

Abstract
OBJECT
Our previous studies have suggested that thymosin beta 4 (Tβ4), a major actin-sequestering protein, improves functional recovery after neural injury. N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP) is an active peptide fragment of Tβ4 and its effect as a treatment of traumatic brain injury (TBI) has not been investigated. Thus, this study was designed to determine whether AcSDKP treatment improves functional recovery in rats after TBI.
METHODS
Young adult male Wistar rats with TBI were randomly divided into the following groups: (1) Sham group (no injury); (2) Vehicle group (0.01N acetic acid); (3) AcSDKP (0.8 mg/kg/day). TBI was induced by controlled cortical impact over the left parietal cortex. AcSDKP or vehicle was administered subcutaneously starting at 1 hour post injury and continuously for 3 days using an osmotic minipump. Sensorimotor function and spatial learning were assessed using a modified neurological severity score and Morris water maze tests, respectively. Some of the animals were sacrificed 1 day after injury and their brains were processed for measurement of fibrin accumulation and neuroinflammation signaling pathways. The remaining animals were sacrificed 35 days after injury and brain sections were processed for measurement of lesion volume, hippocampal cell loss, angiogenesis, neurogenesis and dendritic spine remodeling.
RESULTS
Compared to the vehicle treatment, AcSDKP treatment initiated 1 hour post injury significantly improved sensorimotor functional recovery (Days 7–35, p<0.05) and spatial learning (Days 33–35, p<0.05), reduced cortical lesion volume, and hippocampal neuronal cell loss, reduced fibrin accumulation and activation of microglia/macrophages, enhanced angiogenesis and neurogenesis, and increased the number of dendritic spines in the injured brain (p<0.05). AcSDKP treatment also significantly inhibited the transforming growth factor β1/nuclear factor-kappa B signaling pathway.
CONCLUSIONS
AcSDKP treatment initiated 1 hour post injury provides neuroprotection and neurorestoration after TBI, indicating that this small tetrapeptide has promising therapeutic potential for treatment of TBI. Further investigation of the optimal dose and therapeutic window of AcSDKP treatment for TBI and the associated underlying mechanisms, are therefore warranted.
Keywords: angiogenesis, N-acetyl-seryl-aspartyl-lysyl-proline, functional outcome, controlled cortical impact, neurogenesis, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a serious public health problem worldwide.30 In the US, according to estimates from the Centers for Disease Control and Prevention (CDC), there are approximately 2.5 million TBI related emergency department (ED) visits each year, hospitalizations, and deaths. Among these persons, approximately 87% were treated and released from EDs, 11% were hospitalized and discharged, and 2% died.59 An estimated 5.3 million Americans live with TBI-related disability.37 Although significant efforts have been devoted to the development of neuroprotective agents, all these efforts have failed to demonstrate efficacy in clinical trials of TBI.52,68,83 Clinical trials in TBI have mainly focused on targeting a single pathophysiological pathway.47 However, successful therapy may require targeting multiple injury pathways.77
N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP), a small tetrapeptide, inhibits inflammation,14 fibrosis,14 primitive hematopoietic cell proliferation,38 and promotes endothelial cell proliferation and angiogenesis.14,43,78 It was originally purified from fetal calf bone marrow,38 and its protein precursor is thymosin β4 (Tβ4).27 AcSDKP was first reported as an endogenous hematopoiesis regulator that reversibly inhibited, in vitro and in vivo, the entry of hematopoietic pluripotent stem cells and normal early progenitors into the S phase, maintaining them in the G0 phase and thereby obstructing the DNA synthesis.25,38,49 Angiotensin I-converting enzyme (ACE) inhibitors selectively inhibit AcSDKP degradation and result in an increased plasma level of AcSDKP.6,34,62 Increased AcSDKP level dramatically restrains cardiac and renal fibrosis in hypertensive rats without affecting blood pressure or organ hypertrophy, which suggest that the anti-fibrotic effect of ACE inhibitors maybe related to their ability to increase the level of AcSDKP in plasma.56 ACE inhibitors applied 2 hours prior to induction of stroke in the rat reduced infarct volume.57 Although several studies indicate that ACE inhibitors improve the recovery after cerebral ischemia,41,70,79 a previous study on TBI rats treated with the ACE inhibitors, prior to injury and 7 days thereafter, showed that ACE inhibitor treatment exacerbated motor deficits following TBI, which may be related to its potential effect of increasing neuropeptide substance P activity.29 Administration of AcSDKP prevents the development of diabetic cardiomyopathy and renal damage, which underscores the end-organ protective effects of AcSDKP.12,13,67 However, the biological function of AcSDKP in the brain is unclear. We have recently demonstrated that treatment with AcSDKP initiated 1 hour after stroke onset effectively reduced infarct volume and neurological deficits in adult rats, which provides the first evidence that AcSDKP has a neuroprotective effect.88 However, the effect of AcSDKP on functional recovery in TBI rats has not been investigated. In this study, we subcutaneously infused AcSDKP to rats subjected to TBI induced by controlled cortical impact, and we investigated cognitive and sensorimotor functional recovery as well as the potential mechanisms underlying therapeutic effects of AcSDKP.
