

Abstract
Tricyclic wyosine derivatives are found at position 37 of eukaryotic and archaeal tRNAPhe. In Archaea, the intermediate imG-14 is targeted by three different enzymes that catalyze the formation of yW-86, imG, and imG2. We have suggested previously that a peculiar methyltransferase (aTrm5a/Taw22) likely catalyzes two distinct reactions: N1-methylation of guanosine to yield m1G; and C7-methylation of imG-14 to yield imG2. Here we show that the recombinant aTrm5a/Taw22-like enzymes from both Pyrococcus abyssi and Nanoarchaeum equitans indeed possess such dual specificity. We also show that substitutions of individual conservative amino acids of P. abyssi Taw22 (P260N, E173A, and R174A) have a differential effect on the formation of m1G/imG2, while replacement of R134, F165, E213, and P262 with alanine abolishes the formation of both derivatives of G37. We further demonstrate that aTrm5a-type enzyme SSO2439 from Sulfolobus solfataricus, which has no N1-methyltransferase activity, exhibits C7-methyltransferase activity, thereby producing imG2 from imG-14. We thus suggest renaming such aTrm5a methyltransferases as Taw21 to distinguish between monofunctional and bifunctional aTrm5a enzymes.
Keywords: tRNA modification, Archaea, evolution, wyosine, bifunctional enzyme
INTRODUCTION
Post-transcriptional modifications are essential for the maintenance of structure and function of transfer RNA. While some modifications are relatively simple, those present in the anticodon region are often chemically complex, and their formation usually involves multiple enzymatic steps (Machnicka et al. 2013). One group of such complex compounds is represented by the wyosine derivatives that are composed of tricyclic imidazopurines and are found exclusively at position 37 of tRNAPhe from eukaryotes and Archaea (Urbonavičius et al. 2009). Wyosine derivatives have been shown to be involved in stabilization of codon–anticodon interactions (Konevega et al. 2004) and in prevention of ribosomal −1 frameshifting (Waas et al. 2007).
In the case of Saccharomyces cerevisiae, the enzymatic pathway leading to the formation of wybutosine (yW) in cytoplasmic tRNAPhe has been elucidated using ribonucleome analysis (Noma et al. 2006). It has been shown that the tRNAPhe maturation pathway consists of five distinct sequential steps, with the first step occurring in the nucleus (Ohira and Suzuki 2011). In fungi Torulopsis utilis, wyosine (imG) is the final product of such biosynthetic pathways. In certain other eukaryotes, such as the tench Tinca tinca, the silkmoth Bombyx mori, and the fruit fly Drosophila melanogaster, only m1G is present, since the genes for the biosynthesis of wyosine are absent from their genomes (White and Tener 1973; Mazabraud 1979; Keith and Dirheimer 1980). Recently, the imG nucleotide was found in mitochondria of protist Trypanosoma brucei (Sample et al. 2015).
Depending on species and growth conditions, a mix of wyosine derivatives is often found at position 37 of tRNAPhe in Archaea (de Crécy-Lagard et al. 2010; Urbonavičius et al. 2014 and references therein). However, in euryarchaeon Haloferax volcanii, only the m1G precursor is present in mature tRNAPhe (Gupta 1984). In many other Euryarchaeota, 7-aminocarboxypropyl-demethylwyosine (yW-86), which has been considered to be only an intermediate of the eukaryotic pathway, is found (for review, see Urbonavičius et al. 2014). The formation of yW-86 is catalyzed by Taw2, the archaeal counterpart of yeast Tyw2 that catalyzes the addition of the α-amino-α-carboxypropyl (“acp”) group of S-adenosine-l-methionine (AdoMet) to imG-14 intermediate (Umitsu et al. 2009). In Thermococcales, yW-86 is further methylated (Fig. 1), most likely by archaeal Taw3 methyltransferase (homolog of yeast Tyw3), to yield yW-72 (Phillips and de Crécy-Lagard 2011). Notably, no other intermediates or any corresponding enzymes that lead to the formation of wybutosine in S. cerevisiae have been detected in Archaea. Instead, two unique wyosine derivatives have been identified, namely isowyosine (imG2) and 7-methylwyosine (mimG) (McCloskey et al. 2001; Zhou et al. 2004), that both are present in Thermococalles and several Crenarchaeota (de Crécy-Lagard et al. 2010; Urbonavičius et al. 2014 and references therein). Thus, it has been suggested that imG2 is the likely intermediate of mimG biosynthesis (Fig. 1; Phillips and de Crécy-Lagard 2011; Urbonavičius et al. 2014). Moreover, the biosynthesis of mimG likely depends on the Taw3 methyltransferase, since in Methanosarcina acetovorans, which lacks Taw3, imG2 but not mimG is found (de Crécy-Lagard et al. 2010; Urbonavičius et al. 2014).
FIGURE 1.

Putative enzymatic pathway leading to the formation of wyosine derivatives in Archaea. The specific functions of each enzyme, as well as the alternative action of either bifunctional aTrm5a = Taw22a methyltransferase or monofunctional aTrm5b/c and aTrm5a = Taw21 methyltransferases, are described in the text.
Previously, we demonstrated that the occurrence of imG2 in different Archaea also correlates with the presence of archaeal Trm5a, a member of the archaeal Trm5a/b/c family of enzymes involved in the biosynthesis of the wyosine derivatives. Comparative analysis of the distribution of genes encoding homologs of the Trm5 protein, the AdoMet-dependent tRNA:m1G37 methyltransferase in yeast (Björk et al. 2001), within 62 archaeal genomes, has allowed the division of the family aTrm5 into three subfamilies (Fig. 2): aTrm5a (in this work, further divided into Taw21 and Taw22), aTrm5b, and aTrm5c (de Crécy-Lagard et al. 2010; Urbonavičius et al. 2014). While the enzymes belonging to these subfamilies do not significantly differ in their AdoMet-binding site, small differences have been observed within the NPPY motif, which, in certain amino-methyltransferases, is involved in the positioning of the target nitrogen atom (Fig. 2; Bujnicki 2000). In contrast, the N-terminal sequences of the aforementioned enzymes differ substantially, e.g., a small conservative domain called D1 is present in aTrm5b and aTrm5c but absent in most of the aTrm5a proteins. Notably, while the aTrm5b variant is almost ubiquitous in Euryarchaeota, it is absent in Crenarchaeota, whereas in Sulfalobales and Desulfurococcales, both the aTrm5a and aTrm5c variants are present. It has been suggested previously (de Crécy-Lagard et al. 2010) that the enzymes belonging to the aTrm5b and aTrm5c families can only catalyze the methylation of N1 of G37 to yield m1G37. The presence of both aTrm5c and aTrm5a in Sulfalobales as well as Desulfurococcales suggests that aTrm5c catalyzes the formation of m1G, whereas aTrm5a likely methylates the imG-14 to yield imG2. However, in certain Archaea, such as Thermoproteales (order Crenarchaeota), Euryarchaeon Nanoarchaeum equitans, as well as species belonging to the Korarchaeota phylum, aTrm5a is the only aTrm5-type protein present. This finding suggests that, in the case of the aforementioned organisms, aTrm5 has to display a dual tRNA:m1G/imG2 methyltransferase activity, catalyzing both the formation of m1G and imG2. Similar bifunctional methyltransferases exist in Thermococalles, where aTrm5 is present along with the aTrm5b protein (Urbonavičius et al. 2014).
