

Summary
Herpesviruses suppress cell death to assure sustained infection in their natural hosts. Murine cytomegalovirus (MCMV) encodes suppressors of apoptosis as well as M45-encoded viral inhibitor of RIP activation (vIRA) to block RIP homotypic interaction motif (RHIM)-signaling and recruitment of RIP3 (also called RIPK3), to prevent necroptosis. MCMV and human cytomegalovirus encode a viral inhibitor of caspase (Casp)8 activation to blocks apoptosis, an activity that unleashes necroptosis. Herpes simplex virus (HSV)1 and HSV2 incorporate both RHIM and Casp8 suppression strategies within UL39-encoded ICP6 and ICP10, respectively, which are herpesvirus-conserved homologs of MCMV M45. Both HSV proteins sensitize human cells to necroptosis by blocking Casp8 activity while preventing RHIM-dependent RIP3 activation and death. In mouse cells, HSV1 ICP6 interacts with RIP3 and, surprisingly, drives necroptosis. Thus, herpesviruses have illuminated the contribution of necoptosis to host defense in the natural host as well as its potential to restrict cross-species infections in nonnatural hosts.
Keywords: programmed necrosis, FADD, cFLIP, serine/threonine protein kinase, ribonucleotide reductase
Introduction
Necroptosis is a form of programmed cell death leading to membrane leakage independent of caspases, orchestrated by the activity of receptor interacting protein (RIP)3 kinase and its target, a pseudokinase called mixed lineage kinase-like (MLKL). Hallmark insights using vaccinia virus raised the specter of RIP3 kinase-dependent necroptosis contributing to antiviral host defense [1]. The identification of a specific viral inhibitor of RIP activation (vIRA) encoded by the herpesvirus murine cytomegalovirus (MCMV) demonstrated that RIP3-mediated necroptosis is a bona fide cell autonomous host defense pathway subjected to a specific viral countermeasure [2,3]. Necroptosis has been implicated in additional viral [4,5] as well as bacterial [6,7] infections and is probably a default pathway when caspase (Casp)8 activity is compromised in cells with sufficient levels of RIP3 [8,9]. Many viruses, including both poxviruses and herpesviruses, inhibit caspase (Casp)8 activity [10] and therefore have the potential to unleash this alternate death pathway. For example, the betaherpesvirus-conserved viral inhibitor of Casp8 activation (vICA) naturally suppresses virus-induced apoptosis in macrophages [11–15]. MCMV relies on M45-encoded vIRA to prevent necroptosis and sustain infection. The M45 protein is an enzymatically inactive homolog of the large subunit (R1) of ribonucleotide reductase (RNR) common across the herpesviruses. Based on studies of MCMV, we have posited that an evolutionary dialogue has gone on between host cell death pathways and virus-encoded cell death suppressors that is evident in mammals [9,16], as depicted in Figure 1. Mitochondrial, or cell-intrinsic cell death is an ancient form of host defense as well as a common target of virus-encoded suppressors that provided the selective pressure for Casp8 extrinsic apoptosis to side-step mitochondria and directly activate the executioner caspases, Casp3 and Casp7. The ability of a self-activating caspase to execute cells prompted the acquisition by viruses of specific Casp8 inhibitors [10]. This selected for host adaptation of necroptosis as a trap door to eliminate viruses that suppress Casp8, built upon established RIP homotypic interaction motif (RHIM) signaling pathways. RHIM-competitors encoded by MCMV, HSV1 and HSV2 thus represent most recent evolutionary adaptation in this ancient pathogen-host stand-off [9,16].
Figure 1. Evolutionary relationships in cell autonomous death pathways and virus-encoded countermeasures.

Programmed cell death (PCD) pathways include the mitochondrial (or cell-intrinsic) death pathway that eliminates cells during development and homeostatic turnover as well as in host defense [9,10,53,100]. Viral (v)Bcl2 homologs and other virus-encoded mitochondrial suppressors block mitochondrial PCD, establishing conditions that selected for a Casp8-like activity to directly trigger effector caspases, bypassing mitochondrial Bcl2 function. Viruses adaptation included Casp8-targeted cell death suppressors, some specific, like herpesvirus vFLIPs, betaherpesvirus vICA, and some nonspecific, like the poxvirus serpin, CrmA. RIP3-dependent necroptosis via RHIM-dependent interaction with RIP1, TRIF or DAI, enables the host to bypass the impact of virus-encoded Casp8 inhibitors. The examples of MCMV vIRA and HSV1/HSV2 R1 RHIM signaling competitors are the first examples of viral suppressors of necroptosis [2,3,5,20]. Note that HSV1 ICP6 inhibits necroptosis in human cells but promotes necroptosis in mouse cells [98,99]. This depiction is adapted from [9].
