

Abstract
Heme-containing proteins have recently attracted increasing attention for their ability to promote synthetically valuable transformations not found in nature. Following the recent discovery that engineered variants of myoglobin can catalyze the direct conversion of organic azides to aldehydes, we investigated the azide oxidative deamination reactivity of a variety of hemoproteins featuring different heme coordination environments. Our studies show that although several heme-containing enzymes possess basal activity in this reaction, an engineered variant of the bacterial cytochrome P450 CYP102A1 constitutes a particularly efficient biocatalyst for promoting this transformation, exhibiting a broad substrate scope along with high catalytic activity (up to 11,300 TON), excellent chemoselectivity, and enhanced reactivity toward secondary alkyl azides to yield ketones. Mechanistic studies and Michaelis-Menten analyses provided insights into the mechanism of the reaction and the impact of active site mutations on the catalytic properties of the P450. Altogether, these studies demonstrate that engineered P450 variants represent promising biocatalysts for the synthesis of aryl aldehydes and ketones via the oxidative deamination of alkyl azides under mild reaction conditions.
Keywords: azide oxidative deamination, aldehyde synthesis, P450 BM-3, protein engineering, hemoprotein
Graphical abstract
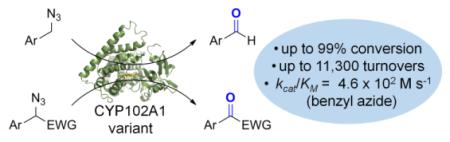
New reaction for P450s: An engineered variant of CYP102A1 efficiently catalyzes the oxidative deamination of primary alkyl azides to yield aldehydes, exhibiting a broad substrate scope along with high catalytic activity and excellent chemoselectivity. The enhanced reactivity of this enzyme also enabled the conversion of selected secondary alkyl azides to ketones.
1. Introduction
Cytochrome P450s constitutes a superfamily of iron-dependent, heme-containing oxygenases which play an important role in drug metabolism, biodegradation, and biosynthesis of secondary metabolites.[1] These enzymes have received significant attention for their ability to hydroxylate aliphatic and aromatic C–H bonds, a challenging reaction to achieve by chemical means[2] Furthermore, P450s are known to promote a variety of other oxidative transformations including heteroatom dealkylation, carbon-carbon bond cleavage, rearrangement reactions, and Baeyer-Villiger reactions.[3] More recently, the scope of cytochrome P450s has been extended to a number of important, 'non-native' reactions useful for the construction of carbon–carbon,[4] carbon–nitrogen,[5] and nitrogen-sulfur[6] bonds.
Our group has recently reported that the heme-containing protein myoglobin (Mb) constitutes a promising scaffold for promoting a variety of synthetically useful transformations mediated by metal-carbenoid and -nitrenoid species.[7] In particular, previous work showed that engineered variants of this hemoprotein can catalyze the oxidative deamination of organic azides to generate aldehydes.[8] In comparison to classical methods involving alcohol oxidation with toxic chromium-based reagents, this transformation provides a convenient approach to the synthesis of aldehydes starting from a non-oxygenated functional group and using readily accessible alkyl azides. Furthermore, synthetic catalysts (e.g., MoO2(S2CNEt2)2) currently available to promote this reaction suffer from poor catalytic efficiency (20-200 turnovers) and require harsh reaction conditions (reflux in toluene/water mixture).[9] In the interest of comparing and contrasting the reactivity of hemoproteins featuring different heme coordination environments in the context of non-native reactions, we investigated and report here the azide oxidative deamination reactivity of a panel of different heme-containing enzymes, including a catalase, a peroxidase, and wild type and engineered variants of a bacterial cytochrome P450. These studies led to the identification of an engineered variant of CYP102A1 as a superior biocatalyst for the conversion of a broad range of aryl-substituted alkyl azides to the corresponding aldehydes and ketones. In addition, insights into the mechanism and catalytic properties of this enzyme were gained via a combination of kinetic studies and isotopic labeling experiments.
