

Abstract
A key factor in the development of obesity is the overconsumption of food calorically high in fat. Overconsumption of food high in fat not only promotes weight gain but elicits changes in reward processing. No studies to date have examined whether consumption of a high-fat (HF) diet alters structural plasticity in brain areas critical for reward processing, which may account for persistent changes in behavior and psychological function by reorganizing synaptic connectivity. To test whether dietary fat may induce structural plasticity we placed rats on one of three dietary conditions: ad libitum standard chow (SC), ad libitum 60% HF (HF-AL), or calorically matched 60% HF (HF-CM) for 3 weeks and then quantified dendritic spine density and type on basal and apical dendrites of pyramidal cells in layer V of the medial prefrontal cortex (mPFC) and medium spiny neurons (MSNs) of the nucleus accumbens. Our results demonstrate a significant reduction in the density of thin spines on the apical and basal segments of dendrites within the infralimbic, but not prelimbic, mPFC.
Keywords: high-fat, prefrontal cortex, plasticity, dendritic spines
Introduction
At present, more than one third of the U.S. adult population is classified as obese, making obesity a substantial and relevant health concern [1]. Obesity contributes to and/or exacerbates a host of health problems, including heart disease [2], diabetes [2], stroke [3] and certain types of cancers [4], all of which could be mitigated by reducing the prevalence of obesity [5]. The development of obesity is likely a consequence of diverse factors that broadly include genetics [6], lifestyle [7] inactivity [8], pregnancy [9], unhealthy diet [10], socioeconomic status [11], and environmental influences [12]. Although there are numerous causes contributing to the obesity epidemic what is ubiquitous among most theories is that excessive consumption of foods calorically high in fat results in weight gain and obesity [13].
A hallmark of obesity is continued unhealthy eating behaviors despite knowledge of negative social and physiological consequences [14]. It has been hypothesized that exposure to dietary high-fat (HF) results in a decrease in natural reward sensitivity and therefore contributes to compensatory hedonic overeating behaviors (i.e., eating in the absence of an energetic demand) [15, 16]. In the rodent model, overconsumption of diets high in fat has consistently and efficiently been shown to induce obesity and induce a number of neurobiological changes within the mesolimbic dopamine reward system [17, 18]. For example, obese rats have a reduction in striatal dopamine D2 receptors (D2Rs) and knockdown of D2Rs potentiates compulsive food seeking to HF food, suggesting a deficit in brain reward processing [19]. Additionally, rats fed a diet consisting of Crisco, cheddar cheese and peanut butter for 15 weeks have lower extracellular dopamine (DA) levels within the nucleus accumbens (NAc) compared to standard chow fed animals [20]. Consistent with adaptations in reward processing, rats susceptible to diet-induced obesity are more prone to food-cue triggered motivation and show increased sensitivity to cocaine, suggesting basal differences in the function of the mesolimbic circuits [21, 22].
In addition to rodent studies, clinical research has shown evidence of dysregulated D2R signaling within obese human populations. Specifically, Wang et al., (2001) showed that striatal D2R availability is attenuated in obese individuals and that this decrease is negatively correlated with body mass index (BMI) [23]. It has also been demonstrated that the down regulation of striatal D2Rs in morbidly obese patients is linked with a decrease in brain glucose metabolism, a measure of brain function, within the prefrontal cortex (PFC) [24].
As demonstrated, previous reports of reward circuit modulation have focused on changes in dopaminergic plasticity, particularly in regards to DA and D2R levels but little is known about the structural plasticity that may underlie those changes. One way to examine structural change is to quantify dendritic spine density and spine type. Dendritic spines are considered to be the primary postsynaptic structures where excitatory synaptic inputs, primarily glutamatergic, are integrated [25]. Activity within the PFC is shaped by several factors. Dopamine in particular has been implicated in mediating primary PFC cognitive functions, including memory and reward [26], and the interaction between DA and glutamate has been studied as a major mechanism underlying neuronal excitability [27, 28] and may subserve plasticity. The influence of DA on glutamate function in the cortex is partially dependent upon what DA receptor is expressed on the neuron [29]. In the PFC, it has been demonstrated that D2 and D1 receptors decrease and increase pyramidal cell excitability, respectively [30]. Despite the well-established relationship between DA and glutamate and the relatively large amount of literature examining DA plasticity in obesity, very little research has aimed to elucidate changes in excitatory transmission.
Functionally, increases in dendritic spine density and shape are hypothesized to be the direct result of long-term potentiation (LTP) [31, 32], particularly on dendritic segments where spine density is already low [33], and can shrink in size following long-term depression (LTD) [34]. As such, changes in spine density and shape are thought to reflect synaptic activity and neuronal remodeling. Dendritic spine plasticity has been extensively studied in response to pathological and non-pathological paradigms. For example, postmortem studies in human patients diagnosed with mental retardation [35] or schizophrenia [36] show a marked decrease in spine density in the CA1 region of the hippocampus and on pyramidal neurons of the PFC, respectively. Alternatively, an increase in spine density has been observed in the rat hippocampal dentate gyrus following consolidation of water maze training, a spatial learning task [37]. Within the reward circuit, an increase in spine density has been demonstrated in NAc shell (NAc-Sh) and mPFC following exposure to the psychostimulants amphetamine and cocaine [38]. Stress too has been reported to have a positive effect on spine density in the NAc-Sh, prelimbic PFC (PL-PFC) and orbitofrontal cortex (OFC) [39]. In contrast, morphine [40] and sucrose [41] elicit the opposite effect on spine density within the NAc-Sh and OFC, respectively.
