

Abstract
The majority of the targeted therapeutic agents in clinical use target proteins and protein function. Although DNA and RNA analyses have been used extensively to identify novel targets and patients likely to benefit from targeted therapies, these are indirect measures of the levels and functions of most therapeutic targets. More importantly, DNA and RNA analysis is ill-suited for determining the pharmacodynamic effects of target inhibition. Assessing changes in protein levels and function is the most efficient way to evaluate the mechanisms underlying sensitivity and resistance to targeted agents. Understanding these mechanisms is necessary to identify patients likely to benefit from treatment and to develop rational drug combinations to prevent or bypass therapeutic resistance. There is an urgent need for a robust approach to assess protein levels and protein function in model systems and across patient samples. While “shot gun” mass spectrometry can provide in-depth analysis of proteins across a limited number of samples, and emerging approaches such as multiple reaction monitoring have the potential to analyze candidate markers, mass spectrometry has not entered into general use because of the high cost, requirement of extensive analysis and support, and relatively large amount of material needed for analysis. Rather, antibody-based technologies, including immunohistochemistry, radio immunoassays, ELISAs and more recently protein arrays, remain the most common approaches for multiplexed protein analysis. Reverse-phase protein array (RPPA) technology has emerged as a robust, sensitive, cost-effective approach to the analysis of large numbers of samples for quantitative assessment of key members of functional pathways that are affected by tumor-targeting therapeutics. The RPPA platform is a powerful approach for identifying and validating targets, classifying tumor subsets, assessing pharmacodynamics, and identifying prognostic and predictive markers, adaptive responses and rational drug combinations in model systems and patient samples. Its greatest utility has been realized through integration with other analytic platforms such as DNA sequencing, transcriptional profiling, epigenomics, mass spectrometry, and metabolomics. The power of the technology is becoming apparent through its use in pathology laboratories and integration into trial design and implementation.
Introduction
Targeted therapy has demonstrated marked activity in a number of diseases. However, for most diseases and most agents, targeted therapy has not delivered on its initial promise: favorable treatment responses have been limited to subsets of patients who have the predicted biomarkers, and often have been of short duration. Some of the apparently limited efficacy of targeted therapy likely arises from an unrealistic expectation that monotherapy would be broadly active in complex and heterogeneous diseases such as solid tumors.
The basic precepts of pharmacokinetics and pharmacodynamics in drug development have too often been ignored in the implementation of targeted therapy. The role of pharmacodynamic analysis in oncology is to determine both the early effects of drug inhibition on the target and downstream signaling, and the longer-term adaptation of the tumor to the effects of the drug. This is limited by the challenges of obtaining and assessing tumor tissue at the appropriate time points after the delivery of a therapeutic agent. Furthermore, biopsy tissues are often small and of diverse tumor and stromal composition; thus, applicable proteomic approaches to effectively analyze the samples are elusive. The objective of such approaches is to broadly determine the effects of the targeted agent (expected and unexpected effects) on the target and on downstream signaling events, cross-talk, and feedback loops. Delayed adaptive responses to the therapeutic agent can inform analytic approaches that can then be used to determine resistance mechanisms and to facilitate the choice of rational combination therapies to prevent resistance and convert what are often cytostatic effects of single agents into cytotoxic effects.
The failure to identify methods to effectively assess early pharmacodynamic responses (whether to use peak inhibition, the area under the curve, or the trough levels of target inhibition as the key determinants of patient response) obviously contributes to the low success rate of current targeted therapy trials. Indeed, for most agents, we do not know which of these criteria indicate an effective response. Perhaps a “hit and run” approach of maximal target inhibition that induces cell death or, conversely, chronic inhibition, will provide the optimal patient benefit. This remains unknown for most agents. Although a systems biology approach allows us to generate predictions through in vitro and animal model studies combined with mathematical modeling, the implementation of these approaches in humans is limited by several challenges. These include accurately measuring the pharmacodynamics of target inhibition, understanding the pharmacokinetics and off-target activity of current targeted agents, and working with a narrow therapeutic index of target inhibition between tumor and normal tissue for many drugs. A careful evaluation of the mechanisms of drug resistance (pre-existing, acquired and adaptive resistance) will be necessary to design rational combination therapies that can prevent the emergence of resistance or overcome established resistance. Indeed, adaptive resistance, the ability of the tumor to rewire signaling networks to bypass the effects of the targeted therapy, may represent the major mechanism of targetable resistance.
In general, targeted therapy is designed to capitalize on the vulnerabilities of tumor cells that arise from the rewiring of functional networks as a consequence of the genomic and epigenetic changes in the tumor or their effects on the tumor microenvironment. Targeted agents typically inhibit, or in rare instances stimulate, protein function. Thus, in order to determine the consequences of target engagement, we need to develop technology that can assess target inhibition as well as the resulting functional changes to the signaling networks. The ability to quantitate RNA levels has rapidly matured; however, the correlations between RNA and protein levels and protein function vary markedly for different proteins, ranging from very high to very limited correlations and thus very limited predictive ability.1, 2 Furthermore, transcriptional analysis, RNA-Seq in particular, is sufficiently complex that it is challenging even under the best circumstances to impute the treatment effects on protein networks and signaling functions. Thus, there is a need to directly assess the effects of the targeted agents on hundreds of different proteins, both predicted and unexpected.
Pharmacodynamic assays for large-scale protein level determination
Two technologies have emerged to fulfill these criteria, each with different strengths and weaknesses. The first technology is mass spectrometry, which can assess thousands of proteins and post-translational events (such as phosphorylation or methylation) that can change function in a single assay.2, 3 Mass spectrometry can unambiguously identify and quantify both wild-type and mutant proteins, identify expected and unexpected proteins and post-translational modifications, and determine the presence of splice variants. Mass spectrometry is, however, limited in its ability to detect rare events, such as proteins or post-translational modifications that are present at low levels, due to a bias toward common molecules such as actin or albumin. This challenge can be partially overcome by new mass spectrometry technologies such as SRM, MRM and SWATH; however, even at their most effective implementation, these technologies lack the sensitivity of a high affinity antibody. The necessity to generate protein fragments that will “fly” in the mass spectrometer also limits the ability to identify post-translational modifications to those with convenient proteolytic cleavage sites and an appropriate charge. Indeed, in a recent analysis of human ovarian and breast cancer xenograft tissue,3 only about 60% of phosphorylation sites identified by parallel antibody-based approaches could be detected, and fewer sites could be quantitated by mass spectrometry. Furthermore, for deep analysis, mass spectrometry requires significant amounts of starting material, expensive equipment, and specialized operators and analytical approaches, all of which limit its utility to a few centers. Nevertheless, mass spectrometry assays designed to assess patient samples have become commercially available and have been implemented in CLIA laboratories.