To investigate the scope of hemoprotein-catalyzed azide-to-aldehyde oxidation, we initially selected a panel of different heme-containing enzymes consisting of bovine catalase (Cat), horseradish peroxidase (HRP), and the bacterial cytochrome CYP102A1[10] (also known as P450BM3)[11]. These enzymes feature a significantly different heme coordination environment compared to each other and to the previously investigated myoglobin (Mb). In Cat and CYP102A1, the amino acid ligand involved in coordinating the heme iron is a tyrosine and cysteine residue, respectively[12], as opposed to histidine in Mb[13]. The heme group in HRP is also bound through a histidine residue,[14] but a strong H-bonding interaction between this proximal His and a neighboring aspartic acid residue confers a considerable anionic character to the former, a structural feature that is not present in Mb. Finally, hemoglobin (Hb) was also considered to evaluate the effect of tetrameric structure on the target reactivity as opposed to the monomeric structure of Mb.
As a model reaction, the conversion of benzyl azide 1 to benzaldehyde 2a under anaerobic conditions in phosphate buffer (pH 7.0) and in presence of sodium dithionite (Na2S2O4) as a reductant (Figure 1a) was used to assess the relative efficiency of these hemoproteins in promoting azide oxidative deamination. Under these conditions, Cat, HRP and Hb show only low to moderate reactivity, supporting about 5, 63 and 195 turnovers (TON), respectively (Figure 1b-c). These TON values are lower or comparable to those achieved with free hemin under similar reaction conditions (~100 turnovers).[8] The poor performance of Hb was somewhat unexpected in light of the high reactivity of the structurally related Mb in this transformation (1,650 TON)[8] and it could stem from unfavorable allosteric effects in the case of the oligomeric protein. In contrast, wild-type CYP102A1 show considerably higher azide oxidation activity, supporting 1,400 catalytic turnovers for formation of the desired product 2a and providing a product conversion ratio of 18% (Figure 1b-c) The reaction also produced small amounts of benzylamine 2b (0.5%) and N-benzyl-benzylimine (2c, <3%), as observed in the Mb-catalyzed reaction.[8] Formation of the benzylamine 2b byproduct was ascribed to an unproductive pathway resulting from decomposition of the azide followed by reduction and protonation of the resulting nitrene-heme intermediate, whereas 2c originates from condensation of 2b with the aldehyde product.[8] Importantly, these initial results clearly evidenced the impact of the heme coordination environment on the reactivity of the different hemoproteins toward organic azides, thereby expanding previous findings from our group.[7e] Indeed, while HRP was previously found to promote the intramolecular C–H amination of arylsulfonyl azides with comparable or higher activity than P450s or Mb,[5a, 5b, 7e] this enzyme shows significantly reduced activity toward azide-to-aldehyde conversion compared to the latter hemoproteins. Conversely, the data in Figure 1b-c suggested that, despite the inherent differences between their active sites, the thiolate-bound heme in the P450 is equally efficient as the histidinyl-bound heme cofactor in Mb in promoting the oxidative deamination of alkyl azide 1 (1,400 TON vs. 1,650 TON for wild-type Mb[8]).
Figure 1.

Catalytic activity of various hemoproteins in the oxidative deamination of benzyl azide 1 to benzaldehyde 2a (a). (b-c) Reactions (400 μL) were conducted under anaerobic conditions with 10 mM BnN3, 10 mM Na2S2O4, and 1 μM protein for 24 hours at room temperature. TON = nmol aldehyde / nmol catalyst. Product yield is based on conversion of 1 to 2a as determined by gas chromatography. (d) Reaction rates as determined over the first 10 minutes of the reaction. (e) TON vs. pH plot for FL#62 reaction.