The aim of our study was to examine whether exposure to dietary HF would induce changes in dendritic spine density within the PFC and NAc similarly to other appetitive stimuli. Rats were maintained on either ad libitum standard chow (SC), ad libitum 60% HF (HF-AL) or 60% HF calorically matched (HF-CM) diets for 3 weeks. Following 3 weeks of feeding on SC, HF-CM, or HF-AL diets, dendritic spine density and type were analyzed within the PL-PFC, infralimbic prefrontal cortex (IL-PFC), the NAc-Sh and NAc core (NAc-C) using DiI staining to visualize the spines.
Methods
Animals
Eighteen adult (PND 60) male Sprague-Dawley rats obtained from our breeding colony and housed in clear plastic cages in a temperature-controlled room with a 12:12-h light-dark cycle with lights on at 0700 were utilized for experimental procedures. Rats were 285-397 g at the beginning of the experiment. All experimental procedures and protocols for animal studies were approved by the University of Wyoming Institutional Animal Care and Use Committee in accordance with international guidelines on the ethical use of animals.
Maintenance diets
Rats were exposed to one of three dietary conditions in their home cages for three weeks: standard chow (SC, n=6), 60% high-fat (HF-AL, n=6), or 60% calorically-matched high-fat (HF-CM, n=6). The nutritional content of the SC diet included ∼29% protein, ∼58% carbohydrate, and ∼13% fat by kilocalorie with a total fuel value of 3.36 kcal/g (Laboratory Rodent Diet 5001, St. Louis, MO). The nutritional content of the HF diet included ∼20% protein, ∼20% carbohydrate, and ∼60% fat by kilocalorie with a gross fuel value of 5.24 kcal/g (Research Diets, New Brunswick, NJ). Animals in the HF-CM group were also fed the 60% HF diet but were food restricted such that their daily caloric intake, and therefore weight gain, did not significantly differ from the SC controls at the time of sacrifice (Figure 1B, Table 1). Each day, food consumption was measured and averaged for each dietary condition. The total amount of food consumed by the SC group was then multiplied by the caloric value of the diet (3.36 kcal/g) in order to obtain an average caloric intake. The average caloric intake was then divided by the caloric value of the HF diet (5.24 kcal/g) to estimate the amount of food to be given to the HF-CM group. This method ensured that the caloric intake of both the SC and HF-CM groups were the same. To validate that this procedure produced similar weight gain in the SC and HF-SM groups, all animals were weighed every other day. Furthermore, following sacrifice, epididymal fat was dissected and weighed as a proxy for overall fat composition. This gonadal fat deposit is among the largest adipose deposits in the rat and is therefore often used in diet studies to determine whether the dietary manipulation induced fat accumulation, in addition to changes in body weight [42].
Fig 1. Ad Libitum exposure to a high-fat diet for 3 weeks increases weight gain and body-fat composition.
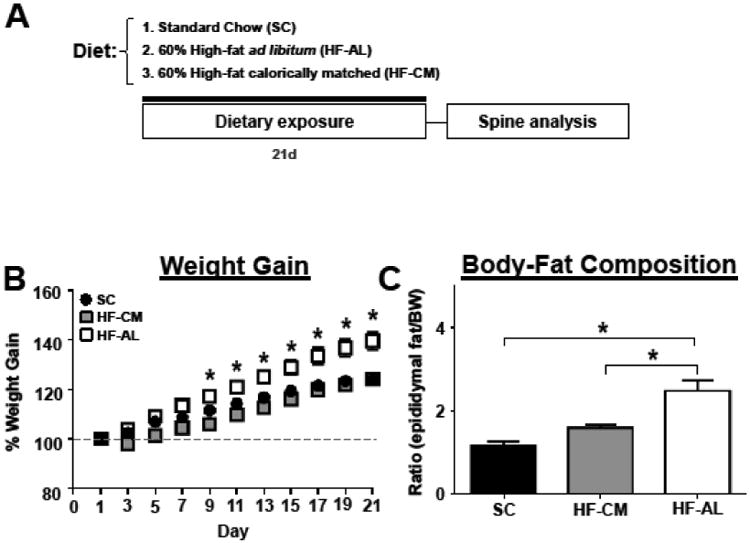
(A) Experimental timeline. (B) Percent weight gain throughout dietary exposure. (C) Body-fat composition measured as the ratio of epididymal fat to body weight. Values represent the mean ± S.E.M. * indicates significant increase in HF-AL from SC and HF-CM fed rats.
Table 1. Dietary compositions and percent weight gain for rats exposed to SC, HF-AL, and HF-CM dietary conditions for 3 weeks.
| % Calories | % Weight Δ | |||
|---|---|---|---|---|
| Protein | Carbohydrates | Fat | 3 wk | |
| Standard Chow | 28.5 | 58.0 | 13.5 | 124.1 ± 0.96 |
| High-Fat Calorically Matched | 20.0 | 20.0 | 60.0 | 124.4 ± 2.16 |
| High-Fat Ad Libitum | 20.0 | 20.0 | 60.0 | 139.7 ± 3.65* |
indicates significant weight gain from SC and HF-CM fed rats.
