

Abstract
Background
In many cancers, microRNAs (miRs) contribute to metastatic progression by modulating phenotypic reprogramming processes such as epithelial-mesenchymal plasticity. This can be driven by miRs targeting multiple mRNA transcripts, inducing regulated changes across large sets of genes. The miR-target databases TargetScan and DIANA-microT predict putative relationships by examining sequence complementarity between miRs and mRNAs. However, it remains a challenge to identify which miR-mRNA interactions are active at endogenous expression levels, and of biological consequence.
Methods
We developed a workflow to integrate TargetScan and DIANA-microT predictions into the analysis of data-driven associations calculated from transcript abundance (RNASeq) data, specifically the mutual information and Pearson’s correlation metrics. We use this workflow to identify putative relationships of miR-mediated mRNA repression with strong support from both lines of evidence. Applying this approach systematically to a large, published collection of unique melanoma cell lines – the Ludwig Melbourne melanoma (LM-MEL) cell line panel – we identified putative miR-mRNA interactions that may contribute to invasiveness. This guided the selection of interactions of interest for further in vitro validation studies.
Results
Several miR-mRNA regulatory relationships supported by TargetScan and DIANA-microT demonstrated differential activity across cell lines of varying matrigel invasiveness. Strong negative statistical associations for these putative regulatory relationships were consistent with target mRNA inhibition by the miR, and suggest that differential activity of such miR-mRNA relationships contribute to differences in melanoma invasiveness. Many of these relationships were reflected across the skin cutaneous melanoma TCGA dataset, indicating that these observations also show graded activity across clinical samples. Several of these miRs are implicated in cancer progression (miR-211, -340, -125b, −221, and -29b). The specific role for miR-29b-3p in melanoma has not been well studied. We experimentally validated the predicted miR-29b-3p regulation of LAMC1 and PPIC and LASP1, and show that dysregulation of miR-29b-3p or these mRNA targets can influence cellular invasiveness in vitro.
Conclusions
This analytic strategy provides a comprehensive, systems-level approach to identify miR-mRNA regulation in high-throughput cancer data, identifies novel putative interactions with functional phenotypic relevance, and can be used to direct experimental resources for subsequent experimental validation.
Computational scripts are available: http://github.com/uomsystemsbiology/LMMEL-miR-miner
Electronic supplementary material
The online version of this article (doi:10.1186/s12943-016-0554-y) contains supplementary material, which is available to authorized users.
Keywords: Systems biology, Cancer, micro-RNA, Melanoma, Phenotypic switching, Statistical association, TargetScan, DIANA-microT CDS
Background
Phenotypic switching is an important process that facilitates melanoma progression, metastasis, and resistance to therapy [1–6]. Despite the neural crest (i.e. non-epithelial) origin of melanocytes, melanomas display cadherin-switching and functional behaviours that resemble epithelial-to-mesenchymal plasticity (EMP). Melanomas often harness lineage-specific molecular pathways from more primitive (less-differentiated) states in order to switch between proliferative, minimally-invasive (i.e. ‘epithelial-like’, or ‘E-like’) and invasive, minimally-proliferative (i.e. ‘mesenchymal-like’, or ‘M-like’) phenotypes [7–9]. Several transcription factors, including ZEB1/2, SNAIL1/2, TWIST1/2, MITF and JUN, have been shown to play key roles in melanoma EMP-like processes [4, 7, 9, 10]; however, the mechanisms by which internal and micro-environmental signals are integrated to modulate transcriptional activity are not fully understood.
Micro-RNAs (miRs) are short (18–24 nucleotide) non-coding RNAs which play an important role regulating the activity of other RNA transcripts. They have been implicated in the oncogenesis and progression of several cancers, as reviewed thoroughly elsewhere [11, 12]. Some miRs directly target important transcription factors, such as miR-200 regulation of ZEB in epithelial carcinomas [13, 14], while in melanoma, miR-148 mediated dysregulation of MITF [15] and miR-125b control of JUN [16] have been noted. In melanoma, a network of miRs, including miR-211 and miR-222 has been shown to mediate some effects of oncogenic BRAF signalling [17]. ArrayCGH studies suggest miR-29b copy number increases in melanoma and a number of miRs have been implicated in melanocyte transformation [18, 19], melanoma progression [20–23], modulating the extent and mode of melanoma cell invasiveness [17, 24, 25], and switching of cellular phenotype (i.e. epithelial-mesenchymal plasticity) in a number of epithelial cancers [14, 26, 27].
Identifying endogenous miRs that play a key role in oncogenesis remains challenging for a number of reasons. Firstly, within mammalian cells, miRs exert their effects through multiple mechanisms that are difficult to observe experimentally utilising any single current method. Multiple lines of evidence have shown that miRs regulate protein abundance by modulating protein translation [28], influencing mRNA transcript stability [29, 30] or through both effects [31]. These effects may not be entirely independent [32, 33], and it has been argued that changes in mRNA stability can be attributed to repression of translation [33]. A recent review has detailed the molecular mechanisms mediating target transcript sequestration or degradation, and translational inhibition [32].
Secondly, there is extensive ‘crosstalk’ and broader, coordinated targeting by miRs [27, 34], by virtue of the mapping of miRs to multiple potential transcript targets with potentially different binding affinities. One common effect which influences miR activity is variation in the abundance of alternative RNA targets within the intracellular milieu – although the effect of protein-coding competitive endogenous RNAs (ceRNAs) is thought to be minimal [35], non-coding ceRNAs [36] and circular RNAs [37, 38] have been shown to act as miRNA “sinks” which exert biological effects in a combinatorial fashion. Together, these effects make it difficult to identify individual miR-mRNA interactions which are consistent across many experimental systems and conditions.
Finally, well-known differences between cellular processes active in transformed cell lines and in primary tissues taken from patients lead to a differing repertoire of miRs, transcripts, and metabolic intermediates. As a direct consequence, experimental results observed in one model system, particularly in cell lines, may not be observable in or relevant to patient tumours in vivo [39]. Overcoming these challenges requires an integrative systems analysis relating miR abundance to target mRNA abundance, as well as to a relevant phenotype. Here we report such an analysis using the Ludwig Melbourne melanoma (LM-MEL) cell line panel [40], an experimental resource designed for the identification and verification of molecular mechanisms contributing to the heterogeneity observed across melanoma tumours.
The LM-MEL panel data were collected from 57 established melanoma cell lines [40] which display a range of phenotypes across the epithelial-like to mesenchymal-like spectrum [41]. The original LM-MEL molecular data were supplemented with miR transcript abundance data (Additional file 1), and information on invasiveness through matrigel (Additional file 2), which allowed us to identify putative interactions between miRs and mRNAs associated with differences in the functional phenotypes of these melanoma cell lines.
We applied statistical measures of association (Pearson’s correlation [rP] and mutual information [MI]) together [42] to identify miRs and mRNAs which showed a strong inverse association across the LM-MEL dataset. Mutual information is an information-theoretic metric that measures divergence from statistical independence [43]. It contains a term that tends to zero with statistical independence, where P(A, B) = P(A)P(B). Mutual information and MI-based metrics have been successfully applied for the analysis of large biological data sets [44–46] (further details within Methods/Statistical associations). Conversely, Pearson’s correlation purely measures the tendency towards a linear association, although it does provide directionality through positive/negative values.
This report describes a systems approach to identify putative regulatory miR-mRNA relationships across related cell lines and clinical samples, applying measures of statistical association and refining the results through the principled inclusion of knowledge from specific relevant databases. Our computational workflow identifies a novel role for miR-29b-3p in regulating melanoma invasiveness, and the results improve our understanding of how miR activity can influence melanoma phenotype switching and EMP.
Results
Changes in miR abundance were predicted to regulate mRNA transcript stability across a panel of melanoma cell lines
Given the evidence implicating miRs in cancer progression and invasiveness, we hypothesised that miRs with differential activity across the LM-MEL cell line panel may drive post-transcriptional regulatory changes that influence cell phenotype. To identify relationships where miRs may regulate mRNA transcripts through reduced stability, we examined miR and mRNA transcript abundance across the LM-MEL cell line panel and selected those with a strong, negative (or inverse) statistical association between miR and mRNA abundance (Fig. 1a).
Fig. 1.
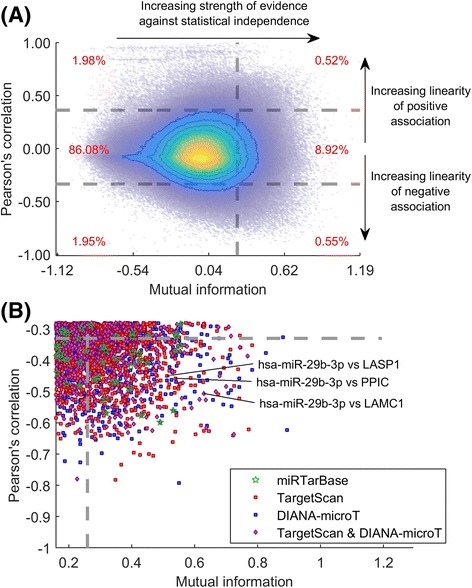
Systems analysis of database-enriched statistical associations between miR and mRNA transcripts. a Density and contour plot for mutual information and Pearson’s correlation between transcript abundance of all miR and mRNA pairs across the LM-MEL cell line panel (n = 57). Associations within the top 10% of mutual information values (vertical dashed line) and bottom (most negative) 2.5% of Pearson’s correlation values (horizontal dashed lines) were selected as those with strong negative associations (relative fraction of associations in red). b These associations (a, bottom right) were matched against high-confidence, predicted relationships from TargetScan and DIANA-microT, and previously validated relationships from miRTarBase. Relationships between hsa-miR-29b-3p and LAMC1, LASP1, and PPIC are examined below, Additional file 3 lists all putative relationships
Numerous known and predicted miR-mRNA interactions show evidence of post-transcriptional regulation across the LM-MEL cell line panel
We integrated TargetScan [47–49] and DIANA-microT [50, 51] (Additional file 1) to limit the search space for putative miR-mRNA interactions. Numerous strong, negative associations across the LM-MEL data were supported by sequence-based predictions with a relatively high confidence (TargetScan top 15th percentile; and/or DIANA-microT top 30th percentile; details in Methods/Databases). Matching putative relationships to miRTarBase (using ‘strong evidence’ experimental methods), only a small sub-set of interactions appear to have been previously validated (Fig. 1b). A full list of predicted relationships is given in Additional file 3 with further details.
Putative regulatory associations contain a number of previously validated associations
Selected relationships are shown in Fig. 2 using in vitro LM-MEL data with phenotypic annotations (top of panel; red, high matrigel-invasiveness cell lines; blue, low invasiveness), and in vivo TCGA [52] Skin Cutaneous Melanoma (SKCM) data (bottom of panel).
Fig. 2.
