

Abstract
Substitutions of Arg838 in the dimerization domain of a human retinal membrane guanylyl cyclase 1 (RetGC1) linked to autosomal dominant cone-rod degeneration type 6 (CORD6) change RetGC1 regulation in vitro by Ca2+. In addition, we find that R838S substitution makes RetGC1 less sensitive to inhibition by retinal degeneration-3 protein (RD3). We selectively expressed human R838S RetGC1 in mouse rods and documented the decline in rod vision and rod survival. To verify that changes in rods were specifically caused by the CORD6 mutation, we used for comparison cones, which in the same mice did not express R838S RetGC1 from the transgenic construct. The R838S RetGC1 expression in rod outer segments reduced inhibition of cGMP production in the transgenic mouse retinas at the free calcium concentrations typical for dark-adapted rods. The transgenic mice demonstrated early-onset and rapidly progressed with age decline in visual responses from the targeted rods, in contrast to the longer lasting preservation of function in the non-targeted cones. The decline in rod function in the retina resulted from a progressive degeneration of rods between 1 and 6 months of age, with the severity and pace of the degeneration consistent with the extent to which the Ca2+ sensitivity of the retinal cGMP production was affected. Our study presents a new experimental model for exploring cellular mechanisms of the CORD6-related photoreceptor death. This mouse model provides the first direct biochemical and physiological in vivo evidence for the Arg838 substitutions in RetGC1 being the culprit behind the pathogenesis of the CORD6 congenital blindness.
Keywords: cyclic GMP (cGMP), eye, guanylate cyclase (guanylyl cyclase), photoreceptor, phototransduction, retina, retinal degeneration, vision, GCAP, calcium-binding proteins
Introduction
Retinal membrane guanylyl cyclase (RetGC)2 is one of the key enzymes in vertebrate visual signaling. Rods and cones respond to light by closing cGMP-gated plasma membrane channels in the outer segment as cGMP becomes hydrolyzed by phosphodiesterase PDE6 and then re-open the channels, after PDE6 activity becomes quenched and cGMP becomes re-synthesized by RetGC (1–5). Two RetGC isozymes expressed in photoreceptors, RetGC1 and RetGC2 (6–8), bind calcium-sensor proteins, GCAPs (7–12), which decelerate the cyclase activity at high intracellular calcium concentrations in the dark and accelerate it in the light, when the influx of calcium through the cGMP-gated channels is shut off (13–19). RetGC also binds retinal degeneration 3 (RD3) protein (20–21), which prevents cyclase activation by GCAPs (22–23) and promotes accumulation of RetGC1 in the outer segments (21, 24, 25). RetGC1 (human gene GUCY2D) accounts for most of the cGMP synthesis in mammalian photoreceptors (26, 27), therefore mutations in RetGC1 that disable the cyclase activity (28–33) cause recessive blindness at birth, Leber congenital amaurosis type 1 (LCA1) (33), mostly a non-degenerative or partially degenerative (31, 32) loss-of-function hereditary retinal disease. Different from the LCA1 type of severe congenital blindness linked to the RetGC1 gene, cone-rod dystrophy type 6 (CORD6), is a progressive early-onset loss of photoreceptors (34–35). The most frequent mutations in GUCY2D found in the CORD6 patients of various ethnic groups are substitutions of the Arg838 (34–45). Studies of the recombinant RetGC1 heterologously expressed in cultured non-photoreceptor cells (44–46) indicate that the CORD6-related substitutions in Arg838 do not inactivate the cyclase, but rather alter its regulation by calcium-sensor proteins, in particular guanylyl cyclase activating protein 1 (GCAP1). However, no experimental evidence has been available to date to demonstrate that the CORD6-linked mutations actually alter cGMP synthesis regulation in living photoreceptors of the retina and cause loss of visual function. In the present study, we describe a new transgenic model that expresses a CORD6-linked variant R838S RetGC1 in mouse photoreceptors and demonstrate that this mutation when expressed in vivo in the living retina alters the biochemistry of the cyclase regulation and disables the physiological function of the retina by causing photoreceptor death.
Results
R838S Desensitizes Negative Regulation of RetGC1 by GCAP1 and RD3
The RetGC1 encoded by the cDNA fragment intended for the transgenic expression in the retina was first tested for Ca2+ sensitivity, apparent affinity for GCAP1, and sensitivity to inhibition by RD3, by being expressed in HEK293 cells under control of a CMV promoter (46) (Fig. 1). Consistently with the original in vitro observations by Ramamurthy et al. (45), and later by Peshenko et al. (46), the half-maximal deceleration of the GCAP1-activated cyclase by calcium was shifted toward nearly 2.5-fold higher concentrations (Fig. 1A). The apparent affinity of the R838S RetGC1 for the Mg2+-liganded activator form of GCAP1 (19) became nearly 5-fold higher than normal (KMgGCAP1 327 ± 10 nm (S.D.) versus 1585 ± 80 nm in wild type) (Fig. 1B). In addition to its being the main reason for less effective deceleration by Ca2+ (44–46), observed in Fig. 1A, the higher affinity for GCAP1 proportionally correlated with a reduced sensitivity of the GCAP1-activated RetGC1 to the inhibition by RD3: the K½ RD3 rose from 2.6 ± 1.2 versus 11.4 ± 0.4 nm, respectively (p < 0.01), in the R838S RetGC1 (Fig. 1C).
FIGURE 1.
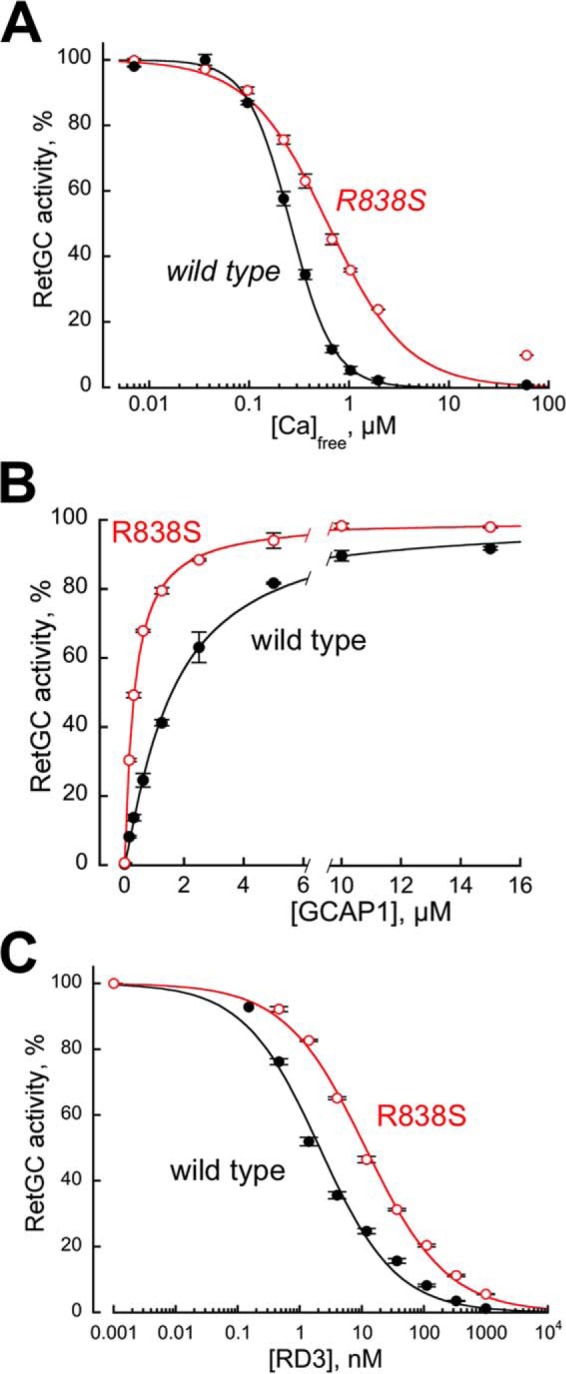
Altered biochemical characteristics of a recombinant CORD6 R838S RetGC1. RetGC1 was expressed in HEK293 cells and tested in vitro prior to making it into the transgene-expression construct. A, the calcium sensitivity of wild type (closed symbols) and R838S (R838S+, open symbols) RetGC1. The cyclase activity was assayed at varied concentrations of free Ca2+ set by Ca2+/Mg2+/EGTA buffers at a constant 0.9 mm free Mg2+. The data (mean ± S.D.) from two independent sets of experiments were normalized as the percentage of maximal activity in each cyclase preparation and fitted using the Hill function, A = (Amax)/(1 + ([Ca]/K½Ca)H), where Amax% is the respective maximal activity, [Ca] is the free Ca2+ concentration; K½Ca is the free Ca2+ concentrations causing 2-fold inhibition of the maximal activity, and H is the Hill coefficient. The respective K½Ca for wild type and R838S RetGC1 heterlogously expressed in HEK293 cells was 257 ± 14 and 610 ± 56 nm (p = 0.0051; here and further the p values are from Student's t test). B, the affinity of the R838S RetGC1 for its activator, Mg2+GCAP1, becomes drastically increased. The cyclase activity was assayed in the presence of 2 mm EGTA and saturating 10 mm MgCl2. The data were normalized in two independent experiments as %% of maximal activity in each case and fitted using the Hill function, A = Amax/(1 + ([GCAP1]/K½GCAP)−H), where [GCAP1] is the concentration of Mg2+ GCAP1, K½GCAP is the GCAP1 concentration required for the half-maximal stimulation, and H is the Hill coefficient. The K½GCAP1 for the wild type and R838S RetGC1 were 1585 ± 80 and 327 ± 10 nm, respectively (p < 0.03). C, activated R838S RetGC1 becomes more resistant to inhibition by RD3 protein. The cyclase assays were conducted in the presence of 2 mm EGTA, 10 mm MgCl2, 1.5 μm GCAP1, and varying concentrations of human recombinant RD3. The data from three independent sets of experiments were fitted using the Hill function, A = Amax/(1 + ([RD3]/K½RD3)H), where K½RD3 is the concentration of RD3 required for 50% inhibition of the RetGC1/GCAP1 complex activity. The K½RD3 for the wild type and the R838S RetGC1 were 2.6 ± 1.2 and 11.4 ± 0.4 nm, respectively (p < 0.01).
R838S RetGC1 Expressed in Mouse Rods Alters Ca2+ Sensitivity of the cGMP Synthesis in the Retina
To express the human CORD6 cyclase mutation in mouse rods, cDNA coding for the R838S RetGC1 was transferred from the expression vector used in HEK293 cells (Fig. 1) and inserted under the control of rod opsin promoter (47) in the transgene construct (Fig. 2A) injected in mouse eggs (see “Experimental Procedures” for details). Although CORD6 affects both rods and cones, we chose the promoter that is efficient only in rods to keep the non-expressing cones as a negative control group of photoreceptors in the same transgenic retinas and thus verify that any physiological changes we may observe specifically originate from the transgene-targeted photoreceptors. Founders screened for the presence of the transgenic construct (Fig. 2A) and passing the transgene to the progeny were then bred to wild type C67B6J mice for at least 4 consecutive generations to establish the R838S RetGC1 expressing lines (R838S+) 379 and 362 for subsequent detailed analysis. As will be demonstrated further in this article, the line 379 developed more gradually deterioration (arbitrarily dubbed “slower” line) and the line 362, a more severe phenotype (“faster” line). The properties of those two mouse lines will be described in that order in the following sections.