Values represent the mean ± S.E.M
Quantification of dendritic spine density
After dietary exposure, rats were euthanized via cardiac perfusion (200 mL, 0.9% saline followed by 300 mL, 1.5% paraformaldehyde (in 0.1M phosphate buffer (PBS)). After washing in PBS, brains were coronally sectioned into 200 μm slices with a Leica VT1200S vibratome (Leica, Buffalo Grove, IL) and briefly collected in PBS. Slices were fixed in 4% paraformaldehyde in PBS for 20 minutes, incubated with Vybrant-DiI cell-labeling solution (1:200, Invitrogen, USA) for 1 hour at room temperature and placed in PBS at 4°C for 48 hours to allow dye diffusion within membranes. Finally, slices were mounted on glass slides with Vectashield (Vector, Burlingame, CA) and imaged using a Zeiss confocal microscope. For cortical pyramidal cells, dendritic spines were quantified on the terminal tips of third or fourth order basal dendrites and second or third order apical dendrites in layer V of the PL-PLC and IL-PFC. For medium spiny neurons, spines on third order or greater terminal tips were quantified in the NAc-Sh and NAc-C [43]. In all regions of interest, visible spines located within the terminal 10 μm were manually counted using 40 × magnification. In addition to total density, spine type was also analyzed following parameters previously described [44]. Briefly, spines were identified as thin type if the ratio of head diameter to neck diameter was greater than 1.1 and maximum head diameter was less than 0.4 μm. Spines were identified as mushroom type if the ratio of head to neck diameter was greater than 1.1 and the maximum head diameter was greater than 0.4 μm. Spines with a head to neck ratio of less than 1.1 were classified as stubby and those that bifurcated above the connection between spine and dendrite were classified as cup shaped. Filopodium were identified as long, thin protrusions lacking a head. Within each animal 4-8 dendrites were selected from distinct cells for analysis in each brain region and counts from these dendrites were then averaged. Dendrites were only selected if staining of the cell was complete enough that the branch order and differentiation of spine types could be determined. In total, 32-40 dendrites were analyzed per group for each brain region. Slides were coded so that the person responsible for cell selection and analysis was blind to the experimental condition. Statistical analyses were performed by averaging dendritic spine counts across animals per brain region. Group differences were assessed using two-way ANOVA and any post hoc comparisons were completed with Tukey's multiple comparisons test (p<0.05).
Results
To determine whether dietary manipulation may alter structural plasticity within the reward circuitry of the brain we quantified changes in dendritic spine density within the PFC and NAc, brain regions known to play a vital role in reward processing [45, 46]. There was a significant dietary effect on weight gain (F(2, 15) = 14.40, p<0.01), a significant day effect (F(10, 150) = 352, p<0.01), and significant dietary and day interaction (F(20, 150) = 9.50, p<0.01). Post-hoc analysis revealed that rats in the HF-AL group showed significant weight gain compared to HF-CM and SC fed rats days 9-21 (Figure 1B). In addition to gaining significant weight, rats maintained on the ad libitum high-fat diet showed a significant increase in estimated body fat composition measured by the ratio of epididymal fat to body weight (SC: 1.18 ± 0.09, HF-CM: 1.6 ± 0.08, HF-AL: 2.49 ± 0.25, Figure 1C). Basal and apical spine density was quantified from pyramidal cells within the PFC-PL and the PFC-IL regions of the prefrontal cortex (Figures 2-3, Table 2) and spines were quantified from medium spiny neurons (MSNs) in the shell and core of the NAc (Figure 4, Table 2). To our surprise the dietary exposure had no significant effect on the total spine density or spine type of MSNs within the NAc-C (Table 2). There was also no dietary effect on the spine density or spine type of pyramidal cells within the PL-PFC (Table 2). However, there was a significant dietary effect within the IL-PFC on the total spine density for both apical and basal dendrites (Apical: SC: 11.43 ± 0.09, HF-CM: 9.89 ± 0.16, HF-AL: 9.91 ± 0.22; F(2, 10) =57.34, p<0.01; Basal: SC: 12.00 ± 0.16, HF-CM: 10.08 ± 0.12, HF-AL: 10.05 ± 0.14, F(2, 10) = 74.22, p<0.01). An analysis of the spine type revealed that this reduction was due largely to an attenuation of thin spines in the IL-PFC. There was a significant dietary and spine type interaction (F(8, 150) = 14.82, p<0.01). Post-hoc analysis revealed that rats in both the HF-AL and HF-CM groups showed a significant attenuation in thin spines (Apical: SC: 7.24 ± 0.20, HF-CM: 5.39 ± 0.19, HF-AL: 5.50 ± 0.09; Basal: SC: 7.37 ± 0.25, HF-CM: 5.33 ± 0.28, HF-AL: 5.64 ± 0.11, Figure 3B). Interestingly, we also observed an increase in stubby type spines on apical dendrites in the HF-CM group compared to SC (SC: 1.16 ± 0.14, HF-CM: 1.69 ± 0.14) and a decrease in mushroom type spines on apical dendrites in the HF-CM group compared to SC (SC: 2.75 ± 0.14, HF-CM: 2.28 ± 0.12).
Fig 2. Exposure to a high-fat diet for 3 weeks has no effect on spine density within the prelimbic region of the prefrontal cortex.