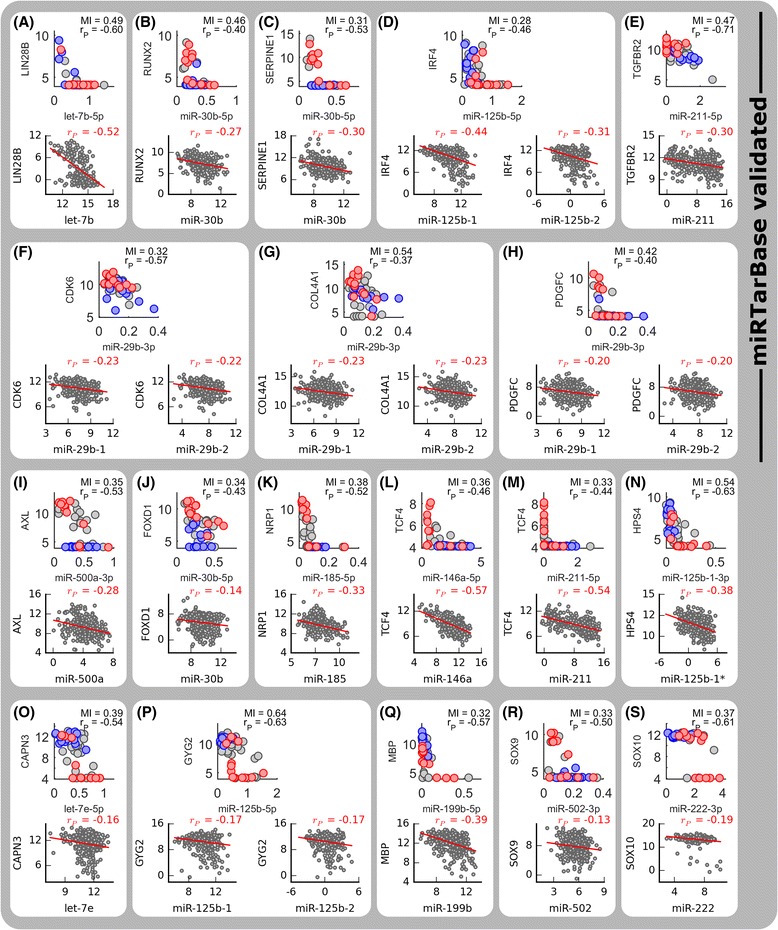
Many putative miR-mRNA relationships involve mRNAs previously implicated in EMP and melanoma phenotype switching. LM-MEL and TCGA (top and bottom subplots, respectively) transcript abundance data for (a-h) selected, previously validated relationships (miRTarBase, ‘strong experimental evidence’; see Methods/Databases), and (i-q) putative relationships regulating mRNAs implicated in melanoma phenotype switching by Widmer et al. [3]. Further details are given in Additional file 4. Predicted regulatory interactions involving SOX transcription factors integral to melanoma phenotype switching are shown (r-s).
Several putative relationships have been validated across different human cell systems (Fig. 2a-h; ‘miRTarBase validated’), and were supported by TCGA data (further details within Additional file 4). In parallel, a number of putative relationships emerged which have not been previously observed within human cell lines, and many of these potentially novel relationships involved mRNA transcripts implicated in melanoma phenotype switching [3] and invasive behaviours (Fig. 2i-q; Additional file 4).
Within the unvalidated interactions, the predicted regulatory interactions between the transcription factors SOX9 and miR-502-3p (Fig. 2r; LM-MEL rP = −0.50, MI = 0.33; TCGA rP = −0.13), and SOX10 and miR-222-3p (Fig. 2s; LM-MEL rP = −0.61, MI = 0.37; TCGA rP = −0.19), is particularly interesting. In melanoma, SOX10 functions both independently and in cooperation with MITF to promote more differentiated and/or proliferative cellular states [53, 54]. A SOX10-low state is associated with reduced cell proliferation and engagement of EMT-like processes in melanoma to promote more invasive phenotypes [55] - a state maintained, in part, through mutual-antagonism with the closely related transcription factor SOX9 [56]. SOX10 suppression contributes to BRAF- and/or MEK-inhibitor resistance in BRAF mutated melanoma, by activating TGFβ signalling to upregulate EGFR and PDGFRB [57], whilst increasing SOX9 transcript abundance has been observed in breast cancer EMT [58]. SOX9-high LM-MEL cell lines are also enriched for an invasive phenotype (Fig. 2r) and there is a distinct subset of SOX10-low, high-invasive LM-MEL cell lines (Fig. 2s) which appears to be recapitulated within the TCGA data.
A number of miRs implicated in the progression of melanoma and other cancers were enriched for relationships with differential regulatory activity
As detailed earlier, miRs can drive phenotypic change through the coordinated regulation of several mRNA targets. To examine this we calculated the relative enrichment of ‘active associations’ (Fig. 1b) for each miR across the LM-MEL data. The top five miRs when using high confidence TargetScan lists were miR-211-5p, miR-340-5p, miR-125b-1-3p, miR-221-3p and miR-29b-3p (Fig. 3a, top row). High confidence DIANA-microT target lists also suggested differential activity for miR-100-5p across the LM-MEL cell line panel (Fig. 3a, second row).
Fig. 3.
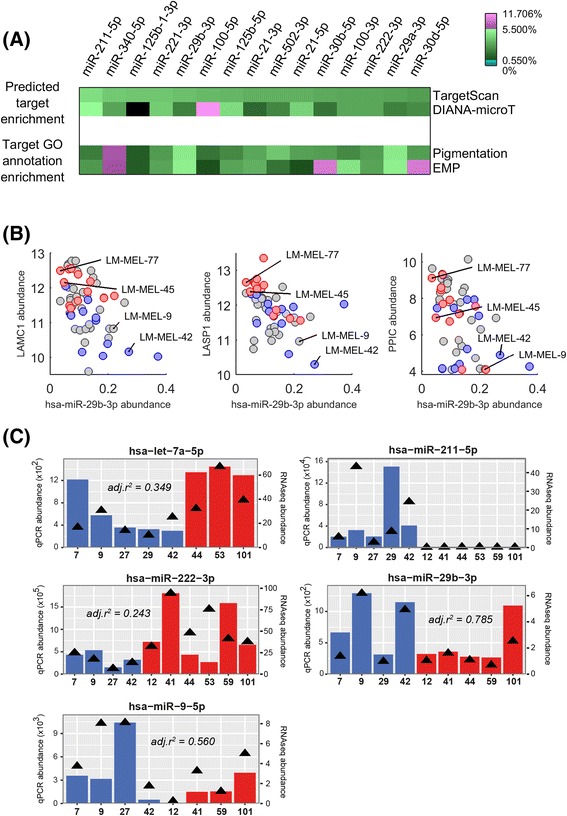
a Putative target list enrichment for individual miRs using TargetScan and DIANA-microT target lists (first and second row, respectively). Relative enrichment of combined miR target lists are also shown for GO terms associated with pigmentation or EMP. b Relationships between miR-29b-3p and LAMC1 (left), LASP1 (centre) and PPIC (right) within the LM-MEL data; for cell lines with matrigel invasiveness measurements, these data are overlaid (red, high invasiveness; blue, low invasiveness). c miR transcript abundances measured by quantitative PCR (qPCR; bar graph) for selected miRs and LM-MEL cell lines (indicated by number along the x-axis), plotted against high-throughput LM-MEL panel data (black triangles); adjusted regression coefficients between these datasets are indicated (negative for miR-211-5p due to multiple non-expressing cell lines)
The top four micro-RNAs identified have been shown previously to regulate melanoma cell proliferation, migration and/or invasiveness. As mentioned above, expression levels of miR-211 are inversely related to melanoma cell migration and invasion, and it has been shown to function as a tumour-suppressor through target genes including IGF2R, TGFBR2 (Fig. 2e) and NFAT5 [59, 60]. Similarly, miR-340 downregulation promotes melanoma progression [61] through de-repression of drug transporters [62], RAS-RAF-MAPK signalling components [63] and the key melanocytic transcription factor MITF [64]. The role of miR-125b in cancer progression has also been studied extensively – in melanoma miR-125b over-expression reduces cell proliferation and migration, mediated partly by the direct suppression of c-JUN mRNA translation [16]; however, much of the literature has examined miR-125b-5p (the dominant mature form; Fig. 3a, seventh column), rather than the -3p forms. Finally, miR-221 has a well-studied role in melanoma progression [20, 65] and circulating miR-221 has been proposed as a melanoma biomarker [66].
Enrichment analyses implicate miR-29b-3p in melanoma phenotype switching
The role of the fifth ranked micro-RNA, miR-29b-3p, in melanoma has not yet been explored. In other cancer types, members of the miR-29a/b/c family influence EMP [67–69], and act as tumour suppressors [70–72]. In cutaneous melanoma, down-regulation of miR-29c has been associated with an adverse prognosis, attributed in part to its regulation of transcripts for the DNA methyltransferases DNMT3A/DNMT3B [73].
Given roles for phenotype switching [4, 74, 75] and pigmentation changes [76] in melanoma progression, we extracted Gene Ontology database terms associated with melanogenesis or pigmentation (‘Pigmentation’), or epithelial-mesenchymal plasticity (‘EMP’). Next, we examined the relative enrichment of these ontological classifications within high-confidence miR target lists from TargetScan and/or DIANA-microT (Fig. 3a, at right). Consistent with its role in melanoma progression [61–63], miR-340-5p had a particularly strong loading for both “pigmentation” and “EMP” categories, whilst miR-29b-3p was also enriched around 10-fold (~5.5%), providing further support that miR-29b-3p may be regulating melanoma cell invasiveness through a diverse repertoire of regulatory interactions.
TargetScan predicts hsa-miR-29-3p family-mediated regulation of Peptidylprolyl Isomerase C (Cyclophilin C; PPIC), Laminin, γ1 (LAMC1) and LIM and SH3 Protein 1 (LASP1), and DIANA-microT also predicts miR-29b-3p regulation of LAMC1 (Fig. 1b). All three predicted targets show a strong negative association across the LM-MEL cell line panel (Fig. 3b) in a manner suggesting that reduced expression of miR-29b-3p and increased expression of these targets is associated with more invasive cell lines (Fig. 3b).
Validation of miR and mRNA transcript abundances across a subset of LM-MEL cell lines
We performed quantitative PCR (qPCR) on selected LM-MEL cell lines to validate the high-throughput panel expression data for interactions of interest. Using miRNA-specific qPCR, transcript abundance of hsa-let-7a-5p, hsa-miR-211-5p, hsa-miR-222-3p, hsa-miR-29b-3p and hsa-miR-9-5p within total RNA showed reasonable concordance with sequencing-derived abundance data from the LM-MEL panel, and expected trends were apparent for divergent miR abundances between cell lines classified as E-like or M-like (Fig. 3c). Expression levels of target gene transcripts assessed using standard qPCR confirmed measurable levels of ATP2A2, CPEB1, TCF4, NRP1, ADAM19, CAV2, LAMC1, LASP1 and PPIC in the cell lines studied, again with trends for divergent levels between E- and M-like cell lines consistent with microarray-derived data (Figure AF5.1 within Additional file 5).
hsa-miR-29b-3p regulates LAMC1, LASP1 and PPIC within several LM-MEL cell lines
As shown in Fig. 2 (panels F-H), targets of hsa-miR-29b-3p (miR-29b) include several molecules with documented roles in extracellular matrix formation, sensing, signalling or modulation, consistent with a role in invasiveness or EMT-like processes. To elucidate the role of miR-29b in melanoma invasiveness, we examined the effect of miR-29b perturbation on the transcript abundance of several putative targets in a subset of LM-MEL cell lines. Cells were transiently transfected at high efficiency with either miR-29b inhibitor or mimic (Figure AF5.2A & B within Additional file 5). MiR-specific qPCR demonstrated a 2-3-log increase in detectable miR-29b levels following transfection with miR-29b mimic at 1nM (Figure AF5.2C within Additional file 5), whilst transfection of miR-29b inhibitor did not appreciably alter qPCR-detectable mature miR-29b abundance.