FIGURE 2.



Amino acid sequence alignment of Trm5a/b/c/ family of proteins. To distinguish between monofunctional and bifunctional aTrm5a enzymes, the appropriate methyltransferases were designated as Taw21 and Taw22, respectively. Conserved residues are highlighted in red boxes. M. jannaschii Trm5b secondary structure elements (α-helices [α], β-strands [β], and 310-helices [η]) are shown at the top. D1 domain as well as motifs A and NPPY are indicated below the alignment. Residues discussed in the text are indicated by red circles. The sequences were aligned using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/), and the figure was generated using ESPript (http://espript.ibcp.fr/ESPript/ESPript/).
Previously, we have demonstrated that the aTrm5a-like protein PAB2272 of Pyrococcus abyssi displays a dual methyltransferase activity (de Crécy-Lagard et al. 2010). While the tRNAPhe:m1G methyltransferase function of PAB2272 has been shown, the exact product resulting from the second methyltransferase activity has not been demonstrated, even though it has been suggested that PAB2272 may also catalyze the methylation of imG-14 to yield imG2. Based on these findings, we have proposed to rename aTrm5a homologs as Taw22 (Urbonavičius et al. 2014).
In this work, to demonstrate the enzymatic activities of aTrm5a proteins, several homologous genes from P. abyssi, N. equitans, and Sulfolobus solfataricus have been cloned and expressed in Escherichia coli. The corresponding recombinant proteins have been purified and their enzymatic activities have been investigated in vitro. In addition, based on the alignment of the aTrm5 proteins and the crystal structure of the Methanocaldococcus jannaschii Trm5b protein MJ0883, we have replaced several respective conservative amino acids of PAB2272 by alanines (except for Pro260, which was replaced by asparagine). The enzymatic activities of recombinant mutant PAB2272 proteins were measured in vitro.
RESULTS
Both P. abyssi PAB2272 and N. equitans NEQ228 proteins display a dual tRNAPhe:m1G/imG2 methyltransferase activity
The recombinant NEQ228 and PAB2272 proteins were purified to near homogeneity (Supplemental Fig. S1A). They were tested for methyltransferase activity using two different types of substrates. First, bulk tRNA, isolated from S. enterica trmD27 mutant containing the unmodified G37 nucleotide, was used. After incubation of the respective purified enzymes with mutant tRNA and [methyl-14C]AdoMet, recovery of tRNA, and digestion to 5′-monophosphate nucleosides by nuclease P1, the resulting hydrolyzate was analyzed by 2D TLC followed by autoradiography. In both cases, incorporation of the radioactive methyl groups was observed leading to the formation of pm1G (Figs. 3A, 6A). No corresponding spots were evident in the absence of any protein (Figs. 3B, 6B). When the recombinant PAB2272 and NEQ228 proteins were incubated under identical conditions with tRNA, which was isolated from the S. cerevisiae tyw2 mutant that contains the imG-14 wyosine derivative, an additional spot, likely corresponding to the previously suggested pimG2pA dinucleotide (de Crécy-Lagard et al. 2010), was detected (Figs. 3C, 7A). Such a dinucleotide may result from incomplete digestion of tRNA with the nuclease P1 and has a migration profile similar to that of YpAp (wybutosine dinucleotide (Droogmans and Grosjean 1987) and “Y” (possibly pYpA [Grosjean et al. 1990]). Minor spots corresponding to pm1G, likely resulting from the small amounts of G37-containing tRNAPhe present in the bulk tRNA isolates from the Scetyw2 mutant, were also observed. No formation of pimG2pA and/or pm1G was observed in the absence of any protein (Fig. 3D, 7B). These results demonstrate that both PAB2272 and NEQ228 proteins catalyze the transfer of a methyl group from the [methyl-14C]AdoMet onto two different nucleosides at position 37 of tRNAPhe, yielding, respectively, m1G from G and, most likely, imG2 from imG-14.
FIGURE 3.
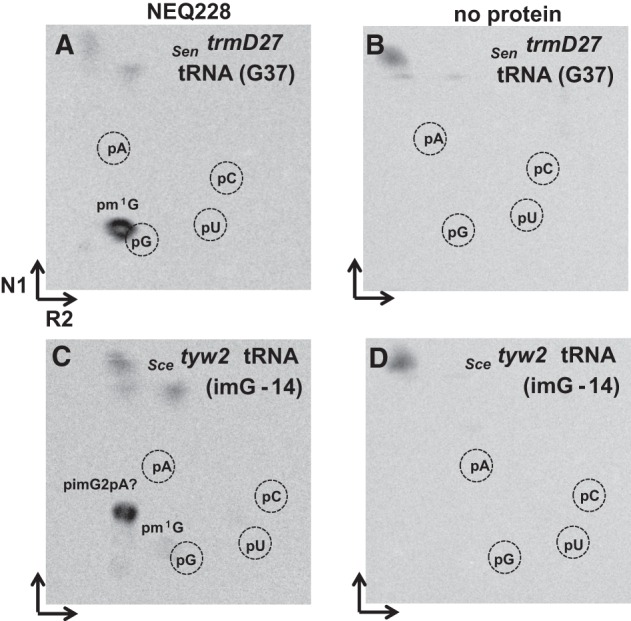
Methylation of tRNA, isolated from the S. enterica trmD27 and S. cerevisiae tyw2 mutants, by the NEQ228 protein. Autoradiograms of the TLC plates (2D-TLC), corresponding to the bulk mutant tRNA incubated for 30 min at 50°C with the respective protein and digested with nuclease P1. (A) NEQ228 + SentrmD27 tRNA(G37). (B) SentrmD27 tRNA(G37), incubated without any protein. (C) NEQ228 + Scetyw2 tRNA(imG-14). (D) Scetyw2 tRNA(imG-14), incubated without any protein. Positions of 5′-monophosphate nucleoside markers (pA, pG, pC, and pU), as detected by their UV shadowing, are indicated by dashed circles. The “pimG2pA?” is expected to be the pimG2pA dinucleotide. Note small amounts of pm1G in panel C, likely resulting from the small amounts of G37-containing tRNAPhe present in the bulk tRNA isolated from the Scetyw2 mutant. The spots on the top of TLC plates probably result from the nonspecific degradation of [methyl-14C] AdoMet. N1 and R2 correspond to solvents used in each dimension (the solvent composition is given in Materials and Methods).