FIGURE 2.
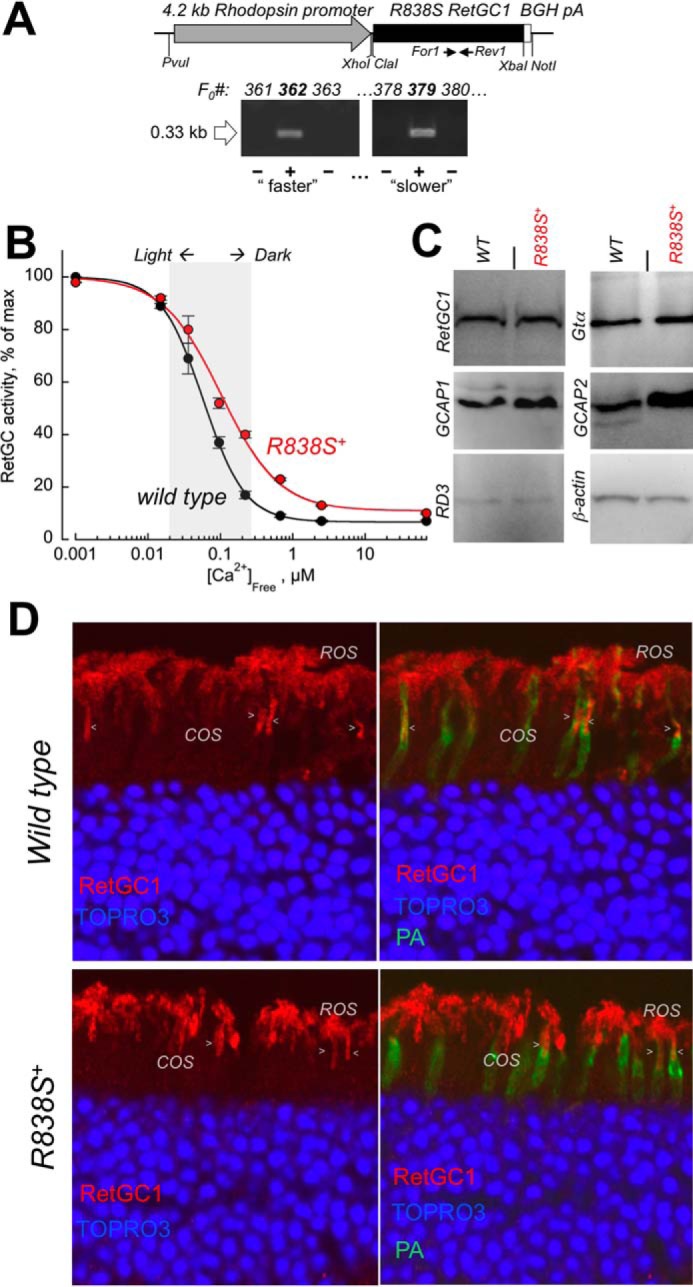
Transgenic expression of human GUCY2D-CORD6 R838S RetGC1 in vivo alters calcium sensitivity of cGMP synthesis in a mouse retina. A, schematics of the R838S RetGC1 transgene expressing construct. The 3.6-kbp R838S RetGC1 cDNA was inserted between the 4.2-kbp mouse rod opsin gene fragment harboring the promoter region and the 0.3-kb BGH polyadenylation signal-containing fragment as described under “Experimental Procedures.” The presence of the transgenic construct in the tail DNA samples from F0 mice was tested by PCR amplification of the transgene-specific 0.33-kb product, between the positions of the For1 and Rev1 primers as indicated in the upper panel and described under “Experimental Procedures.” Lines developing progressive blindness (slower in line 379 and faster in line 362) were derived from the two positive founders and analyzed as described further under “Results.” B, Ca2+ sensitivity of the cGMP synthesis rate in the dark-adapted retinas extracted from 3-week-old transgene-positive (R838S+, red symbols) and transgene-negative (wild type, black symbols) littermate retinas of the slower R838S RetGC1 line 379, assayed at the indicated free Ca2+ concentrations as described under “Experimental Procedures.” The data (mean ± S.D.) were normalized as the percent of maximal catalytic activity of the cyclase in each preparation and fitted with the Hill function, A = (Amax − Amin)/(1 + ([Ca]/K½Ca)H) + Amin, where Amax and Amin are the respective maximal and minimal activity, [Ca], free Ca2+ concentration; K½Ca, free Ca2+ concentration causing 2-fold inhibition of the cyclase activity; and H, the Hill coefficient. The average K½Ca in five independently tested transgenic mice (R838S+) increased to 110 ± 26 from 59 ± 14 nm in four wild type siblings (p < 0.03). The shaded area approximates the physiological range of the free Ca2+ concentrations in photoreceptors between light-adapted and dark-adapted states (73). C, Western immunoblotting of the retinas from 3-week-old wild type (WT) and transgene-positive (R838S+) siblings probed for RetGC1, GCAP1, GCAP2, RD3, Gtα, and β-actin. D, transgenic expression of the CORD6 RetGC1 mutant does not change the overall pattern of RetGC1 immunofluorescence in the R838S+ retina. Paraformaldehyde-fixed retina sections from 3-week-old littermates, either wild type (upper panels) or transgene-positive (bottom panels), were probed with anti-RetGC1 antibody (red) and peanut agglutinin (PA, green); the nuclei were counterstained with TOPRO3 iodide (pseudo-blue). The arrowheads indicate anti-RetGC1 fluorescence in cone outer segments (COS), below the rod outer segment (ROS) layer, identified by the surrounding PA-stained cone sheaths superimposed on the anti-RetGC1 fluorescence (right panels). The samples were processed and stained in parallel and the images were recorded in the same experiment, using identical settings for the anti-RetGC1 fluorescence imaging.
At the age of 3 weeks, RetGC activity tested in the retinas of the R838S+ mice (slower line 379) was markedly shifted outside the physiological levels of the intracellular Ca2+ when compared with the retinas from their wild type non-transgenic littermates (K½Ca 110 versus 59 nm, respectively) (Fig. 2B). However, only a very slight, if any, increase in the total RetGC1 content was detectable by Western immunoblotting in the young transgenic mice, indicating that the expression of mutant cyclase hardly exceeded 30% of the total RetGC1 levels (Fig. 2C). Proteins related to RetGC1 regulation, GCAP1, GCAP2, and RD3, were also present in the R838S+ retinas. Immunofluorescence of RetGC1 in the slower line 379 retinas, already containing differentiated rods and cones but not yet affected at that age by the subsequent massive retinal degeneration (described in the following sections), also resembled that of the wild type (Fig. 2D). The RetGC1 immunofluorescence signal was present both in rods and cones of the R838S+ retinas and the wild type, and may have appeared slightly brighter in some R838S+ rods (Fig. 2D), the difference from the wild type was by far not obvious. Although the overall expression pattern argued that there would be no obvious abnormalities in the localization of the transgenic CORD6 variant, the presence of the endogenous mouse RetGC1 proved to be a major obstacle in attempts to estimate how much of the human transgene product was expressed and where it was located.
To better estimate the level of human CORD6 RetGC1 expression and localization, we transferred the transgene expression into a genetic background lacking the endogenous mouse RetGC1, by consecutively crossing the R838S+ mice from the slower line 379 with the Gucy2e (mouse ortholog of RetGC1, Ref. 48) gene knock-out mice (49) (Fig. 3A). Semi-quantitative estimates by luminescence of the R838S RetGC1 band in Gucy2e−/−R838S+ retinas (Fig. 3A, right panel) indicated that under half the normal levels of R838S RetGC1 were sufficient to shift Ca2+ sensitivity of cGMP synthesis in the presence of the endogenous mouse RetGC1 (Fig. 2B). No less important, having interference from the endogenous RetGC1 eliminated (Fig. 3B) also made it possible to more clearly establish the R838S RetGC1 localization in Gucy2e−/−R838S+ mice by immunofluorescence staining (Fig. 3C). In contrast to the endogenous RetGC1, the transgenically expressed R838S RetGC1 was present in rods but not in cones (Fig. 3C), consistently with the expected activity of the rod opsin promoter used for its expression (47). Except for being absent in cones, the R838S RetGC1 signal in rods was similar to the wild type albeit not uniform between different rods. Most importantly, the R838S RetGC1 was found in the rod outer segments, with no indication of being retained in the inner segment (Fig. 3, C and D).
FIGURE 3.