MiR-29b inhibitor treatment of the E-like cell lines LM-MEL-9 and LM-MEL-42 (high baseline levels of miR-29b) induced dose-dependent increases in transcript abundance of ADAM19, CAV2, LAMC1, PPIC, and to a lesser degree, LASP1 (Figure AF5.3 within Additional file 5). Treatment of the M-like cell lines LM-MEL-45, −57 and −77 with a miR-29b mimic markedly reduced transcript abundance of ADAM19, LAMC1, and PPIC, with more modest effects on CAV2 and LASP1 (Fig. 4a and Figure AF5.3 within Additional file 5). All three genes selected for further investigation (LAMC1, LASP1 and PPIC) exhibited a dose-dependent reduction in transcript level following miR-29b mimic transfection, although this effect was near-maximal at 1nM.
Fig. 4.
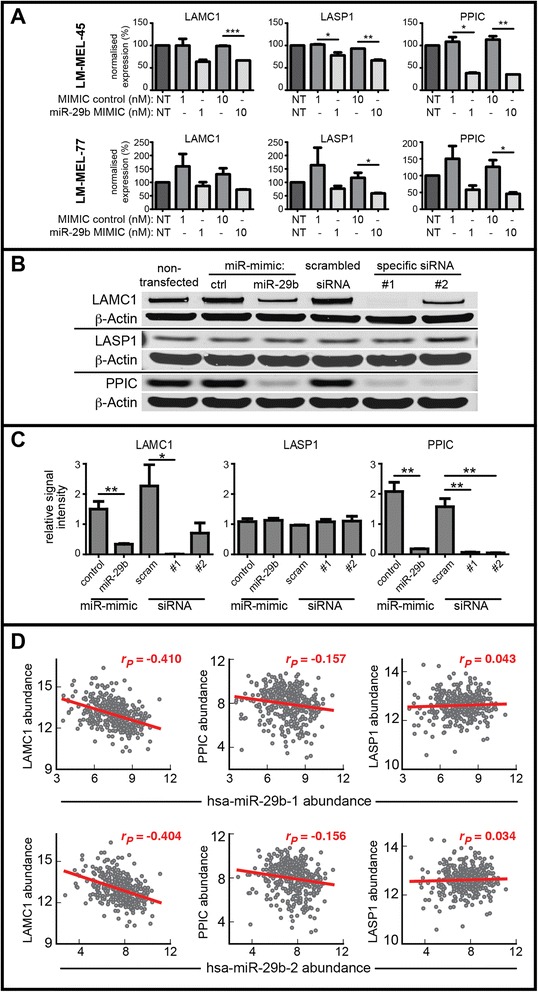
The effects of hsa-miR-29b transfection on mRNA transcript and protein levels for putative targets: LAMC1, LASP1 and PPIC. a For LM-MEL-45 and −77 (upper and lower row, respectively), two mesenchymal-like cell lines, mRNA transcript levels were measured following transfection with hsa-miR-29b mimic. Expression has been normalised to non-transfected (NT) controls. Data represent mean + SD from independent biological duplicates performed in technical triplicates. b Representative Western blots and (c) quantified densitometry for LM-MEL-45 cell lysate harvested 96 h post-transfection with 10nM of miR-29b mimic, miR mimic negative control (ctrl), siRNA scrambled (scram) control, or specific siRNA to the indicated mRNA target. Densitometry data represent mean + SEM signal intensities for independent triplicates normalised against a β-actin loading control, and shown relative to non-treated cells. Pairwise one-tailed t-test statistics are indicated by * p < 0.05, ** p < 0.01, *** p < 0.001. d Relationships between miR-29b-1 and miR-29b-2 (immature -3p precursors), and LAMC1 (at left) LASP1 (at centre) and PPIC (at right) within TCGA SKCM data. Pearson’s correlations and the line of best fit are shown in red
We assessed the effect of miR-29b overexpression on protein levels for putative targets using Western blotting for LAMC1, LASP1 and PPIC in the prototypical M-like cell line LM-MEL-45. Marked reductions in LAMC1 and PPIC protein levels were seen at 96 h following transient transfection with a miR-29b mimic, consistent with the effects of specific siRNAs (Fig. 4b & c). Contrasting transcript abundance changes, LASP1 protein levels were unaffected by either the miR-29b mimic or specific siRNA treatment, suggesting high protein stability and minimal LASP1 protein turnover within the duration of the assay (Fig. 4b & c).
To examine whether these putative relationships are present in vivo, we examined matched miR and mRNA transcript abundances within TCGA skin and cutaneous melanoma (SKCM) data. The abundances of mature hsa-miR-29b-1 and hsa-miR-29b-2 transcripts were examined against mRNA abundances for LAMC1, PPIC, and LASP1 (Fig. 4d). Within the TCGA data there was a relatively strong negative association between LAMC1 and miR-29b, no association between miR-29b and LASP1, and a weak negative association between miR-29b and PPIC.
hsa-miR-29b-3p reduces melanoma cell motility and invasiveness through mechanisms beyond single-gene targets
Mild effects were observed for miR-29b-3p mimic in reducing LM-MEL-45 outgrowth survival at 21 days, with little change in cellular proliferation observed at 72 h (Figure AF5.4 & AF5.5 within Additional file 5).
To better assess the effect of miR-29b on melanoma cell motility, cultured LM-MEL-45 cells were allowed to migrate into a central detection zone of the Oris plate assay system (without a chemotactic gradient) following treatment with miR-29b mimic or specific siRNA for either LAMC1 or PPIC. LASP1 was not examined on the basis that we did not demonstrate reduced levels of protein within the duration of this assay. The density of cells migrating into the central detection zone was measured after 48 h. Following all three treatments, LM-MEL-45 cells demonstrated a comparable reduction in migration, compared with control-treated cells (Fig. 5a).
Fig. 5.
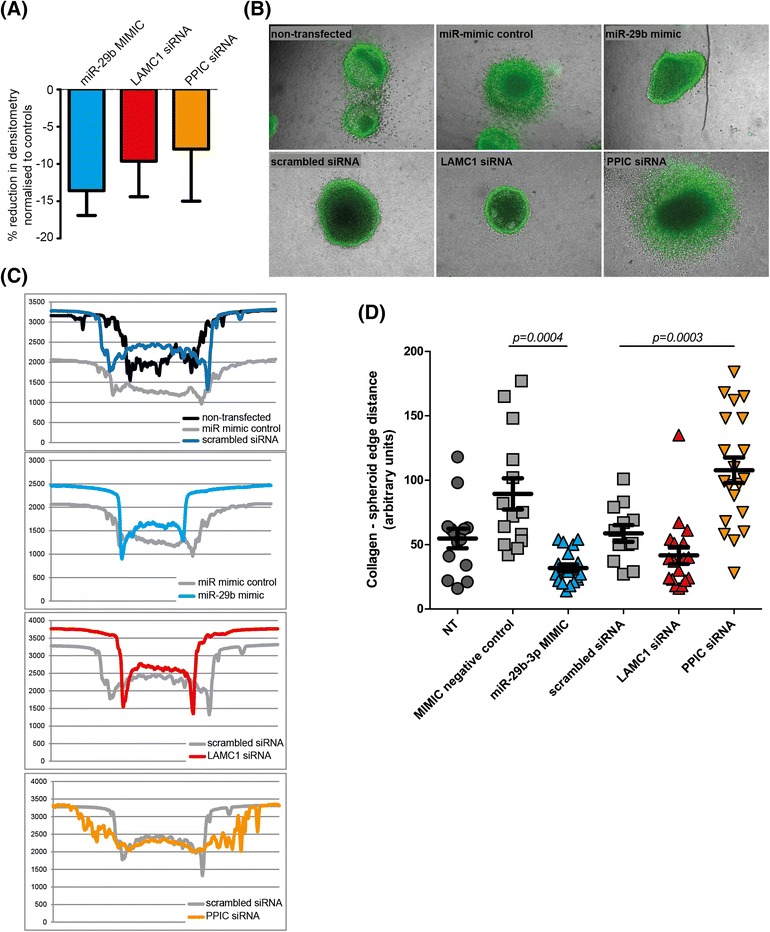
Effects of miR-29b, LAMC1 and PPIC perturbation on motility and invasion of LM-MEL-45. a In the absence of a chemotactic gradient, migration of LM-MEL-45 cells into a central detection zone was decreased by treatment with a miR-29b-3p mimic, and partially phenocopied by siRNA-mediated knock-down of either LAMC1 or PPIC (data represent mean + SEM relative to controls of independent triplicates, all treatments at 10nM). b Following reverse transfection with the indicated agents (final concentrations all 10nM), and overnight culture on agarose, LM-MEL-45 spheroids were embedded into a bovine collagen type I matrix and allowed to invade over 24 h prior to staining with a green-fluorescent viable cell dye and imaging (representative spheroids shown). Treatment with a miR-29b-3p mimic virtually ablated any invasion of cells into surrounding collagen, whilst siRNA-mediated knockdown of PPIC led to marked invasion and/or lack of cohesion, seen as a pronounced spheroid halo. c Cross-sectional cellular density measurements of representative spheroids (see also Figure AF5.6 within Additional file 5) confirm relative lack of invasive cells at the spheroid surface in miR-29b-3p mimic treated cells (second panel) and, to a lesser extent, in LAMC1 knockdown cells (third panel). PPIC knockdown cells displayed a broad spheroid cell density profile consistent with extensive cellular egress into surrounding collagen (bottom panel). d Quantitation of the invasion distance from spheroid edge confirmed significant decrease in collagen invasion following miR-29b-3p overexpression, and increase following PPIC knockdown. Error bars show mean ± SEM of at least seven spheroids per treatment
Next, spheroid collagen invasion assays were performed to compare the same treatments in a three-dimensional matrix-embedded setting. Spheroids were imaged following staining for viable cells. As predicted, miR-29b treatment reduced LM-MEL-45 cellular invasion into surrounding collagen almost entirely (representative spheroids, Fig. 5b). Invasiveness was generally less in siRNA-treated cells, with minimal difference seen for LAMC1 knock-down. Cross-sectional cellular density profiles (Fig. 5c; method illustrated by Figure AF5.6 within Additional file 5) and quantitation of the collagen invasion distance (Fig. 5d) confirmed sharp transitions between relatively acellular surrounding collagen matrix and cell spheroid following miR-29b mimic and LAMC1 transfection (Fig. 5c), consistent with reduced invasion of cells into surrounding collagen.