The second technology is antibody-based analysis, including flow cytometry and its mass spectrometry-based CyTOF variant, multiplexed immunohistochemistry, and forward- and reverse-phase protein arrays. Bar coding of antibodies can allow for concurrent detection of nucleotides and proteins, which facilitates the analysis of DNA, RNA and proteins in a single assay.4 In terms of the analysis of signaling networks, reverse-phase protein arrays (RPPAs) have emerged as a cost-effective, robust, sensitive, and tissue-sparing technology that can assess hundreds of different signaling molecules in a single assay.5–8 This technology is limited by the need for high-quality monospecific antibodies, which is being met by the development of antibodies in commercial and academic laboratories. In addition, large-scale efforts to validate the utility of antibodies to a broad spectrum of targets are being conducted through the Human Proteome Atlas and the National Cancer Institute and research centers with RPPA platforms.9 However, even antibodies predicted to be highly specific can be plagued by unexpected off-target activity, resulting in spurious results. Indeed, the demonstration of a single dominant band of the correct size on Western blotting that correlates in expression with RPPA is a minimal requirement for antibody utility. Multiple antibodies that perform well on Western blotting do not perform well in RPPA because of the essential dot-blot characteristics of the RPPA assay, in which materials that do not enter or run through the Western blot gel are present in the “dot” and can interact with the antibodies. Additional information can increase the confidence that the antibody is indeed faithfully reporting the protein or phosphoprotein levels on RPPAs. Correlations with mRNA levels in the same samples provide “one-way” confidence as translational and post-translational controls can result in markedly different mRNA and protein levels. That is, if RNA and protein levels are highly correlated, this adds to the confidence; if they are not, they are non-informative. For phosphoproteins, the demonstration of increased phosphorylation of the specific site in the presence of growth factors and decreased phosphorylation by phosphatases in the RPPA analysis increases our confidence in the antibody. In cases where the identification of the target of the antibody remains unclear, immunoprecipitation followed by mass spectrometry with confirmation of the target by other approaches may be necessary. Together, these factors indicate that the single antibody, dot-blot nature of the RPPA makes it paramount to validate the antibody targeted in the RPPA assay, itself.
The implementation of RPPAs requires a suite of robotic platforms for printing, staining and imaging; however, these instruments are much less expensive than the equivalent mass spectrometry equipment. Due to the limited availability of high-quality antibodies, as well as the cost and technology constraints, most RPPA centers limit assessments to a range of 300 different targets, with a mixture of total and post-translationally modified antibodies being analyzed. The antibodies used in the assay can be selected to represent key elements of the signaling pathways or cellular functions of interest, which greatly improves the utility of the analysis.1 Of importance, as each antibody is analyzed on a separate slide, it is possible to add additional antibodies to the analysis at any time. Indeed, once denatured and printed on a slide, proteins and phosphoproteins are remarkably stable, with virtually identical results being obtained on slides stained years apart.
Advantages and challenges of RPPA
RPPA, which is essentially a high-throughput “dot blot” enhanced by the use of serial dilutions to improve quantification, was initially popularized by the team of Liotta and Petricoin (Figure 1).5, 10, 11 RPPA has many characteristics that make it a highly attractive approach to multiplex protein analysis. Sample preparation is similar to that used in Western blotting, which is familiar to most laboratories. The ability to rapidly extract and denature proteins decreases protein degradation and protects labile post-translational events such as phosphorylation. Most high-quality antibodies are currently produced against peptides or phosphopeptides, which makes the denaturation conditions used in RPPA optimal for sensitivity and specificity. The technology is sufficiently sensitive to detect femtograms of proteins in nanograms of starting material. This makes RPPAs useful for small sample amounts, including those from needle or core biopsies, rare populations of cells isolated from the tumor or stroma, or different tumor and stromal populations isolated by laser capture microdissection. As over 1,000 samples can be assayed on a single array, this allows for the comparison of many different samples and conditions in a single study, which decreases the complexity of normalization and analysis across assays. However, approaches such as replicate-based normalization, which prints multiple controls across different slides, allows for merging data across experiments.1 The ability to array many samples in dilution series and to perform multiplex analysis facilitates the assessment of proteins with diverse dynamic ranges. Indeed, protein levels of interest within the cell vary by over ten orders of magnitude. Quantification is robust and accurate due to serial dilution of the samples. Although the analysis usually generates relative levels of protein expression and modification (phosphorylation, methylation, or cleavage based on antibody specificity), these can be converted to absolute levels by comparison to protein standards.12 RPPA technology can be applied to human normal tissue or tumor tissue, murine and other animal models, and to cultured cells. The analysis of biomarkers in blood and other fluids with RPPAs has also been demonstrated.13 RPPA technology complements both mass spectrometry and tissue microarrays by measuring total protein and phosphoprotein or cleaved-protein levels as well as other modifications for which there are high-quality antibodies.
Figure 1.
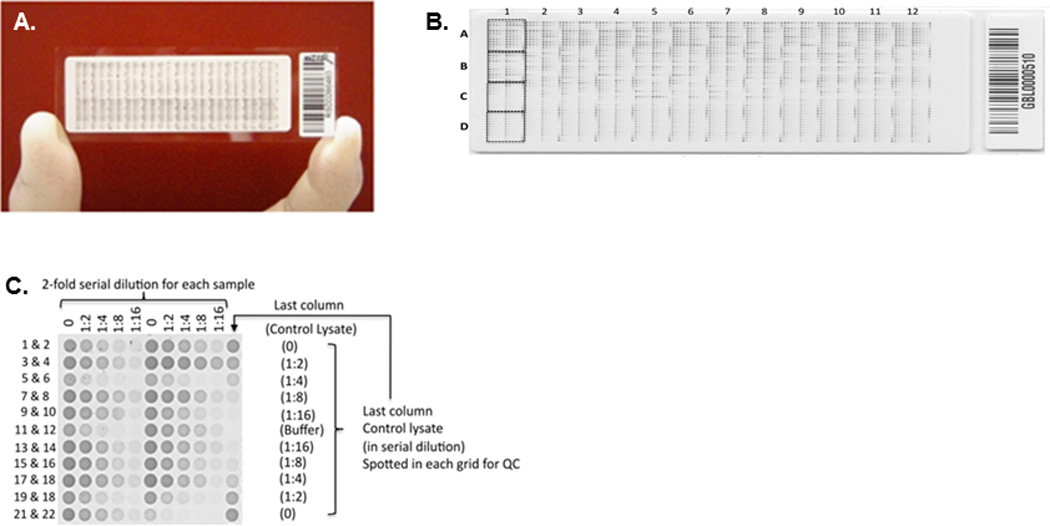
(A) An RPPA slide for measuring a single protein in a set of samples. (B) Magnified view of the slide showing its layout based on the MD Anderson RPPA core specifications. Each slide can accommodate up to 1056 samples and 96 controls. (C) The samples and controls are printed in a 5-step dilution series, with 2-fold dilution at each step, in grids of 11 × 11 spots. Each of the 48 grids can accommodate up to 22 samples and 2 controls. (Reproduced from Tabchy et. al48. Copyright © 2011–2014 Prous Science, S.A.U. or its licensors. All rights reserved.)