MCMV vIRA employs an amino-terminal RHIM to interrupt signal transduction leading to cell death and cytokine activation. In mice or humans, RIP1, DNA-induced activator of interferon (DAI) and TIR-domain-containing adapter-inducing interferon β (TRIF) can partner with RIP3, as depicted in Figure 2 [2,3,17–19]. The MCMV-encoded RHIM competitor, vIRA prevents activation of RIP3 kinase activity, the subsequent recruitment of MLKL, and the later steps leading to cell leakage [19]. vIRA blocks all consequences of RHIM-dependent signaling, including activation of NF-κB [17,20,21] even though the impact on this transcription factor does not appear to impact infection in mouse cells or mice. Despite the crucial role that MCMV M45-encoded vIRA plays in cell death suppression, and the likely preservation of this mechanism in other rodent betaherpesviruses, the HCMV UL45 is inactive [22,23]. Nevertheless, HCMV blocks necroptosis during infection by employing a strategy that subverts a later step in the pathway (Omoto et al., J Biol Chem, manuscript submitted). The M45 homologs of herpes simplex virus (HSV)1 and HSV2 (UL39 gene products, ICP6 and ICP10, respectively) have recently been shown to employ a RHIM competitor mechanism in human cells [5]. Although analogous to MCMV vIRA, HSV cell death suppressors ICP6 and ICP10 make use of an anti-apoptotic domain separate from the RHIM to interact directly with Casp8 death effector domains [24], a function mediated by the carboxyl-terminal RNR domain [5]. Here, we will focus on examples of virus-encoded RHIM-signaling inhibitors and the contribution of Casp8 inhibition to unleashed necroptosis.
Figure 2. Canonical and noncanonical RHIM-dependent activation of RIP3 kinase.
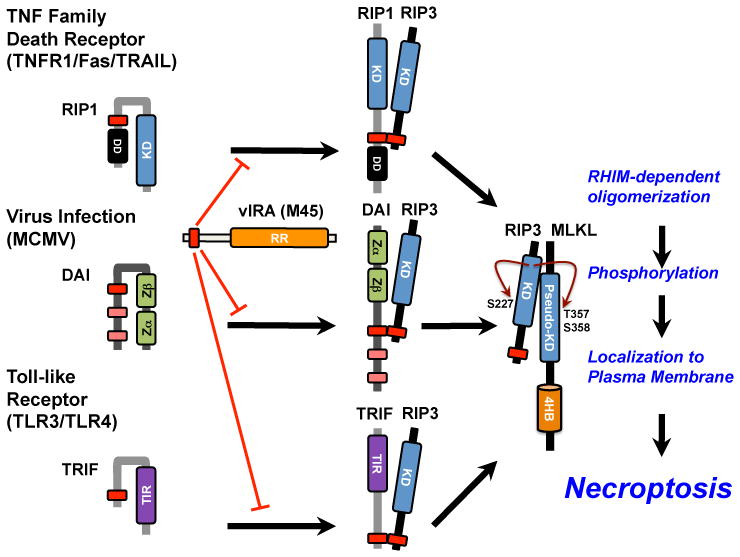
Canonical activation of necroptosis via RIP1 recruitment and activation RIP3 downstream of TNF family death receptors (TNFR1, Fas, TRAIL), highlighting the known and predicted conformational changes that accompany this pathway [28]. Noncanonical activation of necroptosis via DAI downstream of MCMV infection or via TRIF downstream of TLR3 and TLR4 activation, driving recruitment and activation of RIP3, with predicted conformational changes. All forms of RHIM-dependent activation of RIP3 are blocked by MCMV-encoded vIRA and converge on the RIP3-dependent recruitment and phosphorylation events leading to plasma membrane leakage and other outcomes characteristic of necroptosis.
Activation and suppression of necroptosis
Necroptosis hinges on RIP3 RHIM-mediated signal transduction that was initially characterized in cells undergoing cell death following stimulation with TNF in the presence of caspase 8 inhibitor (Figure 3A) or in cells that lack Casp8 [1,25,26], and is the subject of up-to-date reviews [27,28]. RIP3 is a serine-threonine kinase that recruits and phosphorylates the pseudokinase MLKL, controlling oligomerization events at membranes that lead to cellular changes including leakage of cytosolic contents [29–33]. RHIM-dependent interaction of RIP1 and RIP3 characterizes the canonical form of this death pathway [1,25,26] and results in conformational changes and amyloid-like oligomerization of a RIP1-RIP3 complex [28,34]. Canonical TNFR1-mediated necroptosis requires the protein kinase activity of RIP1 in addition to RHIM interaction with RIP3. RIP3 activation and execution of necroptosis may proceed independent of RIP1. DAI as well as TRIF can naturally partner with RIP3 and trigger a noncanonical necroptosis, independent of RIP1 RHIM and kinase activities [2,3,19]. Interestingly, experiments have substantiated the noncanonical pathway even downstream of death receptors (Figure 1) [35–37]. Regardless of the involvement of RIP1, RIP3 must autophosphorylate itself to facilitate interaction with and subsequent phosphorylation of MLKL [19,28,38] (Figure 2). A cytosolic Casp8-containing complex, called Complex IIB [39] or the ripoptosome [28,40–42], forms downstream of TNFR1 or of TRIF-dependent signaling pathways controlled by TLR3 or TLR4. This complex is increasingly recognized to control apoptotic as well as necroptotic outcomes [28,43] after assembling from preexisting cytosolic components in a manner that is independent of pro-apoptotic caspase activity as well as mitochondrial function [44]. The ripoptosome is assembled around Casp8-cFLIPL heterodimer in association with FADD to which RIP1 and RIP3 are recruited through well-established protein-protein homotypic interaction domains depicted in Figure 3A. This assembly oligomerizes has been the subject of many reviews [27,28,45]. MLKL is recruited and phosphorylated by RIP3 kinase within a necrosome complex [31,33,46–48] resulting in MLKL conformational changes and oligomerization that result in membrane association, as well as the pattern of disruption and leakage [27,28].