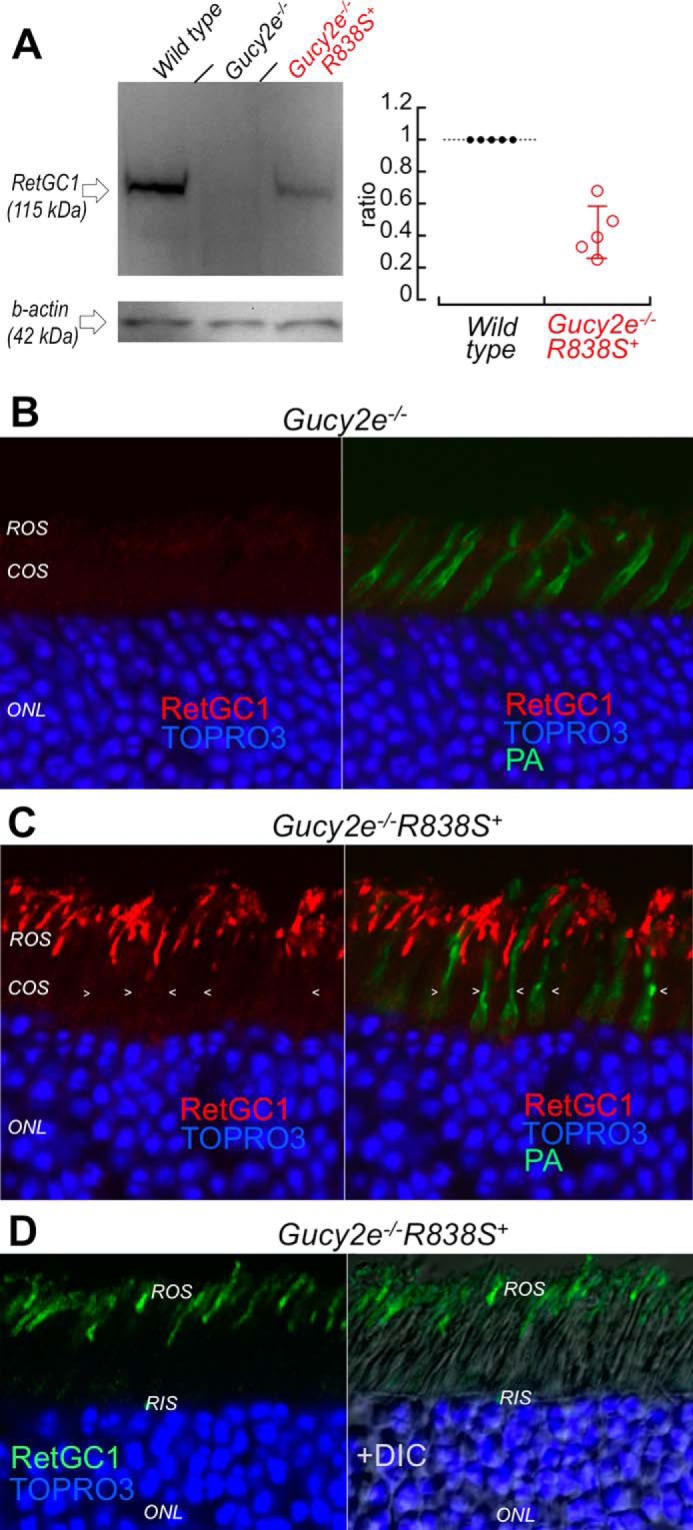
The R838S RetGC1 in the slower line 379 expressed at lower than the wild type mouse RetGC1 level localizes to rod outer segments. A, left: Western immunoblotting (left panel) of RetGC1 in 3-week-old wild type (left lane), Gucy2e−/− (middle) and the Gucy2e−/−R838S+ (right lane) mice. Right: integrated luminescence density of the R838S RetGC1 band in Gucy2e−/−R838S+ normalized by that of the endogenous mouse RetGC1 in wild type retinas on the same immunoblots semi-quantitatively estimates the transgene expression at 0.43 ± 0.16 mouse RetGC1 levels (error bar, standard deviation). B and C, R838S RetGC1 immunofluorescence in Gucy2e−/−R838S+ retinas. Paraformaldehyde-fixed retina sections from 3-week-old littermates were probed with anti-RetGC1 antibody (red) and peanut agglutinin (green fluorescence, superimposed in right panels), as described in the legend to Fig. 2D. Although RetGC1 is undetectable in Gucy2e−/− photoreceptors (panel B), the CORD6 RetGC1 appears in rods (ROS) but not in cones (COS, arrowheads) of their Gucy2e−/−R838S+ littermates (panel C). The samples in panels B and C were processed and stained in parallel and the images were recorded in the same experiment using identical settings for the anti-RetGC1 fluorescence imaging. D, the anti-RetGC1 fluorescence (green), superimposed on differential interference contrast (DIC, right panel), demonstrates that the R838S RetGC1 in the transgenic rods accumulates in the outer (ROS) rather than inner (RIS) segment.
Expression of the R838S RetGC1 Causes Progressive Loss of Visual Function in Living Mice
Rod and cone function in the transgenic animals were assessed using electroretinography (ERG) (Figs. 4 and 5). Despite the modest level of CORD6 RetGC1 expression, scotopic ERG in dark-adapted R838S+ mice of the slower line 379 demonstrated a decline in their rod responses as early as 1 month of age (Fig. 4, A and B). The average maximal scotopic a-wave (negative voltage deflection by hyperpolarization of the rod photoreceptors in response to a bright flash) was reduced in the R838S+ mice to half the normal amplitude (305 ± 81 versus 573 ± 37 μV in wild type, p < 0.0001), but the flash strength producing a half-maximal response was not changed (3.5 × 103 ± 1.5 × 103 versus 3.8 × 103 ± 1.3 × 103 R*/rod, p = 0.63) (Fig. 4B). In a similar fashion, rod-driven scotopic b-wave (positive deflection by depolarization of bipolar cells in the inner retina) was reduced in amplitude by one-third (710 ± 179 versus 1055 ± 115 μV, p < 0.0001), without a change in half-maximum flash strength (266 versus 261 R*/rod) (Fig. 4, A and B). This suggested that rod-based vision in the R838S mice was affected by reduction of the rod numbers, rather than loss of light sensitivity by individual rods.
FIGURE 4.

Deficiency in the R838S RetGC1 rod vision develops from early age. A, waveforms of dark-adapted (scotopic) ERG evoked in R838S+ (red traces) mice from the slower line 379 and their wild type littermates (black traces) by 505-nm flashes of increasing strength (top to bottom): 1.9, 6.6, 17, 133, 1.1 × 103, 4 × 103, and 1.1 × 106 R*/rod; recorded at 1, 3, or 6 months of age as indicated. B, amplitudes of scotopic a-wave (left) and b-wave (right) in 10 1-month-old R838S+ (red symbols) and 8 wild type littermates (black symbols), plotted as a function of the flash strength: 1.9, 3.3, 6.6, 10, 17, 133, 1.1 × 103, 4.3 × 103, 34 × 103 and 1.1 × 106 R*/rod. The asterisks indicate a statistically significant difference between the two genotypes at various flash strengths: p < 0.05 (*), p < 0.01 (**), and p < 0.0001 (***).
FIGURE 5.
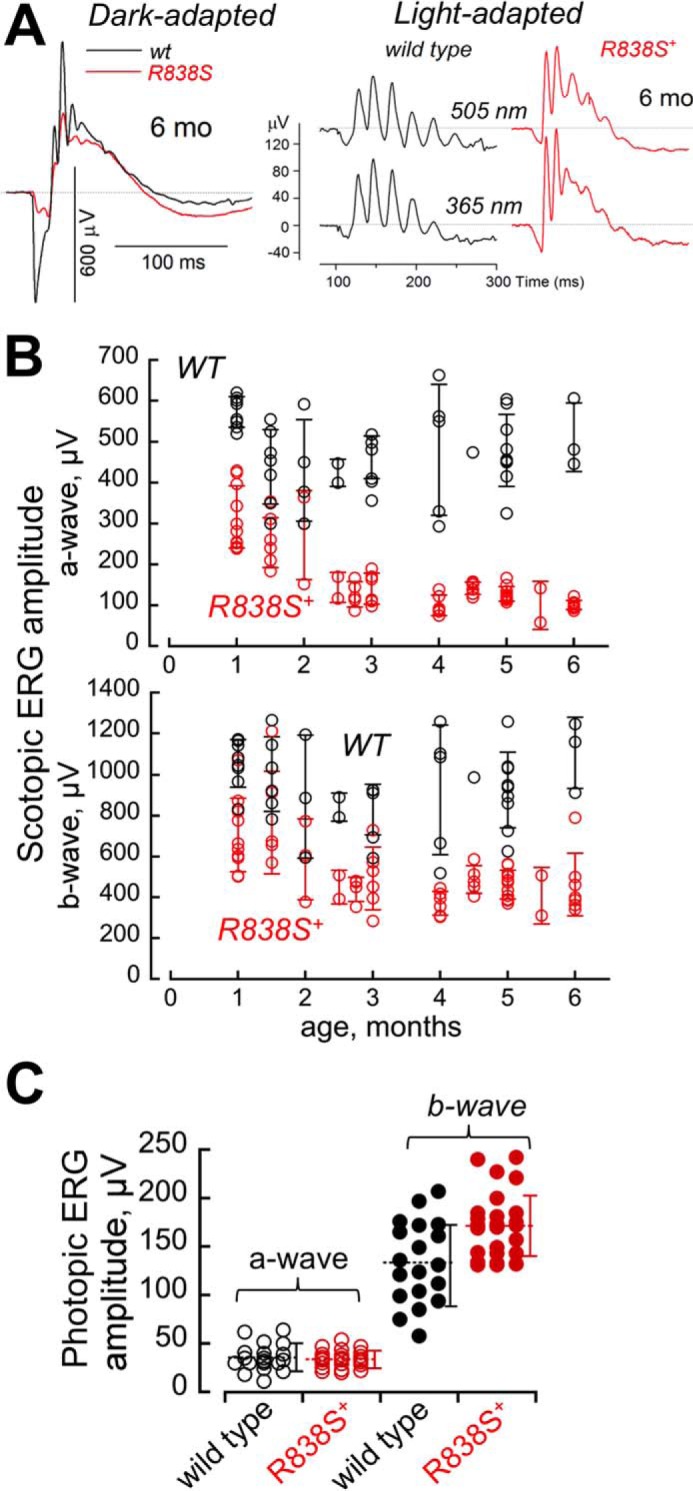
The R838S+ rod ERG in the line 379 becomes strongly reduced by 6 months of age, whereas cones remain functional. A, waveforms of ERG in response to a standard 1.1 × 103 R*/rod bright flash, recorded from dark-adapted (scotopic) wild type mice (black) and R838S+ (red) slower line 379 littermates at 6 months of age (same as in Fig. 4A), shown on the left. The waveforms of light-adapted (photopic) ERG, shown on the right, was recorded from the same pair of mice in response to 6 × 105 R*/rod 505- and 365-nm flashes in a background of a constant light producing 33 × 103 R*/rod/s. Note the remaining strong cone responses in comparison with the suppressed rod response in the R838S+ mouse. B and C, maximal amplitudes of the ERG. B, amplitudes of the dark-adapted (scotopic) a-wave (upper panel) and b-wave (lower panel) to a bright flash of 1.1 × 103 R*/rod in the R838S+ (red symbols) mice become progressively suppressed by 6 months of age compared with the wild type littermates (black symbols); each symbol shows response from different animals, error bars, standard deviation per each age group. C, cones remain responsive as rod vision becomes severely impaired. Averaged S cone a-wave (open symbols) and b-wave (closed symbols) amplitudes to a 6 × 105 R*/rod 365-nm flash at a constant 505-nm 33 × 103 R*/rod/s background light in a combined group of 25 transgenic mice aged 4 to 6 months (red symbols) was not significantly different from 20 wild type (black symbols) littermates (34 ± 9 versus 35 ± 14 μV, respectively; p = 0.7), and the b-wave amplitude even appeared slightly elevated (172 ± 34 and 132 ± 42 μV; p < 0.002); dashed horizontal line, mean average for each group; error bars, standard deviation.
The rod vision in line 379 further declined between 1 and 3 months of age (Fig. 5). For the combined age group of 3 to 6 months, the maximal scotopic a-wave amplitude was already suppressed 4-fold (121 ± 29 μV, n = 38) compared with the wild type littermates (479 ± 94 μV, n = 23; p < 0.0001), and the b-wave more than 2-fold (453 ± 109 versus 931 ± 206 μV, respectively, p < 0.0001). Reduction of the response amplitudes in the whole R838S+ group combining all tested ages (Fig. 5B) was also significant (p < 0.0001) for both a- and b-waves.