To confirm that the radioactive spot obtained after incorporation of the [methyl-14C] group into the bulk tRNA from S. cerevisiae Δtyw2, and visualized after 2D TLC analysis corresponds to the pimG2pA dinucleotide, an in vitro tRNA methylation assay using the PAB2272 enzyme and nonradioactive AdoMet was performed. The resulting products of the methylation reaction were analyzed by HPLC-MS. It was demonstrated (Fig. 4) that upon the incubation with PAB2272, the level of the imG2 product increases with concomitant decrease in that of the imG-14 substrate (cf. Fig. 4B [no enzyme] with Fig. 4C [enzyme added]). The previous phylogenetic analyses (de Crécy-Lagard et al. 2010; Urbonavičius et al. 2014) in conjunction with TLC and HPLC-MS data allow us to conclude that both the PAB2272 and NEQ228 proteins show a dual tRNAPhe:m1G/imG2 methyltransferase activity.
FIGURE 4.
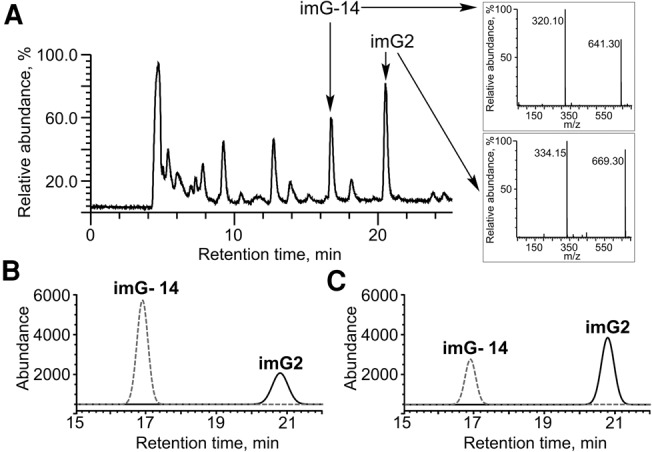
Methylation assay using the PAB2272 protein and tRNA isolated from S. cerevisiae tyw2 mutant. (A) Mass spectrometry analysis of imG-14 and imG2 standards coinjected with tRNA P1/AP hydrolyzate. The mass spectrometry data were acquired in negative ionization mode. Extracted ion chromatograms of m/z = 320 (dashed line) and m/z = 334 (solid line) from a representative negative ion mode chromatogram, obtained after digestion of bulk mutant tRNA with nucleases P1/AP, without (B) or with (C) incubation with PAB2272 protein for 30 min at 50°C. The conversion of imG-14 into imG2 is presented. In B and C, a Gaussian filter was applied to reduce the noise.
Sulfolobus solfataricus SSO2439 protein exhibits a tRNAPhe:imG2, but not tRNAPhe:m1G, methyltransferase activity
Previously (de Crécy-Lagard et al. 2010; Urbonavičius et al. 2014), we suggested that, in Crenarcheota S. solfataricus as well as in other Sulfalobales and Desulfurococcales, two different tRNAPhe methyltransferases are involved in the biosynthesis of mimG, catalyzing the formation of m1G (Trm5c) and imG2 (Trm5a) at position 37 in tRNAPhe, respectively. To verify this hypothesis experimentally, the SSO2439 protein (predicted Trm5a-like enzyme) was purified to near homogeneity (Supplemental Fig. S1A), and its enzymatic activity was tested in the same way as described above. Figure 5 demonstrates incorporation of the [methyl-14C] group into bulk yeast tRNA isolated from S. cerevisiae Δtyw2 (containing imG-14 in tRNAPhe), but not into that from the S. enterica trmD27 (containing G37) mutant. No spots corresponding to m1G or imG2 were detected in the absence of SSO2439 (data not shown). Since the migration profile for likely pimG2pA on TLC plates was the same as that obtained for PAB2272 and NEQ228, we concluded that the SSO2439 protein exhibits the tRNAPhe:imG2, but not the tRNAPhe:m1G, methyltransferase activity.
FIGURE 5.
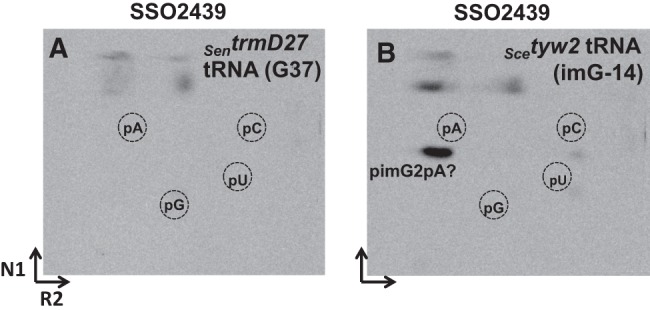
Methylation assay using SSO2439 protein and tRNA isolated from S. enterica trmD27 and S. cerevisiae tyw2 mutants. Autoradiograms of the TLC plates (2D-TLC), corresponding to the bulk mutant tRNA incubated for 30 min at 50°C with the respective protein and digested with nuclease P1. (A) SSO2439 + SentrmD27 tRNA(G37). (B) SSO2439 + Scetyw2 tRNA(imG-14). The “pimG2pA?” likely corresponds to the pimG2pA dinucleotide. The spots on the top of TLC plates probably result from the nonspecific degradation of [methyl-14C] AdoMet. N1 and R2 correspond to solvents used in each dimension (the solvent composition is given in Materials and Methods).
Amino acid residues of PAB2272 that are important for the formation of m1G and imG2 derivatives
A previously conducted bioinformatic analysis (de Crécy-Lagard et al. 2010) in conjunction with the structural work on Methanocaldococcus jannashii Trm5b (MJ0883) and Taw2 (MJ1157) as well as Pyrococcus horikoshii PH0793 enzyme Taw2 (Goto-Ito et al. 2008, 2009; Umitsu et al. 2009) allowed identification of several amino acid residues, critical for the recognition of the S-AdoMet cofactor and the G/imG-14 substrate at position 37 in tRNAPhe. Here, we performed the bioinformatic analysis of the archaeal Trm5a/b/c family of enzymes (Fig. 2) to identify amino acids that are critical for the activity of methyltransferases representing different subfamilies. Consequently, the role of several conservative residues of PAB2272 (those likely involved in the recognition of G37/imG-14 substrates and AdoMet cofactor) in the activity of the enzyme was investigated. Residues R134 (corresponding to R145 of MJ0883 and R76 of PH0793), P260 (corresponding to N265 of MJ0883 and G199 of PH0793), F165 (corresponding to Y177 of MJ0883 and M107 of PH0793), and P262 (corresponding to P267 of MJ0883 and V201 of PH0793) were chosen since respective residues of MJ0883 and PH0793 had been previously demonstrated to contact the G37/imG-14 substrates of tRNA (Goto-Ito et al. 2009; Umitsu et al. 2009). Similarly, R174 (corresponding to R186 of MJ0883 and R116 of PH0793), E213 (corresponding to D223 of MJ0883 and E155 of PH0793), and P260 (corresponding to N265 of MJ0883 and G199 of PH0793) were selected since corresponding residues had been demonstrated to contact the AdoMet cofactor. In addition, E173 was tested since the corresponding E185 residue of MJ0883 had been demonstrated to act as a general base that accepts the proton from G37-N1 during the catalysis (Christian et al. 2010). The respective recombinant PAB2272 mutant proteins were purified to near homogeneity (Supplemental Fig. S1B), and their relative enzymatic activity, compared to that of the wild-type PAB2272, is summarized in Table 1. Mutational analysis revealed that mutants PAB2272_R134A and PAB2272_P262A produced only negligible amounts of m1G and imG2, whereas PAB2272_F165A and PAB2272_E213A produced neither m1G nor imG2 (Figs. 6C,D,G,I, 7C,D,G,I, for the assays on control; see Figs. 6A,B, 7A,B). The other amino acid substitutions tested had differential effects on the formation of m1G and imG2. As seen in Figures 6E,F, 7E,F, the PAB2272_E173A mutant retained 9% of wt PAB2272 tRNAPhe:m1G-methyltransferase activity and 26% of wt PAB2272 tRNAPhe:imG2-methyltransferase activity, whereas PAB2272_R174A 8% and 69%, respectively. Surprisingly, the PAB2272_P260N protein completely lost tRNAPhe:m1G-, but not tRNAPhe:imG2-methyltransferase activity (Figs. 6H, 7H).