Quality control and quantification
As with all technologies, rigorous implementation of standard operating procedures with stringent quality control is necessary to ensure high-quality results. One of the key challenges with RPPAs is the conversion of the serial dilutions into a single relative concentration for each sample. The first step to overcome this challenge is to determine the amount of assayable protein loaded onto each spot, which is usually achieved by using the median of a large number of antibodies and by ensuring that an adequate amount of protein is present to prevent over correction when loading the samples. Software such as SuperCurve or RPPanalyzer14, 15 inputs the spot values for a dilution series of a sample, and outputs a single value that corresponds to the relative protein concentration of the sample. The strength of SuperCurve is that it not only uses the curve obtained from one sample, but superimposes all the curves from all the samples on a slide to obtain a single unified curve that is much more robust to measurement noise and errors (Figure 2). Although SuperCurve outputs relative protein amounts, it is possible to obtain an absolute concentration value by titrating samples with known protein concentrations.12
Figure 2.
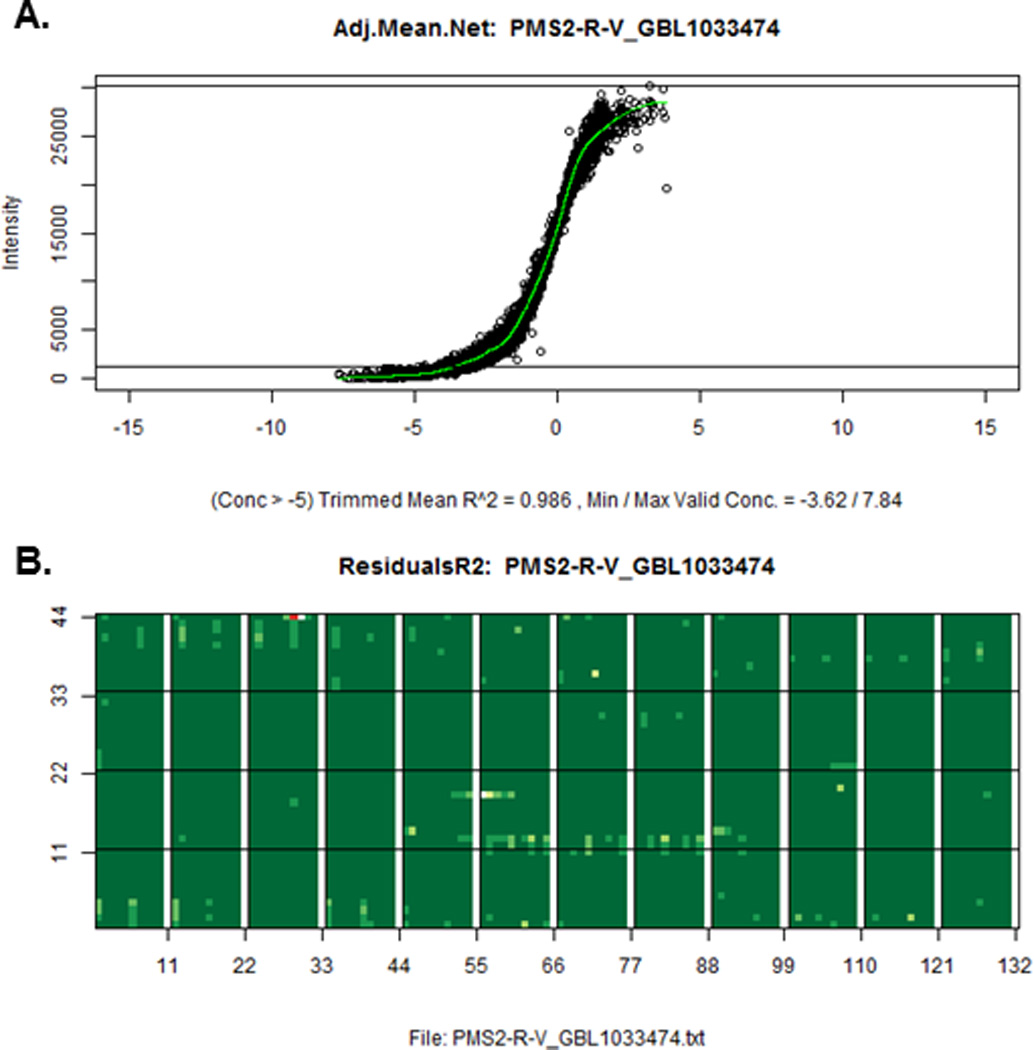
(A) A single unified intensity vs. relative concentration curve output from SuperCurve that uses spots from all the samples. (B) A map of the slide showing spot residual values that deviate from the curve in part A. Dark green represents near zero residual; white represents large residuals.