In stark contrast to the rod vision, response from both midwave (505 nm) and shortwave (365 nm) sensitive cones in light-adapted mice (photopic ERG) remained strong at the age when their rod a-wave was already reduced to rudimentary (Fig. 5, A and C). The averaged S-cone (Fig. 5C) and M-cone (data not shown) a-wave amplitudes in the R838S+ mice between 4 and 6 months of age showed neither substantial nor statistically significant reduction compared with the non-transgenic littermates. Likewise, the cone b-wave amplitude showed no evidence for reduction compared with the wild type (Fig. 5C).
The R838S RetGC1 Causes Progressive Retinal Degeneration in Transgenic Mice
The decline in rod photoreceptor function in R838S+ mice coincided with the progressive degeneration of the photoreceptor cell layer (Fig. 6). Erosion of the rod outer segments (ROS) and reduction of the total count of the photoreceptor nuclei (164 ± 27 versus 233 ± 6 per 100-μm retina section length in wild type; p = 0.043) in the outer nuclear layer (ONL) of the retina was evident in mice aged 6 weeks and rapidly progressed with age, so that by the age of 3 to 4 months less than one-half and by 6 months, only one-third of the photoreceptor nuclei remained in the retina (76 ± 17 versus 247 ± 22 per 100 μm in wild type littermates; p < 0.0001) (Fig. 6B). Because only 3% of all photoreceptors in a mouse retina are cones (50), the major reduction of the ONL nuclei count mostly reflected a rapid loss of the transgenic rods.
FIGURE 6.
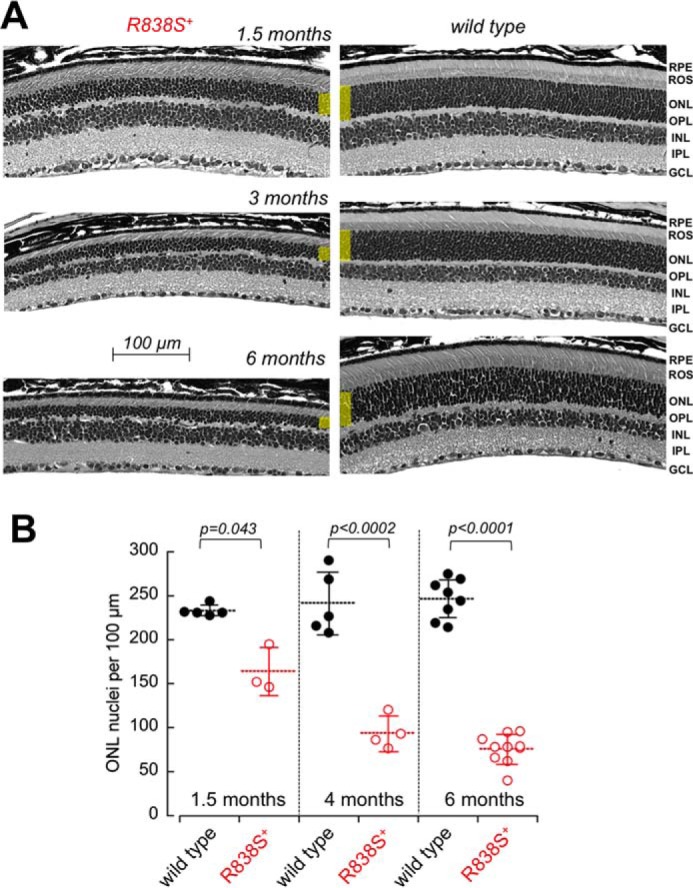
Degeneration of the photoreceptor layer in R838S+ mice. A, the cross-sections of the retinas from the slower line 379 R838S+ at 1, 3, and 6 months of age (left), shown next to their non-transgenic littermates (right). Note the deterioration of the rod morphology and the reduction in their outer nuclear layer thickness (highlighted yellow); RPE, retinal pigment epithelium; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. B, the progressive loss of average photoreceptor nuclei count per 100-μm retina length in the R838S RetGC1-positive mice (open symbols) and their wild type littermates (closed symbols); dashed horizontal bars, mean average for each group; error bars, standard deviation.
Stronger Degenerative Phenotype Accompanies a Larger Shift in Ca2+ Sensitivity of the Retinal Guanylyl Cyclase
In the transgenic retinas from the faster line 362, the right-shift in Ca2+ sensitivity of the cGMP synthesis measurable at 3 weeks of age was larger than in the slower line 379 (to K½Ca 148 nm) (Fig. 7A). Although the expression level in that line was difficult to quantify by immunoblotting due to an early onset of severe retinal degeneration in both Gucy2e+/+ and Gucy2e−/− genetic backgrounds (data not shown), based on the change in Ca2+ sensitivity the level of R838S RetGC1 expression in the line 362 was at least twice the amount expressed in the slower line 379.
FIGURE 7.
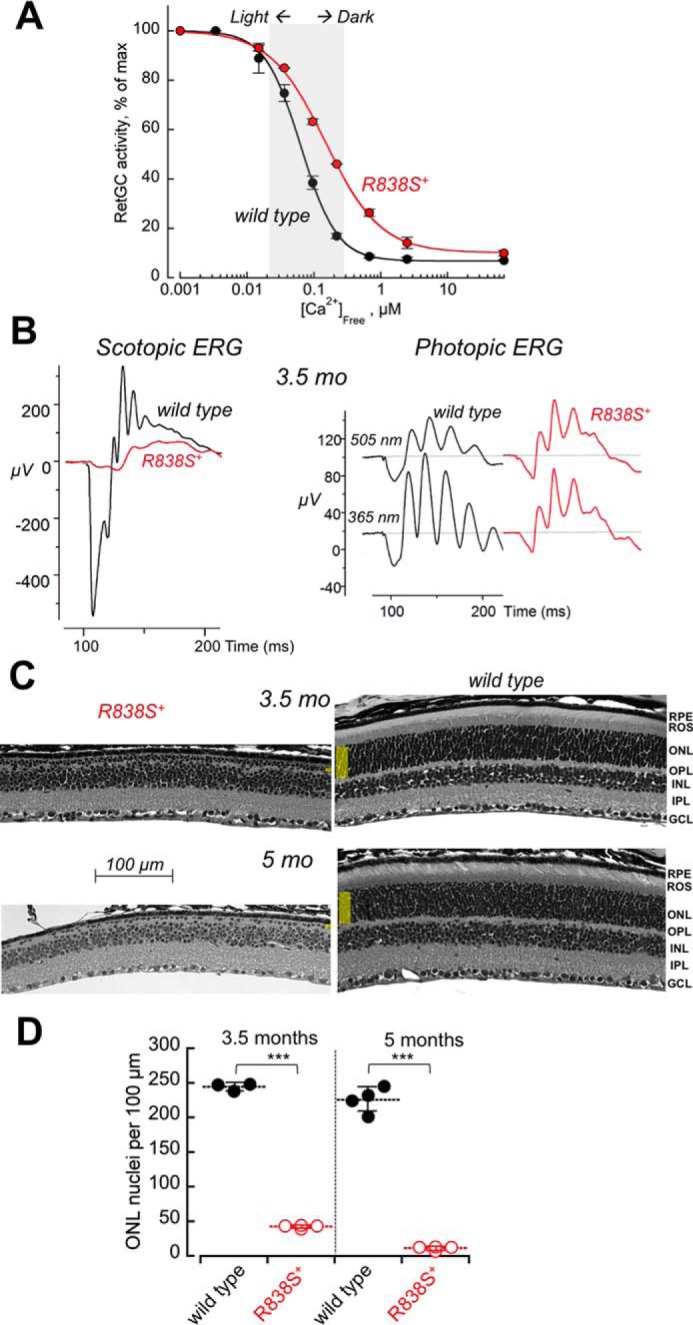
A stronger shift in Ca2+ sensitivity of RetGC1 exacerbates the degenerative phenotype in R838S RetGC1-positive mice. A, calcium sensitivity of cGMP synthesis measured in the retinas from R838S RetGC1 positive (red symbols) and negative (black symbols) littermates of the fast line 362, aged 3 weeks. The assays were conducted as described in the legend to Fig. 2B; the K½Ca from the Hill fit in two independently tested transgenic mice was 148 ± 2 nm compared with the 63 ± 10 nm in four wild type siblings (p < 0.001). B, the waveforms of scotopic (left panel) and photopic (right panel) ERG in 3.5-month-old transgenic (red traces) and wild type (black traces) littermates. C, retinal morphology in line 362 at 3.5 and 5 months of age. Note the extent of reduction of the outer nuclear layer compared with the wild type littermates. D, the loss of photoreceptors in the R838S RetGC1-positive mice (open symbols) and their wild type littermates (closed symbols); horizontal bars, mean average for each group. The average photoreceptor nuclei count per 100 μm length in the transgene-positive retinas was 42 ± 2 compared with the 244 ± 6 in wild type after 3.5 months, and 11 ± 3 versus 226 ± 18 after 5 months (p ≤ 0.0001).
The R838S+ mice from the faster line 362 demonstrated a more severe loss of rod function: the amplitude of scotopic ERG a-wave became negligible and the b-wave rudimentary in comparison with the wild type at 3.5 months of age (Fig. 7B, left panel). The residual activity evoked by the bright flash in dark-adapted animals most likely originated from cones, because both M and S cones remained functional and produced photopic ERG responses (Fig. 7B, right panel). Retinal degeneration was also more pronounced (Fig. 7C): less than 20% of photoreceptor nuclei remained in the outer nuclear layer after 3 months of age and less than 6% could be identified in the single remaining incomplete row of the nuclei after 5 months (Fig. 7D).
Discussion
The Arg838 Mutation in RetGC1 Alters Physiology of Photoreceptors and Triggers Degeneration in a Living Retina
CORD6 is typically diagnosed as an early-onset blinding disorder, frequently by 7 years of age, first as impaired central and color vision (cone function), later involving rods, by progressing to the peripheral visual field with age (34, 35). Among the genes linked to CORD6 in multiple genetic studies (34, 45, 51, 52), GUCY2D coding for a human RetGC1 has been found most frequently affected by substitutions in codon 838 replacing Arg838 with Cys, Gly, His, Pro, or Ser. Three of these substitutions, Cys838, His838 and Ser838, were shown to cause a prominent shift in Ca2+ sensitivity of the cyclase regulation by GCAP1 when heterologously expressed in cultured HEK293 (44–46). However, the physiological effects of the Arg838 substitutions in RetGC1 have not been demonstrated in mammalian photoreceptors. One attempt to heterologously express the CORD6 RetGC1 variant in vivo using zebrafish larvae documented changes in cone morphology detectable in the central retina, but did not impair visual responses in the fish (53).