Figure 3. Formation and disruption of the ripoptosome and necrosome by HSV1 ICP6 and HSV2 ICP10.
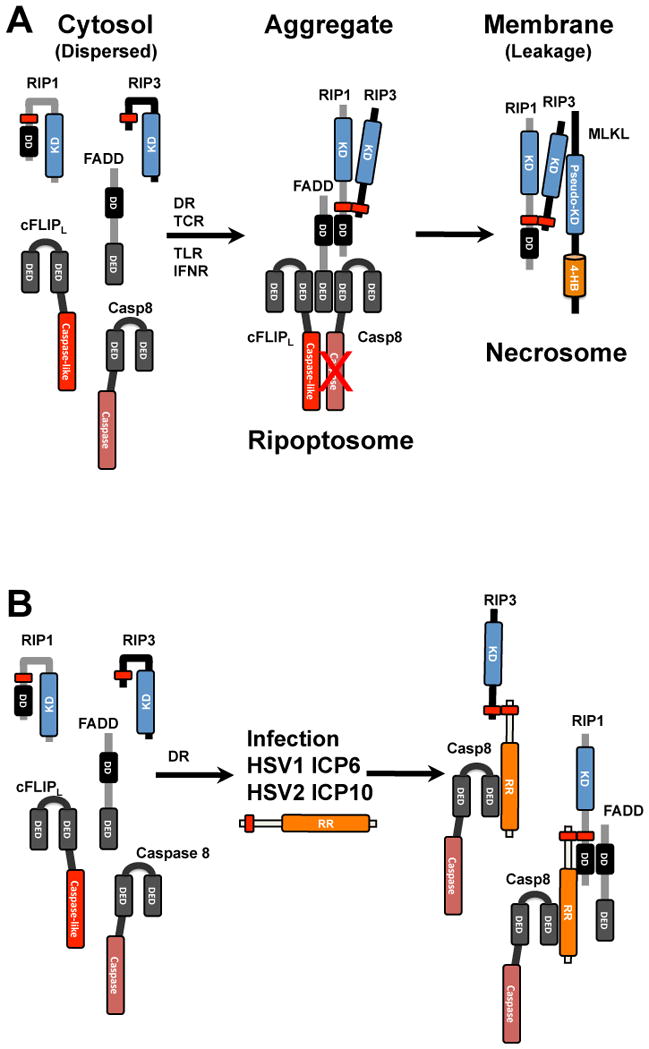
A. The ripoptosome forms from cytosolic components. FADD becomes activated downstream of TNF family death receptor (DR) activation, as well as following T cell receptor (TCR), TLR and interferon receptor (IFNR) activation, to recruit Casp8 and cFLIPL heterodimer via homotypic death effector domain (DED) as well as RIP1 via a homotypic death domain (DD) interactions. RIP1 recruits RIP3 via a common RHIM (shown as a red rectangle) and, when Casp8 activity is inhibited, triggers oligomerization that leads to an amyloid-like complex that recruits MLKL into a necrosome that localizes to membranes and directs the final steps in necroptosis leading to membrane leakage. B. Interaction of HSV1 ICP6 and HSV2 ICP10 with ripoptosome components in human cells. Cytosolic ripoptosome components form HSV R1-dependent complexes via RHIM and Casp8 interaction domains discussed in the text.
Casp8 inhibition has long been recognized as a key factor in the induction of necrotic death [49,50] and is a crucial feature in terms of how this pathway is unleashed in virus-infected cells [9,10]. Besides suppression of apoptosis, inhibition of Casp8 also fundamentally affects the availability of key ripoptosome components. The susceptibility of RIP1 [51] and RIP3 [52] to Casp8 cleavage-dependent inactivation of pro-necrotic kinase activity suggests that both of these pro-necrotic kinases are silenced by Casp8 [27,28]. Although the heterodimeric complex of Casp8 and cFLIPL elaborates a basal protease activity that may be sufficient to inactivate RIP1 and/or RIP3, details of this regulatory process have not yet been worked out. Whereas Casp8 suppression by vICA is needed during natural infection to block apoptosis in macrophages [14], MCMV-infected cells become sensitized to necroptosis such that vIRA is necessary to suppress the RHIM-dependent recruitment of RIP3 by DAI in all infected host tissues [2,3]. Without an intact RHIM in vIRA, MCMV cannot sustain infection in wild-type mice. However, normal infection is restored in RIP3- or DAI-deficient strains of mice. Mice carrying kinase inactive RIP3 also normalize replication of vIRA-deficient MCMV [43], consistent with the essential contribution of this pro-necrotic kinase activity to host defense. The investigation of cell death suppressor mutants of MCMV has revealed the importance of deflecting both Casp8-mediated apoptosis and RIP3-mediated necroptosis [53]. Necroptosis is triggered in tissue-resident cells regardless of whether the same cells are sensitive to apoptosis. Importantly, studies with vIRA-deficient MCMV have revealed that necroptosis may be unleashed in any virus-infected cell; however, mouse or human cells placed into culture vary in their levels of RIP3 and therefore vary in their susceptibility to this death pathway. The reasons why cultured cells tend to lose RIP3 (as well as MLKL) expression is presently unknown, but certainly suggests that cell culture selects against the retention of necroptotic machinery. In addition to herpesviruses, this alternate death is likely to be modulated by poxviruses and other DNA viruses known to block Casp8 activity [10,54].