Given the promising activity of wild-type CYP102A1, we next extended our investigations to two engineered variants of this enzyme, namely CYP102A1(F87A) and FL#62. The former variant was chosen because of the well documented ability of the F87A mutation in expanding the substrate profile of CYP102A1 toward non-native substrates in the context of monooxygenation reactions.[11, 15] In addition to possessing a broad substrate profile,[16] the FL#62 variant, which carries 6 active site mutations and 10 additional mutations within its heme domain, was chosen because of its high reactivity in the context of the C–H amination of azide-based reagents.[5a, 5b] As shown by the data summarized in Figure 1, CYP102A1(F87A) showed only slightly improved TON values for the conversion of benzyl azide 1 into benzaldehyde 2a compared to the wild-type P450 (1,600 vs. 1,400 turnovers). In contrast, FL#62 was found to catalyze this transformation with considerably higher efficiency (Figure 1b-c), supporting 9,600 catalytic turnovers and providing nearly quantitative product conversion (92%) at a catalyst loading of merely 0.001 mol%. At lower catalyst loadings (0.0005 mol%), a TON value as high as 11,300 was measured for this enzyme. Notably, the FL#62 reaction was found to proceed also with excellent chemoselectivity, leading to the clean formation of 2a without producing detectable amounts of the 2b or 2c byproducts. Altogether, these properties make FL#62 not only a considerably more efficient catalytic system for azide-to-aldehyde conversion than synthetic Mo-based catalysts (50-99% conversions using 10 mol% at 100°C in water/toluene mixture) [9a], but also a superior biocatalyst compared to the best engineered Mb variant (Mb(H64V,V68A)) identified in the course of our previous studies.[8]
Further characterization studies revealed a noticeable effect of pH on FL#62 azide oxidative deamination activity, with maximal TON values being achieved around neutral pH (Figure 1e). A similar pH dependence was observed for the Mb-catalyzed reaction.[8] From time course experiments, the FL#62-catalyzed conversion of 1 to 2b was determined to proceed with a rate of approximately 14 turnovers min−1 over the first 10 minutes (Figure 1d). This rate is lower than that observed for the Mb-based catalysts (250 min−1)[8], but the FL#62-catalyzed reaction proceeds with higher selectivity (i.e., no 2b/2c formation) in addition to the higher TON. The initial product formation rates for FL#62 and the other hemoproteins investigated also correlated well with the TON values observed in the corresponding reactions (Figure 1b and 1d).
Next, the substrate scope of the FL#62 biocatalyst was investigated using a wide panel of different organic azides. As shown in Table 1, the majority of these compounds could be readily oxidized to the corresponding aldehydes in good to excellent GC yields (49-99%). In these reactions, the P450 was determined to support from 4,650 to 9,660 TON. In addition, the reaction proceeds with high selectivity (>90-95%) in particular for the alkyl azides most efficiently processed by the enzyme. The viable substrates include variously mono- and disubstituted benzyl azide derivatives (3a, 4a, 6a, 8a, 10a, 11a), (hetero)aryl-substituted primary alkyl azides (17a, 18a), and secondary alkyl azides (15a ,16a), which support the broad substrate scope of the FL#62 catalyst. Modest (20-30%) to low (<20%) product conversions were observed instead with benzyl azide derivatives that carry one or two substituents at the ortho position(s) (i.e., 7b, 9b and 13b). These results suggest a negative effect on the enzymatic transformation of increased steric hindrance in proximity to the azido group. Whereas the conversion yields of these P450-catalyzed reactions are consistently higher than those achieved with engineered myoglobins,[8] particularly striking is the considerably higher efficiency of the P450 catalyst in the synthesis of ketones 15b and 16b from the secondary alkyl azides 15a and 16a, respectively. Indeed, while this reaction proceeds inefficiently in the presence of the Mb catalyst (280-400 TON, <2% conversion)[8], it is effectively catalyzed by the engineered P450 (4,650-7,470 TON), resulting in much improved product conversions (49-76%).
Table 1.
Substrate scope for FL#62-catalyzed oxidative deamination of alkyl azides. Reaction conditions: 10 mM azide, 1 μM FL#62, 10 mM Na2S2O4.
| |||||
|
| |||||
| Prod. | Structure | R1 | R2 |
% conv.