TABLE 1.
Enzymatic activity of PAB2272 mutant proteins
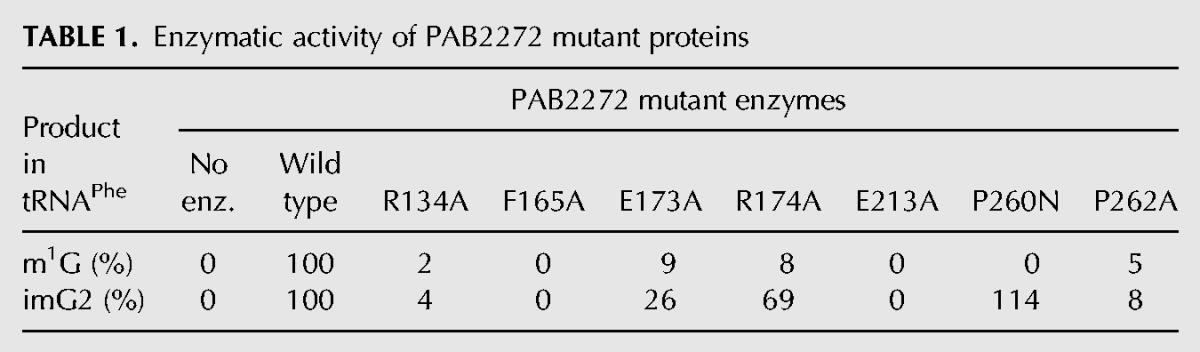
FIGURE 6.

Methylation assay using mutant PAB2272 proteins and tRNA isolated from the S. enterica trmD27 mutant. Autoradiograms of the TLC plates (2D-TLC), corresponding to the bulk mutant tRNA incubated for 30 min at 50°C with the respective protein and digested with nuclease P1. (A) PAB2272 + SentrmD27 tRNA(G37). (B) No protein + SentrmD27 tRNA(G37). (C) PAB2272_R134A + SentrmD27 tRNA(G37). (D) PAB2272_F165A + SentrmD27 tRNA(G37). (E) PAB2272_E173A + SentrmD27 tRNA(G37). (F) PAB2272_R174A + SentrmD27 tRNA(G37). (G) PAB2272_E213A + SentrmD27 tRNA(G37). (H) PAB2272_P260N + SentrmD27 tRNA(G37). (I) PAB2272_P262A + SentrmD27 tRNA(G37).The “pimG2pA?” is probably the pimG2pA dinucleotide. The spot “X” on the top of TLC plates, which likely results from the nonspecific degradation of [methyl-14C] AdoMet, was used as an internal standard for the spot intensity calculations. N1 and R2 correspond to solvents used in each dimension (the solvent composition is given in Materials and Methods).
FIGURE 7.

Methylation assay using mutant PAB2272 proteins and tRNA isolated from S. cerevisiae tyw2 mutants. Autoradiograms of the TLC plates (2D-TLC), corresponding to the bulk mutant tRNA incubated for 30 min at 50°C with the respective protein and digested with nuclease P1. (A) PAB2272 + Scetyw2 tRNA(imG-14). (B) No protein + Scetyw2 tRNA(imG-14). (C) PAB2272_R134A + Scetyw2 tRNA(imG-14). (D) PAB2272_F165A + Scetyw2 tRNA(imG-14). (E) PAB2272_E173A + Scetyw2 tRNA(imG-14). (F) PAB2272_R174A + Scetyw2 tRNA(imG-14). (G) PAB2272_E213A + Scetyw2 tRNA(imG-14). (H) PAB2272_P260N + Scetyw2 tRNA(imG-14). (I) PAB2272_P262A + Scetyw2 tRNA(imG-14). The “pimG2pA?” is probably the pimG2pA dinucleotide. The spot “X” on the top of TLC plates, which likely results from the nonspecific degradation of [methyl-14C] AdoMet, was used as an internal standard for the spot intensity calculations. N1 and R2 correspond to solvents used in each dimension (the solvent composition is given in Materials and Methods).
DISCUSSION
In this work, we have investigated the enzymatic activity of several archaeal proteins previously predicted to be involved in the formation of the imG2 intermediate of the mimG biosynthesis pathway. Based on our results, the aTrm5a proteins of Euryarchaeota P. abyssi (PAB2272) and N. equitans (NEQ228) display a dual tRNAPhe:m1G/imG2 activity (Figs. 3A,C, 6A, 7A). While the ability of PAB2272 to catalyze the formation of the m1G nucleoside from G has been demonstrated previously (de Crécy-Lagard et al. 2010), the conversion of imG-14 into imG2, catalyzed by the same enzyme, has been proposed, yet has not been formally demonstrated in that work. Here, using the chemically synthesized imG-14 and imG2 nucleosides as the internal standards for the HPLC-MS analysis, we demonstrate that PAB2272, in addition to a tRNAPhe:m1G-methyltransferase, also possesses tRNAPhe:imG2-methyltransferase activity. The latter activity has been demonstrated using yeast bulk tRNA instead of the purified tRNAPhe, but it is well known that in S.cerevisiae, wyosines (wybutosine and other derivatives in different tyw mutants) are present only at position 37 in this particular tRNA (Noma et al. 2006; Juhling et al. 2009). This strongly supports our conclusion that the imG2-methyltransferase is specific for the tRNAPhe substrates.
The PAB2272 protein differs from NEQ228 by the absence of the D1 domain (de Crécy-Lagard et al. 2010), yet both enzymes clearly demonstrate the dual specificity, suggesting that this domain is not responsible for the dual activity of the aTrm5a proteins. Similar observations have been made for the aTrm5b-type enzyme MJ0883 (Goto-Ito et al. 2008), where D1 was shown to be dispensable for the methylation of the G37 tRNA substrate. Nevertheless, one cannot exclude that the D1 domain is important at physiological temperatures above 50°C, which was used for the methylation assays during our experiments.
