183. [DOI] [PubMed] [Google Scholar]
- 14. Levin LR, Buck J. Physiological roles of acid‐base sensors. Annu Rev Physiol 2015;77:347–362. [DOI] [PubMed] [Google Scholar]
- 15. Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR, et al. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 2000;289:625–628. [DOI] [PubMed] [Google Scholar]
- 16. Mardones P, Medina JF, Elferink RP. Activation of cyclic AMP signaling in Ae2‐deficient mouse fibroblasts. J Biol Chem 2008;283:12146–12153. [DOI] [PubMed] [Google Scholar]
- 17. Buck J, Sinclair ML, Schapal L, Cann MJ, Levin LR. Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. Proc Natl Acad Sci USA 1999;96:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Braun T, Dods RF. Development of a Mn2+‐sensitive, “soluble” adenylate cyclase in rat testis. Proc Natl Acad Sci USA 1975;72:1097–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geng W, Wang Z, Zhang J, Reed BY, Pak CY, Moe OW. Cloning and characterization of the human soluble adenylyl cyclase. Am J Physiol Cell Physiol 2005;288:C1305–C1316. [DOI] [PubMed] [Google Scholar]
- 20. Strazzabosco M, Fiorotto R, Melero S, Glaser S, Francis H, Spirli C, et al. Differentially expressed adenylyl cyclase isoforms mediate secretory functions in cholangiocyte subpopulation. Hepatology 2009;50:244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Steegborn C. Structure, mechanism, and regulation of soluble adenylyl cyclases—similarities and differences to transmembrane adenylyl cyclases. Biochim Biophys Acta 2014;1842:2535–2547. [DOI] [PubMed] [Google Scholar]
- 22. Jaiswal BS, Conti M. Calcium regulation of the soluble adenylyl cyclase expressed in mammalian spermatozoa. Proc Natl Acad Sci USA 2003;100:10676–10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Litvin TN, Kamenetsky M, Zarifyan A, Buck J, Levin LR. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J Biol Chem 2003;278:15922–15926. [DOI] [PubMed] [Google Scholar]
- 24. Kumar S, Kostin S, Flacke JP, Reusch HP, Ladilov Y. Soluble adenylyl cyclase controls mitochondria‐dependent apoptosis in coronary endothelial cells. J Biol Chem 2009;284:14760–14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Acin‐Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab 2009;9:265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Di Benedetto G, Scalzotto E, Mongillo M, Pozzan T. Mitochondrial Ca2+ uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab 2013;17:965–975. [DOI] [PubMed] [Google Scholar]
- 27. Zippin JH, Chen Y, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA, et al. Compartmentalization of bicarbonate‐sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J 2003;17:82–84. [DOI] [PubMed] [Google Scholar]
- 28. Zippin JH, Farrell J, Huron D, Kamenetsky M, Hess KC, Fischman DA, et al. Bicarbonate‐responsive “soluble” adenylyl cyclase defines a nuclear cAMP microdomain. J Cell Biol 2004;164:527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang JC, Oude‐Elferink RP. Role of the bicarbonate‐responsive soluble adenylyl cyclase in pH sensing and metabolic regulation. Front Physiol 2014;5:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tresguerres M, Levin LR, Buck J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int 2011;79:1277–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Appukuttan A, Kasseckert SA, Micoogullari M, Flacke JP, Kumar S, Woste A, et al. Type 10 adenylyl cyclase mediates mitochondrial Bax translocation and apoptosis of adult rat cardiomyocytes under simulated ischaemia/reperfusion. Cardiovasc Res 2012;93:340–349. [DOI] [PubMed] [Google Scholar]
- 32. Appukuttan A, Kasseckert SA, Kumar S, Reusch HP, Ladilov Y. Oxysterol‐induced apoptosis of smooth muscle cells is under the control of a soluble adenylyl cyclase. Cardiovasc Res 2013;99:734–742. [DOI] [PubMed] [Google Scholar]
- 33. Kumar S, Appukuttan A, Maghnouj A, Hahn S, Peter Reusch H, Ladilov Y. Suppression of soluble adenylyl cyclase protects smooth muscle cells against oxidative stress‐induced apoptosis. Apoptosis 2014;19:1069–1079. [DOI] [PubMed] [Google Scholar]
- 34. Grubman SA, Perrone RD, Lee DW, Murray SL, Rogers LC, Wolkoff LI, et al. Regulation of intracellular pH by immortalized human intrahepatic biliary epithelial cell lines. Am J Physiol Gastrointest Liver Physiol 1994;266:G1060–G1070. [DOI] [PubMed] [Google Scholar]
- 35. Webster CR, Usechak P, Anwer MS. cAMP inhibits bile acid‐induced apoptosis by blocking caspase activation and cytochrome c release. Am J Physiol Gastrointest Liver Physiol 2002;283:G727–G738. [DOI] [PubMed] [Google Scholar]
- 36. Kim BJ, Ryu SW, Song BJ. JNK‐ and p38 kinase‐mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem 2006;281:21256–21265. [DOI] [PubMed] [Google Scholar]
- 37. Hunt GR, Jawaharlal K. A 1H‐NMR investigation of the mechanism for the ionophore activity of the bile salts in phospholipid vesicular membranes and the effect of cholesterol. Biochim Biophys Acta 1980;601:678–684. [DOI] [PubMed] [Google Scholar]
- 38. Combettes L, Berthon B, Doucet E, Erlinger S, Claret M. Characteristics of bile acid‐mediated Ca2+ release from permeabilized liver cells and liver microsomes. J Biol Chem 1989;264:157–167. [PubMed] [Google Scholar]
- 39. Oelberg DG, Wang LB, Sackman JW, Adcock EW, Lester R, Dubinsky WP. Bile salt‐induced calcium fluxes in artificial phospholipid vesicles. Biochim Biophys Acta 1988;937:289–299. [DOI] [PubMed] [Google Scholar]
- 40. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011;476:336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher‐Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011;476:341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Banales JM, Saez E, Uriz M, Sarvide S, Urribarri AD, Splinter P, et al. Up‐regulation of microRNA 506 leads to decreased Cl‐/HCO3 ‐ anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology 2012;56:687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Malbon CC, Tao J, Shumay E, Wang HY. AKAP (A‐kinase anchoring protein) domains: beads of structure‐function on the necklace of G‐protein signalling. Biochem Soc Trans 2004;32:861–864. [DOI] [PubMed] [Google Scholar]
- 44. Kamenetsky M, Middelhaufe S, Bank EM, Levin LR, Buck J, Steegborn C. Molecular details of cAMP generation in mammalian cells: a tale of two systems. J Mol Biol 2006;362:623–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sayner SL, Alexeyev M, Dessauer CW, Stevens T. Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circ Res 2006;98:675–681. [DOI] [PubMed] [Google Scholar]
- 46. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL‐2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014;15:49–63. [DOI] [PubMed] [Google Scholar]
- 47. Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N, et al. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem 2004;279:21085–21095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28550/suppinfo.
Supporting Information