(% select.) |
TON |
|
| |||||
| 3b |
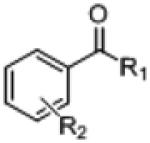
|
H | p-CH3 | 58 (94) | 6,000 |
|
|
|
||||
| 4b | H |
p- OCH3 |
99 (99) | 9,440 | |
|
|
|
||||
| 5b | H |
p- NO2 |
25 (84) | 2,670 | |
|
|
|
||||
| 6b | H | p-CF3 | 69 (93) | 6,820 | |
|
|
|
||||
| 7b | H | o-CH3 | 22 (79) | 2,000 | |
|
|
|
||||
| 8b | H | o-F | 96 (98) | 9,660 | |
|
|
|
||||
| 9b | H |
o- NO2 |
1 (18) | 60 | |
|
|
|
||||
| 10b | H |
m- NO2 |
67 (94) | 6,600 | |
|
|
|
||||
| 11b | H |
o,p- di-F |
51 (90) | 5,850 | |
|
|
|
||||
| 12b | H |
m,m- di- CH3 |
36 (88) | 3,540 | |
|
|
|
||||
| 13b | H |
o,o- di-Cl |
7 (72) | 720 | |
|
|
|
||||
| 14b | CH3 | Ph | 5 (51) | 490 | |
|
|
|
||||
| 15b | CF3 | Ph | 76 (98) | 7,470 | |
|
|
|
||||
| 16b | CO2Me | Ph | 49 (91) | 4,650 | |
|
| |||||
| 17b |
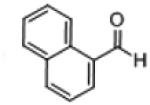
|
70 (94) | 7,480 | ||
|
| |||||
| 18b |
|
97 (98) | 9,500 | ||
|
| |||||
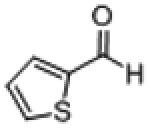
| 19b |
|
19 (85) | 1,970 | ||
|
| |||||
To further evaluate the synthetic utility of FL#62 in the context of this reaction, a larger scale reaction with benzyl azide 1 (50 mg, 0.38 mmol) was carried out in the presence of 0.05 mol% FL#62 in phosphate buffer (pH 7.0) at room temperature. After a simple extraction and purification step, the desired benzaldehyde product 2a was obtained in 87% isolated yield (35 mg, 0.33 mmol), thus supporting the scalability of this biocatalytic transformation.
In terms of mechanism, we previously proposed that this reaction involves heme-catalyzed decomposition of the heme-bound alkyl azide to give an imido-iron(IV) intermediate. The latter then undergoes a tautomerization to an imine-iron(II) complex, followed by hydrolysis of the imine (in either free or heme-bound form) to yield the aldehyde product (Figure 2). Experimental evidence in support of the imine tautomerization step included the observation of a significant primary kinetic isotope effect upon H→D substitution at the level of the α carbon in the azide substrate.[8] Interestingly, a related imine intermediate was recently invoked in the azide-to-nitrile conversion catalyzed by non-heme iron-dependent dioxygenases.[17] Furthermore, 18O labelling experiments indicated that the oxygen atom in the aldehyde product derives from a hydrolytic process.[8] In light of the higher catalytic activity (TON) and slower rate of the P450-catalyzed reaction versus the Mb-catalyzed one, we thus wondered whether these biocatalytic transformations shared an analogous mechanism or not. To this end, we measured the KIE for the FL#62-catalyzed oxidative deamination of benzyl azide (1) from competition experiments in the presence of an equimolar concentration of the deuterated analog, d2−1. LC-MS analysis of this reaction (SI Figure S3a) yielded a KIE value (kH/kD) of 1.9 ± 0.2 at 22°C, which is comparable to that observed in the presence of Mb(H64V,V68A) as the catalyst (kH/kD = 1.7 ± 0.3)[8]. In addition, the FL#62-catalyzed conversion of benzyl azide (1) in the presence of 18O-labeled water resulted in the formation of 18O-labeled aldehyde product 2a(18O) (SI Figure S3b), as observed for the Mb-catalyzed reaction. Altogether, these results hint at important mechanistic similarities between the two systems, although differences between them are also apparent. Indeed, the negligible formation of the amine byproduct in the FL#62 reactions across all the tested azide substrates suggests that the unproductive pathway leading to this reduction reduction is largely disfavored in the case of the P450 system. In addition, the significantly higher (10- to 15-fold) conversions and TON values measured for the FL#62-catalyzed formation of 15b and 16b versus 14b (Table 1) suggest that the P450 catalyst is significantly more sensitive to an increase in the acidity of the alpha protons compared to the Mb catalyst (3- to 4-fold higher TON for 15b and 16b, respectively, compared to 14b).[8]
Figure 2.