Our results show that the S. solfataricus Trm5a (SSO2439) protein exhibits only the tRNAPhe:imG2, but not tRNAPhe:m1G, methyltransferase activity (Fig. 5). This is not unexpected, since in Desulforococcales and Sulfolobales, the aTrm5c variant, likely responsible for the formation of m1G at position 37 in all tRNA species of these microorganisms, is present. Previously (Urbonavičius et al. 2014), we proposed to rename the tRNAPhe:m1G/imG2 methyltransferases aTrm5a as Taw22. Our present work demonstrates that the situation regarding the substrate specificity of aTrm5a methyltrasferases is even more complicated: In Thermococcales, an aTrm5b tRNA:m1G methyltransferase implicated in the formation of m1G in all tRNA species bearing G37 is present. Apparently it makes the aTrm5 = Taw22 enzyme to be specific only toward tRNAPhe but not toward other tRNA species in these organisms. However, since the aTrm5b (and c) enzymes are absent in N. equitans as well as in the Thermoproteales (de Crécy-Lagard et al. 2010), aTrm5a is, apparently, singularly responsible not only for the formation of m1G37 in all tRNA species bearing G37, but also for the formation of imG2 in tRNAPhe . The aTrm5a in S. solfataricus and probably also in other Sulfolobus, Sulfolobales, and Desulforococalles clearly differs from aTrm5a = Taw22 (like PAB2272 and NEQ228) by possessing only a single enzymatic activity. Therefore, we propose to rename such aTrm5a variants as Taw21.
Our results suggest that at least two aTrm5a enzymes exhibit a tRNAPhe:m1G/imG2 methyltransferase activity, whereas S. sulfolobus aTrm5a (and, likely, other related enzymes) acts as tRNAPhe:imG2 methyltransferase only. One could imagine that, after emergence of the Taw1 enzyme that catalyzes the formation of the imG-14 intermediate of the mimG pathway, the N1-methylating enzyme may have evolved to methylate the C7-atom of imG-14 as well. Such intrinsically relaxed specificity may be created by the existence of the mesomeric forms of the imG-14 substrate, allowing the C7-atom to become sufficiently negatively charged to act as a nucleophile (Lin 2011; Urbonavičius et al. 2014). After the subsequent gene duplication events, each gene may have evolved differently: It may encode either the bifunctional enzyme (e.g., aTrm5a/Taw22 enzymes PAB2272 and NEQ228), G37-specific N1-methyltransferase (aTrm5b, c, e.g., PAB0505), imG-14-C7-methyltransferase (Taw21, as in the case of SSO2439), or imG-14-C7-3-amino-3-carboxypropyltransferase (Taw2, e.g., MJ1557, PH0793 [Umitsu et al. 2009]). For all possible enzymatic modifications of imG-14, see Figure 1. Previously, it was demonstrated (de Crécy-Lagard et al. 2010) that aTrm5b/c methyltransferases contain Asn/Asp residues, whereas aTrm5a proteins have Pro/Thr residues in their NPPY motif that are important for positioning of the target nitrogen atoms (Bujnicki 2000). Here, we show that in the case of the aTrm5/Taw22 enzyme PAB2272, the P260N substitution within the NPPY motif abolishes the formation of m1G37 but not that of imG2 (Table 1; Figs. 6H, 7H). Our results suggest that, in this mutant, imG-14, but not G37, is still correctly positioned for methylation. Notably, replacement of E173 (corresponding to E185 of MJ0883, a general base that accepts the proton during catalysis) with alanine demonstrated a differential effect on the formation of m1G/imG2 (yielding 9% and 26% of the respective products; see Table 1; Figs. 6E, 7E). This finding suggests that for the methylation reaction, the proton abstraction at the N1-atom of G37 by E173 is likely more important than that at the C7-atom of imG-14. A similar differential effect was observed in the case of R174, where substitution with alanine yielded, respectively, 8% and 69% of m1G/imG2 (Table 1; Figs. 6F, 7F), suggesting that, in the case of G37, the interaction of this particular amino acid with the AdoMet cofactor is extremely important. As expected, replacement by alanine of R134, F165, E213, and P262 residues, all of which have been suggested to be involved in the positioning of AdoMet toward the G/imG-14 substrate, abolished the formation of both m1G and imG2 (for more detail, see Figs. 6, 7, as well as Table 1).
As mentioned above, several evolutionally related enzymes (Taw2, Taw22, and Taw21) that, together with Trm5b (and c), belong to the same family of PACE25/COG2520 proteins (Tatusov et al. 1997; Matte-Tailliez et al. 2000) target imG-14 of tRNAPhe. Interestingly, while the crystal structure of aTrm5b (MJ0883) has been solved using either sinefungin (analog of S-AdoMet) or S-AdoMet as a methyl donor, and tRNACys or tRNALeu as a substrate (Goto-Ito et al. 2008, 2009), no crystal structure of any enzyme in complex with tRNAPhe containing imG-14 as a substrate has been solved thus far. The recognition mechanism of imG-14 in tRNAPhe was inferred only for Taw2 (PH0793) of Pyrococcus horikoshii using a docking model (Umitsu et al. 2009). At first glance it is not evident what structural features distinguish Taw22, which converts both G and imG-14 substrates, from Taw21, which uses only imG-14 as a substrate. Previously, the sequence motif MXSX2NX3R/K (Motif A, Fig. 2) has been shown to be responsible for the differences in enzymatic activity of Trm5b and Taw2 (Umitsu et al. 2009). It has also been shown that Arg186 (MJ0883 numbering) of Motif A interacts with the carboxyl group of AdoMet. In the Taw22 but not the Taw21 proteins, this residue is replaced by histidine, suggesting that the sequence YEH (Tyr-Glu-His) in motif A distinguishes the Taw21 family of proteins from Trm5b/c/Taw2/Taw22 (Fig. 2). To improve our understanding on how different paralogs of the COG2520/PACE25 family are functioning, biochemical and structural studies, similar to those already performed for aTrm5b and Taw2 (Christian et al. 2006; Goto-Ito et al. 2008, 2009; Umitsu et al. 2009; Sakaguchi et al. 2012), have been initiated.