Unexpectedly, siRNA-mediated knockdown of PPIC dramatically increased LM-MEL-45 cell invasiveness, and on cross-sectional spheroid cell density analysis, no clear transition point was observed in spheroids treated with PPIC siRNA (Fig. 5b & c), indicating diffuse cellular invasion into surrounding collagen. Within the limitations of the assay system used, it could not be determined whether this diffuse invasion of PPIC-knocked-down cells represented enhanced cellular invasiveness, decreased cell-cell cohesion, or a mixture of both processes. The effect of PPIC knockdown was opposite to that predicted following the observation that miR-29b-3p overexpression, which reduces PPIC expression, led to a decrease in spheroid collagen invasion. This contradictory effect is entirely consistent with the notion that microRNAs exert their overall observable effects through the summation of effects on multiple individual gene targets. Such individual effects may be concordant, or discordant, with the overall effect. In this case, specific PPIC knockdown appears to be pro-invasive, but when combined with the totality of perturbations induced by miR-29b-3p overexpression, the pro-invasive effect of PPIC knockdown is more than negated, leading to a net reduction in invasion. This finding demonstrates that the effects of miR-29b-3p on melanoma cell migration are not accurately replicated by perturbation of any single mRNA target in isolation.
Discussion
Integrated analysis of matched microRNA and mRNA abundance data across a large panel of melanoma cell lines identified several putative regulatory relationships influencing melanoma phenotypic plasticity. We demonstrate a novel modulatory effect for miR-29b-3p activity on melanoma cell invasiveness, most likely mediated by the combined effects on multiple gene targets. Dysregulation of miR-29b has been observed in a range of carcinomas of gastrointestinal [69, 71], breast [68], gynaecologic [77] and prostatic origin [78], with a more specific role in suppression of tumour growth and metastasis in colorectal cancer cells shown to involve blockade of epithelial-to-mesenchymal transition [69]. In breast cancer models, it has been shown that miR-29b is induced by, and mediates the EMT-inhibitory effect of GATA3 via repression of tissue microenvironment remodelling factors such as MMP and VEGFA [68, 79]. The functions of miR-29b relevant to phenotypic plasticity in melanoma remain comparatively very poorly-defined. In a study of an IFN-γ-STAT1-miR-29 family interaction circuit, a minority of patient-derived primary melanomas were found to have markedly elevated miR-29b and miR-29a levels relative to benign nevi and normal skin, whilst metastatic samples had marginal or no elevation in miR-29a/b abundance [80]. In a small panel of melanoma cell lines an inverse relationship between miR-29b level and melanoma cell line proliferation was observed, seemingly at odds with the higher miR-29a ~ b1 cluster abundance in some primary melanomas, and our finding that high miR-29b levels are associated with a more proliferative, epithelial-like phenotype. The confluence-based assay used to determine proliferation in the former study, together with markedly higher/combinatorial doses of miR-29 mimics/inhibitor (up to 150nM) likely contribute to these different findings.
We examined LAMC1 and PPIC in detail as potential mediators of miR-29b-3p effects across our melanoma cell models. Laminins, including LAMC1, are abundant glycoproteins within basement membranes. Consistent with our findings in melanoma, LAMC1 suppression by miR-29a/b/c has been shown to influence prostate cancer cell migration and invasion [70]. PPIC, also known as cyclophilin C, is a peptidyl-prolyl cis-trans isomerase (PPIase) which influences protein folding. PPIases are substrates for cyclosporin A and may be secreted in response to cyclosporin exposure; PPIC plays a key role in endoplasmic reticulum redox homeostasis, together with PPIB [81]. Little is known about PPIC in cancer, however PPIA and another PPIase PIN1 have been shown to interact with key growth and signalling proteins including cyclin D1, CDK10, cdc27, and PLK1, with diverse downstream effects on MAPK signalling [82–85]. Further, PIN1 participates in EMT processes within drug-resistant breast cancer [86], and drives invasiveness and tumorigenicity of A375 melanoma cells in murine models [87]. PPIC has been used as a marker of circulating tumour cells (CTCs) in epithelial ovarian cancer [88]. Breast cancer studies suggest a more complicated network of extracellular matrix-mitogenic signalling interactions involving PPIC, osteopontin, CD147 and AKT [89], warranting further exploration in the melanoma setting given prior studies demonstrating that CD147 is a clear driver of melanoma cell proliferation and metastasis in murine models [90].
The inability of LAMC1 and/or PPIC siRNA to recapitulate the effects of a miR-29b-3p mimic within the collagen spheroid assay (Fig. 5d) is likely attributable to several aspects of miR biology. Firstly, miRs have multiple potential targets but only a subset of this repertoire will be active in a given cell; for example, a miR cannot post-transcriptionally regulate a target gene that is not expressed. Secondly, the absolute abundances of miR and target gene influence the likelihood of a physical interaction, which is a pre-requisite for a regulatory interaction. Importantly, the abundances of both miR and targets may vary over time, thus creating a “moving network” of interactions. Finally, the magnitude of effect exerted by a miR on a target gene is not easily predicted and may influence the functional outcome. Taken together, these factors imply that the overall functions of a miR may be difficult to predict by perturbing any single target in isolation.
Individual miR-29b family members display variable expression patterns across tumour types and tumour stage, despite all ultimately coding for mature miRs with equivalent target repertoires [71, 72]. This apparently independent regulation of expression of miRs from distinct genomic locations suggests that coordination of miR function may be intricately linked to the expression of other genomic features, possibly linked by transcription factor binding sites, co-location, and local epigenetic chromatin modification, all of which may combine with other regional genomic effects to produce distinct biological consequences.
Conclusions
Two key features of micro-RNA biology are their ability to target and downregulate a large number of mRNAs [27], and the ability of many micro-RNAs to target a single mRNA, resulting in a network architecture for regulatory interactions [26]. We studied pairwise associations in a large panel of melanoma cell lines informed by empirical phenotypic features. A defining feature of this analysis is the examination of mutual information together with Pearson’s correlation, allowing us to identify strong associations which may are not necessarily linear, before the principled inclusion of sequence-based miR-mRNA predicted interactions. Our approach should provide a useful framework to guide experimental work and elucidate the role of miRs in controlling cell phenotype across a number of cancers.
Our findings implicate miR-29b as a regulator of cellular phenotype in melanoma through proteins including LAMC1 and PPIC within a network of post-transcriptionally regulated genes. It is likely, however, that a complex interplay between these and other as yet undetermined miR-29b targets combine to define the overall cellular behaviours observed. Future work will focus on predicting and modelling more complex transcriptome-wide interactions. Such studies will need to address synchronous and competitive interactions, as well as sequential multi-step interactions such as those mediated by transcription factors. Experimentally validating such networks may be technically challenging, particularly under conditions simulating physiological levels of miR and mRNA transcript abundance. Nonetheless, combinatorial perturbations informed by computational modelling will enhance our understanding of summative biological effects relevant to key cell state transitions in cancer, as typified by EMP.
Methods
Overview
Figure 6 provides an illustrative overview of our analytical pipeline that integrates various data sources (Additional file 1), with additional technical detail given below. For the LM-MEL melanoma cell line panel [40], published mRNA and novel miR transcript data were collected and processed (Fig. 6a-d). Statistical associations were calculated and filtered (Fig. 6e-f), before integrating the TargetScan [47–49], DIANA-microT CDS [50, 51] and miRTarBase [91] databases to identify a list of putative relationships (Fig. 6g-h). We examine and discuss putative miR-mRNA associations in Fig. 2 (Fig. 6i), then explore the relative enrichment of putative relationships for each miR, and the functional annotations of putative miR targets (Fig. 6j-k). Phenotypic invasiveness assays for 24 cell lines were used for functional annotations associated with each putative relationship (Fig. 6l-n), and TCGA melanoma samples were used to consider the potential for these associations within clinical tumour data (Fig. 6o-q).
Fig. 6.
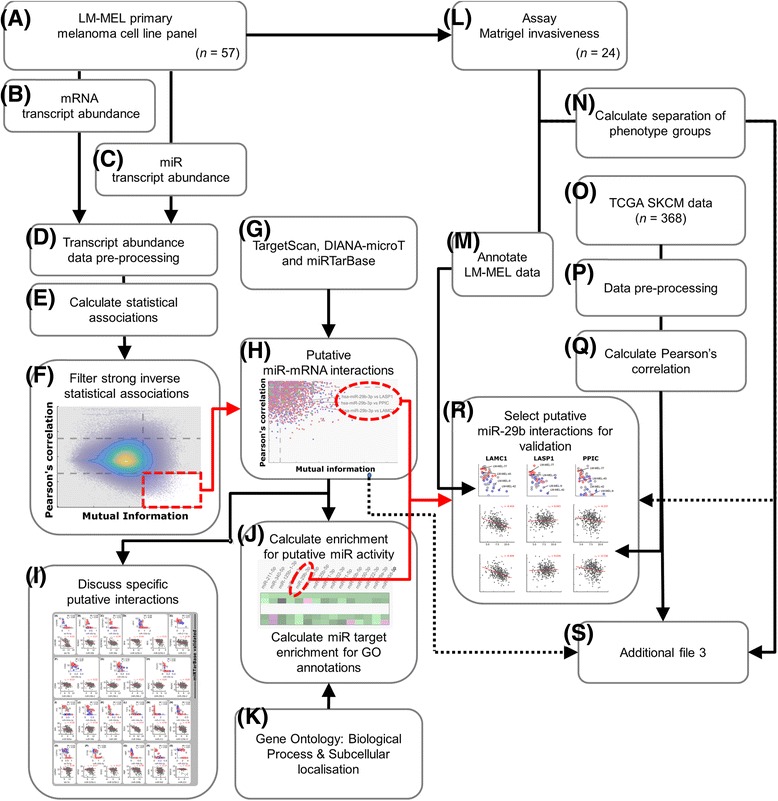
Computational workflow overview. a 57 melanoma cell lines derived (b) underwent mRNA and c miR transcript abundance profiling. d The data were processed and (e) measures of statistical association were calculated between miR and mRNA transcripts, across the LM-MEL panel data. f Strong, negative (inverse) associations (Fig. 1a) were filtered and (g) predicted interactions from TargetScan and/or DIANA-microT, and validated interactions from miRTarBase (h) were matched to provide a list of putative melanoma-relevant associations (Fig. 1b). i A number of interesting putative relationships were examined in further detail (Fig. 2). Enrichment of (j) miR-target associations within our list, and (k) Gene Ontology annotations associated with epithelial-mesenchymal plasticity or melanogenesis/pigmentation were calculated (Fig. 3a). l Independent phenotypic (invasiveness) data were used to (m) annotate results and (n) identify putative miR-mRNA relationships which separated invasiveness groups. o Samples from the TCGA melanoma data were (p) processed and matched allowing (q) Pearson’s correlation between miRs and mRNAs in vivo, to be calculated. r miR-29b-3p appeared to have a novel role regulating melanoma invasiveness, and several putative relationships were validated experimentally (Figs. 3b & 4). s All putative relationships are listed within Additional file 3. Please refer to the main text for details
Evidence suggested that miR-29b-3p influences melanoma invasiveness through regulating several mRNA transcripts (Fig. 6r), thus these putative relationships were experimentally investigated. All putative miR-mRNA relationships are listed in Additional file 3 (Fig. 6s).