Cell death outcomes are strongly influenced by posttranslational modification that acts as a scaffold such as those provided by K63 or M1 (linear) polyubiquitination [55,56]. RIP1 seems to be the most important target of these modifications given its contribution to balancing death outcomes emanating from the ripoptosome [42,56–58]. Compromised K63 or linear polyubiquitination increases susceptibility of cells to apoptotic as well as necroptotic death [9]. Inhibitors and mutants affecting either of two distinct E3 ubiquitin ligase complexes, cellular inhibitor of apoptosis (cIAP)1/cIAP2 [55] or linear ubiquitination chain assembly complex (LUBAC) [56] have been employed to show the importance of polyubiquitination in dictating sensitivity to death signaling [25,40,41,59–61]. Although the polyubiquitination machinery is not a known target of mammalian viruses cell death suppressors, the first IAP ever identified emerged from studies in baculoviruses where virus-encoded IAPs suppress cell death to sustain infection [62,63]. To date only large DNA viruses infecting arthropods encode IAP-like suppressors of apoptosis [64]. Interestingly, the noncanonical induction of necroptosis that occurs in MCMV M45 mutant virus infected cells is insensitive to IAP1/IAP2 antagonists, consistent with known RIP1 independent characteristics of the pathway [2,3]. Only time and additional investigation will reveal whether mammalian and insect cells differ in some fundamental way in harnessing IAP function for host defense.
Uncontrolled necroptosis undermines development and tissue homeostasis
The discovery that midgestational death in Casp8 or FADD-deficient mice results from unrestricted RIP3- and RIP1-dependent death [65–67] completely revised thinking about ripoptosome function in development and inflammatory disease. These investigations, together with incisive observations on Casp8- or FADD-deficient T cells [68,69] revealed how dysregulation of death pathways during development as well as during the T response to antigen impact cell fate. The complex dialogue between FADD/Casp8 and pro-necrotic kinases is further bolstered by investigation of FADD-deficient keratinocytes [70] and intestinal epithelium [71] as well as in a number of other settings [10,16,27,28,72]. Thus, it is now widely understood that midgestational death due to germ line deficiency in Casp8 (or FADD) is due to canonical RIP1-RIP3 necroptosis, particularly because the phenotype is reversed by either kinase-inactive RIP3 [43,73] or kinase-inactive RIP1 [57]. Both RIP1 and RIP3 kinase activities contribute directly to fetal demise. Consistent with the observations on Casp8 and FADD, serious defects are observed with germ line deficiency in other ripoptosome components. cFLIP-deficient [74] or RIP1-deficient mice [57,75,76] die during development but are fully rescued by the combined elimination of apoptotic (Casp8 or FADD) and necroptotic (RIP3 or MLKL) machinery. Thus, mice deficient in multiple complementary components are fully viable, fertile, and where examined [57], immunocompetent and able to clear virus infection. The picture that emerges from these studies and from a growing number of examples where disruption of Casp8 is performed in a tissue-specific manner [27,28] that the balance of Casp8-dependent apoptosis and RIP3-dependent necroptosis is crucial to sustain tissue homeostasis [16,28]. The common ability of death receptors, TLRs, T cell receptors, genotoxic stress and interferon receptors as well as virus infection to drive innate immune cell death as well as innate immune cytokine activation indicates that the ripoptosome may act as a pathogen supersensor to guard mammalian cells from insult [9]. Perturbation of the ripoptosome undermines development even though its function is dispensable for either development or tissue homeostasis.
It is important to note that developmental consequences of mutant ripoptosome components only show modest dependence on TNF signaling despite the fact that canonical, RIP1-dependent activation of RIP3 kinase activity and RHIM signal transduction are crucial to the demise of mutant mice [1,19,25,26,65,66]. The ripoptosome forms under a wide variety of innate signaling conditions, executing either apoptosis or necroptosis [9]. The recent demonstration that the ripoptosome is also triggered by conformational changes in RIP3 independent of its pronecrotic kinase activity [43] reveals a remarkable capacity of this protein kinase to regulate caspase activation in addition to necroptosis. RIP3 deficiency itself does not cause any developmental problems [77] but reveals modest innate immune response dysregulation [78]. The potential for RIP3 kinase domain mutants as well as small molecule kinase inhibitors to trigger apoptosis dependent on RIP1 and RIP3 scaffold function independent of activity of these kinases [43,73] expanded the settings where the ripoptosome controls cell fate. Further dissection of the scaffold versus the enzymatic roles of RIP1 as well as RIP3 will provide further insights into the mechanism(s) underlying alternate death outcomes.