Methods
All study procedures were approved by the Institutional Animal Care and Use Committee of Henry Ford Health System. To prevent potential biases of performance and detection, the persons who performed the experiments, collected data, and assessed outcome were blinded throughout the course of the experiments and were unaware of the treatment allocation.
Animal Model and Experimental Groups
The controlled cortical impact rat model of TBI was employed for this study.20 Adult male Wistar rats (2–3 months old) weighing 300 to 335 g (Charles River Laboratories) were anesthetized with chloral hydrate (350 mg/kg body weight) intraperitoneally. Rectal temperature was maintained at 37°C ± 5°C by using a feedback-regulated water heating pad. Rats were placed in a stereotactic frame. Two 10-mm-diameter craniotomies were performed adjacent to the central suture, midway between the Lambda and Bregma. The second craniotomy allowed for lateral movement of cortical tissue. The dura mater was kept intact over the cortex. Cortical injury was delivered by impacting the left (ipsilateral) cortex with a pneumatic piston containing a 6-mm-diameter tip at a rate of 4 m/sec and 2.5 mm of compression. Velocity was measured with a linear velocity displacement transducer. For the present study, rats were randomly divided into 3 groups (n=15/group): Group 1, sham (craniotomies only, no injury); Group 2, TBI + vehicle (0.01N acetic acid,); and Group 3, TBI+ AcSDKP (0.8 mg/kg/day, Abbiotec). AcSDKP or an equal volume of 0.01N acetic acid was administered 1 hour after injury subcutaneously with an osmotic pump (Alzet model 1003D) for 3 days. The dose of AcSDKP used in the present study was chosen based on our previous study in rats after stroke, which was shown to provide potent neuroprotection.88 To label proliferating cells after injury, 100 mg/kg 5-bromo-2′-deoxyuridine (BrdU)86 was injected intraperitoneally into rats daily for 10 days, starting 1 day after injury. Seven animals from each group were sacrificed 1 day after injury for Western blot analysis (n=3) and immunostaining for neuroinflammation (n=4). Remaining rats were allowed to survive for 35 days after injury (n=8/group).
Evaluation of Neurological Outcome
Modified Neurological Severity Score Test (mNSS)
To evaluate neurological functional recovery, the modified Neurological Severity Score (mNSS) test15 was performed prior to injury and at 1, 7, 14, 21, 28, and 35 days after injury, which was graded from 0–18 (normal score 0; maximal deficit score 18). Our previous studies utilized the mNSS to test the motor (muscle status, abnormal movement), sensory (visual, tactile, and proprioceptive), and reflex reactions.45 For each abnormal behavior or for lack of an expected reaction, 1 point is awarded, which means that the higher the score the more severe the injury.
Foot-Fault Test
To evaluate sensorimotor function, the foot-fault test was employed prior to injury and at 1, 7, 14, 21, 28, and 35 days after injury. With each weight-bearing step walking on a grid, a paw might fall or slip from the wires. Each fall or slip was recorded as a foot fault.8 A total of 50 steps were recorded for the right forelimb.
Morris Water Maze (MWM) Test
To detect spatial learning functional recovery, a modified version of the MWM test was used.16 From Day 31 to Day 35 after injury, animals were tested with 4 trials per day consecutively. A swimming pool for rats (1.8 m in diameter) was located in a room with many clues (such as pictures on the walls, lamps, and a camera on the ceiling) which were visible to rats from the pool for spatial orientation. The position of the cues remained fixed throughout the experiment. Data were collected by the Human Visual Image (HVS) Image 2020 Plus Tracking System (US HVS Image).46 The swimming pool was divided into 4 equal quadrants formed by imaging lines. Throughout the test period, the platform was located in the northeast (NE) quadrant 2 cm below water in a randomly changing position during 4 trials every day, including locations against the wall, toward the middle of the pool, or off-center but always within the target quadrant. At each trial, the rat was placed at one of four fixed starting points (designated North, South, East and West), facing toward the wall of pool. The animal was allowed 90 seconds to find the submerged and nonvisible transparent platform. Once found, rats were allowed to remain on the platform for 10 seconds. If the animal failed to find the platform within 90 seconds, it was placed on the platform for 10 seconds, and the trial was terminated and 90 seconds was assigned as the score. The percentage of time the animals spent swimming within the target quadrant relative to the total amount of time swimming in the maze before reaching the hidden platform or within 90 seconds for those rats that failed to find the platform was recorded for statistical analysis.