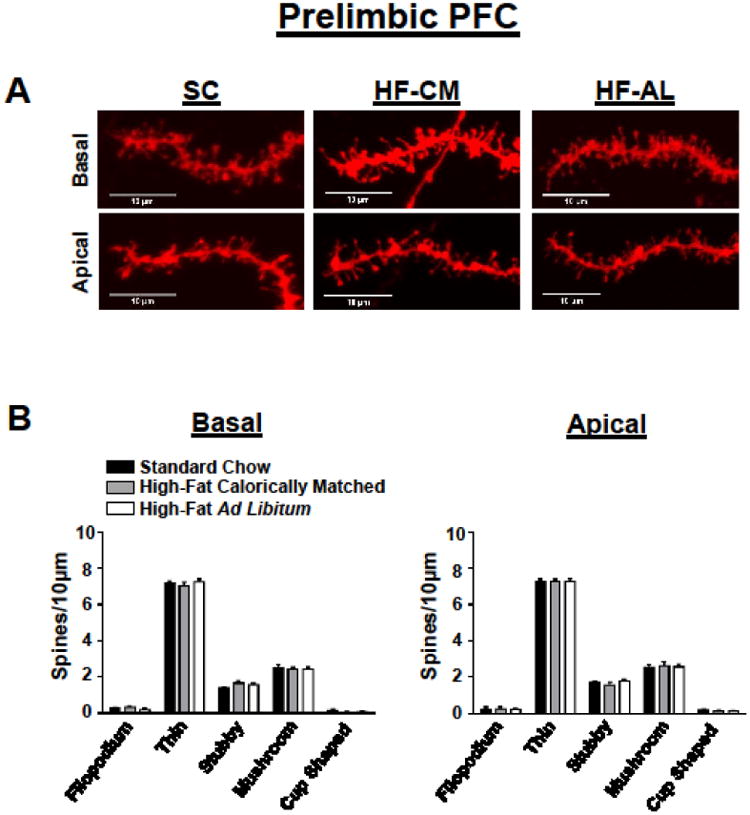
(A) Representative prelimbic PFC apical (bottom) and basal (top) DiI-stained dendrites from each dietary condition. (B) Quantification of spine type from the basal (left) and apical (right) dendrites of pyramidal cells in the PL-PFC. Values represent the mean ± S.E.M.
Fig 3. Exposure to a high-fat diet for 3 weeks decreases spine density within the infralimbic prefrontal cortex.
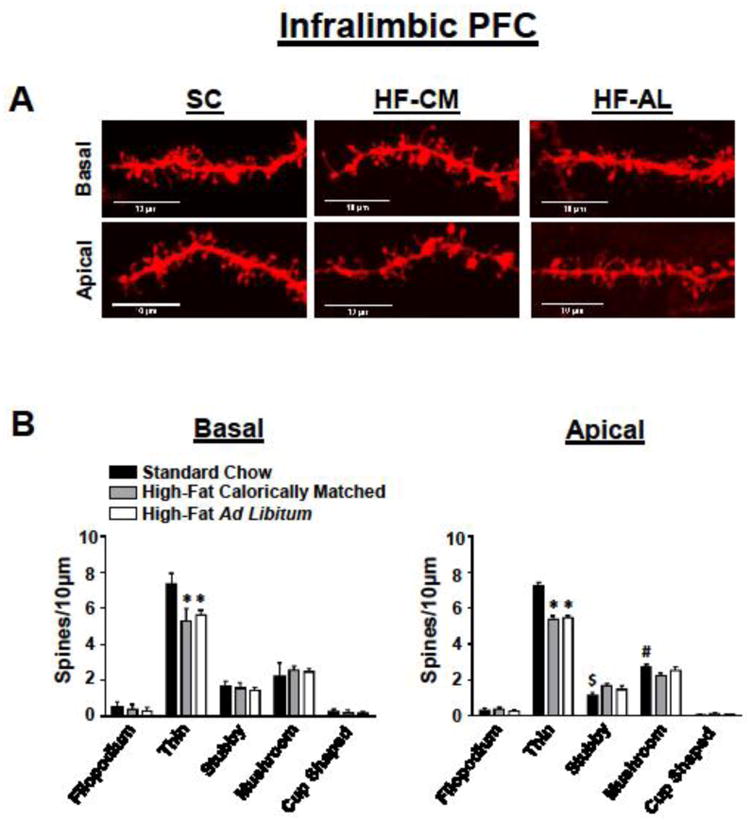
(A) Representative IL-PFC apical (bottom) and basal (top) DiI-stained dendrites from each dietary condition. (B) Quantification of spine type from the basal (left) and apical (right) dendrites of pyramidal cells in the IL-PFC. Values represent the mean ± S.E.M.
Table 2. Summary of dendritic spine quantification.