Evolutionary adaptation of innate immune programmed cell death in host defense
Evolutionary relationships of Casp8-mediated apoptosis and RIP3-mediated necroptosis pathways in response to viral suppressors have been reviewed [9] and are illustrated in Figure 1. Our work with MCMV brought to light the importance of RIP3-dependent necroptosis as a host defense mechanism [2]. Many diverse innate and adaptive immune mechanisms ranging from interferons to antibodies and T cells mediate control over pathogens, but only necroptosis has been shown to completely prevent a herpesvirus such as MCMV from infecting its natural host, even when high doses of virus are used. This can be attributed to the unique ability of necroptosis to eliminate virus-infected cells before production of progeny can occur, thereby shutting down the infection. Because the MCMV vIRA completely blocks the elaboration of necroptosis during MCMV infection, this remarkably potent host defense pathway becomes effectively neutralized. This absolutely fascinating pathogen-host relationship extends to human cell settings where necroptosis is targeted by HCMV as well as HSV gene products [5]. In all of these settings, we hypothesize that caspase (Casp)8, together with its partners, FADD and cFLIPL, forms a pathogen supersensor [9] and triggers cell autonomous defense pathways that include cell death and cytokine production [16], although, in mice, death pathways predominate innate signaling and play out completely independent of interferon and inflammatory cytokine function. The subversion tactics by herpesviruses have codified the importance of neutralizing these pathways in pathogenesis.
Suppression of necroptosis by HSV
HSV1 suppresses programmed cell death induced by a wide variety of insults [79], relying on the regulatory protein ICP4 as well as protein kinase US3 [80,81]. In addition to US3 [82,83], early viral gene products gD [84,85] and, in particular, R1 [24,86–89] are able to inhibit apoptosis, albeit in a cell-type specific manner [90,91]. HSV1 also manipulates apoptosis as a component of latency in rodent neurons [92,93]. The HSV1 and HSV2 R1 proteins, ICP6 and ICP10, suppress death receptor-dependent apoptosis by interacting with death effector domains of Casp8 via a conserved C-terminal RNR domain [24]. MCMV vIRA known to block apoptosis as well as neroptosis [2,3,20], but does not bind to Casp8 [5]. HSV R1-mediated inhibition of Casp8 function may be analogous to betaherpesvirus vICA [12,13]. Until recently, little attention was given to necroptosis, although there have been longstanding clues that caspase-independent pathways may dominate under certain conditions in HSV-infected cells and tissues [91,94–96]. Intriguingly, ICP6 and ICP10 exhibit sequence homology with the N-terminal RHIM of MCMV M45 and cellular RHIM-containing proteins [97]. This RHIM-like amino acid sequence is located in the amino-terminal extension of all R1 homologs encoded by the primate alphaherpesviruses and rodent betaherpesviruses. Recent comprehensive investigation of HSV1 ICP6 reveals an ability of this RHIM-like region to interact with RIP1 and RIP3 in human cells [5] as well as in mouse cells [98,99] (Figure 3B); however, the RHIM-dependent interaction yields apparently opposite biological consequences in human versus mouse cells. Both HSV1 ICP6 and HSV2 ICP10 function as RHIM competitors in necroptosis-sensitive human cells [5], behavior that is reminiscent of MCMV vIRA [2,3]. This behavior is consistent with a role in counteracting host defense. In contrast to the behavior in human cells, HSV1 ICP6 promotes necroptosis in mouse cells, where the RHIM-like region activates the pro-necrotic kinases [98,99]. This is most dramatically demonstrated by comparing infection of parental WT HSV1 with an ICP6 mutant (ΔICP6) HSV1. Infection with ΔICP6 HSV1 mutant renders human cells highly sensitive to TNF due to the induction of apoptosis as well as necroptosis [5]; whereas, infection with this mutant virus eliminates virus-induced necroptosis in mouse cells [98,99] (Figure 3B). These findings raise important questions about the role of death pathways in modulating cross-species infections as well as about the reliance on rodents as surrogate hosts in studies of HSV1 pathogenesis and latency. It remains to be determined whether HSV2 ICP10 acts similarly to HSV1 ICP6 in mice. Importantly, and in contrast to the species-specific behavior of HSV1 ICP6, ability of MCMV vIRA to block either death receptor or virus-induced necroptosis function is conserved in human as well as mouse cells [5]. Indeed, a swap of the M45 RHIM into HSV1 ICP6 converted the latter from an inducer to a suppressor of necroptosis in mouse cells [98].
The ability of HSV1 ICP6 and HSV2 ICP10 to bind Casp8 is integral to their suppression activity against necroptosis in human cells [5]. Early [24] and recent studies[5] demonstrate that Casp8 binding is mediated by the large carboxyl-terminal region of HSV R1 proteins, a region that is also responsible for ribonucleotide reductase activity. The amino-terminal region carrying the RHIM-like element is sufficient for interaction with RIP1 or RIP3 in a RHIM-dependent fashion, but the ability to block necroptosis in infected cells requires interaction with both the RIP1/RIP3 and Casp8-binding regions. Importantly, HSV1-infected cells are effectively sensitized to TNF-induced necroptosis by the Casp8-binding domain of ICP6 or ICP10. Importantly, ICP6mutRHIM virus-infected cells die prematurely following TNF treatment due to the suppression of Casp8 activity in the absence of RHIM competitor activity [5]. Curiously, mouse cells are rescued from ICP6-induced necroptosis when infected with this same virus, confirming the contribution of RHIM signaling to the induction of the pathway in a species-specific fashion [98]. From these recent studies, HSV1 ICP6 and HSV2 ICP10 emerge as important suppressors of necroptosis in cells from their natural host species, where Casp8 suppression sensitizes to necroptosis. This prevents RHIM signal competition, activation of RIP3 and recruitment of MLKL into a necrosome complex.