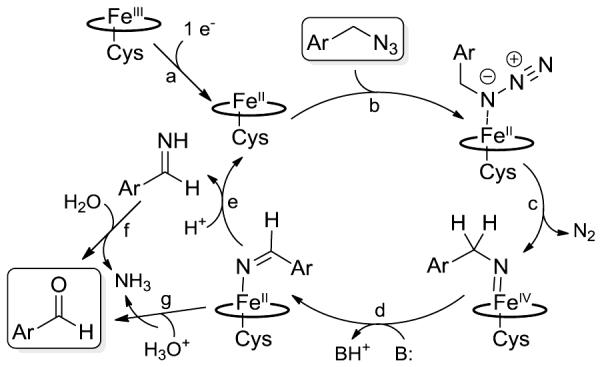
Proposed mechanism and catalytic steps for the P450-catalyzed oxidation of alkyl azides to aldehydes.
Kinetic experiments with wild-type CYP102A1, CYP102A1(F87A), and FL#62 were carried out to gain further insights into the catalytic properties of these enzymes. In the reaction with model substrate 1, all of these P450s were found to follow Michaelis-Menten kinetics (Figure 3). Similar kinetic parameters (KM, kcat) were determined for the parent enzyme and its single mutant variant, CYP102A1(F87A), which is in line with their similar performance in terms of TON and product conversion (Table 1). Conversely, the superior catalytic performance of FL#62 is reflected by an 8-fold higher catalytic efficiency (kcat/KM = 4.6 × 102 M−1 s−1) compared to the parent enzyme and F87A variant, which derives as a result of a two-fold lower KM and a four-fold faster turnover number, kcat (Table 2). While the catalytic efficiency of FL#62 in this non-native reaction is significantly lower than that of CYP102A1 for the hydroxylation of fatty acids (kcat/KM ~ 105 M s−1)[18]], but it falls in the same range of those exhibited by P450s involved in drug and hormone metabolism (e.g., P4501B1/retinol, kcat/KM ~ 7 × 103 M s−1)[19] For all the P450s, incubation with benzyl azide resulted in a small (<5-10%) but detectable heme spin shift (SI Figure S4), which is indicative of the displacement of the axial water ligand on the heme by the bound substrate. Titration experiments revealed a higher binding affinity of FL#62 for benzyl azide as compared to wild type CYP102A1 and CYP102A1(F87A) (KD of 68 μM vs. 93 and 84 μM, respectively; Table 2; SI Figure S4).
Figure 3.
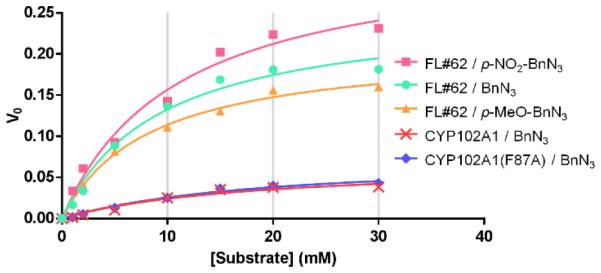
Overlay plot of initial velocity (V0) versus substrate concentration for the reactions of CYP102A1 and its variants with selected alkyl azide substrates. The corresponding kinetic parameters (kcat, KM, kcat/KM) are reported in Table 2.
Table 2.