| A Prelimbic PFC | |||||||
|---|---|---|---|---|---|---|---|
| Basal | Apical | ||||||
| SC | HF-CM | HF-AL | SC | HF-CM | HF-AL | ||
| Filopodium | 0.27 ± 0.11 | 0.29 ± 0.09 | 0.23 ± 0.09 | Filopodium | 0.28 ± 0.08 | 0.36 ± 0.09 | 0.22 ± 0.08 |
| Thin | 7.30 ± 0.13 | 7.32 ± 0.10 | 7.29 ± 0.16 | Thin | 7.21 ± 0.12 | 7.06 ± 0.20 | 7.27 ± 0.16 |
| Stubby | 1.74 ± 0.08 | 1.56 ± 0.15 | 1.79 ± 0.10 | Stubby | 1.40 ± 0.03 | 1.68 ± 0.16 | 1.60 ± 0.06 |
| Mushroom | 2.54 ± 0.14 | 2.64 ± 0.16 | 2.59 ± 0.14 | Mushroom | 2.53 ± 0.15 | 2.44 ± 0.11 | 2.43 ± 0.14 |
| Cup Shaped | 0.22 ± 0.06 | 0.16 ± 0.04 | 0.16 ± 0.03 | Cup Shaped | 0.19 ± 0.05 | 0.08 ± 0.03 | 0.09 ± 0.04 |
| Total | 12.07 ± 0.19 | 12.01 ± 0.22 | 12.07 ± 0.23 | Total | 11.62 ± 0.24 | 11.61 ± 0.24 | 11.62 ± 0.11 |
| B Infralimbic PFC | |||||||
|---|---|---|---|---|---|---|---|
| Basal | Apical | ||||||
| SC | HF-CM | HF-AL | SC | HF-CM | HF-AL | ||
| Filopodium | 0.54 ± 0.12 | 0.40 ± 0.12 | 0.31 ± 0.08 | Filopodium | 0.31 ± 0.12 | 0.39 ± 0.07 | 0.28 ± 0.05 |
| Thin | 7.37 ± 0.25 | 5.33 ± 0 28* | 5.64 ± 0.11* | Thin | 7.24 ± 0.20 | 5.39 ± 0.19* | 5.50 ± 0.09* |
| Stubby | 1.67 ± 0.13 | 1.58 ± 0.12 | 1.45 ± 0.07 | Stubby | 1.16 ± 0.14 | 1.69 ±0.14$ | 1.50 ± 0.20 |
| Mushroom | 2.20 ± 0.33 | 2.57 ± 0.10 | 2.48 ± 0.07 | Mushroom | 2.75 ± 0.14 | 2.28 ± 0.12 # | 2.56 ± 0.19 |
| Cup Shaped | 0.24 ± 0.07 | 0.21 ± 0.06 | 0.16 ± 0.04 | Cup Shaped | 0.05 ± 0.03 | 0.14 ± 0.05 | 0.09 ± 0.04 |
| Total | 12.01 ± 0.16 | 10.08 ± 0.12 | 10.05 ± .14 | Total | 11.43 ± 0.09 | 9.89 ± 0.16 | 9.91 ± 0.22 |
| C Nucleus Accumbens | |||||||
|---|---|---|---|---|---|---|---|
| Core | Shell | ||||||
| SC | HF-CM | HF-AL | SC | HF-CM | HF-AL | ||
| Filopodium | 0.41 ± 0.05 | 0.37 ± 0.12 | 0.23 ± 0.02 | Filopodium | 0.32 ± 0.09 | 0.30 ± 0.12 | 0.24 ± 0.08 |
| Thin | 9.45 ± 0.16 | 9.41 ± 0.25 | 9.4 ± 0.07 | Thin | 9.44 ± 0.13 | 9.36 ± 0.10 | 9.37 ± 0.17 |
| Stubby | 1.68 ± 0.06 | 1.91 ± 0.09 | 1.91 ± 0.15 | Stubby | 1.53 ± 0.14 | 1.67 ± 0.23 | 1.72 ± 0.09 |
| Mushroom | 2.67 ± 0.06 | 2.60 ± 0.08 | 2.55 ± 0.19 | Mushroom | 2.65 ± 0.12 | 2.52 ± 0.11 | 2.53 ± 0.19 |
| Cup Shaped | 0.29 ± 0.04 | 0.13 ± 0.05 | 0.19 ± 0.05 | Cup Shaped | 0.15 ± 0.07 | 0.16 ± 0.11 | 0.12 ± 0.05 |
| Total | 14.50 ± 0.08 | 14.42 ± 0.21 | 14.41 ± 0.24 | Total | 14.08 ± 0.18 | 13.99 ± 0.20 | 13.96 ± 0.23 |
indicates significant decrease in HF-AL and HF-CM from SC fed rats.
indicates significant increase in HF-CM from SC fed rats.
indicates significant decrease in HF-CM from SC fed rats.
Values represent the mean ± S.E.M
Fig 4. Exposure to a high-fat diet for 3 weeks has no effect on spine density within the nucleus accumbens.
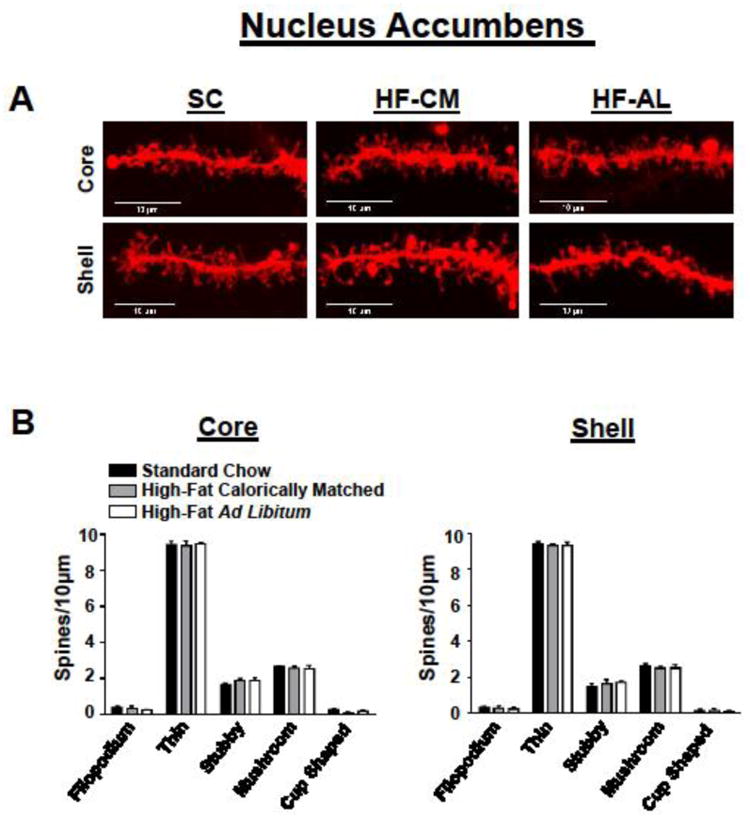
(A) Representative nucleus accumbens core (top) and shell (bottom) DiI-stained dendrites from each dietary condition. (B) Quantification of spine type from medium spiny neurons of the core (left) and shell (right). Values represent the mean ± S.E.M
Discussion
Our study looked to expand upon previous research that has shown neurobiological changes as a result of exposure to dietary HF. To the best of our knowledge, no laboratories have examined whether dietary HF induces changes in dendritic spine density within the reward circuit. In the present study, rats exposed to dietary HF independent of weight gain had attenuation in spine density on both apical and basal dendrites of the IL-PFC compared to SC controls (Figure 3). This reduction in spine density can be mostly attributed to a loss of thin spines. This is not surprising given that thin spines are the most numerous comparatively and form and disappear more rapidly in the context of varying levels of synaptic activity, suggesting that they are more susceptible to plasticity [47]. Thin spines have been widely characterized under a multitude of circumstances. For example, a loss of thin spines has been observed in the monkey prefrontal cortex following aging [48] while an increase of thin spines in this region has been demonstrated subsequent of seven days forced abstinence from cocaine self-administration in rats [49]. The loss of thin spines