Perspectives
Pathogenesis studies in mice have unambiguously identified the cognate pathways targeted by virus-encoded cell death suppressors in natural pathogen-host settings, such as infection with MCMV, underscoring the importance of specific host defense mechanisms, including programmed cell death. Using MCMV mutants M45mutRHIM, both RIP3 and DAI were found to be the cognate host cell targets of RHIM competitor activity. When they associate, necroptosis is activated to eliminate infected cells in all exposed tissues [2,3,20]. These studies opened the way to show that RIP3 necroptosis underlies the phenotype of germ line Casp8-deficient mice [66] and that perinatal lethality due to combined effects of Casp8-dependent apoptosis and RIP3-dependent necroptosis underlies RIP1-deficiency [57]. Along the way, we showed that the combined elimination of Casp8 and RIP3 programmed cell death pathways are remarkably dispensable for mammalian development, viability, fertility and the ability to mount a fully developed T cell response to control viral infection [57]. The key components of the ripoptosome become dispensable for development when eliminated in precise combinations that respect the ability of single mutants to dysregulate cell death by unleashing alternate cell death pathways, pathways that are elaborated in mammals to control pathogens. Recent demonstration that TNF-dependent necroptosis plays out against HSV1 in human cells and is blocked by the HSV1 and HSV2 R1 proteins adds significantly to the growing understanding of the antiviral role of necroptosis. These observations reveal viral evasion strategies that have the potential to prevent cross-species infections. The specter of necroptosis as a cross-species restriction factor, however, must be pursued further. Necroptosis emerges as a way to eliminate viruses capable of inhibiting Casp8, as illustrated initially by the behavior of vaccinia as well as by a growing body of data on MCMV infection and HSV1 infection, strategies that are likely to be usurped by other mammalian viruses.
Research Highlights.
Necroptosis and apoptosis are alternate programmed cell death pathways
Necroptosis, like apoptosis, provides cell autonomous host defense
Herpesviruses block necroptosis by encoding RHIM signaling competitors
Herpesvirus suppression of caspase 8 unleashes necroptosis
Acknowledgments
Work performed in our laboratories is supported by NIH grants to ESM (R01 AI020211) and WJK (DP1 OD012198), although the content is solely the responsibility of the authors and not the NIH or PHS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cho YS, Challa S, Moquin D, Genga R, Ray TD, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Upton JW, Kaiser WJ, Mocarski ES. DAI (ZBP1/DLM-1) complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger AK, Danthi P. Reovirus activates a caspase-independent cell death pathway. MBio. 2013;4:e00178–00113. doi: 10.1128/mBio.00178-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo H, Omoto S, Harris PA, Finger JN, Bertin J, et al. Herpes simplex virus suppression of necroptosis in human cells. Cell Host Microbe. 2015;17:243–251. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, et al. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U S A. 2014;111:7391–7396. doi: 10.1073/pnas.1403477111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Upton JW, Chan FK. Staying alive: Cell death in antiviral immunity. Mol Cell. 2014;54:273–280. doi: 10.1016/j.molcel.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaiser WJ, Upton JW, Mocarski ES. Viral modulation of programmed necrosis. Curr Opin Virol. 2013;3:296–306. doi: 10.1016/j.coviro.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol. 2011;12:79–88. doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menard C, Wagner M, Ruzsics Z, Holak K, Brune W, et al. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J Virol. 2003;77:5557–5570. doi: 10.1128/JVI.77.10.5557-5570.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCormick AL, Skaletskaya A, Barry PA, Mocarski ES, Goldmacher VS. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology. 2003;316:221–233. doi: 10.1016/j.virol.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, et al. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci U S A. 2001;98:7829–7834. doi: 10.1073/pnas.141108798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cicin-Sain L, Ruzsics Z, Podlech J, Bubic I, Menard C, et al. Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J Virol. 2008;82:2056–2064. doi: 10.1128/JVI.01803-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCormick AL, Roback L, Livingston-Rosanoff D, St Clair C. The human cytomegalovirus UL36 gene controls caspase-dependent and -independent cell death programs activated by infection of monocytes differentiating to macrophages. J Virol. 2010;84:5108–5123. doi: 10.1128/JVI.01345-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mocarski ES, Kaiser WJ, Livingston-Rosanoff D, Upton JW, Daley-Bauer LP. True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J Immunol. 2014;192:2019–2026. doi: 10.4049/jimmunol.1302426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009;10:916–922. doi: 10.1038/embor.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3 and MLKL. J Biol Chem. 2013;288:31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Upton JW, Kaiser WJ, Mocarski ES. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem. 2008;283:16966–16970. doi: 10.1074/jbc.C800051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol. 2008;181:6427–6434. doi: 10.4049/jimmunol.181.9.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hahn G, Khan H, Baldanti F, Koszinowski UH, Revello MG, et al. The human cytomegalovirus ribonucleotide reductase homolog UL45 is dispensable for growth in endothelial cells, as determined by a BAC-cloned clinical isolate of human cytomegalovirus with preserved wild-type characteristics. J Virol. 2002;76:9551–9555. doi: 10.1128/JVI.76.18.9551-9555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patrone M, Percivalle E, Secchi M, Fiorina L, Pedrali-Noy G, et al. The human cytomegalovirus UL45 gene product is a late, virion-associated protein and influences virus growth at low multiplicities of infection. J Gen Virol. 2003;84:3359–3370. doi: 10.1099/vir.0.19452-0. [DOI] [PubMed] [Google Scholar]