Substrate binding affinity and Michaelis-Menten parameters corresponding to CYP102A1, CYP102A1(F87A), FL#62 and selected alkyl azide substrates. Equilibrium dissociation constants (KD) were calculated from substrate-induced heme spin shift experiments (SI Figure S4). The kinetic parameters (kcat, KM, kcat/KM) were calculated via non-linear fitting of the V0 vs. concentration curves (Figure 3) to the Michaelis-Menten equation.
| Enzyme | Substr ate |
KD (μM) |
KM (M) |
kcat (min−1) |
kcat/KM (M−1·s−1) |
|---|---|---|---|---|---|
|
| |||||
| CYP102A1 | BnN3 (1) | 93 (± 9) |
1.8 × 10−2
(± 0.7) |
66 (± 13) |
61 (± 27) |
|
| |||||
| CYP102A1 (F87A) |
BnN3
(1) |
84 (± 13) |
1.9 × 10−2
(± 0.5) |
75 (± 11) |
66 (± 20) |
|
| |||||
| FL#62 | BnN3
(1) |
68 (± 10) |
9.2 × 10-3
(± 1.9) |
255 (± 20) |
4.6 × 102 (± 1.0) |
|
| |||||
| FL#62 |
p-OMe- BnN3 (4a) |
107 (± 19) |
8.3 × 10−3
(± 1.0) |
209 (± 9) |
4.2 × 102
(± 0.5) |
|
| |||||
| FL#62 |
p-NO2- BnN3 (5a) |
259 (± 36) |
1.1 × 10−2
(± 0.2) |
329 (± 29) |
5.0 × 102
(± 1.0) |
Another interesting result emerging from our substrate scope investigations with FL#62 is the noticeable difference between the TON values associated with the synthesis of electrondeficient benzyl aldehydes (5b; 2,670 TON) as compared to electronrich ones (4b; 9,440). To elucidate the basis of this trend, we extended the substrate binding experiments and Michaelis-Menten analyses to the FL#62-catalyzed oxidative deamination of p-nitro-benzyl azide (5a) and p-methoxy-benzyl azide (4a). While p-nitro-benzyl azide has weaker affinity for FL#62 compared to benzyl azide and p-methoxy-benzyl azide (Table 2), no significant differences were found among the kinetic parameters (KM, kcat) and catalytic efficiency (kcat/KM) of the enzyme for the transformation of these substrates, suggesting that other factors must be at the basis of the less efficient conversion of 5a as compared to 4a and 1.
To further examine this aspect, we performed product inhibition experiments in which reaction mixtures containing FL#62 and 1, 4a, or 5a as the substrates were spiked with increasing concentrations (0-10 mM) of the corresponding aldehyde products. Interestingly, the addition of para-nitro-benzaldehyde (5b) significantly reduced the conversion of 5a (Figure 4a), evidencing the occurrence of product inhibition. In contrast, no product inhibition was observed within the concentration range tested for both benzyl azide (1) (SI Figure S5) and para-methoxy-benzyl azide (4b) (Figure 4b), thus explaining the greater yields observed in the presence of these substrates (Table 1).
Figure 4.

Product inhibition experiments for (a) the reaction of FL#62 and p-nitro-benzyl azide (5a) in the presence of added p-nitro-benzaldehyde (5b) and (b) the reaction of FL#62 and p-methoxy-benzyl azide (4a) in the presence of added p-methoxy-benzaldehyde (4b). See caption of SI Figure S5 for further details.
In conclusion, our results demonstrate that cytochrome P450s are promising catalysts for the synthesis of aromatic aldehydes via the oxidative deamination of alkyl azides. In particular, we established that the engineered CYP102A1 variant FL#62 represents a superior biocatalyst for this transformation compared to other heme-containing enzymes and the previously reported Mb catalysts. In addition to excellent chemoselectivity and high catalytic activity (up to 11,300 TON) across a broad range of alkyl azides, the enhanced reactivity of this P450 enabled extension of this transformation to the synthesis of ketones from secondary azides. These studies contributed valuable insights into the differential reactivity of hemoproteins, and engineered variants thereof, in the context of non-native reactions, which is expected to aid the future development of biocatalysts for a growing number of synthetically useful transformations not found in nature.
Supplementary Material
Acknowledgements
This work was supported by the U.S. National Institute of Health grant GM098628. MS instrumentation was supported by the U.S. NSF grant CHE-0946653.