in response to HF exposure may underlie previous reports that HF consumption impairs learning and memory, evidenced by performance on both hippocampal and frontal-dependent tasks [50]. While there were also significant changes between SC and HF-CM groups in mushroom and stubby spines in the IL-PFC, it is unlikely a dietary effect since the HF-AL group did not also show this trend. It is however possible that this finding may be attributed to food restriction given the nature of the feeding regimen in the HF-CM group, but future studies are required to elucidate this. It is further hypothesized that a lack of change in spine type or density in the nucleus accumbens may be due to a lack of ability to discriminate between D1 and D2 dopamine receptor-containing neurons, given their differential change in spine density following treatments such as withdrawal from cocaine [51].
Quantification of spine density is a common approach used to examine experience-induced modifications to synaptic organization [35] [36] [37]. Although it is an indirect measure, providing information about changes to the postsynaptic cell exclusively, electron microscopy (EM) studies have demonstrated that changes in spine density correlate with sites of synaptic contact [52, 53]. Specifically, time-lapse two-photon imaging studies within the hippocampus shows an increased spine density following induction of long-term potentiation (LTP), a phenomenon known to increase synaptic strength [25]. Similarly, following long-term depression (LTD), or loss of synaptic efficacy, shrinkage in spine size is observed [34] suggesting that changes in spine dynamics is correlated with synaptic activity, or lack thereof.
To examine a potential neurobiological consequence of the HF diet, we quantified dendritic spine density in the PFC (IL and PL) and NAc (core and shell), key reward areas long characterized as being involved in reward seeking and reward reinforcement, respectively [38, 54, 55]. The PL-PFC has been known to initiate reward-seeking, as found in cocaine studies demonstrating that inhibition of this region blocks several forms of reinstated drug seeking [56-58]. The significance of the IL-PFC is presently less established but is suggested to play an opposing role in reward related behavior. Specifically, it has been demonstrated that the IL-PFC is responsible for the inhibition of cocaine seeking in extinguished rats, as measured by an increase in cocaine reinstatement following inactivation of this region [59]. Similarly, inactivation of the IL-PFC immediately following extinction impairs the maintenance of consolidation of extinction after cocaine self-administration [60]. The above findings suggest that while the PL-PFC facilitates reward seeking, the IL-PFC is an integral component of inhibitory control [61].
Our results demonstrate that exposure to HF decreases dendritic spine density and type in the IL-PFC within both the HF-AL and HF-CM groups compared to SC controls, while no differences between groups were observed in the PL-PFC or NAc. This is the first report of morphological changes following exposure to a HF diet. Given the proposed role of the IL-PFC in inhibitory control of drug reward seeking, we hypothesize that the decrease in spine density may underlie challenges in dieting behavior after exposure to fatty foods. This finding is supported by other reports of hypofunctionality in the PFC, notably the decrease in glucose metabolism in obese patients [14]. Future studies will expand upon these findings to study the downstream consequence of altering excitatory input and output from the IL-PFC in controlling feeding behaviors.
Acknowledgments
The authors would like to thank Dr. Zhaojie Zhang, the director of the Neuroscience microscopy facility at the University of Wyoming, for his help and guidance imaging the spine data. We would also like to thank Kevin Schlidt and Morgan Deters for their assistance with animal care. We are also grateful for the support contributed by NIGMS grant P30 GM103398, and the College of Health Sciences Seed Grant from the University of Wyoming.