Tissue Preparation
Western blot analysis and immunostaining for neuroinflammation: On day 1 after injury, rats were anesthetized with 4% chloral hydrate intraperitoneally and then perfused with saline solution transcardially. The ipsilateral cortical tissue from the lesion boundary zone was dissected, frozen in liquid nitrogen, and stored at −80°C until use. Immunohistochemistry analysis: On day 35, rats were anesthetized with chloral hydrate intraperitoneally and then perfused with saline solution transcardially, followed by 4% paraformaldehyde in 0.1 M PBS (pH 7.4). After perfusion, rat brains were removed and fixed in 4% paraformaldehyde for 2 days and then each forebrain was cut into 2-mm-thick coronal blocks for a total of 7 blocks.55 All blocks were embedded in paraffin and were cut into a series of 6-μm-thick sections.
Lesion Volume Analysis
To measure the lesion volume after injury, hematoxylin and eosin staining was used to demarcate the lesion area of each coronal section. To measure lesion volume after injury, one 6-mm-thick section from each of 7 coronal blocks was traced by a microcomputer imaging device (MCID, Imaging Research), as described previously.87 The formula to calculate the percentage of cortical lesion volume was: [(contralateral cortical volume - ipsilateral cortical volume)/ (contralateral cortical volume)] × 100%.71
Immunohistochemical Studies
Antigen retrieval was performed by boiling sections in 10 mM citrate buffer (pH 6) for 10 minutes. After washing with PBS, sections were incubated with 0.3% H2O2 in PBS for 10 minutes, blocked with 1% bovine serum albumin containing 0.3% Triton X-100 at room temperature for 1 hour, and incubated with either anti–endothelial barrier antigen (EBA, 1:1000; Covance), anti-CD68 (1:200; AbD, Serotec, Kidlington, UK) or anti–glial fibrillary acidic protein (GFAP, 1:1000; Dako) or anti-NeuN monoclonal antibody (1:300, MAB 377, Chemicon) or anti-synaptophysin antibody (1:800; MAB5258, Millipore) or anti-fibrin antibody (1:1000, Acc Chem) at 4°C overnight. For negative controls, primary antibodies were omitted. After washing, sections were incubated with biotinylated anti–mouse or anti–rabbit antibodies (1:200; Vector Laboratories, Inc.) at room temperature for 30 minutes. After an additional washing, sections were incubated with an avidin-biotin peroxidase system (ABC kit; Vector Laboratories, Inc.), visualized with diaminobenzidine (Sigma), and counterstained with hematoxylin.
Immunofluorescent Staining
Double immunostaining was performed to identify newly generated endothelial cells (BrdU/EBA+) and newly formed mature neurons (BrdU/NeuN+) 35 days after injury. Briefly, after being deparaffinized and rehydrated, brain sections were boiled in 10 mM citric acid buffer (pH 6) for 10 minutes. After washing with PBS, sections were incubated in 2.4 N HCl at 37°C for 20 minutes. Sections were incubated with 1% bovine serum albumin containing 0.3% Triton X-100 in PBS. Sections were then incubated with mouse anti-NeuN antibody (1:200; Chemicon) or anti-EBA at 4°C overnight. For negative controls, primary antibodies were omitted. Fluorescein isothiocyanate–conjugated anti–mouse antibody (1:400; Jackson ImmunoResearch) was added to sections at room temperature for 2 hours. Sections were then incubated with rat anti-BrdU antibody (1:200; Dako) at 4°C overnight, and subsequently incubated with Cy3-conjugated goat anti–rat antibody (1:400; Jackson ImmunoResearch) at room temperature for 2 hours. Each of the steps was followed by three 5-minute rinses in PBS. Tissue sections were mounted with Vectashield mounting medium (Vector Laboratories).
Golgi-Cox Staining
To measure the number of neuronal dendritic spines, the FD Rapid Golgi Stain kit (FD NeuroTechnologies) was used to perform Golgi staining following the vendor’s protocol. Briefly, the freshly dissected brains were immersed in impregnation solution (made by mixing equal volumes of Solutions A and B) and stored at room temperature for 2 weeks in the dark. The brains were then transferred into solution C and kept for 48 hours at 4°C in the dark. Vibratome sections (100 μm) were cut and stained using standard staining procedures.