Our present study provides the first direct biochemical and physiological evidence that a CORD6-related mutation alters photoreceptor physiology in a manner consistent with the in vitro studies of heterologously expressed Arg838 mutants of RetGC1 (44–46). We demonstrate that R838S RetGC1 shifts Ca2+ sensitivity of the cyclase regulation in living rods of the mouse retina and also, by triggering degeneration of photoreceptors, causes early-onset progressive blindness, even at the levels of expression lower than the endogenous RetGC1 (Fig. 3). The photoreceptor death caused by the R838S RetGC1 (Figs. 5–7) in our studies is a striking contrast to the restoration of vision by transgenic overexpression of wild type RetGC1, rescuing rods and cones lacking functional guanylyl cyclase (54–57).
Notably, the effect of R838S RetGC1 is specific for the targeted photoreceptors, because the non-targeted cones remain functional even at times when severe degeneration of the transgenic rods has already occurred and most of the rod visual function has already become impaired (Figs. 5 and 7). Although half of the R838S+ rods in the slower line 379 died by the age of 3 months, the degeneration did not progress to eliminate all remaining rods within the following three months. It is likely that transgene expression under the rhodopsin promoter in that line was not uniform among all rods (47) and those of them who expressed the CORD6 cyclase died out more quickly, whereas those with lower levels of expression sustained lesser damage and lingered for much longer. In the faster line 362, where the average transgene expression was higher, more strongly affecting Ca2+ sensitivity of cGMP production, all but a few photoreceptors degenerated within 6 months (Fig. 7). The massive death of rods in the line 362 eventually provoked the secondary loss of cones as well, because after 5 months virtually no ERG responses were detected (data not shown).
Relevance and Limitations for the Mouse Model in Studying GUCY2D CORD6 Mechanisms
CORD6 is a dominant congenital disease diagnosed at a young age as a progressive loss of cone and then rod function (34–38). Consistently with the dominant character of the disease, heterozygous CORD6 RetGC1 transgenes in our model alter physiology and trigger degeneration of rods, whereas their non-transgenic siblings retain visual function and normal retinal morphology at all tested ages. The loss of the rod function in transgenic mice becomes clearly detectable after 1 month (Fig. 4) and progresses with age, which is also generally consistent with the clinical manifestation of CORD6 in humans. At the same time, the design of our model does not mimic all clinical symptoms of the human CORD6. Unlike in human patients, cones do not express the mutated cyclase in our model, by its design, and therefore remain functional for much longer than rods. However, we reason that this mouse model can be adequately used for studying photoreceptor death related to CORD6. Rods account for nearly 97% of all photoreceptors in a mouse retina (50), therefore, the vast majority of photoreceptors undergoing physiological changes and apoptotic death caused by the CORD6 RetGC1 constitute a fair model in further studies of cellular mechanisms of CORD6 blindness and testing potential therapy. Having easily been distinguishable by their electrophysiology cones as a small non-diseased photoreceptor population should be beneficial, because it provides an additional means to assess nonspecific side effects of the potential therapy applications.
GUCY2D CORD6 presents an even bigger challenge for gene therapy than recessive GUCY2D LCA1. In animal models for the LCA1, vision can be sustainably restored by expression of a normal RetGC1 allele in dysfunctional rods and cones, when delivered via adeno-associated virus (54–57). Consequently, such gene therapy is expected to be perspective in the treatment of a human GUCY2D LCA1 (31). In contrast, to suppress expression of a dominant disease-causing GUCY2D allele in CORD6, more complex approaches would likely be required, such as CRISPR-mediated destruction of the CORD6 allele or suppression of the CORD6-specific transcripts by short interference RNA. Development of these approaches in treating CORD6 would require rigorous evaluation of their efficiency and specificity. For these types of studies, the availability of a GUCY2D CORD6 mouse model would be highly beneficial, provided that such a model is comprehensively characterized in-depth and the mechanisms of photoreceptor degeneration are fully understood.
What Processes Downstream of the Mutated CORD6 RetGC1 Cause Photoreceptor Degeneration?
Although our study decisively demonstrates that the mutation of Arg838 in RetGC1 causes blindness in vivo, the pathway(s) leading to photoreceptor death remains to be delineated. At present, several non-mutually exclusive scenarios that could, separately or by acting in concert, cause the photoreceptor death need to be considered as follows.
Deregulation of RetGC Activity in the Dark
Negative Ca2+ feedback regulates RetGC1 via Ca2+-sensor proteins (1, 5, 10), predominantly by GCAP1 (58, 59). The biochemical studies of CORD6 mutations in ReGC1 pioneered by J. Hurley's group (44, 45) demonstrated that Arg838 substitutions alter Ca2+ sensitivity of RetGC regulation by affecting coiled-coil interactions in the cyclase dimerization domain. Although RetGC1 is not a calcium-sensitive protein, the dimerization domain is part of the GCAP1 and GCAP2 binding interface (59–61), therefore, change in its structure unbalances RetGC1 affinities for two competing with each forms of GCAP1, Mg2+-liganded (activator) and Ca2+-liganded (inhibitor) (46). Because the mutant cyclase binds the activator form of GCAP1 with higher affinity (44–46), it now takes higher Ca2+ concentrations, converting a larger fraction of GCAP1 into the inhibitor state, to outcompete the activator form of GCAP1 and decelerate the cyclase (46).
Deregulation of RetGC1 is the main reason for photoreceptor death in mouse models expressing Y99C or E155G GCAP1 mutants (58, 62, 63). When RetGC1 deceleration by Ca2+ in the dark becomes less sensitive, a larger fraction of cGMP-gated channels in the plasma membrane maintains Ca2+ influx in the dark, eventually provoking the onset of apoptosis (62, 63). Stimulation of cGMP-dependent protein kinase activity can also lead to mouse photoreceptor death independently of the Ca2+ pathway (64, 65). In our present study, faster rod degeneration accompanied a larger shift in Ca2+ sensitivity of the cGMP synthesis in the retina (Fig. 7), and this supports RetGC1 deregulation as the most probable cause of GUCY2D CORD6. However, the extent to which the cyclase reduces its sensitivity in the R838S+ retinas (Figs. 2B and 7A) appears less drastic as in the mouse models expressing Y99C or E155G GCAP1 (58, 63). Therefore, it would be premature at this point to conclude that the change in Ca2+ sensitivity of the CORD6 cyclase alone is fully sufficient to cause photoreceptor death in our experimental model.
Reduced Affinity for RD3
One should also consider for future studies a new intriguing observation, that the R838S mutation results in a much weaker affinity of the GCAP1-activated RetGC1 for RD3 (Fig. 1C). RD3 evidently plays a dual functional role in photoreceptors (66): it enhances the efficiency of RetGC expression and accumulation in rod and cone outer segments and blocks RetGC1 activity, most probably up until the cyclase reaches the outer segment. The lack of RD3 causes retinal degeneration in the affected humans and in the rd3/rd3 mouse strain (20). Evidently, both proposed functions of RD3 contribute to photoreceptor survival, because photoreceptors in mice lacking RD3 degenerate even faster than in mice completely devoid of both RetGC1 and RetGC2 (23). We find that R838S RetGC1 reaches the outer segment in a seemingly normal fashion (Fig. 3), however, the reduced affinity of the activated RetGC1 for RD3 (Fig. 1C) would undoubtedly affect the proposed function of RD3 to transiently block premature cyclase activation in the inner segment (22, 66).
Unfolded Protein Response
Mutations in some proteins have shown propensity to provoke photoreceptor death by causing endoplasmic reticulum (ER) stress triggering apoptotic unfolded protein response (65, 67). We find that the CORD6 mutant of RetGC1 is functional in vivo (Figs. 2 and 6) and properly accumulates in the rod outer segments (Fig. 3, C and D) rather than the inner segments where the ER is located. Therefore, the unfolded protein response scenario seems less likely for the GUCY2D CORD6. Yet it cannot be completely ruled out, because RetGC1 is a membrane protein and therefore could produce ER stress if some of it is improperly incorporates in the ER membranes.
Further studies will determine which of these scenarios take place in the diseased photoreceptors. Detailed characterization of the model will need to address the potential impact of the R838S RetGC1 on single-cell electrophysiology and/or other cGMP-based signaling pathways potentially altered by the CORD6-linked mutation.
Experimental Procedures
Animals
All animal work was conducted in accordance with the Public Health Service guidelines and approved by the Institutional Animal Care and Use Committee. The wild type C57B6 strain was purchased from JAX Research/Jacksons Laboratories. The mouse Gucy2e knock-out line (49), from Dr. David Garbers laboratory (University of Texas), was maintained heterozygous, by crossing to the C57B6 for 10 generations, before breeding into a homozygous Gucy2e−/− state. Mice of a mixed SJL/C57B6 background were used to produce founders for the R838S RetGC1 transgenic mice. Both male and female mice were used in experiments.
Transgenic Mice
The expression construct was assembled in a Stratagene pBlueScript plasmid as follows. Bovine growth hormone (BGH) gene 0.3-kb fragment containing the polyadenylation signal was PCR-amplified from Invitrogen pRCCMV-based plasmid harboring wild type RetGC1 cDNA from previously published studies (46) using a forward 5′-GTGGGTGAAATAAAGGCATACTGTCTTCC and a reverse 5′-AAAAAGCGGCCGCCAGCTGGTTCTTTCCGCCTCAGA primer and inserted into the XbaI/NotI sites of pBlueScript, yielding the intermediate construct “pBlue-BGH.” A 4.2-kb KpnI/XhoI rod opsin gene promoter-containing gene fragment (47) was transferred from the pRho-mp3 construct (14, 47) into the KpnI/XhoI sites of the pBlue-BGH construct, thus yielding intermediate construct “pRho-BGH” containing both rod opsin promoter and the BGH polyadenylation signal fragment. A HindIII/XbaI RetGC1 cDNA fragment including the ribosome-recognition motif and the translation initiation codon, were subcloned from the pRCCMV construct (46, 68) into the HindIII/XbaI restriction sites of pBlueScript, excised using the ClaI/XbaI digest, and inserted into the ClaI/XbaI sites of the pRho-BGH vector. To generate the final construct, pRho-R838S-BGH, the BssHII/XbaI fragment of the RetGC1 cDNA was replaced by that containing the 2512C → A transversion coding for the R838S substitution, excised from the construct (46) previously used for the R838S RetGC1 expression in HEK293 cells (Fig. 1). The pRho-R838S-BGH construct (Fig. 2A) was verified by restriction nuclease digest and then by DNA sequencing on both strands. A 8.3-kb PvuI/NotI fragment excised from the pRho-R838S-BGH construct and purified from the agarose gel was injected in fertilized mouse egg pronuclei (service of the University of Michigan Transgenic Animal Model Core). The tail DNA samples from the F0 founders and later of their progeny from a series of consecutive breeding to C57B6J mice were tested for the presence of the transgene by PCR using a forward primer (For1), 5′-CGATCCAAACAACATCTGCGGT, and a reverse primer (Rev1), 5′-CAGCCAAACCCTGTCTCCCTCAT, amplifying a 0.33-kb transgene-specific region (Fig. 2A). Three lines were initially established from the founders designated by numbers 362, 377, and 379, but the phenotype of the line 377 in the initial testing (not shown) was similar to that of line 379, and therefore only lines 379 and 362 were selected for the subsequent detailed analysis described under “Results.” After consecutive breeding of the transgene-positive mice to C57B6J, the comparative study was conducted on the R838S+ mice from the lines 379 and 362 using their transgene-negative littermates as the wild type control.