Computational scripts
The miR-mRNA interaction pipeline for the LM-MEL data (Fig. 6d-n, r & s) was developed using MATLAB (R2015a) and a selection of data-processing scripts were developed using R (v3.2.3; Fig. 6b, c, o & p) and python (v3.0; Fig. 6i [TCGA plots], q & r). All computational scripts developed for this project are freely available under an MIT license, from: https://github.com/uomsystemsbiology/LMMEL-miR-miner
To reproduce these results without MATLAB and extensive system configuration (e.g. installation of python packages), a Virtual Reference Environment [92] is available; however, users must accept the MATLAB Compiler Runtime Libraries License displayed during installation: https://github.com/uomsystemsbiology/LMMEL-miR-miner_reference_environment
LM-MEL panel mRNA data
The Ludwig Melbourne melanoma (LM-MEL) panel comprised of 57 cell lines derived from mostly metastatic melanoma tumours (Fig. 6a) has previously undergone mRNA transcript abundance profiling with the Illumina HT12 beadchip microarray platform, version 4 [40]. Data were downloaded from ArrayExpress (Additional file 1) and underwent background correction, quantile normalisation and log2-transformation within R, retaining multiple probes as distinct observations (Fig. 6b).
LM-MEL panel miR data
MicroRNA profiling was performed across the LM-MEL panel of cell lines by small RNA sequencing at the Australian Genome Research Facility (AGRF) on the Illumina HiSeq platform using total RNA, including small RNAs, purified from cell line pellets using the Qiagen miRNEasy isolation kit, following the manufacturer’s recommendations (Qiagen, Chadstone, Victoria, Australia). Library preparation and 5’-barcode multiplexing were performed prior to sequencing; each sample was run in 3–4 sequencing lanes as required to achieve adequate sequencing depth. Initial read quality assessment and filtering were performed by AGRF. De-multiplexed raw read data and quality scores were provided in fastq format. All reads for each sample were concatenated using the UNIX command line and collapsed to single fasta format files using the FASTX-Toolkit (v. 0.0.13) command-line tool FASTQ Collapser. Collapsed reads were processed through the miRanalyzer webserver [93] to map reads to the genome using the hg18 build of the UCSC Homo sapiens genome, followed by mapping of miRs to miRBase [94–99]. Raw and processed miR abundance data were deposited on the Gene Expression Omnibus (GEO; dataset GSE89438; Additional file 1). These data underwent normalisation and a log2-transformation was performed (Fig. 6c).
LM-MEL phenotypic invasiveness data
Invasiveness through matrigel-coated Boyden chambers (8 μm pore size) was profiled for 24 LM-MEL cell lines (Fig. 6l); cell lines were subsequently grouped as high- or low-invasiveness (Additional file 1) and used for annotation (Fig. 6m) and further analysis (Fig. 6n).
TCGA miR and mRNA data
Matched miR and mRNA sequencing data from TCGA skin cutaneous melanoma samples were downloaded (Additional file 1), comprised of 368 samples from 366 individual patients (Fig. 6o). R scripts were developed to use the dataset’s native directory structure for extracting and compiling normalised mature microRNA isoform read counts (including ‘star’ [*] forms), and RNAseq normalised read counts from all samples (Fig. 6p). Samples were matched across the microRNA and RNA data according to the unique TCGA patient/sample barcodes.
LM-MEL data pre-processing
To reduce the computational burden of the statistical analysis, the LM-MEL mRNA and miR abundance data were pre-processed (Fig. 6d) to remove RNAs with a very low abundance or dynamic range, as these features can indicate a poor signal-to-noise ratio for the data. The 10th-percentiles of mRNA and miR abundance were identified, and any miRs or mRNAs with less than 25% of observations (i.e. across the 14/57 cell lines) above their respective thresholds were excluded. Similarly, miRs which did not have a range greater than the 90th percentile of the miR abundance data and mRNA probes which did not have a range greater than 10% of the total mRNA data range, were also filtered. This pre-processing retained 198/2592 miRs (note that 916 of the miRs had no reads across any cell line) and 16482/47231 mRNA probes within the LM-MEL data.
Statistical associations
Pearson’s correlation and mutual information were calculated between miR and mRNA abundance, across all pairwise combinations of miRs and mRNAs which passed pre-processing (Fig. 6e). The intrinsic ‘corr’ function within MATLAB (R2015a; Statistics and Machine Learning Toolbox) was used to calculate the Pearson’s correlation.
As noted earlier, mutual information is a measure of statistical association which is formulated such that it will tend towards zero in the case of statistical independence between the miR and mRNA abundance. Mutual information was calculated using the Java Information Dynamics Toolkit (JIDT) [43] with the ‘multivariate Kraskov 2 estimator’ (i.e. miR and mRNA transcript abundance data were treated as continuous variables and the Kraskov-Stoegbauer-Grassberger estimator was used to calculate the MI), implemented within the MATLAB Network Analysis and Inference Libraries [42]. Note that the JIDT corrects for mutual information over-estimation [46], and this subtraction can lead to negative mutual information values (Fig. 1a & b).
For the TCGA data analysed within python, correlations were calculated (Fig. 6q) using the numpy package [100] and plots (Figs. 6I & 7r) were produced using the matplotlib package [101].
Under the hypothesis that miR-mediated degradation of target mRNA transcripts would manifest as a negative association, we filtered for the top 10% of associations when ranked by mutual information (mutual information > 0.259), and the most negative 2.5% of associations when ranked by Pearson’s correlation (Pearson’s correlation < −0.330; Figs. 1a & 7f).
Databases
Ensembl BioMart [102] was used to match identifiers between different databases as necessary. To reduce the search domain of putative interactions and filter indirect associations which may arise through the modulation of an intermediate regulatory component (e.g. a transcription factor), we filtered for predicted miR-mRNA relationships with a relatively high confidence. Specifically, we took the top 15th percentile of TargetScan v7.0 (context + score < −0.286) [47–49], and/or the top 30th percentile of DIANA-microT CDS (miTG-score > 0.634) [50, 51] (Fig. 6g). The miR-mRNA relationships which had been experimentally validated with ‘strong evidence’ on miRTarBase [91] (‘Luciferase reporter assay’; 'qRT-PCR' or 'Western blot') were also extracted (Fig. 6g) and used to annotate the selected statistical associations. This allowed us to identify a set of putative miR-mRNA relationships (Figs. 1b & 7 h) which may show graded levels of activity across different melanoma samples, contributing to differences in cell phenotype.
Micro-RNAs can regulate large phenotypic changes through distributed regulation of targets with a related function [27]. Annotations were downloaded from the Gene Ontology [103] database [104] (Fig. 6k) and mRNAs were matched to GO annotations using strings related to epithelial-mesenchymal plasticity (‘epith’,’mesench’) or pigmentation/melanogenesis (‘pigment’, ‘melan’; Fig. 6j, at bottom). GO categories were excluded if they had less than 5, or more than 500, gene members, and all categories are listed in Additional file 6.
Invasiveness-separation metric
A metric was created to quantify the relative separation of low- and high-invasive cell line groups over the miR abundance ([miR]) versus mRNA abundance ([mRNA]) association (Fig. 6n). Calculating the centroid/geometric mean of the low- and high-invasive groups from mRNA and miR abundances, we defined:
The percentile rank of dSep,norm was calculated for all miR-mRNA associations, where the miR and mRNA association passed the data pre-processing, and is listed with corresponding relationships within Additional file 3.
Cell line culture
Melanoma cell lines were obtained from institutional stocks, as previously described [40]. LM-MEL-7, −9, −42, −45 and −77 were used for in vitro experiments; identity was confirmed by STR profiling and HLA-matching to the documented patient of origin. All cell lines were maintained in adherent culture incubated in 5% atmospheric CO2 at 37°C in RPMI 1640 media (Gibco®, Life Technologies™, Mulgrave VIC, Australia) supplemented with 10% fetal bovine serum (Sigma, St Louis MO, USA), 1% glutamine (glutaMAX™) and 1% penicillin/streptomycin (both from Gibco®, Life Technologies™).
Transient transfections with miR inhibitor/mimic or siRNA
Cells were seeded into 6, 12 or 96 well plates as required for planned downstream assays. Transfections were performed when cells were approximately 70% confluent, using the Lipofectamine® RNAiMAX transfection reagent (Invitrogen™, Life Technologies™) at 0.3, 2 or 3 μL per well of 96, 12 or 6 well plates, respectively, diluted in OptiMEM® reduced serum medium (Gibco®, Life Technologies™) and mixed 1:1 with microRNA/siRNA construct (also in OptiMEM®) to a final volume producing a 1:5 final dilution into growth media. For microRNA transfections, cells were forward transfected with mirVana™ miRNA inhibitor negative control #1 or miRNA-29b-3p inhibitor, miRNA mimic negative control #1 or miRNA-29b-3p mimic (all Ambion™, Life Technologies™, Cat. Nos. 4464076, 4464084, 4464058, 4464066) at final concentrations as indicated in results. For gene target knockdown studies, cells were transfected with siRNA scrambled control or specific siRNAs targeting LAMC1, LASP1 or PPIC (all OriGene Trilencer-27 Human siRNA, Cat. Nos. SR30004, SR302649, SR302655, SR303664; Rockville, MD, USA), at doses indicated in results. For spheroid assays, cells were reverse transfected using identical reagent mixtures at the time of seeding onto agar.
Real time-PCR
Total RNA was collected from treated/untreated cell pellets using RNeasy (standard gene qPCR only) or miRNeasy (total RNA including small RNA) kits (Qiagen, Melbourne, Victoria, Australia) and cDNA formed using the high-capacity cDNA reverse transcription kit (Applied Biosystems™, Life Technologies™, Cat. No. 4368814) or TaqMan® microRNA reverse transcription kit (Cat. No. 4366596), respectively, according to the manufacturer’s protocol. MicroRNA qPCR was performed using TaqMan® Universal PCR Master Mix, no AmpErase® UNG (Cat. No. 4324018) and TaqMan® microRNA assays (all Cat. No. 4427975) specific for hsa-miR-211 (ID 000514), hsa-miR-125b-1* (ID 002378), hsa-miR-221* (ID 002096), hsa-miR-9 (ID 000583), hsa-miR-23b (ID 000400), hsa-miR-29b (ID 000413), hsa-miR-222 (ID 002276), hsa-let-7a (ID 000377), or controls RNU44 (ID 001094) and RNU24 (ID 001001). Target gene qPCR was performed using SensiFAST™ SYBR® Lo-ROX mastermix (Bioline, Alexandria, NSW, Australia) with PCR primers designed as per Table AF5.1 in Additional file 5.
Data were collected as ∆∆CT with melt curve inspection using the ViiA™ 7 Real-Time PCR System and accompanying software (Applied Biosystems™, Life Technologies™) based on technical triplicates. Mean CT from 2 to 3 biological replicates was expressed as copies per 10,000 copies of a reference gene (β-actin, RNU24 or RNU44).
In vitro proliferation assays
Cells were seeded at 5000 cells per well in flat-bottomed 96-well tissue culture plates prior to transfection with microRNA or siRNA agents as described above. Cellular proliferation was measured at baseline (i.e. pre-transfection) and at 24 and 72 h following transfection by incubation for 1.5 h in MTS reagent (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega, Madison WI, USA) diluted 1 in 6 in culture media, followed by measurement of the absorbance at 490 nm and background correction using a no-cell control.
Low-density seeding assays
Transfected cells were re-harvested and plated in 6-well tissue-culture treated plates at a density of 2000 cells per well, in 2 mL culture medium. Cells were monitored visually each day and allowed to grow for 21 days prior to fixation in 2% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) and staining with 0.01% crystal violet/10% ethanol. Plates were imaged in the 680 nm infrared channel using the Odyssey scanner (LI-COR® Biosciences, Lincoln, NE, USA) prior to subsequent quantitation of cellular outgrowth using the Colony Area plugin for ImageJ (version 1.47) [105, 106].