- 24.Dufour F, Sasseville AM, Chabaud S, Massie B, Siegel RM, et al. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFalpha- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis. 2011;16:256–271. doi: 10.1007/s10495-010-0560-2. [DOI] [PubMed] [Google Scholar]
- 25.He S, Wang L, Miao L, Wang T, Du F, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 26.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 27.Sun L, Wang X. A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends Biochem Sci. 2014;39:587–593. doi: 10.1016/j.tibs.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Chan FK, Luz NF, Moriwaki K. Programmed Necrosis in the Cross Talk of Cell Death and Inflammation. Annu Rev Immunol. 2014 doi: 10.1146/annurev-immunol-032414-112248. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun L, Wang H, Wang Z, He S, Chen S, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 30.Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109:5322–5327. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 32.Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem. 2013;288 doi: 10.1074/jbc.M112.435545. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111:15072–15077. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moujalled DM, Cook WD, Okamoto T, Murphy J, Lawlor KE, et al. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis. 2013;4:e465. doi: 10.1038/cddis.2012.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McQuade T, Cho Y, Chan FK. Positive and negative phosphorylation regulates RIP1 and RIP3-induced programmed necrosis. Biochem J. 2013;456:409–415. doi: 10.1042/BJ20130860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho Y, McQuade T, Zhang H, Zhang J, Chan FK. RIP1-Dependent and Independent Effects of Necrostatin-1 in Necrosis and T Cell Activation. PLoS One. 2011;6:e23209. doi: 10.1371/journal.pone.0023209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu J, Huang Z, Ren J, Zhang Z, He P, et al. Mlkl knockout mice demonstrate the indispensable role of MLKL in necroptosis. Cell Res. 2013;23:994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 40.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, et al. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, et al. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feoktistova M, Geserick P, Panayotova-Dimitrova D, Leverkus M. Pick your poison: The ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle. 2012;11:460–467. doi: 10.4161/cc.11.3.19060. [DOI] [PubMed] [Google Scholar]
- 43.Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tait SW, Oberst A, Quarato G, Milasta S, Haller M, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013;5:878–885. doi: 10.1016/j.celrep.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Raam BJ, Salvesen GS. Proliferative versus apoptotic functions of caspase-8 hetero or homo: the caspase-8 dimer controls cell fate. Biochim Biophys Acta. 2012;1824:113–122. doi: 10.1016/j.bbapap.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Sun L, Su L, Rizo J, Liu L, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 48.Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–981. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 49.Holler N, Zaru R, Micheau O, Thome M, Attinger A, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 50.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng S, Yang Y, Mei Y, Ma L, Zhu DE, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19:2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 53.McCormick AL, Mocarski ES. Cell death pathways controlled by cytomegaloviruses. In: Reddehase MJ, editor. Cytomegaloviruses: From Molecular Pathogenesis to Intervention. Norfolk, United Kingdom: Caister Scientific Press; 2013. pp. 263–276. [Google Scholar]
- 54.Hedrick SM, Ch’en IL, Alves BN. Intertwined pathways of programmed cell death in immunity. Immunol Rev. 2010;236:41–53. doi: 10.1111/j.1600-065X.2010.00918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silke J, Meier P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a008730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walczak H. TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol Rev. 2011;244:9–28. doi: 10.1111/j.1600-065X.2011.01066.x. [DOI] [PubMed] [Google Scholar]
- 57.Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U S A. 2014;111:7753–7758. doi: 10.1073/pnas.1401857111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. Mol Cell. 2014;56:469–480. doi: 10.1016/j.molcel.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;2:e230. doi: 10.1038/cddis.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 61.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 62.Crook NE, Clem RJ, Miller LK. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J Virol. 1993;67:2168–2174. doi: 10.1128/jvi.67.4.2168-2174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Birnbaum MJ, Clem RJ, Miller LK. An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J Virol. 1994;68:2521–2528. doi: 10.1128/jvi.68.4.2521-2528.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clem RJ. The role of apoptosis in defense against baculovirus infection in insects. Curr Top Microbiol Immunol. 2005;289:113–129. doi: 10.1007/3-540-27320-4_5. [DOI] [PubMed] [Google Scholar]