References
- [1].a) Guengerich FP. Nature Rev. Drug Discov. 2002;1:359–366. doi: 10.1038/nrd792. [DOI] [PubMed] [Google Scholar]; b) Kumar S. Expert Opin. Drug Metab. Toxicol. 2010;6:115–131. doi: 10.1517/17425250903431040. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem. Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]; d) Poulos TL. Biochem. Biophys. Res. Commun. 2005;338:337–345. doi: 10.1016/j.bbrc.2005.07.204. [DOI] [PubMed] [Google Scholar]
- [2].a) Fasan R. ACS Catal. 2012;2:647–666. [Google Scholar]; b) Lewis JC, Coelho PS, Arnold FH. Chem. Soc. Rev. 2011;40:2003–2021. doi: 10.1039/c0cs00067a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chefson A, Auclair K. Mol. Biosys. 2006;2:462–469. doi: 10.1039/b607001a. [DOI] [PubMed] [Google Scholar]; d) Urlacher VB, Girhard M. Trends Biotechnol. 2012;30:26–36. doi: 10.1016/j.tibtech.2011.06.012. [DOI] [PubMed] [Google Scholar]; e) Gillam EM, Hayes MA. Curr. Top. Med. Chem. 2013;13:2254–2280. doi: 10.2174/15680266113136660158. [DOI] [PubMed] [Google Scholar]; f) Grogan G. Curr. Opin. Chem. Biol. 2011;15:241–248. doi: 10.1016/j.cbpa.2010.11.014. [DOI] [PubMed] [Google Scholar]
- [3].a) Isin EM, Guengerich FP. Biochim. Biophys. Acta. 2007;1770:314–329. doi: 10.1016/j.bbagen.2006.07.003. [DOI] [PubMed] [Google Scholar]; b) Ortiz de Montellano PR, Nelson SD. Arch. Biochem. Biophys. 2011;507:95–110. doi: 10.1016/j.abb.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Guengerich FP, Munro AW. J. Biol. Chem. 2013;288:17065–17073. doi: 10.1074/jbc.R113.462275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]; b) Wang ZJ, Renata H, Peck NE, Farwell CC, Coelho PS, Arnold FH. Angew. Chem. Int. Ed. 2014;53:6810–6813. doi: 10.1002/anie.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Singh R, Bordeaux M, Fasan R. ACS Catal. 2014;4:546–552. doi: 10.1021/cs400893n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Singh R, Kolev JN, Sutera PA, Fasan R. ACS Catal. 2015;5:1685–1691. doi: 10.1021/cs5018612. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR, Arnold FH. Angew. Chem. Int. Ed. 2013;52:9309–9312. doi: 10.1002/anie.201304401. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hyster TK, Farwell CC, Buller AR, McIntosh JA, Arnold FH. J. Am. Chem. Soc. 2014;136:15505–15508. doi: 10.1021/ja509308v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Farwell CC, McIntosh JA, Hyster TK, Wang ZJ, Arnold FH. J. Am. Chem. Soc. 2014;136:8766–8771. doi: 10.1021/ja503593n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Bordeaux M, Tyagi V, Fasan R. Angew. Chem. Int. Ed. 2015;54:1744–1748. doi: 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tyagi V, Bonn RB, Fasan R. Chem. Sci. 2015;6:2488–2494. doi: 10.1039/c5sc00080g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sreenilayam G, Fasan R. Chem. Commun. 2015;51:1532–1534. doi: 10.1039/c4cc08753d. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tyagi V, Fasan R. Angew. Chem. Int. Ed. 2016;55:2512–2516. doi: 10.1002/anie.201508817. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Bordeaux M, Singh R, Fasan R. Bioorg. Med. Chem. 2014;22:5697–5704. doi: 10.1016/j.bmc.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Giovani S, Singh R, Fasan R. Chem. Sci. 2016;7:234–239. doi: 10.1039/c5sc02857d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Maddani M, Prabhu KR. Tetrahedron Lett. 2008;49:4526–4530. [Google Scholar]; b) Zhang HP, Dai YZ, Tao LM. J. Chem. Res. 2011:720–722. [Google Scholar]