Besides aTrm5a = Taw22, other bifunctional enzymes acting on RNA exist in Nature. One such example is Cfr methyltransferase from E. coli, capable of methylating 2- and 8-carbon of A2503 of 23S rRNA to form m2,8A (Giessing et al. 2009). Also, a bifunctional methyltransferase NS5 of flavoviruses catalyzes methylation of both the N7-atom of guanosine and the 2′-hydroxyl group of the neighboring adenosine, resulting in type 1 cap m7GpppAm of viral RNA (Dong et al. 2008a,b). Such sequential cap methylations (GpppA-RNA → m7GpppA-RNA → m7GpppAm-RNA) have also been shown in West Nile (WNV), Dengue (DENV), and yellow fewer viruses (YFV) (Zhou et al. 2007). The consecutive methylation events may proceed by two alternative mechanisms: either AdoMet replenishment/RNA repositioning or RNA dissociation/reassociation (Dong et al. 2014). It is possible that more bifunctional methyltransferases exist in the organisms with reduced genomes, where protein functional complexity is required to compensate for the gene loss (Kelkar and Ochman 2013). During the course of evolution or under certain conditions, two methylation events might be decoupled and act independently. In our case, this may be illustrated by aTrm5a (=Taw22), where the methylation events have been split between aTrm5c and aTrm5a (=Taw21) (Fig. 1). Our work also demonstrates that, more often than not, one's attempts to infer the enzymatic activity of homologous proteins based solely on in silico analysis may prove to be quite spurious. In addition, the results presented here provide a glimpse into events that lead to the development of sequential metabolic pathways.
MATERIALS AND METHODS
DNA manipulations
The pET101D-topo (Invitrogen) expression vector, containing the P. abyssi PAB2272 gene (pSBTN-AD18), was obtained from Jean Armengaud (CEA, Bagnols-sur-Cèze, France). The pJexpress401 vector with the synthetic N. equitans NEQ228 gene optimized for E. coli codon usage was purchased from DNA 2.0. The S. solfataricus SSO2439 gene was cloned into the pET21d expression vector (Novagen). For this purpose, SSO2439-fwd and SSO2439-rev primers were used for the PCR reaction (Supplemental Table S1). The S. solfataricus P2 chromosomal DNA was obtained from David Prangishvili (Institut Pasteur, Paris, France), and the PCR reaction was performed using Pfu DNA polymerase (Thermo Fisher Scientific). The resulting PCR product (∼800 bp) was cloned into the pJET1.2 vector (Thermo Fisher Scientific) and then recloned via NcoI and XhoI restriction sites of pET21d. The sequence of the insert was verified at Macrogen Europe.
To construct PAB2272_R133A, PAB2272_F165A, PAB2272_ E173A, PAB2272_R174A, PAB2272_E213A, PAB2272_P260N, and PAB2272_P262A mutant proteins, in vitro mutagenesis was carried out using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) with the pSBTN-AD18 plasmid, containing the PAB2272 gene as the template and respective mutagenic primers (Supplemental Table S1). Thus, the recombinant plasmids pSBTN-AD18_Ala133, pSBTN-AD18_Ala165, pSBTN-AD18_Ala173, pSBTN-AD18_Ala174, pSBTN-AD18_ Ala213, pSBTN-AD18_Asn260, and pSBTN-AD18_Ala262 were obtained, and the presence of the desired mutation was verified by sequencing at Macrogen Europe.
Overexpression and purification of recombinant archaeal enzymes
E. coli BL-21(DE3) (Invitrogen) was used as the host strain for the overexpression of His×6-tagged proteins. For this purpose, cells harboring a respective plasmid (pSBTN-AD18, pJexpress401_ NEQ228, pET21d-SSO2439, pSBTN-AD18_Ala133, pSBTN-AD18_Ala165, pSBTN-AD18_Ala173, pSBTN-AD18_Ala174, pSBTN-AD18_ Ala213, pSBTN-AD18_Asn260, or pSBTN-AD18_ Ala262) were grown in a standard LB medium supplemented with 50 µg/mL ampicillin (except for pJexpress401_NEQ228, where 50 µg/mL kanamycin was used). In total, 50 mL of medium was inoculated with 0.5 mL of the corresponding overnight culture and incubated at 30°C until an OD600 of 0.6–0.8 was reached. Protein expression was then induced by adding IPTG to a final concentration of 0.5 mM, and the incubation was continued for an additional 18 h. The cells were harvested by centrifugation for 15 min at 4000g (at 4°C), resuspended in 50 mM Tris–HCl buffer (pH 8.0), containing 0.5 M KCl and 10% (w/w) glycerol (buffer A), and disrupted by sonication at 22 kHz for 1 min, using 50% amplitude. The cell lysates were centrifuged at 10,000g for 20 min at 4°C to remove the debris.
The purification of PAB2272 recombinant protein and its respective mutants was performed as follows. After centrifugation, each cellular extract was subjected to a 20 min heat treatment at 70°C and 65°C, respectively. Cell extracts with aggregated proteins were immediately centrifuged at 10,000g for 20 min at 4°C, and resulting supernatants were loaded onto a 1 mL HiTrap Chelating HP column (GE Healthcare) equilibrated with buffer A. The recombinant proteins were eluted using a linear gradient of 0–500 mM imidazole in buffer A, and the appropriate fractions were analyzed by SDS electrophoresis, pooled, and desalted by gel filtration on a 5 mL HiTrap Desalting column (GE Healthcare) equilibrated with 50 mM Tris–HCl buffer (pH 8.0), containing 50 mM KCl. The purification of SSO2439 and NEQ228 proteins was performed as described above except that the heat-treatment step was omitted.
Two additional purification steps following the Ni-affinity purification were devised for PAB2272. In this case, the desalted protein sample was loaded at a flow rate of 0.5 mL/min onto a 1 mL Resource S column (GE Healthcare) equilibrated with 50 mM Tris–HCl buffer, pH 7.5. After washing the column with five column volumes, PAB2272 was eluted over a linear gradient comprising 0–500 mM NaCl. The fractions corresponding to the major peak eluted from the column were pooled, saturated with solid ammonium sulfate up to 0.8 M concentration, and applied onto a 1 mL phenyl-sepharose column (GE Healthcare) previously equilibrated with 25 mM Tris–HCl (pH 8.0), and containing 0.8 M ammonium sulfate. The PAB2272-containing fractions were eluted over a linear gradient comprising 0.8–0 M ammonium sulfate, and the fractions corresponding to the major peak eluted from the column were desalted by gel filtration on a 5 mL HiTrap Desalting column, previously equilibrated with 50 mM Tris–HCl buffer (pH 8.0), containing 10% (w/w) glycerol.
The homogeneity of all recombinant proteins was verified by SDS–PAGE. Protein concentrations were determined by the Lowry method using bovine serum albumin as the standard. Purified protein samples were flash-frozen and stored at −80°C until further use.
Chemical synthesis of imG-14 and imG2 wyosine derivatives
The imG-14 and imG2 wyosine derivatives were synthesized by carrying out a ring closure reaction (from guanosine to form N1–N2 carbon bridge) according to the procedures described previously by Zhou et al. (2004), with slight modifications. Chloroacetone was used instead of bromoacetone for the synthesis of imG-14, and the ratios of reagents as well as a purification step of the crude mixtures were optimized. The obtained respective wyosine derivatives were purified by column chromatography using silica gel and chloroform/methanol mixtures as an eluent. The identity and purity of the obtained compounds were confirmed by HPLC-MS and NMR analyses.