References
- 1.Ogden CL, et al. Prevalence of overweight and obesity in the United States, 1999-2004. JAMA. 2006;295(13):1549–55. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 2.Wild SH, Byrne CD. ABC of obesity. Risk factors for diabetes and coronary heart disease. BMJ. 2006;333(7576):1009–11. doi: 10.1136/bmj.39024.568738.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurth T, et al. Body mass index and the risk of stroke in men. Arch Intern Med. 2002;162(22):2557–62. doi: 10.1001/archinte.162.22.2557. [DOI] [PubMed] [Google Scholar]
- 4.Calle EE. Obesity and cancer. BMJ. 2007;335(7630):1107–8. doi: 10.1136/bmj.39384.472072.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finkelstein EA, et al. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff (Millwood) 2009;28(5):w822–31. doi: 10.1377/hlthaff.28.5.w822. [DOI] [PubMed] [Google Scholar]
- 6.Yamada Y, et al. Genetic factors for obesity. Int J Mol Med. 2006;18(5):843–51. [PubMed] [Google Scholar]
- 7.Shigeta H, et al. Lifestyle, obesity, and insulin resistance. Diabetes Care. 2001;24(3):608. doi: 10.2337/diacare.24.3.608. [DOI] [PubMed] [Google Scholar]
- 8.Spence D. Inactivity and obesity. BMJ. 2011;343:d5093. doi: 10.1136/bmj.d5093. [DOI] [PubMed] [Google Scholar]
- 9.Yogev Y, Catalano PM. Pregnancy and obesity. Obstet Gynecol Clin North Am. 2009;36(2):285–300. viii. doi: 10.1016/j.ogc.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Hensrud DD. Diet and obesity. Curr Opin Gastroenterol. 2004;20(2):119–24. doi: 10.1097/00001574-200403000-00012. [DOI] [PubMed] [Google Scholar]
- 11.Sobal J, Stunkard AJ. Socioeconomic status and obesity: a review of the literature. Psychol Bull. 1989;105(2):260–75. doi: 10.1037/0033-2909.105.2.260. [DOI] [PubMed] [Google Scholar]
- 12.Hebebrand J, Hinney A. Environmental and genetic risk factors in obesity. Child Adolesc Psychiatr Clin N Am. 2009;18(1):83–94. doi: 10.1016/j.chc.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Bray GA, Paeratakul S, Popkin BM. Dietary fat and obesity: a review of animal, clinical and epidemiological studies. Physiol Behav. 2004;83(4):549–55. doi: 10.1016/j.physbeh.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 14.Volkow ND, et al. Overlapping neuronal circuits in addiction and obesity: evidence of systems pathology. Philos Trans R Soc Lond B Biol Sci. 2008;363(1507):3191–200. doi: 10.1098/rstb.2008.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cordeira JW, et al. Brain-derived neurotrophic factor regulates hedonic feeding by acting on the mesolimbic dopamine system. J Neurosci. 2010;30(7):2533–41. doi: 10.1523/JNEUROSCI.5768-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiger BM, et al. Evidence for defective mesolimbic dopamine exocytosis in obesity-prone rats. FASEB J. 2008;22(8):2740–6. doi: 10.1096/fj.08-110759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masek J, Fabry P. High-fat diet and the development of obesity in albino rats. Experientia. 1959;15:444–5. doi: 10.1007/BF02157708. [DOI] [PubMed] [Google Scholar]
- 18.Schemmel R, Mickelsen O, Gill JL. Dietary obesity in rats: Body weight and body fat accretion in seven strains of rats. J Nutr. 1970;100(9):1041–8. doi: 10.1093/jn/100.9.1041. [DOI] [PubMed] [Google Scholar]
- 19.Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci. 2010;13(5):635–41. doi: 10.1038/nn.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geiger BM, et al. Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience. 2009;159(4):1193–9. doi: 10.1016/j.neuroscience.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson MJ, et al. Individual Differences in Cue-Induced Motivation and Striatal Systems in Rats Susceptible to Diet-Induced Obesity. Neuropsychopharmacology. 2015;40(9):2113–23. doi: 10.1038/npp.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vollbrecht PJ, et al. Pre-existing differences in motivation for food and sensitivity to cocaine-induced locomotion in obesity-prone rats. Physiol Behav. 2015;152(Pt A):151–160. doi: 10.1016/j.physbeh.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang GJ, et al. Brain dopamine and obesity. Lancet. 2001;357(9253):354–7. doi: 10.1016/s0140-6736(00)03643-6. [DOI] [PubMed] [Google Scholar]
- 24.Volkow ND, et al. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: possible contributing factors. Neuroimage. 2008;42(4):1537–43. doi: 10.1016/j.neuroimage.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399(6731):66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 26.Schultz W. Getting formal with dopamine and reward. Neuron. 2002;36(2):241–63. doi: 10.1016/s0896-6273(02)00967-4. [DOI] [PubMed] [Google Scholar]
- 27.Ferrario CR, et al. Withdrawal from cocaine self-administration alters NMDA receptor-mediated Ca2+ entry in nucleus accumbens dendritic spines. PLoS One. 2012;7(8):e40898. doi: 10.1371/journal.pone.0040898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baldwin AE, Sadeghian K, Kelley AE. Appetitive instrumental learning requires coincident activation of NMDA and dopamine D1 receptors within the medial prefrontal cortex. J Neurosci. 2002;22(3):1063–71. doi: 10.1523/JNEUROSCI.22-03-01063.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- 30.Tseng KY, O'Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci. 2004;24(22):5131–9. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tominaga-Yoshino K, et al. Repetitive activation of protein kinase A induces slow and persistent potentiation associated with synaptogenesis in cultured hippocampus. Neurosci Res. 2002;44(4):357–67. doi: 10.1016/s0168-0102(02)00155-4. [DOI] [PubMed] [Google Scholar]
- 32.Tominaga-Yoshino K, et al. Repetitive induction of late-phase LTP produces long-lasting synaptic enhancement accompanied by synaptogenesis in cultured hippocampal slices. Hippocampus. 2008;18(3):281–93. doi: 10.1002/hipo.20391. [DOI] [PubMed] [Google Scholar]