Pre-analytic challenges
All approaches to assess protein levels and post-translational modifications are limited by pre-analytic challenges. In terms of patient samples, critical considerations include the tumor content, intratumoral heterogeneity, warm and cold ischemia, and spatial changes in these characteristics. Concerns with tumor content and contributions of tissue from different microenvironmental compartments can be alleviated, in part, by using laser capture microdissection.6–8, 16 However, challenges also arise with this technique, including increased cost, sampling bias, difficulty in determining protein loading, potential damage to proteins, and proteins being post-translationally modified during the isolation process. Both mass spectrometry3 and RPPA technology17 have demonstrated that cold ischemia does not alter, to a major degree, the total protein amount or the composition of most phosphoproteins and, unexpectedly, appears to increase the amount of stress-related phosphorylation of particular targets in a predictable manner. Indeed, when an adequate dynamic range and sensitivity are present, mass spectrometry and RPPAs have demonstrated correlation coefficients for changes in the levels of phosphoproteins in the range of 0.75 to 0.83 in complex patient samples.3, 18 However, protein patterns can be markedly different in needle aspirates, core biopsies and tumor samples; thus, careful awareness of the type of samples being analyzed is required.18 While RPPAs (and mass spectrometry) can be utilized on formalin-fixed paraffin-embedded (FFPE) tissues, performance is degraded by the cross-linking effects of formalin, unknown times of penetration of formalin into tissue, and oxidation of proteins over time. Thus, only a subset of proteins that can be analyzed on fresh or frozen tissue can be reliably assessed on FFPE specimens.19, 20
Uses of RPPA technology
As long as care is taken to ensure a high level of quality control, the RPPA platform facilitates robust analysis of cell lines, model organisms (including transgenic and knockout murine models), xenografts, patient-derived xenografts (PDX), and patient samples. Caution must be taken when assessing murine models or xenografts to ensure that the detection approach for the antibodies bound to the RPPA slide, which almost invariably includes an amplification step of secondary antibodies, does not react with murine antibodies that may be present in the tumor. However, there is an ample assortment of high-quality rabbit monoclonal antibodies and polyclonal antibodies from other species that can be used in murine models.
RPPAs have been used extensively in the baseline characterization of cell lines across lineages, and to assess the response of cell lines to chemotherapy and targeted agents or perturbations with siRNA or miRNA. Combining results from RPPAs with other platforms, such as the analysis of DNA and RNA, provides the overall analysis with additional power. These approaches have provided insights into potential biomarkers of sensitivity to drugs and of network structures.1 RPPAs have provided information related to the functional effects of microRNA that was not apparent in the analysis of transcriptomic and seed sequence data.21 Further, RPPAs have helped to unravel the complexities of the action of siRNA.22 The RPPA platform has proven to be a powerful approach for determining the mechanisms underlying the effects of genomic aberrations, which has provided both a new level of understanding and potential therapeutic avenues to explore.23–25 In cell lines, RPPAs have demonstrated that phosphorylation levels are highly predictive of the efficacy of targeted therapies (Figure 3). For example, the IC50 of lapatinib response is inversely correlated with phospho-EGFR expression across 49 lung cancer cell lines (r = −0.465 p = 0.00077). This suggests the efficiency of assessing the functional activity of the EGFR pathway rather than total EGFR levels in the tumor to identify whether a patient is likely to benefit from EGFR-targeted therapy. One of the most cogent observations has been that, compared to non-small cell lung cancer cell lines and tumors, small cell lung cancer cell lines and tumors have elevated levels of PARP, the target of a family of therapeutics classified as PARP inhibitors (PARPi).26 The elevated PARP levels in small cell lung cancer have been associated with increased sensitivity to PARPi, both in vitro and in vivo. Strikingly, these predictions from cell lines have been confirmed in an unexpected degree of responsiveness to PARPi among patients with small cell lung cancers. Subsequent studies have identified potential biomarkers that may identify patients likely to benefit from PARPi.27 Recent patient studies have identified both predictive protein markers and potential pharmacodynamic markers of response to PARPi, albeit in a different tumor system.28
Figure 3.
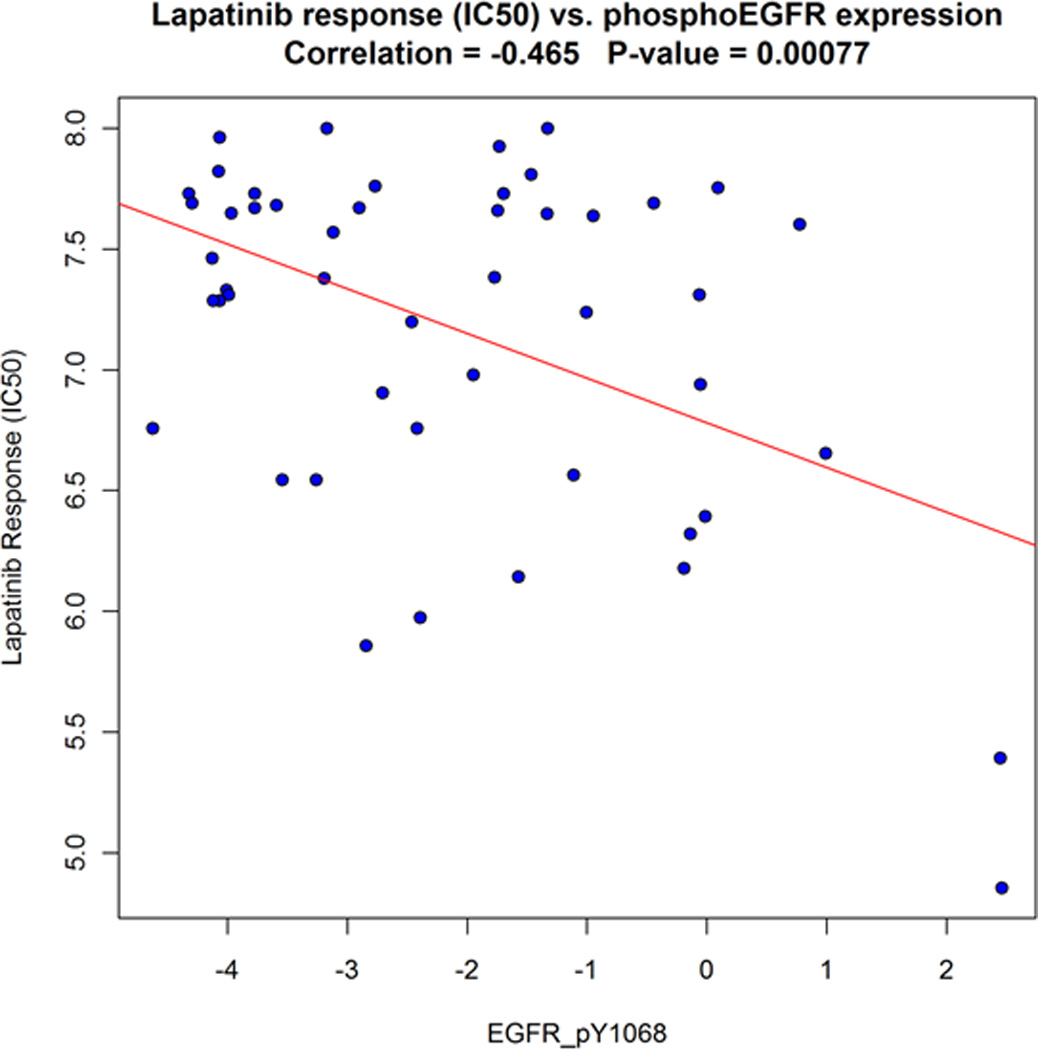
Response to the drug lapatinib (IC50 values capped at 8.0) vs. phospho-EGFR expression in lung cancer cell lines. IC50 is inversely correlated with phospho-EGFR expression (Pearson correlation = −0.465, P-value = 0.00077), indicating that cell lines with higher expression of phospho-EGFR are more sensitive to the drug. The line of best fit is shown in red. Correlation with these previously-known findings further validate the data produced by the RPPA platform and the methods described.