Isolation of bulk tRNA from S. cerevisiae and Salmonella enterica
Bulk (unfractionated) tRNA from S. cerevisiae Δtyw2 (Euroscarf) or S. enterica trmD27 (derivative of S. enterica serovar Typhimurium LT2, obtained from Glenn Björk and Gunilla Jäger, Umeå University, Umeå, Sweden) mutants was isolated essentially as described in de Crécy-Lagard et al. (2010). The cells were grown in standard YEPD (yeast) or LB (bacteria) media overnight, collected by centrifugation and broken with aqueous phenol. After phase separation by centrifugation, the nucleic acids were recovered by ethanol precipitation and dissolved in 50 mM sodium acetate (pH 6.5), 10 mM MgCl2, 150 mM NaCl to which 12 M LiCl was added to reach 2 M final concentration. After incubation for 1 h on ice, the insoluble ribosomal RNA (and remaining DNA) was eliminated by centrifugation, and bulk “soluble” RNA (mainly tRNA) was recovered by ethanol precipitation, washed twice with cold 70:30% ethanol:water, and finally dissolved in double-distilled water. The purity of tRNA was evaluated by agarose gel electrophoresis, and the concentration was determined spectrophotometrically.
In vitro methyltransferase activity assay
To determine the tRNA:m1G and/or tRNA:imG2 methyltransferase activities, purified recombinant PAB2272, PAB2272_Ala133, PAB2272_Ala165, PAB2272_Ala173, PAB2272_Ala174, PAB2272_ Ala213, PAB2272_Asn260, PAB2272_Ala262, NEQ228, or SSO2439 proteins were incubated with the respective tRNA substrates. Routinely, the reaction mixture of 100 µL contained ∼1 µg of the respective purified recombinant protein in 50 mM Tris–HCl buffer (pH 8.0) with 10 mM MgCl2, 0.5 mM DTT, 5% glycerol, 100 nCi of [methyl-14C]-S-AdoMet (PerkinElmer), and 40 µg of tRNA, isolated from either S. enteric trmD27 (containing G37) or S. cerevisiae tyw2 (containing mostly imG-14 in tRNAPhe) mutants. Each mixture was incubated for 30 min at 50°C and then processed as described below.
After incubation, the reaction was stopped by adding 200 µL of cold 0.3 M sodium acetate (pH 5.3) immediately followed by the addition of an equal volume of phenol/chloroform (24:1). Denatured proteins were then removed by centrifugation at 13,000g for 3 min at room temperature, and nucleic acids present in the upper phase were precipitated with ethanol, collected by centrifugation, washed once with 70% ethanol, dried, and finally completely digested into 5′-monophosphate nucleosides by overnight incubation at 37°C with 1U of nuclease P1 (Sigma-Aldrich) in 10 µL of 50 mM ammonium acetate/acetic acid buffer at a pH of 5.3. The resulting hydrolyzates were then analyzed after separation by two-dimensional thin-layer chromatography on 10×10 cm cellulose plates (Merck) using the N1 (Nishimura 1) and R2 (Rajbhandary 2) migration solutions for each dimension and autoradiography. The N1 solution was composed of isobutyric acid/conc. NH4OH/water (66:1:33), containing 1 mM EDTA. The R2 solution was composed of 0.1 M sodium phosphate (pH6.8)/solid (NH4)2SO4/propanol-1 (100:60:2). The necessary reference maps used for such analysis were according to Grosjean et al. (2004). Detection of radioactive spots on the thin-layer plates was performed after the exposure for at least 3 d in a Fujifilm FLA-5100 imaging system. To calculate the intensity of radioactive signals, Multigauge 3.0 program was used. The spot X, likely resulting from the nonspecific degradation of [methyl-14C] AdoMet, was used as an internal standard.
For the HPLC-MS analysis, 5 µg of the PAB2272 protein was incubated with 400 µg of bulk tRNA, isolated from the S. cerevisiae Δtyw2 mutant, at conditions described above, except that 80 µM of nonradioactive S-AdoMet (Sigma-Aldrich) was used instead of the radioactive one. After digestion of tRNA with 10 U of P1 nuclease, the solution was adjusted to pH 8.0 with 0.1 M ammonium bicarbonate buffer and supplemented with 2 U of FAST AP alkaline phosphatase (Thermo Fisher Scientific). The digested extracts were subjected to HPLC-MS analysis. Such analysis was performed using a high-performance liquid chromatography system (CBM-20A controller, two LC-2020AD pumps, SIL-30AC autosampler and CTO-20AC column oven) equipped with an SPD-M20A Prominence photo diode array (PDA) detector and a mass spectrometer (LCMS-2020, equipped with an ESI source), all the equipment are from Shimadzu, Japan. The chromatographic separation was conducted using a Hydrosphere C18 column, 4×150 mm (YMC), or YMC Pack Pro column, 3×150 mm (YMC, Japan) at 40°C and a mobile phase that consisted of 5 mM ammonium acetate (pH 5.3) (solvent A) and acetonitrile (solvent B) delivered in the gradient elution mode. Mass scans were measured from m/z 50 up to m/z 700, at an interface temperature of 350°C, DL temperature of 250°C, and interface voltage of ±4500 V, with neutral DL/Qarray, using N2 as the nebulizing and drying gas. Mass spectrometry data were acquired in both positive and negative ion modes. The data were analyzed using the LabSolutions LCMS software, and the data fitting was performed using the Wolfram Mathematica program.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Henri Grosjean (CNRS, Gif-sur-Yvette, France) for the suggestions on how to improve the manuscript and Audrius Laurynėnas (Vilnius University) for help with data presentation. This work was supported by the Research Council of Lithuania (LMT grant MIP-103/2015) to J.U. and the Lithuanian-French program Gilibert (LMT grant TAP-LZ_14-003) to J.U.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.057059.116.
Freely available online through the RNA Open Access option.