Western blotting
Protein was harvested from cells 96 h following transient transfection with miR-29b agents or specific siRNAs against LAMC1, LASP1 or PPIC. Cell lysates were prepared in 100 μL of RIPA buffer (Thermo Scientific™ Pierce™, Cat. No. 89900) containing the manufacturer’s recommended concentrations of PhosSTOP™ phosphatase inhibitor and cOmplete™ ULTRA EDTA-free protease inhibitor (Roche Diagnostics GmbH, Mannheim, Germany), incubated for 30 min at 4°C prior to manual scraping of cells/lysates. Protein concentrations were estimated using the BCA method (Thermo Scientific™ Pierce™ BCATM Protein Assay Kit, Cat#23225) as per the manufacturer’s directions. Samples were immediately adjusted to uniform final concentrations by the addition of extra lysis buffer as required, prior to the addition of NuPAGE® LDS Sample Buffer (4×) and NuPAGE® sample reducing agent (10×) (both from Novex®, Life Technologies, Cat#NP008 and Cat#NP009). Samples were electrophoresed on NuPAGE® Novex® Bis-Tris 4–12% pre-cast gels prior to transfer to nitrocellulose membrane using the iBlot® Dry Blotting System (Invitrogen™, Life Technologies, Mulgrave, Victoria, Australia). Membranes were blocked in Odyssey Blocking Buffer (LI-COR® Biosciences) for 1 h at room temperature before probing for LAMC1 (apparent MW 178 kDa) using the rabbit anti-laminin gamma 1 monoclonal antibody RabMab [EPR8217] (Abcam, Cat. No. ab134059; Melbourne, Victoria, Australia) at 1:1000, for LASP1 (apparent MW 30 kDa) using the rabbit polyclonal anti-LASP1 – N-terminal antibody (Abcam, Cat. No. ab191022) at 1:1000, or for PPIC using the rabbit monoclonal anti-PPIC – C-terminal antibody RabMab [EPR15355] (Abcam, Cat. No. ab184552) at 1:10,000. Loading was controlled to β-Actin (apparent MW 45 kDa) using the mouse monoclonal anti-β-Actin (8H10D10) antibody (Cell Signaling Technology, Cat. No. 3700; Danvers, MA, USA) at 1:3000. Membranes were incubated with primary antibodies for 2 h at room temperature in a 1:1 mixture of PBS containing 0.05% tween-20 (PBST) and Odyssey Blocking Buffer. Bands were visualised by staining with IRDye® 680RD goat anti-mouse IgG and IRDye® 800CW goat anti-rabbit IgG secondaries at 1:40,000 and 1:20,000 respectively (LI-COR®, Prod. Nos. 926–68070 and 926–32211) for 1 h at room temperature. Air-dried membranes were scanned on the LI-COR® ODYSSEY® Infrared Imaging System and densitometry performed in the Image Studio™ Lite Version 5.0 software (LI-COR®). Densitometry results were normalized to β-Actin and expressed relative to the appropriate control treatment.
Undirected radial migration assays
Transfected cells were re-harvested, counted and delivered at 40,000 cells per well of 96 well optical-bottom plates (Nunc, ThermoFisher Scientific) fitted with Oris™ Cell Migration Assay stoppers (Platypus Technologies, Madison, WI, USA). Cells were allowed to adhere overnight prior to removal of stoppers, leaving a 2 mm diameter detection zone in the centre of each well. After changing to fresh growth media, cells were allowed to migrate for 24–48 h prior to fixation in 2% paraformaldehyde and staining with 0.01% crystal violet/10% ethanol.
Spheroid collagen invasion assays
Collagen-implanted spheroid invasion was performed based on previously-described methods [107]. Spheroids were prepared by seeding 150,000 cells in 2 mL of growth medium (with required transfection reagents) into wells of six well plates on top of pre-formed 0.8% agarose base-layers (Sigma, Cat. No. A9539) diluted from sterilised 3.2% agarose stock with growth medium. After 48 h incubation, spheroids were harvested by gentle aspiration, transferred to sterile collection tubes and allowed to settle by gravity for 30 min prior to manual aspiration of overlying media. Neutralised collagen was prepared from 5 mg/mL bovine collagen type I stock (Trevigen Cultrex®, Cat. No. 3442-050-01, Gaithersberg, MD, USA) by dilution 1:1 with 2× growth medium (2× RPMI 1640, 20% FBS) and pH adjusted by dropwise addition of sterile 1 M NaOH. Acellular collagen base layers were prepared by aliquoting 300 μL of neutralised collagen into wells of a 24 well plate and allowed to gel at room temperature for 30 min. Spheroids were then gently resuspended in 500 μL of neutralised collagen diluted 2:1 with 1× growth medium (final collagen concentration 1.67 mg/mL) and overlaid onto base layers and allowed to gel for 10 min at room temperature before returning to the incubator. Once completely gelled, collagen was overlaid with a further 500 μL of growth media. Spheroids were allowed to invade for 24 h prior to fluorescent viability staining with calcein-AM (live) and EthD-III (dead) using the Live/Dead Cell Staining Kit II (PromoKine, Cat. No. PK-CA707-30002, PromoCell GmbH, Heidelberg, Germany) before imaging.
Acknowledgements
The authors would like to acknowledge researchers who have established and maintain the databases (Additional file 1) which make this analysis feasible. The results published here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
JC would like to thank: Dr C Bracken at the Centre for Cancer Biology, University of South Australia, for his advice on the computational analysis and for providing feedback on the manuscript.
Funding
MCA is supported by a Postgraduate Medical Scholarship from the National Health and Medical Research Council of Australia (#1055456). This work was funded in part by: Ludwig Cancer Research; the Operational Infrastructure Support Program of the Victorian State Government; and the Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (project number CE140100036).
Availability of data and materials
All data used in this study are included within the article and additional files, or made freely available within repositories as specified in Additional file 1.
Authors' contributions
MCA carried out gene and microRNA expression analysis, data processing, and interpretation, in vitro validation studies, and drafting of the manuscript. JC performed transcriptomic analysis, data integration, systems analysis and interpretation, and drafting of the manuscript. DGH participated in transcriptomic and systems analysis and interpretation, and drafting of the manuscript. MA participated in sample acquisition, and generating primary mRNA/microRNA abundance data. AB participated in sample acquisition, cell line and primary data generation, transcriptomic data interpretation, in vitro studies and phenotypic assays. JSC was involved in sample acquisition. JSC and EC participated in data interpretation. All authors were involved in study conception and design, and have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- CTCs
Circulating tumour cells
- EMP
Epithelial-mesenchymal plasticity
- EMT
Epithelial-to-mesenchymal (like) transition
- LAMC1
Laminin, γ1 (Entrez Gene ID 3915)
- LASP1
LIM and SH3 Protein 1 (Entrez Gene ID 3927)
- LM-MEL
Ludwig Melbourne melanoma cell line
- miR
microRNA
- miR-29b
hsa-miR-29b-3p (miRBase Accession: MIMAT0000100)
- PPIC
Peptidylprolyl Isomerase C (Cyclophilin C; Entrez Gene ID 5480)
- TCGA
The Cancer Genome Atlas
Additional files
Data and Computational Tools (.docx) – Contains Table AF1.1, which lists detailed information on the data and computational tools used in this study. (DOCX 14 kb)
Table (.csv) of matrigel invasiveness (classified as invasive/non-invasive) for LM-MEL cell lines which is read into the computational scripts. For details on the experimental protocol please refer to Methods/LM-MEL phenotypic invasiveness data. (CSV 273 bytes)
Table (.csv) output from the computational scripts containing (1041) putative relationships with a strong negative statistical association and support from TargetScan and/or DIANA-microT, or mirTarBase (i.e. the full list of associations from Fig. 1b). mRNA Transcripts which have been annotated with a role in pigmentation or EMP via their GO category (from Fig. 1c) are labelled. mRNA transcripts which have been previously implicated in phenotypic switching in melanoma are labelled [3]. (CSV 77 kb)
miRNA-mRNA Interactions of Interest (.docx) – Contains: Table AF4.1, which lists miRNA-mRNA interactions identified from our analysis that have been previously validated in human cellular contexts. Table AF4.2, which lists novel putative miRNA-mRNA interactions that involve mRNA transcripts implicated in melanoma EMP behaviours and/or invasiveness. (DOCX 86 kb)
Supporting Experimental Data (.pdf) – Contains: Table AF5.1, which lists the PCR primers used. Figure AF5.1 which displays baseline expression for genes of interest in a subset of LM-MEL melanoma cell lines using qPCR. Figure AF5.2, which displays siRNA transfection efficiency; Figure AF5.3, which displays additional qPCR results showing the effects of miR-29b mimic/inhibitor transfection. Figure AF5.4 & AF5.5, which display the effects of miR-29b mimic/inhibitor transfection on LM-MEL-77 cell proliferation and outgrowth. Figure AF5.6, which displays information to aid the reader’s interpretation of melanoma spheroid data. (PDF 3182 kb)
Table (.csv) of Gene Ontology (GO) database terms used for the enrichment analysis of ‘epithelial-mesenchymal plasticity’ (EMP) and pigmentation processes in Fig. 3a, together with the number of genes within each category. As detailed in Methods/Databases, terms were identified by string matching and GO terms were excluded if they had less than five or more than 500 member genes. (CSV 5 kb)
Contributor Information
Miles C. Andrews, Email: miles.andrews@onjcri.org.au
Joseph Cursons, Email: joseph.cursons@unimelb.edu.au.
Daniel G. Hurley, Email: daniel.hurley@unimelb.edu.au
Matthew Anaka, Email: matt.anaka@gmail.com.
Jonathan S. Cebon, Email: jonathan.cebon@onjcri.org.au
Andreas Behren, Email: andreas.behren@onjcri.org.au.