Guanylyl Cyclase Assays
RetGC1 activity was assayed as previously described in detail (26, 69). Briefly, the typical assay mixture contained in 25 μl of 30 mm MOPS-KOH (pH 7.2), 60 mm KCl, 4 mm NaCl, 1 mm DTT, 2 mm Ca2+/EGTA buffer, MgCl2 added to maintain 0.9 mm free Mg2+ (unless indicated otherwise), purified GCAP1, and RD3 at concentrations indicated in the pertinent figure legends, 0.3 mm ATP, 4 mm cGMP, 1 mm GTP, 10 mm creatine phosphate, 0.5 unit of creatine phosphokinase (Sigma), 1 μCi of [α-32P]GTP, 0.1 μCi of [8-3H]cGMP (PerkinElmer Life Sciences), phosphodiesterase inhibitors zaprinast and dipyridamole, and variable concentrations of GCAPs, RD3, and Ca2+. Membrane fractions from transfected HEK293 cell cultures expressing RetGC1 under control of the CMV promoter as previously described in detail (70) were assayed under ambient illumination. The reaction was incubated for 40 min at 30 °C and quenched by heating at 95 °C for 2 min. Mouse retinas for RetGC activity measurements were excised from dark-adapted 3-week-old mice using infrared illumination and a dissecting microscope fitted with an Excalibur infrared goggles as described (26), wrapped in aluminum foil, frozen in liquid N2, and stored at −70 °C prior to their use in cyclase activity assays. The assays containing retinal samples were conducted under infrared illumination using Kodak number 11 infrared filters and Excalibur infrared binocular goggles, and the incubation time for the reaction was 12 min. The [32P]cGMP product of the reaction was separated from the substrate using thin-layer chromatography on fluorescent-back polyethyleneimine cellulose plates (Merk), eluted in 2 m LiCl solution, and quantified by liquid scintillation counting using ScintiSafe scintillation mixture (Fisher Scientific) containing 20% ethanol. Ca2+/EGTA buffers maintaining variable free Ca2+ concentrations at physiological for the photoreceptors (71) 0.9 mm free Mg2+ were prepared using the Tsien and Pozzan method (72) and verified by fluorescent indicator dyes as previously described (73).
ERG
Mice were dark-adapted overnight, their pupils were dilated by 1% tropicamide and 2.5% phenylephrine ophthalmic eye drops under dim red safelight illumination, and the mice were dark-adapted for another 15 min. Full-field electroretinography was performed in the dark using a Maxwellian view projection-based Phoenix Research Laboratories Ganzfeld ERG2 setup. Mice were anesthetized by inhalation of 1.7–1.9% isoflurane (VEDCO), delivered by a Kent Scientific SomnoSuite Small Animal Anesthesia System at the flow rate of 50 ml/min, and remained on a heated pad during the entire procedure to maintain body temperature. To evoke scotopic ERG responses, 0.1–1-ms 505-nm light pulses ranging between 1.9 and 1.1 × 106 photoisomerizations (R*) per rod at the mouse retinal surface were delivered through a infrared camera-guided corneal electrode/LED light source in 1-s to 3-min intervals for different flash strengths, and typically 3–10 traces for each flash strength series were averaged. Constant 505-nm illumination, equivalent of 33 × 103 R*/rod/s, was used as a background light to record photopic (cone) ERG responses (74) to 1-ms 505- and 365-nm pulses, equivalent of 5.8 × 105 R*/rod, delivered at 1-s intervals; traces were averaged from 15 to 20 trials for each animal.
GCAP1 and RD3
Myristoylated bovine GCAP1 D6S was expressed from a Novagen pET11d vector in BLR(DE3) Escherichia coli strain harboring N-myristoyl transferase and purified to ∼95% electrophoretic homogeneity from inclusion bodies using urea extraction, hydrophobic, and size exclusion chromatography as described previously in detail (75, 76). Human RD3 was expressed from pET11d vector in BL21(DE3) CodonPlus E. coli strain (Stratagene/Agilent Technologies), extracted from the inclusion bodies and purified to ∼95% electrophoretic homogeneity as previously described in detail (22, 23).
Retinal Morphology
Mice were anesthetized with a lethal dose of ketamine/xylazine injection, perfused through the heart with phosphate-buffered saline (PBS), and then with 2.5% glutaraldehyde in PBS. The eyes were surgically removed and fixed overnight in 2.5% glutaraldehyde, 2.5% formaldehyde, PBS solution (Electron Microscopy Sciences) at 4 °C. The fixed eyes were washed three times for 15 min each in PBS, soaked in PBS overnight, processed for paraffin embedding, sectioned, and stained with hematoxylin/eosin (AML Laboratories, Saint Augustine, FL). The retinas were photographed using an Olympus Magnafire camera mounted on an Olympus BX21 microscope. The photoreceptor nuclei in the outer nuclear layer of the retina were counted from the 425-μm fragment of the 5-μm thick retina section, between the optic nerve and the periphery, and the density of the nuclei per 100 μm were averaged from several frames for each retina. The average nuclei counts were then typically compared using 3–5 retinas of different genotypes.
Immunofluorescence
Mice were perfused with the fixative solution as described above except that 10% freshly prepared formaldehyde solution in PBS was used instead of glutaraldehyde. The eyes were then dissected and the eyecups were placed into 10% formaldehyde/PBS for 5 min and then 5% formaldehyde/PBS for 1 h at room temperature. The fixed eyecups were impregnated with 30% sucrose solution in PBS for cryoprotection, placed in OCT medium (Electron Microscopy Sciences), and frozen at −70 °C. The cryosections were taken using a Hacker-Bright cryogenic microtome, dried for 1 h at room temperature, and stored in −70 °C. The sections were washed three times in PBS containing 0.1 m glycine (pH 7.4), blocked for 1 h at 30 °C with the same solution containing 5% bovine serum albumin and 0.1% Triton X-100, incubated overnight at 4 °C, and then 1 h at room temperature with anti-RetGC1 rabbit polyclonal antibody against catalytic domain of RetGC1 (68), washed with PBS solution three times for 15 min each, incubated with the 1:400 diluted donkey anti-rabbit Alexa Fluor 568- or Alexa Fluor 488-conjugated antibody (Molecular Probes), and washed four times with PBS for 15 min each at room temperature. Where indicated, fluorescein-labeled peanut agglutinin (Vector Laboratories) was added with the secondary antibody to label cone sheaths, and the sections were covered with TOPRO3 iodide-containing Vectashield mounting medium (Vector Laboratories) to counterstain the nuclei. Confocal images were acquired using an Olympus FV1000 Spectral instrument controlled by FluoView FV10-ASW software, collecting in a sequential mode the images of emission excited by 488-, 543-, and 635-nm lasers, assigned as, respectively, green, red, and pseudo-blue color channels. Where indicated, fluorescence was superimposed on a differential interference contrast image. No changes were made to the original images except for minor gamma correction applied to the whole image for more clear presentation in the print.
Immunoblotting
RD3 polyclonal antibody 10929 was produced in rabbit (22); GCAP1, GCAP2, and RetGC1 rabbit polyclonal antibodies were reported previously (17, 68), Gtα antibody was purchased from Santa Cruz Biotechnology and β-actin antibody from GeneTex. Retinas from 3-week-old mice were excised in 50 μl of 10 mm Tris-HCl (pH 7.4), containing 100 μm phenylmethylsulfonyl fluoride and 10 μg/ml of leupeptin, homogenized in 250 μl of Laemmli SDS-containing sample buffer (Sigma), and subjected to electrophoresis in a 7% polyacrylamide gel containing 0.1% SDS. The volume of the retinal material in R838S RetGC1 versus wild type was corrected for a small decrease in photoreceptor nuclei count at that age. Following the electrophoresis, the proteins were transferred to iBlot PVDF mini stack cassette (Novex) for 22 min using a Invitrogen/Life Sciences iBlot instrument. The membranes were transiently stained by Ponceau S (Sigma) dye solution in 1% acetic acid to mark the positions of molecular mass markers, de-stained by a series of washes in water and Tris-buffered saline (Fisher Scientific) containing 0.5% Tween 20 (TTBS), blocked by Super Block (ThermoFisher) solution in TTBS, probed by rabbit polyclonal anti-RetGC1 antibody, and the luminescence was developed using peroxidase-conjugated goat anti-rabbit polyclonal IgG (Cappel/MP Biomedical) and a Pierce SuperSignal Femto substrate protocol (Thermo Scientific). The images were acquired using a Fotodyne Luminous FX imager and subjected to densitometry using National Institutes of Health ImageJ software.
Statistics
The data are mean average ± S.D. Statistical significance of the differences was evaluated by unpaired/unequal variance Student's t test using Synergy Kaleidagraph 4 software.
Author Contributions
A. M. D. conceived and coordinated the study and wrote the paper, A. M. D. and E. V. O. produced the animal models, A. M. D. recorded ERG, E. V. O. performed morphological and immunofluorescence studies, I. V. P. performed guanylyl cyclase assays, I. V. P. and A. M. D. analyzed the data and prepared figures for publication.
This work was supported, in whole or in part, by National Institutes of Health Grant EY11522 from the NEI and a CURE grant from Pennsylvania Department of Health. The authors declare that they have no conflicts of interest with the content of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- RetGC
- retinal membrane guanylyl cyclase
- GCAP
- guanylyl cyclase activating protein
- CORD6
- cone-rod dystrophy type 6
- ERG
- electroretinography
- RD3
- retinal degeneration 3 protein
- ROS
- rod outer segments
- ONL
- outer nuclear layer
- ER
- endoplasmic reticulum
- BGH
- Bovine growth hormone
- LCA1
- Leber congenital amaurosis type 1.