The ability to analyze a number of key pathways has enabled investigators to identify critical pathways involved in the behavior of newly developed cell lines,29 PDXs,30, 31 and patient tumor samples. The most cogent example of this is the analysis of several thousand patient samples from the Cancer Genome Atlas (TCGA), for which RPPA technology has provided information on signaling pathways (Figure 4) activated by genomic and transcriptomic aberrations, correlations with DNA, RNA, miRNA and methylation, identified new patient subtypes,32 and provided prognostic utility.1 It has also served to identify both disease-specific and pan-cancer therapeutic opportunities.31, 33
Figure 4.
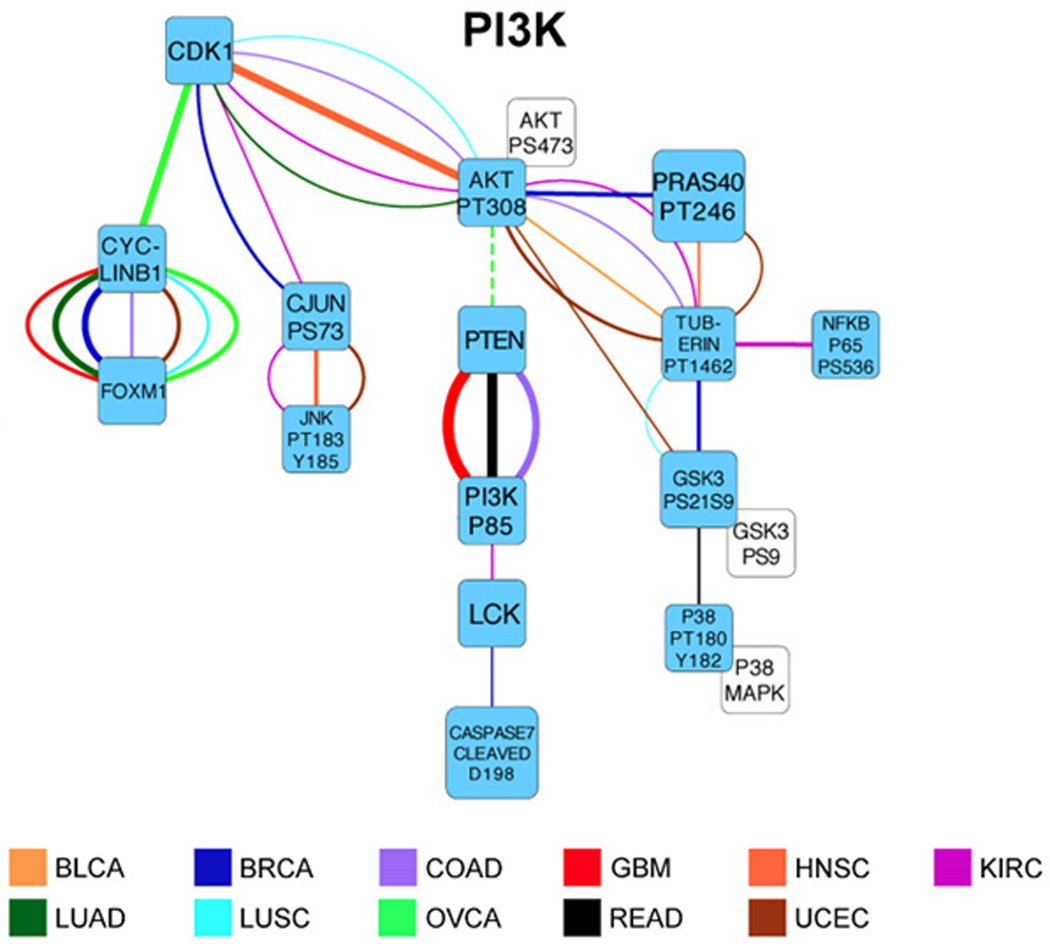
An example of the PI3K signaling network elucidated by RPPA from a TCGA cohort of 3,467 tumor samples across 11 lineages. Interplay between proteins was quantified using scores from a probabilistic graphical model analysis that identified links between proteins. Only the strongest links are shown. The color of a link indicates tumor lineage, which is specified by the standard TCGA disease acronym. Green nodes are individual proteins; white nodes are related proteins that were highly correlated and therefore merged prior to network analysis. Positive (negative) correlations are indicated with continuous (dotted) lines. The graph shows that correlations between proteins and their associated pathways are highly lineage dependent. Only a handful of proteins are shown to be correlated across virtually all tumor lineages (Adapted from Akbani et al.1).
In a comprehensive genomic, transcriptomic, and proteomic analysis of the processes involved in the responsiveness and resistance to BRAF inhibitors in melanoma model systems, RPPAs provided the most useful information, both in terms of mechanisms for bypassing the BRAF inhibitors and therapeutic avenues34 to prevent that bypass. Indeed, in many analyses, RPPAs provided new information on prognosis that was not apparent from an analysis at the DNA or RNA level.35, 36 In ovarian cancer, RPPA analysis has similarly proven to be superior to DNA and RNA analysis at predicting patient outcomes.37 Protein levels assessed through RPPAs have been shown to be powerful predictors in endometrial cancer.38 Even in breast cancer that had been highly characterized by multiple platforms, the RPPA platform was able to identify a new subset of breast cancer (called “Reactive”) with good outcome (Figure 5).32 Subsequent analysis of eleven different cancer lineages demonstrated that the Reactive subtype signature was found in many different cancer lineages where it also predicted patient outcomes.1 Furthermore, Figure 5 shows that the RPPA clusters are strongly associated with PAM50 calls (based on mRNA), HER2 amplification status, TP53 mutations, and PIK3CA mutations, illustrating that RPPA data are correlated with mRNA and DNA, as expected. One of the more novel approaches offered by RPPAs is to characterize individual cancers in a “patient like me” approach, borrowing information from the responses of other patients to help define interesting therapeutic options.