- 65.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, Zhang H, Li J, Rosenberg S, Zhang EC, et al. RIP1-mediated regulation of lymphocyte survival and death responses. Immunol Res. 2011;51:227–236. doi: 10.1007/s12026-011-8249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu JV, Weist BM, van Raam BJ, Marro BS, Srinivas P, et al. Complementary roles of FADD and RIPK3 in T cell homeostasis and antiviral immunity. Proc Natl Acad Sci U S A. 2011;108:15312–15317. doi: 10.1073/pnas.1102779108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med. 2011;208:633–641. doi: 10.1084/jem.20110251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35:572–582. doi: 10.1016/j.immuni.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 71.Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 72.Weinlich R, Dillon CP, Green DR. Ripped to death. Trends Cell Biol. 2011;21:630–637. doi: 10.1016/j.tcb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Newton K, Dugger DL, Wickliffe KE, Kapoor N, Cristina de-Almagro M, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 74.Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, et al. Survival function of the FADD-Caspase-8-cFLIP(L) complex. Cell Rep. 2012;1:401–407. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157:1189–1202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–1188. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 77.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa B signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24:1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moriwaki K, Bertin J, Gough PJ, Chan FK. A RIPK3-Caspase 8 Complex Mediates Atypical Pro-IL-1beta Processing. J Immunol. 2015 doi: 10.4049/jimmunol.1402167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Galvan V, Roizman B. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proc Natl Acad Sci U S A. 1998;95:3931–3936. doi: 10.1073/pnas.95.7.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leopardi R, Roizman B. The herpes simplex virus major regulatory protein ICP4 blocks apoptosis induced by the virus or by hyperthermia. Proc Natl Acad Sci U S A. 1996;93:9583–9587. doi: 10.1073/pnas.93.18.9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leopardi R, Van Sant C, Roizman B. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc Natl Acad Sci U S A. 1997;94:7891–7896. doi: 10.1073/pnas.94.15.7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benetti L, Roizman B. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci U S A. 2004;101:9411–9416. doi: 10.1073/pnas.0403160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Munger J, Roizman B. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc Natl Acad Sci U S A. 2001;98:10410–10415. doi: 10.1073/pnas.181344498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou G, Avitabile E, Campadelli-Fiume G, Roizman B. The domains of glycoprotein D required to block apoptosis induced by herpes simplex virus 1 are largely distinct from those involved in cell-cell fusion and binding to nectin1. J Virol. 2003;77:3759–3767. doi: 10.1128/JVI.77.6.3759-3767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou G, Roizman B. Truncated forms of glycoprotein D of herpes simplex virus 1 capable of blocking apoptosis and of low-efficiency entry into cells form a heterodimer dependent on the presence of a cysteine located in the shared transmembrane domains. J Virol. 2002;76:11469–11475. doi: 10.1128/JVI.76.22.11469-11475.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Perkins D, Yu Y, Bambrick LL, Yarowsky PJ, Aurelian L. Expression of herpes simplex virus type 2 protein ICP10 PK rescues neurons from apoptosis due to serum deprivation or genetic defects. Exp Neurol. 2002;174:118–122. doi: 10.1006/exnr.2001.7849. [DOI] [PubMed] [Google Scholar]
- 87.Perkins D, Pereira EF, Gober M, Yarowsky PJ, Aurelian L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J Virol. 2002;76:1435–1449. doi: 10.1128/JVI.76.3.1435-1449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Langelier Y, Bergeron S, Chabaud S, Lippens J, Guilbault C, et al. The R1 subunit of herpes simplex virus ribonucleotide reductase protects cells against apoptosis at, or upstream of, caspase-8 activation. J Gen Virol. 2002;83:2779–2789. doi: 10.1099/0022-1317-83-11-2779. [DOI] [PubMed] [Google Scholar]
- 89.Chabaud S, Sasseville AM, Elahi SM, Caron A, Dufour F, et al. The ribonucleotide reductase domain of the R1 subunit of herpes simplex virus type 2 ribonucleotide reductase is essential for R1 antiapoptotic function. J Gen Virol. 2007;88:384–394. doi: 10.1099/vir.0.82383-0. [DOI] [PubMed] [Google Scholar]
- 90.Han JY, Miller SA, Wolfe TM, Pourhassan H, Jerome KR. Cell type-specific induction and inhibition of apoptosis by Herpes Simplex virus type 2 ICP10. J Virol. 2009;83:2765–2769. doi: 10.1128/JVI.02088-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aurelian L. HSV-induced apoptosis in herpes encephalitis. Curr Top Microbiol Immunol. 2005;289:79–111. doi: 10.1007/3-540-27320-4_4. [DOI] [PubMed] [Google Scholar]
- 92.Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science. 2000;287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 93.Du T, Zhou G, Roizman B. Induction of apoptosis accelerates reactivation of latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc Natl Acad Sci U S A. 2012;109:14616–14621. doi: 10.1073/pnas.1212661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Galvan V, Brandimarti R, Roizman B. Herpes simplex virus 1 blocks caspase-3-independent and caspase-dependent pathways to cell death. J Virol. 1999;73:3219–3226. doi: 10.1128/jvi.73.4.3219-3226.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Peri P, Mattila RK, Kantola H, Broberg E, Karttunen HS, et al. Herpes simplex virus type 1 Us3 gene deletion influences toll-like receptor responses in cultured monocytic cells. Virol J. 2008;5:140. doi: 10.1186/1743-422X-5-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peri P, Nuutila K, Vuorinen T, Saukko P, Hukkanen V. Cathepsins are involved in virus-induced cell death in ICP4 and Us3 deletion mutant herpes simplex virus type 1-infected monocytic cells. J Gen Virol. 2011;92:173–180. doi: 10.1099/vir.0.025080-0. [DOI] [PubMed] [Google Scholar]
- 97.Lembo D, Brune W. Tinkering with a viral ribonucleotide reductase. Trends Biochem Sci. 2009;34:25–32. doi: 10.1016/j.tibs.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 98.Huang Z, Wu S-Q, Liang Y, Zhou X, Chen W, et al. Targeting HSV-1 protein ICP6 by RIP1 and RIP3 initiates necroptosis to restrict HSV-1 propagation in mice. Cell Host Microbe. 2015;17:229–242. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 99.Wang X, Li Y, Liu S, Yu X, Li L, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A. 2014;111:15438–15443. doi: 10.1073/pnas.1412767111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008;4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]