References
- 1. Koch K. W., and Dell'Orco D. (2015) Protein and signaling networks in vertebrate photoreceptor cells. Front. Mol. Neurosci. 8, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arshavsky V. Y., and Burns M. E. (2012) Photoreceptor signaling: supporting vision across a wide range of light intensities. J. Biol. Chem. 287, 1620–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yau K. W., and Hardie R. C. (2009) Phototransduction motifs and variations. Cell 139, 246–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arshavsky V. Y., Lamb T. D., and Pugh E. N. Jr. (2002) G proteins and phototransduction. Annu. Rev. Physiol. 64, 153–187 [DOI] [PubMed] [Google Scholar]
- 5. Pugh E. N. Jr., Nikonov S., and Lamb T. D. (1999) Molecular mechanisms of vertebrate photoreceptor light adaptation. Curr. Opin. Neurobiol. 9, 410–418 [DOI] [PubMed] [Google Scholar]
- 6. Yang R. B., Foster D. C., Garbers D. L., and Fülle H. J. (1995) Two membrane forms of guanylyl cyclase found in the eye. Proc. Natl. Acad. Sci. U.S.A. 92, 602–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dizhoor A. M., Lowe D. G., Olshevskaya E. V., Laura R. P., and Hurley J. B. (1994) The human photoreceptor membrane guanylyl cyclase, RetGC, is present in outer segments and is regulated by calcium and a soluble activator. Neuron 12, 1345–1352 [DOI] [PubMed] [Google Scholar]
- 8. Lowe D. G., Dizhoor A. M., Liu K., Gu Q., Spencer M., Laura R., Lu L., and Hurley J. B. (1995) Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc. Natl. Acad. Sci. U.S.A. 92, 5535–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Palczewski K., Subbaraya I., Gorczyca W. A., Helekar B. S., Ruiz C. C., Ohguro H., Huang J., Zhao X., Crabb J. W., Johnson R. S., Walsh K. A., Gray-Keller M. P., Detwiller P. B., and Baehr W. (1994) Molecular cloning and characterization of retinal photoreceptor guanylyl cyclase-activating protein. Neuron 13, 395–404 [DOI] [PubMed] [Google Scholar]
- 10. Dizhoor A. M., Olshevskaya E. V., Henzel W. J., Wong S. C., Stults J. T., Ankoudinova I., and Hurley J. B. (1995) Cloning, sequencing, and expression of a 24-kDa Ca2+-binding protein activating photoreceptor guanylyl cyclase. J. Biol. Chem. 270, 25200–25206 [DOI] [PubMed] [Google Scholar]
- 11. Imanishi Y., Yang L., Sokal I., Filipek S., Palczewski K., and Baehr W. (2004) Diversity of guanylate cyclase-activating proteins (GCAPs) in teleost fish: characterization of three novel GCAPs (GCAP4, GCAP5, GCAP7) from zebrafish (Danio rerio) and prediction of eight GCAPs (GCAP1–8) in pufferfish (Fugu rubripes). J. Mol. Evol. 59, 204–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scholten A., and Koch K.-W. (2011) Differential calcium signaling by cone specific guanylate cyclase-activating proteins from the zebrafish retina. PLoS ONE 6, e23117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koch K. W., and Stryer L. (1988) Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature 334, 64–66 [DOI] [PubMed] [Google Scholar]
- 14. Mendez A., Burns M. E., Sokal I., Dizhoor A. M., Baehr W., Palczewski K., Baylor D. A., and Chen J. (2001) Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc. Natl. Acad. Sci. U.S.A. 98, 9948–9953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burns M. E., Mendez A., Chen J., and Baylor D. A. (2002) Dynamics of cyclic GMP synthesis in retinal rods. Neuron 36, 81–91 [DOI] [PubMed] [Google Scholar]
- 16. Sakurai K., Chen J., and Kefalov V. J. (2011) Role of guanylyl cyclase modulation in mouse cone phototransduction. J. Neurosci. 31, 7991–8000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Makino C. L., Peshenko I. V., Wen X. H., Olshevskaya E. V., Barrett R., and Dizhoor A. M. (2008) A role for GCAP2 in regulating the photoresponse: guanylyl cyclase activation and rod electrophysiology in GUCA1B knock-out mice. J. Biol. Chem. 283, 29135–29143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Makino C. L., Wen X. H., Olshevskaya E. V., Peshenko I. V., Savchenko A. B., and Dizhoor A. M. (2012) Enzymatic relay mechanism stimulates cyclic GMP synthesis in rod photoresponse: biochemical and physiological study in guanylyl cyclase activating protein 1 knockout mice. PLoS ONE 7, e47637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dizhoor A. M., Olshevskaya E. V., and Peshenko I. V. (2010) Mg2+/Ca2+ cation binding cycle of guanylyl cyclase activating proteins (GCAPs): role in regulation of photoreceptor guanylyl cyclase. Mol. Cell Biochem. 334, 117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Friedman J. S., Chang B., Kannabiran C., Chakarova C., Singh H. P., Jalali S., Hawes N. L., Branham K., Othman M., Filippova E., Thompson D. A., Webster A. R., Andréasson S., Jacobson S. G., Bhattacharya S. S., et al. (2006) Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am. J. Hum. Genet. 79, 1059–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Azadi S., Molday L. L., and Molday R. S. (2010) RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc. Natl. Acad. Sci. U.S.A. 107, 21158–21163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peshenko I. V., Olshevskaya E. V., Azadi S., Molday L. L., Molday R. S., and Dizhoor A. M. (2011) Retinal degeneration 3 (RD3) protein inhibits catalytic activity of retinal membrane guanylyl cyclase (RetGC) and its stimulation by activating proteins. Biochemistry 50, 9511–9519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2016) Functional study and mapping sites for interaction with the target enzyme in retinal degeneration 3 (RD3) protein. J. Biol. Chem. 291, 19713–19723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Molday L. L., Djajadi H., Yan P., Szczygiel L., Boye S. L., Chiodo V. A., Gregory-Evans K., Sarunic M. V., Hauswirth W. W., and Molday R. S. (2013) RD3 gene delivery restores guanylate cyclase localization and rescues photoreceptors in the Rd3 mouse model of Leber congenital amaurosis 12. Hum. Mol. Genet. 22, 3894–3905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zulliger R., Naash M. I., Rajala R. V., Molday R. S., and Azadi S. (2015) Impaired association of retinal degeneration-3 with guanylate cyclase-1 and guanylate cyclase-activating protein-1 leads to Leber congenital amaurosis-1. J. Biol. Chem. 290, 3488–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peshenko I. V., Olshevskaya E. V., Savchenko A. B., Karan S., Palczewski K., Baehr W., and Dizhoor A. M. (2011) Enzymatic properties and regulation of the native isozymes of retinal membrane guanylyl cyclase (RetGC) from mouse photoreceptors. Biochemistry 50, 5590–5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Helten A., Säftel W., and Koch K. W. (2007) Expression level and activity profile of membrane bound guanylate cyclase type 2 in rod outer segments. J. Neurochem. 103, 1439–1446 [DOI] [PubMed] [Google Scholar]
- 28. Perrault I., Rozet J. M., Calvas P., Gerber S., Camuzat A., Dollfus H., Châtelin S., Souied E., Ghazi I., Leowski C., et al. (1996) Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat. Genet. 14, 461–464 [DOI] [PubMed] [Google Scholar]
- 29. Perrault I., Rozet J. M., Gerber S., Ghazi I., Ducroq D., Souied E., Leowski C., Bonnemaison M., Dufier J. L., Munnich A., and Kaplan J. (2000) Spectrum of retGC1 mutations in Leber's congenital amaurosis. Eur. J. Hum. Genet. 8, 578–582 [DOI] [PubMed] [Google Scholar]
- 30. Peshenko I. V., Olshevskaya E. V., Yao S., Ezzeldin H. H., Pittler S. J., and Dizhoor A. M. (2010) Activation of retinal guanylyl cyclase RetGC1 by GCAP1: stoichiometry of binding and effect of new LCA-related mutations. Biochemistry 49, 709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jacobson S. G., Cideciyan A. V., Peshenko I. V., Sumaroka A., Olshevskaya E. V., Cao L., Schwartz S. B., Roman A. J., Olivares M. B., Sadigh S., Yau K. W., Heon E., Stone E. M., and Dizhoor A. M. (2013) Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum. Mol. Genet. 22, 168–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Milam A. H., Barakat M. R., Gupta N., Rose L., Aleman T. S., Pianta M. J., Cideciyan A. V., Sheffield V. C., Stone E. M., and Jacobson S. G. (2003) Clinicopathologic effects of mutant GUCY2D in Leber congenital amaurosis. Ophthalmology 110, 549–558 [DOI] [PubMed] [Google Scholar]
- 33. Stone E. M. (2007) Leber congenital amaurosis, a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 144, 791–811 [DOI] [PubMed] [Google Scholar]
- 34. Kelsell R. E., Gregory-Evans K., Payne A. M., Perrault I., Kaplan J., Yang R. B., Garbers D. L., Bird A. C., Moore A. T., and Hunt D. M. (1998) Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum. Mol. Genet. 7, 1179–1184 [DOI] [PubMed] [Google Scholar]
- 35. Gregory-Evans K., Kelsell R. E., Gregory-Evans C. Y., Downes S. M., Fitzke F. W., Holder G. E., Simunovic M., Mollon J. D., Taylor R., Hunt D. M., Bird A. C., and Moore A. T. (2000) Autosomal dominant cone rod retinal dystrophy (CORD6) from heterozygous mutation of GUCY2D, which encodes retinal guanylate cyclase. Ophthalmology 107, 55–61 [DOI] [PubMed] [Google Scholar]
- 36. Hunt D. M., Buch P., and Michaelides M. (2010) Guanylate cyclases and associated activator proteins in retinal disease. Mol. Cell Biochem. 334, 157–168 [DOI] [PubMed] [Google Scholar]
- 37. Payne A. M., Morris A. G., Downes S. M., Johnson S., Bird A. C., Moore A. T., Bhattacharya S. S., and Hunt D. M. (2001) Clustering and, frequency of mutations in the retinal guanylate cyclase (GUCY2D) gene in patients with dominant cone-rod dystrophies. J. Med. Genet. 38, 611–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kitiratschky V. B., Wilke R., Renner A. B., Kellner U., Vadalà M., Birch D. G., Wissinger B., Zrenner E., and Kohl S. (2008) Mutation analysis identifies GUCY2D as the major gene responsible for autosomal dominant progressive cone degeneration. Invest. Ophthalmol. Vis. Sci. 49, 5015–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weigell-Weber M., Fokstuen S., Török B., Niemeyer G., Schinzel A., and Hergersberg M. (2000) Codons 837 and 838 in the retinal guanylate cyclase gene on chromosome 17p: hot spots formutations in autosomal dominant cone-rod dystrophy? Arch. Ophthalmol. 118, 300. [DOI] [PubMed] [Google Scholar]