Figure 5.
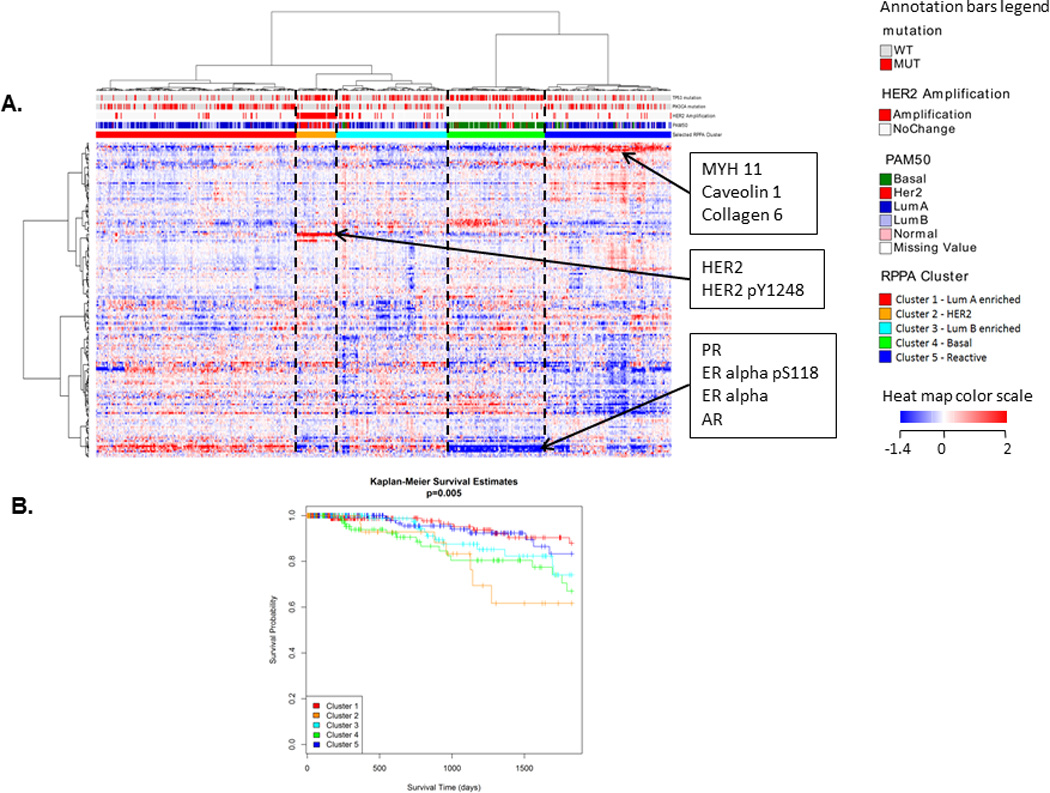
(A) Clustered heat map of TCGA breast cancer RPPA data with samples in columns and proteins in rows. Five different clusters can be seen. The clusters are associated with PAM50 calls, HER2 amplification status, TP53 mutation, and PIK3CA mutation status (p < 0.001, χ2 test). Some known biomarkers can be seen to be differentially expressed between the clusters, such as HER2, phosphoHER2, PR, AR, ER-alpha, and phosphoER-alpha. A newly discovered “Reactive” subtype (cluster 5) based on RPPA data can also be seen with biomarkers MYH11, Caveolin1, and Collagen6. (B) Kaplan-Meier survival curves for the 5 clusters. The Reactive subtype (in dark blue) has good outcome (overall p = 0.005). (Heat map dynamically explorable at: http://bioinformatics.mdanderson.org/TCGA/NGCHMPortal/)
RPPA technology is making the transition to the clinic.39 As mentioned, RPPAs have been used to identify a number of useful biomarkers, therapeutic targets and rational drug combinations to explore in clinical trials. This is contributing to novel therapeutic trial designs. The TheraLink HER Family Assay introduced by Theranostics Health, Inc. (Rockville, MD) is designed to identify tumors that are addicted to the function of cell surface receptors, in particular to HER2, by analyzing the level of the receptor in question, other receptors in the HER2 family, and the activation of the receptors and downstream signaling pathways based on phosphorylation events. Clinical trials are underway to determine whether multiplexed proteomic analysis can identify subsets of patients likely to benefit from specific therapeutic interventions that target activated nodes in the tumor.40
RPPA for pharmacodynamic analyses
RPPA technology is particularly adaptive to the analysis of pharmacodynamics. It can be applied to the small amount of material that is obtained from biopsies, including needle biopsies, as well as to cell populations purified by flow cytometry or laser capture microdissection. It has been used extensively to identify on- and off-target activity of drugs in cell lines, xenografts, and patient samples, and to identify on- and off-target activity within drug compound series.41 The ability to use intact cells to demonstrate that therapeutic compounds are effectively inhibiting the function of the targets provides a ready tool for promoting a therapeutic series.41 Characterizing the immediate consequences of target inhibition aids in determining both the degree and duration of target engagement by therapeutic agents.
The ability to characterize long-term adaptive responses is proving to be a particularly powerful approach to identify patients who are benefiting from a targeted agent prior to changes in the tumor being assessable through imaging. The functional consequences of therapeutic interventions as assessed by RPPAs are providing early signals to identify patients who will benefit from continued treatment with a particular intervention42, 43 and those who may benefit from the addition of another agent or switching to an alternative therapy. One of the most exciting uses of pharmacodynamic analysis of adaptive response is in elucidating feedback and feed-forward loops as well as homeostatic processes.44 The processes by which cells adapt to therapeutic perturbations identify potential targets for rational combination therapies.44 Several of these combinations have been validated and are likely to be tested in therapeutic trials in the near future.