REFERENCES
- Björk GR, Jacobsson K, Nilsson K, Johansson MJ, Byström AS, Persson OP. 2001. A primordial tRNA modification required for the evolution of life? EMBO J 20: 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujnicki JM. 2000. Phylogenomic analysis of 16S rRNA:(guanine-N2) methyltransferases suggests new family members and reveals highly conserved motifs and a domain structure similar to other nucleic acid amino-methyltransferases. FASEB J 14: 2365–2368. [DOI] [PubMed] [Google Scholar]
- Christian T, Evilia C, Hou YM. 2006. Catalysis by the second class of tRNA(m1G37) methyl transferase requires a conserved proline. Biochemistry 45: 7463–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, Hoffmann K, Perona JJ, Hou YM. 2010. Mechanism of N-methylation by the tRNA m1G37 methyltransferase Trm5. RNA 16: 2484–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Crécy-Lagard V, Brochier-Armanet C, Urbonavičius J, Fernandez B, Phillips G, Lyons B, Noma A, Alvarez S, Droogmans L, Armengaud J, et al. 2010. Biosynthesis of wyosine derivatives in tRNA: an ancient and highly diverse pathway in Archaea. Mol Biol Evol 27: 2062–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Ren S, Li H, Shi PY. 2008a. Separate molecules of West Nile virus methyltransferase can independently catalyze the N7 and 2′-O methylations of viral RNA cap. Virology 377: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Ren S, Zhang B, Zhou Y, Puig-Basagoiti F, Li H, Shi PY. 2008b. West Nile virus methyltransferase catalyzes two methylations of the viral RNA cap through a substrate-repositioning mechanism. J Virol 82: 4295–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Fink K, Zust R, Lim SP, Qin CF, Shi PY. 2014. Flavivirus RNA methylation. J Gen Virol 95: 763–778. [DOI] [PubMed] [Google Scholar]
- Droogmans L, Grosjean H. 1987. Enzymatic conversion of guanosine 3′ adjacent to the anticodon of yeast tRNAPhe to N1-methylguanosine and the wye nucleoside: dependence on the anticodon sequence. EMBO J 6: 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giessing AM, Jensen SS, Rasmussen A, Hansen LH, Gondela A, Long K, Vester B, Kirpekar F. 2009. Identification of 8-methyladenosine as the modification catalyzed by the radical SAM methyltransferase Cfr that confers antibiotic resistance in bacteria. RNA 15: 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Ishii R, Muto Y, Bessho Y, Yokoyama S. 2008. Crystal structure of archaeal tRNA(m1G37)methyltransferase aTrm5. Proteins 72: 1274–1289. [DOI] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Kuratani M, Bessho Y, Yokoyama S. 2009. Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat Struct Mol Biol 16: 1109–1115. [DOI] [PubMed] [Google Scholar]
- Grosjean H, Droogmans L, Giege R, Uhlenbeck OC. 1990. Guanosine modifications in runoff transcripts of synthetic transfer RNA-Phe genes microinjected into Xenopus oocytes. Biochim Biophys Acta 1050: 267–273. [DOI] [PubMed] [Google Scholar]
- Grosjean H, Keith G, Droogmans L. 2004. Detection and quantification of modified nucleotides in RNA using thin-layer chromatography. Methods Mol Biol 265: 357–391. [DOI] [PubMed] [Google Scholar]
- Gupta R. 1984. Halobacterium volcanii tRNAs. Identification of 41 tRNAs covering all amino acids, and the sequences of 33 class I tRNAs. J Biol Chem 259: 9461–9471. [PubMed] [Google Scholar]
- Juhling F, Morl M, Hartmann RK, Sprinzl M, Stadler PF, Putz J. 2009. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res 37: D159–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith G, Dirheimer G. 1980. Primary structure of Bombyx mori posterior silkgland tRNAPhe. Biochem Biophys Res Commun 92: 109–115. [DOI] [PubMed] [Google Scholar]
- Kelkar YD, Ochman H. 2013. Genome reduction promotes increase in protein functional complexity in bacteria. Genetics 193: 303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konevega AL, Soboleva NG, Makhno VI, Semenkov YP, Wintermeyer W, Rodnina MV, Katunin VI. 2004. Purine bases at position 37 of tRNA stabilize codon-anticodon interaction in the ribosomal A site by stacking and Mg2+-dependent interactions. RNA 10: 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. 2011. S-Adenosylmethionine-dependent alkylation reactions: when are radical reactions used? Bioorg Chem 39: 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. 2013. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res 41: D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matte-Tailliez O, Zivanovic Y, Forterre P. 2000. Mining archaeal proteomes for eukaryotic proteins with novel functions: the PACE case. Trends Genet 16: 533–536. [DOI] [PubMed] [Google Scholar]
- Mazabraud A. 1979. Deficiency of the peroxy-Y base in oocyte phenylalanine tRNA. FEBS Lett 100: 235–240. [DOI] [PubMed] [Google Scholar]
- McCloskey JA, Graham DE, Zhou S, Crain PF, Ibba M, Konisky J, Soll D, Olsen GJ. 2001. Post-transcriptional modification in archaeal tRNAs: identities and phylogenetic relations of nucleotides from mesophilic and hyperthermophilic Methanococcales. Nucleic Acids Res 29: 4699–4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A, Kirino Y, Ikeuchi Y, Suzuki T. 2006. Biosynthesis of wybutosine, a hyper-modified nucleoside in eukaryotic phenylalanine tRNA. EMBO J 25: 2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira T, Suzuki T. 2011. Retrograde nuclear import of tRNA precursors is required for modified base biogenesis in yeast. Proc Natl Acad Sci 108: 10502–10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips G, de Crécy-Lagard V. 2011. Biosynthesis and function of tRNA modifications in Archaea. Curr Opin Microbiol 14: 335–341. [DOI] [PubMed] [Google Scholar]
- Sakaguchi R, Giessing A, Dai Q, Lahoud G, Liutkevičiūtė Z, Klimašauskas S, Piccirilli J, Kirpekar F, Hou YM. 2012. Recognition of guanosine by dissimilar tRNA methyltransferases. RNA 18: 1687–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample PJ, Koreny L, Paris Z, Gaston KW, Rubio MA, Fleming IM, Hinger S, Horakova E, Limbach PA, Lukes J, et al. 2015. A common tRNA modification at an unusual location: the discovery of wyosine biosynthesis in mitochondria. Nucleic Acids Res 43: 4262–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. 1997. A genomic perspective on protein families. Science 278: 631–637. [DOI] [PubMed] [Google Scholar]
- Umitsu M, Nishimasu H, Noma A, Suzuki T, Ishitani R, Nureki O. 2009. Structural basis of AdoMet-dependent aminocarboxypropyl transfer reaction catalyzed by tRNA-wybutosine synthesizing enzyme, TYW2. Proc Natl Acad Sci 106: 15616–15621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbonavičius J, Droogmans L, Armengaud J, Grosjean H. 2009. Deciphering the complex enzymatic pathway for biosynthesis of wyosine derivatives in anticodon of tRNAPhe. In DNA and RNA modification enzymes: structure, mechanism, function and evolution (ed. Grosjean H), pp. 423–435. Landes Bioscience, Austin, TX. [Google Scholar]
- Urbonavičius J, Meškys R, Grosjean H. 2014. Biosynthesis of wyosine derivatives in tRNAPhe of Archaea: role of a remarkable bifunctional tRNAPhe:m1G/imG2 methyltransferase. RNA 20: 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waas WF, Druzina Z, Hanan M, Schimmel P. 2007. Role of a tRNA base modification and its precursors in frameshifting in eukaryotes. J Biol Chem 282: 26026–26034. [DOI] [PubMed] [Google Scholar]
- White BN, Tener GM. 1973. Properties of tRNAPhe from Drosophila. Biochim Biophys Acta 312: 267–275. [DOI] [PubMed] [Google Scholar]
- Zhou S, Sitaramaiah D, Noon KR, Guymon R, Hashizume T, McCloskey JA. 2004. Structures of two new “minimalist” modified nucleosides from archaeal tRNA. Bioorg Chem 32: 82–91. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Ray D, Zhao Y, Dong H, Ren S, Li Z, Guo Y, Bernard KA, Shi PY, Li H. 2007. Structure and function of flavivirus NS5 methyltransferase. J Virol 81: 3891–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.