- 40. Downes S. M., Payne A. M., Kelsell R. E., Fitzke F. W., Holder G. E., Hunt D. M., Moore A. T., and Bird A. C. (2001) Autosomal dominant cone-rod dystrophy with mutations in the guanylate cyclase 2D gene encoding retinal guanylate cyclase-1. Arch. Ophthalmol. 119, 1667–1673 [DOI] [PubMed] [Google Scholar]
- 41. Udar N., Yelchits S., Chalukya M., Yellore V., Nusinowitz S., Silva-Garcia R., Vrabec T., Hussles Maumenee I., Donoso L., and Small K. W. (2003) Identification of GUCY2D gene mutations in CORD5 families and evidence of incomplete penetrance. Hum. Mutat. 21, 170–171 [DOI] [PubMed] [Google Scholar]
- 42. Ito S., Nakamura M., Ohnishi Y., and Miyake Y. (2004) Autosomal dominant cone-rod dystrophy with R838H and R838C mutations in the GUCY2D gene in Japanese patients. Jpn. J. Ophthalmol. 48, 228–235 [DOI] [PubMed] [Google Scholar]
- 43. Garcia-Hoyos M., Auz-Alexandre C. L., Almoguera B., Cantalapiedra D., Riveiro-Alvarez R., Lopez-Martinez M. A., Gimenez A., Blanco-Kelly F., Avila-Fernandez A., Trujillo-Tiebas M. J., Garcia-Sandoval B., Ramos C., and Ayuso C. (2011) Mutation analysis at codon 838 of the guanylate cyclase 2D gene in Spanish families with autosomal dominant cone, cone-rod, and macular dystrophies. Mol. Vis. 17, 1103–1109 [PMC free article] [PubMed] [Google Scholar]
- 44. Tucker C. L., Woodcock S. C., Kelsell R. E., Ramamurthy V., Hunt D. M., and Hurley J. B. (1999) Biochemical analysis of a dimerization domain mutation in RetGC-1 associated with dominant cone-rod dystrophy. Proc. Natl. Acad. Sci. U.S.A. 96, 9039–9044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ramamurthy V., Tucker C., Wilkie S. E., Daggett V., Hunt D. M., and Hurley J. B. (2001) Interactions within the coiled-coil domain of RetGC-1 guanylyl cyclase are optimized for regulation rather than for high affinity. J. Biol. Chem. 276, 26218–26229 [DOI] [PubMed] [Google Scholar]
- 46. Peshenko I. V., Moiseyev G. P., Olshevskaya E. V., and Dizhoor A. M. (2004) Factors that determine Ca2+ sensitivity of photoreceptor guanylyl cyclase: kinetic analysis of the interaction between the Ca2+-bound and the Ca2+-free guanylyl cyclase activating proteins (GCAPs) and recombinant photoreceptor guanylyl cyclase 1 (RetGC-1). Biochemistry 43, 13796–13804 [DOI] [PubMed] [Google Scholar]
- 47. Lem J., Applebury M. L., Falk J. D., Flannery J. G., and Simon M. I. (1991) Tissue-specific and developmental regulation of rod opsin chimeric genes in transgenic mice. Neuron 6, 201–210 [DOI] [PubMed] [Google Scholar]
- 48. Garbers D. L. (1999) The guanylyl cyclase receptors. Methods 19, 477–484 [DOI] [PubMed] [Google Scholar]
- 49. Yang R. B., Robinson S. W., Xiong W. H., Yau K. W., Birch D. G., and Garbers D. L. (1999) Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. J. Neurosci. 19, 5889–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jeon C. J., Strettoi E., and Masland R. H. (1998) The major cell populations of the mouse retina. J. Neurosci. 18, 8936–8946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao X., Ren Y., Zhang X., Chen C., Dong B., and Li Y. (2013) A novel GUCY2D mutation in a Chinese family with dominant cone dystrophy. Mol. Vis. 19, 1039–1046 [PMC free article] [PubMed] [Google Scholar]
- 52. Lazar C. H., Mutsuddi M., Kimchim A., Zelinger L., Mizrahi-Meissonnier L., Marks-Ohana D., Boleda A., Ratnapriya R., Sharon D., Swaroop A., and Banin E. (2014) Whole exome sequencing reveals GUCY2D as a major gene associated with cone and cone-rod dystrophy in Israel. Invest. Ophthalmol. Vis. Sci. 56, 420–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Collery R. F., Cederlund M. L., and Kennedy B. N. (2013) Transgenic zebrafish expressing mutant human RETGC-1 exhibit aberrant cone and rod morphology. Exp. Eye Res. 108, 120–128 [DOI] [PubMed] [Google Scholar]
- 54. Boye S. E., Boye S. L., Pang J., Ryals R., Everhart D., Umino Y., Neeley A. W., Besharse J., Barlow R., and Hauswirth W. W. (2010) Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS One 5, e11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mihelec M., Pearson R. A., Robbie S. J., Buch P. K., Azam S. A., Bainbridge J. W., Smith A. J., and Ali R. R. (2011) Long-term preservation of cones and improvement in visual function following gene therapy in a mouse model of leber congenital amaurosis caused by guanylate cyclase-1 deficiency. Hum. Gene. Ther. 22, 1179–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boye S. L., Peshenko I. V., Huang W. C., Min S. H., McDoom I., Kay C. N., Liu X., Dyka F. M., Foster T. C., Umino Y., Karan S., Jacobson S. G., Baehr W., Dizhoor A., Hauswirth W. W., and Boye S. E. (2013) AAV-mediated gene therapy in the guanylate cyclase (RetGC1/RetGC2) double knockout mouse model of Leber congenital amaurosis. Hum. Gene. Ther. 24, 189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boye S. L., Peterson J. J., Choudhury S., Min S. H., Ruan Q., McCullough K. T., Zhang Z., Olshevskaya E. V., Peshenko I. V., Hauswirth W. W., Ding X. Q., Dizhoor A. M., and Boye S. E. (2015) Gene therapy fully restores vision to the all-Cone Nrl−/− Gucy2e−/−) mouse model of Leber congenital amaurosis-1. Hum. Gene. Ther. 26, 575–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Olshevskaya E. V., Peshenko I. V., Savchenko A. B., and Dizhoor A. M. (2012) Retinal guanylyl cyclase isozyme 1 is the preferential in vivo target for constitutively active GCAP1 mutants causing congenital degeneration of photoreceptors. J. Neurosci. 32, 7208–7217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2015) Evaluating the role of retinal membrane guanylyl cyclase 1 (RetGC1) domains in binding guanylyl cyclase-activating proteins (GCAPs). J. Biol. Chem. 290, 6913–6924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2015) Dimerization domain of retinal membrane guanylyl cyclase 1 (RetGC1) is an essential part of guanylyl cyclase-activating protein (GCAP) binding interface. J. Biol. Chem. 290, 19584–19596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zägel P., Dell'Orco D., and Koch K. W. (2013) The dimerization domain in outer segment guanylate cyclase is a Ca2+-sensitive control switch module. Biochemistry 52, 5065–5074 [DOI] [PubMed] [Google Scholar]
- 62. Olshevskaya E. V., Calvert P. D., Woodruff M. L., Peshenko I. V., Savchenko A. B., Makino C. L., Ho Y. S., Fain G. L., and Dizhoor A. M. (2004) The Y99C mutation in guanylyl cyclase-activating protein 1 increases intracellular Ca2+ and causes photoreceptor degeneration in transgenic mice. J. Neurosci. 24, 6078–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Woodruff M. L., Olshevskaya E. V., Savchenko A. B., Peshenko I. V., Barrett R., Bush R. A., Sieving P. A., Fain G. L., and Dizhoor A. M. (2007) Constitutive excitation by Gly90Asp rhodopsin rescues rods from degeneration caused by elevated production of cGMP in the dark. J. Neurosci. 27, 8805–8815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xu J., Morris L., Thapa A., Ma H., Michalakis S., Biel M., Baehr W., Peshenko I. V., Dizhoor A. M., and Ding X. Q. (2013) cGMP accumulation causes photoreceptor degeneration in CNG channel deficiency: evidence of cGMP cytotoxicity independently of enhanced CNG channel function. J. Neurosci. 33, 14939–14948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ma H., Butler M. R., Thapa A., Belcher J., Yang F., Baehr W., Biel M., Michalakis S., and Ding X. Q. (2015) cGMP/protein kinase G signaling suppresses inositol 1,4,5-trisphosphate receptor phosphorylation and promotes endoplasmic reticulum stress in photoreceptors of cyclic nucleotide-gated channel-deficient mice. J. Biol. Chem. 290, 20880–20892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Molday L. L., Jefferies T., and Molday R. S. (2014) Insights into the role of RD3 in guanylate cyclase trafficking, photoreceptor degeneration, and Leber congenital amaurosis. Front. Mol. Neurosci. 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gorbatyuk M., and Gorbatyuk O. (2013) Retinal degeneration: focus on the unfolded protein response. Mol. Vis. 19, 1985–1998 [PMC free article] [PubMed] [Google Scholar]
- 68. Laura R. P., Dizhoor A. M., and Hurley J. B. (1996) The membrane guanylyl cyclase, retinal guanylyl cyclase-1, is activated through its intracellular domain. J. Biol. Chem. 271, 11646–11651 [DOI] [PubMed] [Google Scholar]
- 69. Peshenko I. V., and Dizhoor A. M. (2004) Guanylyl cyclase-activating proteins (GCAPs) are Ca2+/Mg2+ sensors: implications for photoreceptor guanylyl cyclase (RetGC) regulation in mammalian photoreceptors. J. Biol. Chem. 279, 16903–16906 [DOI] [PubMed] [Google Scholar]
- 70. Peshenko I. V., and Dizhoor A. M. (2007) Activation and inhibition of photoreceptor guanylyl cyclase by guanylyl cyclase activating protein 1 (GCAP-1): the functional role of Mg2+/Ca2+ exchange in EF-hand domains. J. Biol. Chem. 282, 21645–21652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen C., Nakatani K., and Koutalos Y. (2003) Free magnesium concentration in salamander photoreceptor outer segments. J. Physiol. 553, 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tsien R., and Pozzan T. (1989) Measurement of cytosolic free Ca2+ with Quin2. Methods Enzymol. 172, 230–262 [DOI] [PubMed] [Google Scholar]
- 73. Woodruff M. L., Sampath A. P., Matthews H. R., Krasnoperova N. V., Lem J., and Fain G. L. (2002) Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J. Physiol. 542, 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lyubarsky A. L., Lem J., Chen J., Falsini B., Iannaccone A., and Pugh E. N. Jr. (2002) Functionally rodless mice: transgenic models for the investigation of cone function in retinal disease and therapy. Vision Res. 42, 401–415 [DOI] [PubMed] [Google Scholar]
- 75. Peshenko I. V., and Dizhoor A. M. (2006) Ca2+ and Mg2+ binding properties of GCAP-1: evidence that Mg2+-bound form is the physiological activator of photoreceptor guanylyl cyclase. J. Biol. Chem. 281, 23830–23841 [DOI] [PubMed] [Google Scholar]
- 76. Peshenko I. V., Olshevskaya E. V., Lim S., Ames J. B., and Dizhoor A. M. (2014) Identification of target binding site in photoreceptor guanylyl cyclase-activating protein 1 (GCAP1). J. Biol. Chem. 289, 10140–10154 [DOI] [PMC free article] [PubMed] [Google Scholar]