- [10].Narhi LO, Fulco AJ. J. Biol. Chem. 1986;261:7160–7169. [PubMed] [Google Scholar]
- [11].Whitehouse CJ, Bell SG, Wong LL. Chem. Soc. Rev. 2012;41:1218–1260. doi: 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]
- [12].a) Fita I, Rossmann MG. Proc. Natl. Acad. Sci. USA. 1985;82:1604–1608. doi: 10.1073/pnas.82.6.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li H, Poulos TL. Nat. Struct. Biol. 1997;4:140–146. doi: 10.1038/nsb0297-140. [DOI] [PubMed] [Google Scholar]
- [13].Yang F, Phillips GN., Jr. J. Mol. Biol. 1996;256:762–774. doi: 10.1006/jmbi.1996.0123. [DOI] [PubMed] [Google Scholar]
- [14].Berglund GI, Carlsson GH, Smith AT, Szoke H, Henriksen A, Hajdu J. Nature. 2002;417:463–468. doi: 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- [15].a) Oliver CF, Modi S, Sutcliffe MJ, Primrose WU, Lian LY, Roberts GC. Biochemistry. 1997;36:1567–1572. doi: 10.1021/bi962826c. [DOI] [PubMed] [Google Scholar]; b) Schwaneberg U, Schmidt-Dannert C, Schmitt J, Schmid RD. Anal. Biochem. 1999;269:359–366. doi: 10.1006/abio.1999.4047. [DOI] [PubMed] [Google Scholar]; c) Carmichael AB, Wong LL. Eur. J. Biochem. 2001;268:3117–3125. doi: 10.1046/j.1432-1327.2001.02212.x. [DOI] [PubMed] [Google Scholar]; d) Li QS, Ogawa J, Shimizu S. Biochem. Biophys. Res. Commun. 2001;280:1258–1261. doi: 10.1006/bbrc.2001.4261. [DOI] [PubMed] [Google Scholar]; e) Cirino PC, Arnold FH. Angew. Chem. Int. Ed. 2003;42:3299–3301. doi: 10.1002/anie.200351434. [DOI] [PubMed] [Google Scholar]; f) Lewis JC, Mantovani SM, Fu Y, Snow CD, Komor RS, Wong CH, Arnold FH. Chembiochem. 2010;11:2502–2505. doi: 10.1002/cbic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Weber E, Seifert A, Antonovici M, Geinitz C, Pleiss J, Urlacher VB. Chem. Commun. 2011;47:944–946. doi: 10.1039/c0cc02924f. [DOI] [PubMed] [Google Scholar]; h) Kolev JN, Zaengle JM, Ravikumar R, Fasan R. Chembiochem. 2014;15:1001–1010. doi: 10.1002/cbic.201400060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Zhang K, El Damaty S, Fasan R. J. Am. Chem. Soc. 2011;133:3242–3245. doi: 10.1021/ja109590h. [DOI] [PubMed] [Google Scholar]; b) Zhang KD, Shafer BM, Demars MD, Stern HA, Fasan R. J. Am. Chem. Soc. 2012;134:18695–18704. doi: 10.1021/ja3073462. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kolev JN, O'Dwyer KM, Jordan CT, Fasan R. ACS Chem. Biol. 2014;9:164–173. doi: 10.1021/cb400626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vila MA, Pazos M, Iglesias C, Veiga N, Seoane G, Carrera I. Chembiochem. 2016;17:291–295. doi: 10.1002/cbic.201500653. [DOI] [PubMed] [Google Scholar]
- [18].Noble MA, Miles CS, Chapman SK, Lysek DA, Mackay AC, Reid GA, Hanzlik RP, Munro AW. Biochem. J. 1999;339:371–379. [PMC free article] [PubMed] [Google Scholar]
- [19].Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB. Drug Metab. Dispos. 2004;32:840–847. doi: 10.1124/dmd.32.8.840. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.