Edmund J. Crampin, Email: edmund.crampin@unimelb.edu.au
References
- 1.Zipser MC, Eichhoff OM, Widmer DS, Schlegel NC, Schoenewolf NL, Stuart D, Liu W, Gardner H, Smith PD, Nuciforo P, et al. A proliferative melanoma cell phenotype is responsive to RAF/MEK inhibition independent of BRAF mutation status. Pigment Cell Melanoma Res. 2011;24:326–33. doi: 10.1111/j.1755-148X.2010.00823.x. [DOI] [PubMed] [Google Scholar]
- 2.Hoek KS, Eichhoff OM, Schlegel NC, Dobbeling U, Kobert N, Schaerer L, Hemmi S, Dummer R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68:650–6. doi: 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- 3.Widmer DS, Cheng PF, Eichhoff OM, Belloni BC, Zipser MC, Schlegel NC, Javelaud D, Mauviel A, Dummer R, Hoek KS. Systematic classification of melanoma cells by phenotype-specific gene expression mapping. Pigment Cell Melanoma Res. 2012;25:343–53. doi: 10.1111/j.1755-148X.2012.00986.x. [DOI] [PubMed] [Google Scholar]
- 4.Ramsdale R, Jorissen RN, Li FZ, Al-Obaidi S, Ward T, Sheppard KE, Bukczynska PE, Young RJ, Boyle SE, Shackleton M, et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal. 2015;8:ra82. doi: 10.1126/scisignal.aab1111. [DOI] [PubMed] [Google Scholar]
- 5.Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4:816–27. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, Narayan R, Flaherty KT, Wargo JA, Root DE, Garraway LA. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504:138–42. doi: 10.1038/nature12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J, Hutchinson P, Tse G, et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell. 2013;24:466–80. doi: 10.1016/j.ccr.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 8.Alonso SR, Tracey L, Ortiz P, Perez-Gomez B, Palacios J, Pollan M, Linares J, Serrano S, Saez-Castillo AI, Sanchez L, et al. A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res. 2007;67:3450–60. doi: 10.1158/0008-5472.CAN-06-3481. [DOI] [PubMed] [Google Scholar]
- 9.Hao L, Ha JR, Kuzel P, Garcia E, Persad S. Cadherin switch from E- to N-cadherin in melanoma progression is regulated by the PI3K/PTEN pathway through Twist and Snail. Br J Dermatol. 2012;166:1184–97. doi: 10.1111/j.1365-2133.2012.10824.x. [DOI] [PubMed] [Google Scholar]
- 10.Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, Testori A, Larue L, Goding CR. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20:3426–39. doi: 10.1101/gad.406406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tili E, Michaille JJ, Croce CM. MicroRNAs play a central role in molecular dysfunctions linking inflammation with cancer. Immunol Rev. 2013;253:167–84. doi: 10.1111/imr.12050. [DOI] [PubMed] [Google Scholar]
- 12.Olive V, Jiang I, He L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol. 2010;42:1348–54. doi: 10.1016/j.biocel.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–54. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 14.Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, Morris M, Wyatt L, Farshid G, Lim YY, et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22:1686–98. doi: 10.1091/mbc.E11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haflidadottir BS, Bergsteinsdottir K, Praetorius C, Steingrimsson E. miR-148 regulates Mitf in melanoma cells. PLoS One. 2010;5 doi: 10.1371/journal.pone.0011574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kappelmann M, Kuphal S, Meister G, Vardimon L, Bosserhoff AK. MicroRNA miR-125b controls melanoma progression by direct regulation of c-Jun protein expression. Oncogene. 2013;32:2984–91. doi: 10.1038/onc.2012.307. [DOI] [PubMed] [Google Scholar]
- 17.Couts KL, Anderson EM, Gross MM, Sullivan K, Ahn NG. Oncogenic B-Raf signaling in melanoma cells controls a network of microRNAs with combinatorial functions. Oncogene. 2013;32:1959–70. doi: 10.1038/onc.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Streicher KL, Zhu W, Lehmann KP, Georgantas RW, Morehouse CA, Brohawn P, Carrasco RA, Xiao Z, Tice DA, Higgs BW, et al. A novel oncogenic role for the miRNA-506-514 cluster in initiating melanocyte transformation and promoting melanoma growth. Oncogene. 2012;31:1558–70. doi: 10.1038/onc.2011.345. [DOI] [PubMed] [Google Scholar]
- 19.Schultz J, Lorenz P, Gross G, Ibrahim S, Kunz M. MicroRNA let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Res. 2008;18:549–57. doi: 10.1038/cr.2008.45. [DOI] [PubMed] [Google Scholar]
- 20.Felicetti F, Errico MC, Bottero L, Segnalini P, Stoppacciaro A, Biffoni M, Felli N, Mattia G, Petrini M, Colombo MP, et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res. 2008;68:2745–54. doi: 10.1158/0008-5472.CAN-07-2538. [DOI] [PubMed] [Google Scholar]
- 21.Bonazzi VF, Stark MS, Hayward NK. MicroRNA regulation of melanoma progression. Melanoma Res. 2012;22:101–13. doi: 10.1097/CMR.0b013e32834f6fbb. [DOI] [PubMed] [Google Scholar]
- 22.Dar AA, Majid S, Rittsteuer C, de Semir D, Bezrookove V, Tong S, Nosrati M, Sagebiel R, Miller JR, 3rd, Kashani-Sabet M. The role of miR-18b in MDM2-p53 pathway signaling and melanoma progression. J Natl Cancer Inst. 2013;105:433–42. doi: 10.1093/jnci/djt003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penna E, Orso F, Cimino D, Vercellino I, Grassi E, Quaglino E, Turco E, Taverna D. miR-214 coordinates melanoma progression by upregulating ALCAM through TFAP2 and miR-148b downmodulation. Cancer Res. 2013;73:4098–111. doi: 10.1158/0008-5472.CAN-12-3686. [DOI] [PubMed] [Google Scholar]
- 24.Bell RE, Khaled M, Netanely D, Schubert S, Golan T, Buxbaum A, Janas MM, Postolsky B, Goldberg MS, Shamir R, Levy C. Transcription factor/microRNA axis blocks melanoma invasion program by miR-211 targeting NUAK1. J Invest Dermatol. 2014;134:441–51. doi: 10.1038/jid.2013.340. [DOI] [PubMed] [Google Scholar]
- 25.Elson-Schwab I, Lorentzen A, Marshall CJ. MicroRNA-200 family members differentially regulate morphological plasticity and mode of melanoma cell invasion. PLoS One. 2010;5(10) doi: 10.1371/journal.pone.0013176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bracken CP, Khew-Goodall Y, Goodall GJ. Network-based approaches to understand the roles of miR-200 and other microRNAs in cancer. Cancer Res. 2015;75:2594–9. doi: 10.1158/0008-5472.CAN-15-0287. [DOI] [PubMed] [Google Scholar]
- 27.Bracken CP, Li X, Wright JA, Lawrence DM, Pillman KA, Salmanidis M, Anderson MA, Dredge BK, Gregory PA, Tsykin A, et al. Genome-wide identification of miR-200 targets reveals a regulatory network controlling cell invasion. EMBO J. 2014;33:2040–56. doi: 10.15252/embj.201488641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 29.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–40. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eichhorn SW, Guo H, McGeary SE, Rodriguez-Mias RA, Shin C, Baek D, Hsu SH, Ghoshal K, Villen J, Bartel DP. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol Cell. 2014;56:104–15. doi: 10.1016/j.molcel.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–33. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 33.Cloonan N. Re-thinking miRNA-mRNA interactions: intertwining issues confound target discovery. Bioessays. 2015;37:379–88. doi: 10.1002/bies.201400191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plaisier CL, Pan M, Baliga NS. A miRNA-regulatory network explains how dysregulated miRNAs perturb oncogenic processes across diverse cancers. Genome Res. 2012;22:2302–14. doi: 10.1101/gr.133991.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell. 2014;54:766–76. doi: 10.1016/j.molcel.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez-Mejias A, Tay Y. Competing endogenous RNA networks: tying the essential knots for cancer biology and therapeutics. J Hematol Oncol. 2015;8:30. doi: 10.1186/s13045-015-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–8. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 38.Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160:1125–34. doi: 10.1016/j.cell.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 39.Philippidou D, Schmitt M, Moser D, Margue C, Nazarov PV, Muller A, Vallar L, Nashan D, Behrmann I, Kreis S. Signatures of microRNAs and selected microRNA target genes in human melanoma. Cancer Res. 2010;70:4163–73. doi: 10.1158/0008-5472.CAN-09-4512. [DOI] [PubMed] [Google Scholar]
- 40.Behren A, Anaka M, Lo PH, Vella LJ, Davis ID, Catimel J, Cardwell T, Gedye C, Hudson C, Stan R, Cebon J. The Ludwig institute for cancer research Melbourne melanoma cell line panel. Pigment Cell Melanoma Res. 2013;26:597–600. doi: 10.1111/pcmr.12097. [DOI] [PubMed] [Google Scholar]
- 41.Jayachandran A, Anaka M, Prithviraj P, Hudson C, McKeown SJ, Lo PH, Vella LJ, Goding CR, Cebon J, Behren A. Thrombospondin 1 promotes an aggressive phenotype through epithelial-to-mesenchymal transition in human melanoma. Oncotarget. 2014;5:5782–97. doi: 10.18632/oncotarget.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hurley DG, Cursons J, Wang YK, Budden DM, Print CG, Crampin EJ. NAIL, a software toolset for inferring, analyzing and visualizing regulatory networks. Bioinformatics. 2015;31:277–8. doi: 10.1093/bioinformatics/btu612. [DOI] [PubMed] [Google Scholar]
- 43.Lizier JT. JIDT: An information-theoretic toolkit for studying the dynamics of complex systems. arXiv preprint arXiv:14083270 2014.