Data sharing
A key step in the utilization of technology is in data sharing across the research community. All of our approaches and antibody validation information are freely available to the research community.45 We have made much of our data available through an interactive website (The Cancer Proteome Atlas, tcpaportal.org) that is associated with a number of analytical and visualization tools.46 These include association analysis, next-generation clustered heat maps, and correlations with patient outcomes. The data on over 7,000 TCGA samples can also be obtained from the TCGA website and are available for integrated analysis with DNA and RNA at the cBioPortal (cbioportal.org). Extensive breast cancer cell perturbation data formed the basis for the HPN-DREAM8 breast cancer network inference challenge. RPPA data also featured prominently in DREAM7 challenges.47 These data represent valuable resources that are enriching community efforts to understand signaling networks, patient prognosis and tumor classification.
Conclusion
In most cancers, multiple genetic and epigenetic changes integrate into modified protein networks that manifest as the functional outcomes of cancer. Indeed, deconvoluting the myriad genomic and epigenomic changes that occur in cancer cells remains a key challenge. However, these myriad effects appear to integrate as a much smaller constellation of effects on proteins and protein networks. This provides the exciting possibility that these fewer protein events can be successfully targeted. Although the current results with targeted therapeutics are not fulfilling their initial promise, the potential for this promise yet exists. The objective is to convert the exciting short-term responses to targeted therapy into more durable responses with greater impact through careful analysis of the pharmacodynamic effects, adaptive responses and, in particular, the implementation of rational combinations of targeted agents or targeted agents with chemotherapy, radiation therapy, or immunotherapy. The ability to efficiently measure the effects of targeted therapy on the targets and protein networks through proteomic technologies that include RPPAs has the potential to contribute to the promise of personalized cancer therapy.
Acknowledgments
This work was supported in part by the U.S. National Institutes of Health/National Cancer Institute (NIH/NCI) through the Cancer Center Support Grant (NCI P30CA016672, Bioinformatics Shared Resource); the MD Anderson TCGA Genome Data Analysis Center grant (NCI CA143883 and CA083639); the Ovary SPORE (NCI P50CA083639); the Endometrial SPORE (NCI P50CA098258); ICBP (NCI U54CA112970); LNCS (NCI U54HG008100); the Cancer Prevention Research Institute of Texas grant (CPRIT RP130397); the Adelson Medical Research Foundation; the Mary K. Chapman Foundation; and the Michael & Susan Dell Foundation (honoring Lorraine Dell).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: The authors report no conflict of interest related to this manuscript.
Research Performed At: The University of Texas MD Anderson Cancer Center
References
- 1.Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun. 2014;5:3887. doi: 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics. 2014;13:1690–1704. doi: 10.1074/mcp.M113.036392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ullal AV, Peterson V, Agasti SS, Tuang S, Juric D, Castro CM, et al. Cancer cell profiling by barcoding allows multiplexed protein analysis in fine-needle aspirates. Sci Transl Med. 2014;6:219ra9. doi: 10.1126/scitranslmed.3007361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akbani R, Becker KF, Carragher N, Goldstein T, de Koning L, Korf U, et al. Realizing the promise of reverse phase protein arrays for clinical, translational, and basic research: a workshop report: the RPPA (Reverse Phase Protein Array) society. Mol Cell Proteomics. 2014;13:1625–1643. doi: 10.1074/mcp.O113.034918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sereni MI, Baldelli E, Gambara G, Deng J, Zanotti L, Bandiera E, et al. Functional characterization of epithelial ovarian cancer histotypes by drug target based protein signaling activation mapping: implications for personalized cancer therapy. Proteomics. 2015;15:365–373. doi: 10.1002/pmic.201400214. [DOI] [PubMed] [Google Scholar]
- 7.Baldelli E, Haura EB, Crino L, Cress DW, Ludovini V, Schabath MB, et al. Impact of upfront cellular enrichment by laser capture microdissection on protein and phosphoprotein drug target signaling activation measurements in human lung cancer: Implications for personalized medicine. Proteomics Clin Appl. 2015 doi: 10.1002/prca.201400056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wulfkuhle JD, Berg D, Wolff C, Langer R, Tran K, Illi J, et al. Molecular analysis of HER2 signaling in human breast cancer by functional protein pathway activation mapping. Clin Cancer Res. 2012;18:6426–6435. doi: 10.1158/1078-0432.CCR-12-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 10.Charboneau L, Tory H, Chen T, Winters M, Petricoin EF, 3rd, Liotta LA, et al. Utility of reverse phase protein arrays: applications to signalling pathways and human body arrays. Brief Funct Genomic Proteomic. 2002;1:305–315. doi: 10.1093/bfgp/1.3.305. [DOI] [PubMed] [Google Scholar]
- 11.Espina V, Liotta LA, Petricoin EF., 3rd Reverse-phase protein microarrays for theranostics and patient tailored therapy. Methods Mol Biol. 2009;520:89–105. doi: 10.1007/978-1-60327-811-9_7. [DOI] [PubMed] [Google Scholar]
- 12.Sheehan KM, Calvert VS, Kay EW, Lu Y, Fishman D, Espina V, et al. Use of reverse phase protein microarrays and reference standard development for molecular network analysis of metastatic ovarian carcinoma. Mol Cell Proteomics. 2005;4:346–355. doi: 10.1074/mcp.T500003-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Grote T, Siwak DR, Fritsche HA, Joy C, Mills GB, Simeone D, et al. Validation of reverse phase protein array for practical screening of potential biomarkers in serum and plasma: accurate detection of CA19-9 levels in pancreatic cancer. Proteomics. 2008;8:3051–3060. doi: 10.1002/pmic.200700951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J, He X, Baggerly KA, Coombes KR, Hennessy BTJ, Mills GB. Non-parametric quantification of protein lysate arrays. Bioinformatics. 2007;23:1986–1994. doi: 10.1093/bioinformatics/btm283. [DOI] [PubMed] [Google Scholar]
- 15.Mannsperger HA, Gade S, Henjes F, Beissbarth T, Korf U. RPPanalyzer: Analysis of reverse-phase protein array data. Bioinformatics. 2010;26:2202–2203. doi: 10.1093/bioinformatics/btq347. [DOI] [PubMed] [Google Scholar]
- 16.Mueller C, deCarvalho AC, Mikkelsen T, Lehman NL, Calvert V, Espina V, et al. Glioblastoma cell enrichment is critical for analysis of phosphorylated drug targets and proteomic-genomic correlations. Cancer Res. 2014;74:818–828. doi: 10.1158/0008-5472.CAN-13-2172. [DOI] [PubMed] [Google Scholar]