- 33.Oe Y, et al. Dendritic spine dynamics in synaptogenesis after repeated LTP inductions: dependence on pre-existing spine density. Sci Rep. 2013;3:1957. doi: 10.1038/srep01957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44(5):749–57. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 35.Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10(10):1038–44. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- 36.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57(1):65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 37.O'Malley A, et al. Transient spine density increases in the mid-molecular layer of hippocampal dentate gyrus accompany consolidation of a spatial learning task in the rodent. Neuroscience. 2000;99(2):229–32. doi: 10.1016/s0306-4522(00)00182-2. [DOI] [PubMed] [Google Scholar]
- 38.Robinson TE, Kolb B. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 1999;11(5):1598–604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- 39.Muhammad A, Kolb B. Maternal separation altered behavior and neuronal spine density without influencing amphetamine sensitization. Behav Brain Res. 2011;223(1):7–16. doi: 10.1016/j.bbr.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 40.Diana M, Spiga S, Acquas E. Persistent and reversible morphine withdrawal-induced morphological changes in the nucleus accumbens. Ann N Y Acad Sci. 2006;1074:446–57. doi: 10.1196/annals.1369.045. [DOI] [PubMed] [Google Scholar]
- 41.Crombag HS, et al. Opposite effects of amphetamine self-administration experience on dendritic spines in the medial and orbital prefrontal cortex. Cereb Cortex. 2005;15(3):341–8. doi: 10.1093/cercor/bhh136. [DOI] [PubMed] [Google Scholar]
- 42.Bjorndal B, et al. Different adipose depots: their role in the development of metabolic syndrome and mitochondrial response to hypolipidemic agents. J Obes. 2011;2011:490650. doi: 10.1155/2011/490650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrario CR, et al. Neural and behavioral plasticity associated with the transition from controlled to escalated cocaine use. Biol Psychiatry. 2005;58(9):751–9. doi: 10.1016/j.biopsych.2005.04.046. [DOI] [PubMed] [Google Scholar]
- 44.Bloss EB, et al. Evidence for reduced experience-dependent dendritic spine plasticity in the aging prefrontal cortex. J Neurosci. 2011;31(21):7831–9. doi: 10.1523/JNEUROSCI.0839-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nestler EJ. The neurobiology of cocaine addiction. Sci Pract Perspect. 2005;3(1):4–10. doi: 10.1151/spp05314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162(8):1403–13. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- 47.Holtmaat A, et al. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006;441(7096):979–83. doi: 10.1038/nature04783. [DOI] [PubMed] [Google Scholar]
- 48.Dumitriu D, et al. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J Neurosci. 2010;30(22):7507–15. doi: 10.1523/JNEUROSCI.6410-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rasakham K, et al. Synapse density and dendritic complexity are reduced in the prefrontal cortex following seven days of forced abstinence from cocaine self-administration. PLoS One. 2014;9(7):e102524. doi: 10.1371/journal.pone.0102524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenwood CE, Winocur G. Learning and memory impairment in rats fed a high saturated fat diet. Behav Neural Biol. 1990;53(1):74–87. doi: 10.1016/0163-1047(90)90831-p. [DOI] [PubMed] [Google Scholar]
- 51.Lee KW, et al. Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc Natl Acad Sci U S A. 2006;103(9):3399–404. doi: 10.1073/pnas.0511244103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gray EG. Electron microscopy of synaptic contacts on dendrite spines of the cerebral cortex. Nature. 1959;183(4675):1592–3. doi: 10.1038/1831592a0. [DOI] [PubMed] [Google Scholar]
- 53.Kleim JA, et al. Synaptogenesis and Fos expression in the motor cortex of the adult rat after motor skill learning. J Neurosci. 1996;16(14):4529–35. doi: 10.1523/JNEUROSCI.16-14-04529.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cornish JL, Duffy P, Kalivas PW. A role for nucleus accumbens glutamate transmission in the relapse to cocaine-seeking behavior. Neuroscience. 1999;93(4):1359–67. doi: 10.1016/s0306-4522(99)00214-6. [DOI] [PubMed] [Google Scholar]
- 55.Dong Y, et al. Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci. 2005;25(4):936–40. doi: 10.1523/JNEUROSCI.4715-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McFarland K, Kalivas PW. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2001;21(21):8655–63. doi: 10.1523/JNEUROSCI.21-21-08655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Capriles N, et al. A role for the prefrontal cortex in stress- and cocaine-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 2003;168(1-2):66–74. doi: 10.1007/s00213-002-1283-z. [DOI] [PubMed] [Google Scholar]
- 58.McLaughlin J, See RE. Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology (Berl) 2003;168(1-2):57–65. doi: 10.1007/s00213-002-1196-x. [DOI] [PubMed] [Google Scholar]
- 59.Peters J, LaLumiere RT, Kalivas PW. Infralimbic prefrontal cortex is responsible for inhibiting cocaine seeking in extinguished rats. J Neurosci. 2008;28(23):6046–53. doi: 10.1523/JNEUROSCI.1045-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.LaLumiere RT, Niehoff KE, Kalivas PW. The infralimbic cortex regulates the consolidation of extinction after cocaine self-administration. Learn Mem. 2010;17(4):168–75. doi: 10.1101/lm.1576810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bechara A. Decision making, impulse control and loss of willpower to resist drugs: a neurocognitive perspective. Nat Neurosci. 2005;8(11):1458–63. doi: 10.1038/nn1584. [DOI] [PubMed] [Google Scholar]