- 44.Reshef DN, Reshef YA, Finucane HK, Grossman SR, McVean G, Turnbaugh PJ, Lander ES, Mitzenmacher M, Sabeti PC. Detecting novel associations in large data sets. Science. 2011;334:1518–24. doi: 10.1126/science.1205438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005;37:382–90. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- 46.Budden DM, Crampin EJ. Information theoretic approaches for inference of biological networks from continuous-valued data. BMC Syst Biol. 2016;10:89. doi: 10.1186/s12918-016-0331-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 50.Paraskevopoulou MD, Georgakilas G, Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C, Dalamagas T, Hatzigeorgiou AG. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41:W169–73. doi: 10.1093/nar/gkt393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reczko M, Maragkakis M, Alexiou P, Grosse I, Hatzigeorgiou AG. Functional microRNA targets in protein coding sequences. Bioinformatics. 2012;28:771–6. doi: 10.1093/bioinformatics/bts043. [DOI] [PubMed] [Google Scholar]
- 52.Cancer Genome Atlas Research N. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12:406–14. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 54.Fufa TD, Harris ML, Watkins-Chow DE, Levy D, Gorkin DU, Gildea DE, Song L, Safi A, Crawford GE, Sviderskaya EV, et al. Genomic analysis reveals distinct mechanisms and functional classes of SOX10-regulated genes in melanocytes. Hum Mol Genet. 2015;24:5433–50. doi: 10.1093/hmg/ddv267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng PF, Shakhova O, Widmer DS, Eichhoff OM, Zingg D, Frommel SC, Belloni B, Raaijmakers MI, Goldinger SM, Santoro R, et al. Methylation-dependent SOX9 expression mediates invasion in human melanoma cells and is a negative prognostic factor in advanced melanoma. Genome Biol. 2015;16:42. doi: 10.1186/s13059-015-0594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shakhova O, Cheng P, Mishra PJ, Zingg D, Schaefer SM, Debbache J, Hausel J, Matter C, Guo T, Davis S, et al. Antagonistic cross-regulation between Sox9 and Sox10 controls an anti-tumorigenic program in melanoma. PLoS Genet. 2015;11 doi: 10.1371/journal.pgen.1004877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118–22. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 58.Cursons J, Leuchowius KJ, Waltham M, Tomaskovic-Crook E, Foroutan M, Bracken CP, Redfern A, Crampin EJ, Street I, Davis MJ, Thompson EW. Stimulus-dependent differences in signalling regulate epithelial-mesenchymal plasticity and change the effects of drugs in breast cancer cell lines. Cell Commun Signal. 2015;13:26. doi: 10.1186/s12964-015-0106-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levy C, Khaled M, Iliopoulos D, Janas MM, Schubert S, Pinner S, Chen PH, Li S, Fletcher AL, Yokoyama S, et al. Intronic miR-211 assumes the tumor suppressive function of its host gene in melanoma. Mol Cell. 2010;40:841–9. doi: 10.1016/j.molcel.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jukic DM, Rao UN, Kelly L, Skaf JS, Drogowski LM, Kirkwood JM, Panelli MC. Microrna profiling analysis of differences between the melanoma of young adults and older adults. J Transl Med. 2010;8:27. doi: 10.1186/1479-5876-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mueller DW, Rehli M, Bosserhoff AK. miRNA expression profiling in melanocytes and melanoma cell lines reveals miRNAs associated with formation and progression of malignant melanoma. J Invest Dermatol. 2009;129:1740–51. doi: 10.1038/jid.2008.452. [DOI] [PubMed] [Google Scholar]
- 62.Wozniak M, Sztiller-Sikorska M, Czyz M. Diminution of miR-340-5p levels is responsible for increased expression of ABCB5 in melanoma cells under oxygen-deprived conditions. Exp Mol Pathol. 2015;99:707–16. doi: 10.1016/j.yexmp.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 63.Poenitzsch Strong AM, Setaluri V, Spiegelman VS. MicroRNA-340 as a modulator of RAS-RAF-MAPK signaling in melanoma. Arch Biochem Biophys. 2014;563:118–24. doi: 10.1016/j.abb.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goswami S, Tarapore RS, Poenitzsch Strong AM, TeSlaa JJ, Grinblat Y, Setaluri V, Spiegelman VS. MicroRNA-340-mediated degradation of microphthalmia-associated transcription factor (MITF) mRNA is inhibited by coding region determinant-binding protein (CRD-BP) J Biol Chem. 2015;290:384–95. doi: 10.1074/jbc.M114.590158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Felicetti F, Errico MC, Segnalini P, Mattia G, Care A. MicroRNA-221 and −222 pathway controls melanoma progression. Expert Rev Anticancer Ther. 2008;8:1759–65. doi: 10.1586/14737140.8.11.1759. [DOI] [PubMed] [Google Scholar]
- 66.Kanemaru H, Fukushima S, Yamashita J, Honda N, Oyama R, Kakimoto A, Masuguchi S, Ishihara T, Inoue Y, Jinnin M, Ihn H. The circulating microRNA-221 level in patients with malignant melanoma as a new tumor marker. J Dermatol Sci. 2011;61:187–93. doi: 10.1016/j.jdermsci.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 67.Zhang JX, Mai SJ, Huang XX, Wang FW, Liao YJ, Lin MC, Kung HF, Zeng YX, Xie D. MiR-29c mediates epithelial-to-mesenchymal transition in human colorectal carcinoma metastasis via PTP4A and GNA13 regulation of beta-catenin signaling. Ann Oncol. 2014;25:2196–204. doi: 10.1093/annonc/mdu439. [DOI] [PubMed] [Google Scholar]
- 68.Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol. 2013;15:201–13. doi: 10.1038/ncb2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang B, Li W, Liu H, Yang L, Liao Q, Cui S, Wang H, Zhao L. miR-29b suppresses tumor growth and metastasis in colorectal cancer via downregulating Tiam1 expression and inhibiting epithelial-mesenchymal transition. Cell Death Dis. 2014;5 doi: 10.1038/cddis.2014.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nishikawa R, Goto Y, Kojima S, Enokida H, Chiyomaru T, Kinoshita T, Sakamoto S, Fuse M, Nakagawa M, Naya Y, et al. Tumor-suppressive microRNA-29 s inhibit cancer cell migration and invasion via targeting LAMC1 in prostate cancer. Int J Oncol. 2014;45:401–10. doi: 10.3892/ijo.2014.2437. [DOI] [PubMed] [Google Scholar]
- 71.Gong J, Li J, Wang Y, Liu C, Jia H, Jiang C, Wang Y, Luo M, Zhao H, Dong L, et al. Characterization of microRNA-29 family expression and investigation of their mechanistic roles in gastric cancer. Carcinogenesis. 2014;35:497–506. doi: 10.1093/carcin/bgt337. [DOI] [PubMed] [Google Scholar]
- 72.Yamamoto N, Kinoshita T, Nohata N, Yoshino H, Itesako T, Fujimura L, Mitsuhashi A, Usui H, Enokida H, Nakagawa M, et al. Tumor-suppressive microRNA-29a inhibits cancer cell migration and invasion via targeting HSP47 in cervical squamous cell carcinoma. Int J Oncol. 2013;43:1855–63. doi: 10.3892/ijo.2013.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nguyen T, Kuo C, Nicholl MB, Sim MS, Turner RR, Morton DL, Hoon DS. Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics. 2011;6:388–94. doi: 10.4161/epi.6.3.14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seip K, Fleten KG, Barkovskaya A, Nygaard V, Haugen MH, Engesaeter BO, Maelandsmo GM, Prasmickaite L. Fibroblast-induced switching to the mesenchymal-like phenotype and PI3K/mTOR signaling protects melanoma cells from BRAF inhibitors. Oncotarget. 2016;7(15):19997–20015. [DOI] [PMC free article] [PubMed]
- 75.Hoek KS, Goding CR. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010;23:746–59. doi: 10.1111/j.1755-148X.2010.00757.x. [DOI] [PubMed] [Google Scholar]
- 76.Pinner S, Jordan P, Sharrock K, Bazley L, Collinson L, Marais R, Bonvin E, Goding C, Sahai E. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 2009;69:7969–77. doi: 10.1158/0008-5472.CAN-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Flavin R, Smyth P, Barrett C, Russell S, Wen H, Wei J, Laios A, O'Toole S, Ring M, Denning K, et al. miR-29b expression is associated with disease-free survival in patients with ovarian serous carcinoma. Int J Gynecol Cancer. 2009;19:641–7. doi: 10.1111/IGC.0b013e3181a48cf9. [DOI] [PubMed] [Google Scholar]
- 78.Ru P, Steele R, Newhall P, Phillips NJ, Toth K, Ray RB. miRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol Cancer Ther. 2012;11:1166–73. doi: 10.1158/1535-7163.MCT-12-0100. [DOI] [PubMed] [Google Scholar]
- 79.Melo SA, Kalluri R. miR-29b moulds the tumour microenvironment to repress metastasis. Nat Cell Biol. 2013;15:139–40. doi: 10.1038/ncb2684. [DOI] [PubMed] [Google Scholar]
- 80.Schmitt MJ, Philippidou D, Reinsbach SE, Margue C, Wienecke-Baldacchino A, Nashan D, Behrmann I, Kreis S. Interferon-gamma-induced activation of Signal Transducer and Activator of Transcription 1 (STAT1) up-regulates the tumor suppressing microRNA-29 family in melanoma cells. Cell Commun Signal. 2012;10:41. doi: 10.1186/1478-811X-10-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stocki P, Chapman DC, Beach LA, Williams DB. Depletion of cyclophilins B and C leads to dysregulation of endoplasmic reticulum redox homeostasis. J Biol Chem. 2014;289:23086–96. doi: 10.1074/jbc.M114.570911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Khanal P, Yun HJ, Lim SC, Ahn SG, Yoon HE, Kang KW, Hong R, Choi HS. Proyl isomerase Pin1 facilitates ubiquitin-mediated degradation of cyclin-dependent kinase 10 to induce tamoxifen resistance in breast cancer cells. Oncogene. 2012;31:3845–56. doi: 10.1038/onc.2011.548. [DOI] [PubMed] [Google Scholar]
- 83.Shen M, Stukenberg PT, Kirschner MW, Lu KP. The essential mitotic peptidyl-prolyl isomerase Pin1 binds and regulates mitosis-specific phosphoproteins. Genes Dev. 1998;12:706–20. doi: 10.1101/gad.12.5.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Obchoei S, Sawanyawisuth K, Wongkham C, Kasinrerk W, Yao Q, Chen C, Wongkham S. Secreted cyclophilin A mediates G1/S phase transition of cholangiocarcinoma cells via CD147/ERK1/2 pathway. Tumour Biol. 2015;36:849–59. doi: 10.1007/s13277-014-2691-5. [DOI] [PubMed] [Google Scholar]
- 85.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001;20:3459–72. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim MR, Choi HK, Cho KB, Kim HS, Kang KW. Involvement of Pin1 induction in epithelial-mesenchymal transition of tamoxifen-resistant breast cancer cells. Cancer Sci. 2009;100:1834–41. doi: 10.1111/j.1349-7006.2009.01260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jin J, Zhang Y, Li Y, Zhang H, Li H, Yuan X, Li X, Zhou W, Xu B, Zhang C, et al. RNA-interference-mediated downregulation of Pin1 suppresses tumorigenicity of malignant melanoma A375 cells. Neoplasma. 2013;60:92–100. doi: 10.4149/neo_2013_013. [DOI] [PubMed] [Google Scholar]
- 88.Obermayr E, Castillo-Tong DC, Pils D, Speiser P, Braicu I, Van Gorp T, Mahner S, Sehouli J, Vergote I, Zeillinger R. Molecular characterization of circulating tumor cells in patients with ovarian cancer improves their prognostic significance -- a study of the OVCAD consortium. Gynecol Oncol. 2013;128:15–21. doi: 10.1016/j.ygyno.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 89.Lee J, Kim SS. Current implications of cyclophilins in human cancers. J Exp Clin Cancer Res. 2010;29:97. doi: 10.1186/1756-9966-29-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kanekura T, Chen X. CD147/basigin promotes progression of malignant melanoma and other cancers. J Dermatol Sci. 2010;57:149–54. doi: 10.1016/j.jdermsci.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 91.Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;2016(44):D239–47. doi: 10.1093/nar/gkv1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hurley DG, Budden DM, Crampin EJ. Virtual Reference Environments: a simple way to make research reproducible. Brief Bioinform. 2015;16(5):901–3. doi: 10.1093/bib/bbu043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hackenberg M, Rodriguez-Ezpeleta N, Aransay AM. miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res. 2011;39:W132–8. doi: 10.1093/nar/gkr247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Griffiths-Jones S. miRBase: microRNA sequences and annotation. Curr Protoc Bioinformatics. 2010;Chapter 12:Unit 12 19 11–10. doi: 10.1002/0471250953.bi1209s29. [DOI] [PubMed] [Google Scholar]
- 95.Griffiths-Jones S. miRBase: the microRNA sequence database. Methods Mol Biol. 2006;342:129–38. doi: 10.1385/1-59745-123-1:129. [DOI] [PubMed] [Google Scholar]
- 96.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–4. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–8. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–7. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Van Der Walt S, Colbert SC, Varoquaux G. The NumPy array: a structure for efficient numerical computation. Computing in Science & Engineering. 2011;13:22–30. doi: 10.1109/MCSE.2011.37. [DOI] [Google Scholar]
- 101.Hunter JD. Matplotlib: A 2D graphics environment. Computing in science and engineering. 2007;9:90–5. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 102.Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, et al. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–6. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–56. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.ImageJ. [http://imagej.nih.gov/ij/]
- 106.Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One. 2014;9 doi: 10.1371/journal.pone.0092444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Smalley KS, Lioni M, Noma K, Haass NK, Herlyn M. In vitro three-dimensional tumor microenvironment models for anticancer drug discovery. Expert Opin Drug Discov. 2008;3:1–10. doi: 10.1517/17460441.3.1.1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this study are included within the article and additional files, or made freely available within repositories as specified in Additional file 1.