- 17.Hennessy BT, Lu Y, Gonzalez-Angulo AM, Carey MS, Myhre S, Ju Z, et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin Proteomics. 2010;6:129–151. doi: 10.1007/s12014-010-9055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meric-Bernstam F, Akcakanat A, Chen H, Sahin A, Tarco E, Carkaci S, et al. Influence of biospecimen variables on proteomic biomarkers in breast cancer. Clin Cancer Res. 2014;20:3870–3883. doi: 10.1158/1078-0432.CCR-13-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo H, Liu W, Ju Z, Tamboli P, Jonasch E, Mills GB, et al. An efficient procedure for protein extraction from formalin-fixed, paraffin-embedded tissues for reverse phase protein arrays. Proteome Sci. 2012;10:56. doi: 10.1186/1477-5956-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bader S, Zajac M, Friess T, Ruge E, Rieder N, Gierke B, et al. Evaluation of protein profiles from treated xenograft tumor models identifies an antibody panel for FFPE tissue analysis by reverse phase protein arrays. Mol Cell Proteomics. 2015 doi: 10.1074/mcp.O114.045542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu HS, Llobet-Navas D, Yang X, Chung WJ, Ambesi-Impiombato A, Iyer A, et al. Cupid: simultaneous reconstruction of microRNA-target and ceRNA networks. Genome Res. 2015;25:257–267. doi: 10.1101/gr.178194.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu Y, Muller M, Smith D, Dutta B, Komurov K, Iadevaia S, et al. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene. 2011;30:4567–4577. doi: 10.1038/onc.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang H, Cheung LW, Li J, Ju Z, Yu S, Stemke-Hale K, et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 2012;22:2120–2129. doi: 10.1101/gr.137596.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaluvally-Raghavan P, Zhang F, Pradeep S, Hamilton MP, Zhao X, Rupaimoole R, et al. Copy number gain of hsa-miR-569 at 3q26.2 leads to loss of TP53INP1 and aggressiveness of epithelial cancers. Cancer Cell. 2014;26:863–879. doi: 10.1016/j.ccell.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheung LW, Yu S, Zhang D, Li J, Ng PK, Panupinthu N, et al. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer Cell. 2014;26:479–494. doi: 10.1016/j.ccell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012;2:798–811. doi: 10.1158/2159-8290.CD-12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cardnell RJ, Feng Y, Diao L, Fan YH, Masrorpour F, Wang J, et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res. 2013;19:6322–6328. doi: 10.1158/1078-0432.CCR-13-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JM, Hays JL, Annunziata CM, Noonan AM, Minasian L, Zujewski JA, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst. 2014;106:dju089. doi: 10.1093/jnci/dju089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ince TA, Sousa AD, Jones MA, Harrell JC, Agoston ES, Krohn M, et al. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat Commun. 2015;6:7419. doi: 10.1038/ncomms8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73:4885–4897. doi: 10.1158/0008-5472.CAN-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. Cancer Genome Atlas Research N. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwong LN, Boland GM, Frederick DT, Helms TL, Akid AT, Miller JP, et al. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J Clin Invest. 2015;125:1459–1470. doi: 10.1172/JCI78954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan Y, Van Allen EM, Omberg L, Wagle N, Amin-Mansour A, Sokolov A, et al. Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat Biotechnol. 2014;32:644–652. doi: 10.1038/nbt.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang JY, Yoshihara K, Tanaka K, Hatae M, Masuzaki H, Itamochi H, et al. Predicting time to ovarian carcinoma recurrence using protein markers. J Clin Invest. 2013;123:3740–3750. doi: 10.1172/JCI68509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, Werner H, Li J, Westin S, Lu Y, Salvesen H, et al. A Protein-marker Driven Integrative Prognostic Model for Early-stage Endometrioid Endometrial Carcinoma. Clinical Cancer Research. 2015 doi: 10.1158/1078-0432.CCR-15-0104. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gallagher RI, Espina V. Reverse phase protein arrays: mapping the path towards personalized medicine. Mol Diagn Ther. 2014;18:619–630. doi: 10.1007/s40291-014-0122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pierobon M, Silvestri A, Spira A, Reeder A, Pin E, Banks S, et al. Pilot phase I/II personalized therapy trial for metastatic colorectal cancer: evaluating the feasibility of protein pathway activation mapping for stratifying patients to therapy with imatinib and panitumumab. J Proteome Res. 2014;13:2846–2855. doi: 10.1021/pr401267m. [DOI] [PubMed] [Google Scholar]
- 41.Kim WY, Chang DJ, Hennessy B, Kang HJ, Yoo J, Han SH, et al. A novel derivative of the natural agent deguelin for cancer chemoprevention and therapy. Cancer Prev Res (Phila) 2008;1:577–587. doi: 10.1158/1940-6207.CAPR-08-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindholm EM, Krohn M, Iadevaia S, Kristian A, Mills GB, Maelandsmo GM, et al. Proteomic characterization of breast cancer xenografts identifies early and late bevacizumab-induced responses and predicts effective drug combinations. Clin Cancer Res. 2014;20:404–412. doi: 10.1158/1078-0432.CCR-13-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gungor H, Saleem A, Curry E, Babar S, Dina R, ElpBahrawy M, et al. Dose-Finding PET and predictive biomarker study in platinum resistant ovarian cancer demonstrates tolerability and activity for the pan-AKT inhibitor GSK2141795. 2015 Submitted. [Google Scholar]
- 44.Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21:227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. http://www.mdanderson.org/education-and-research/resources-for-professionals/scientific-resources/core-facilities-and-services/functional-proteomics-rppa-core/index.html.
- 46.Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, et al. TCPA: a resource for cancer functional proteomics data. Nat Methods. 2013;10:1046–1047. doi: 10.1038/nmeth.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Creighton CJ. Widespread molecular patterns associated with drug sensitivity in breast cancer cell lines, with implications for human tumors. PLoS One. 2013;8:e71158. doi: 10.1371/journal.pone.0071158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tabchy A, Hennessy BT, Gonzalez-Angulo AM, Bernstam FM, Lu Y, Mills GB. Quantitative proteomic analysis in breast cancer. Drugs Today (Barc) 2011;47:169–182. doi: 10.1358/dot.2011.47.2.1576695. [DOI] [PubMed] [Google Scholar]