Western Blot Analysis
Brain tissue were washed in phosphate-buffered saline once and sonicated in lysis buffer (20 mM Tris pH 7.6, 100 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 1% deoxycholic acid, 10% glycerol, 1 mM EDTA, 1 mM NaVO3, 50 mM NaF, Protease Inhibitor Cocktail Set I from Calbiochem). Soluble protein was then obtained by centrifugation at 13,000 × g at 4°C for 15 minutes. Bicinchoninic acid (BCA) protein assay (Pierce) was used to measure total protein concentration. Equal amounts of lysate (20 μg protein/lane) were loaded to SDS-polyacrylamide electrophoresis on Novex Trisglycine precast gels (Invitrogen), and separated proteins were then electrotransferred to polyvinylidene fluoride (PVDF) membranes (Millipore). SuperSignal West Pico Chemiluminescent Substrate system (Thermo Scientific) was applied to visualize different proteins by reacting to various antibodies. The band intensity was analyzed using Scion Image software (Scion). Antibodies used for Western blot included anti-tumor transforming growth factor beta1 (TGF-β1) polyclonal antibody (Santa Cruz, SC-146); anti-nuclear factor-kappa B (NFkB) antibody (Abcam, ab7970) and anti-actin (1:5000, Santa Cruz Biotechnology).
Cell Counting and Quantitation
The CD68+ microglia/macrophages, glial fibrillary acidic protein (GFAP)+ astrocytes, neuronal nuclei (NeuN)+ neuronal cells, NeuN/BrdU double stained newborn mature neurons and EBA/BrdU-colabeled newborn endothelial cells were counted in the lesion boundary zone and the DG. For analysis of neurogenesis, we counted BrdU+ cells and NeuN/BrdU-colabeled cells in the DG and its subregions, including the subgranular zone, the granular cell layer and the molecular layer. The fields of interest were digitized under the light microscope (Eclipse 80i, Nikon) at a magnification of either 200 or 400, using a CoolSNAP color camera (Photometrics) interfaced with the MetaMorph image analysis system (Molecular Devices), as described in detail previously.91 Briefly, 5 fields of view in the lesion boundary zone from the epicenter of the injury cavity (Bregma −3.3 mm) and 9 fields of view in the ipsilateral DG were counted in each section. From our previous experience, our inter-rater reliability was greater than 95% when the cell counts were compared between 2 independent trained blinded observers scoring the same sections of an animal. In the present study, one blinded observer performed the cell counting in all brain sections.
Statistical Analysis
All data are presented as the mean ± SD. The analysis if variance (ANOVA) test was used for repeated measurements of spatial performance, sensorimotor function and Western blot analysis. For cell counting, a one-way ANOVA followed by post hoc Tukey’s tests was used to compare the differences between the AcSDKP-treated, acetic acid-treated and sham-injured groups. Differences were considered significant when the p value was < 0.05.
Results
AcSDKP Significantly Reduced Cortical Lesion Volume in Rats after TBI
Lesion volume was measured 35 days post injury. AcSDKP treatment significantly decreased lesion volume compared with the vehicle group (Fig. 1, F2, 21 = 318.50, q = 9.990, p < 0.05), indicating a protective effect of AcSDKP on brain tissue.
Fig. 1.
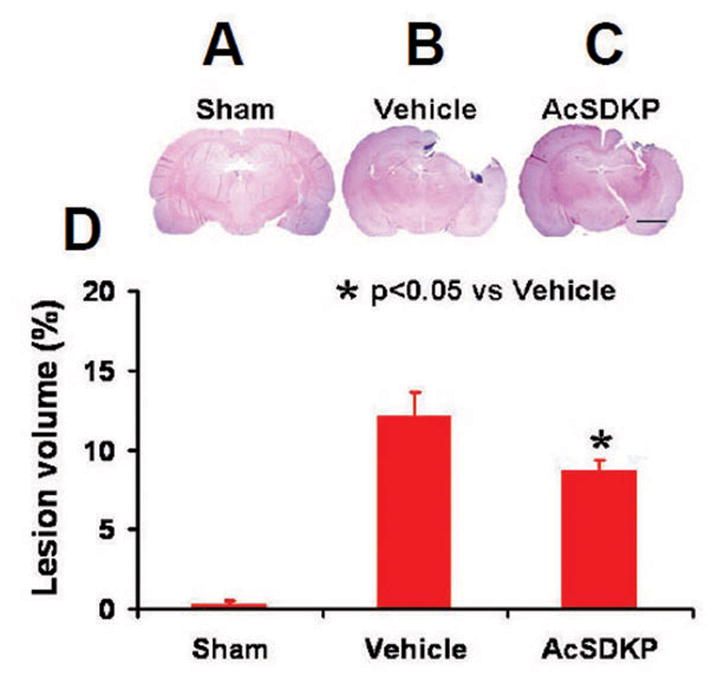
The effect of AcSDKP on cortical lesion volume examined 35 days after TBI. TBI caused significant cortical tissue loss (B, C). AcSDKP treatment significantly reduced lesion volume caused by TBI compared to the saline-treated groups (D). Scale bar = 3 mm. Data represent mean ± SD. There were 8 rats per group.
AcSDKP Significantly Enhanced Spatial Learning in Rats after TBI
MWM tests were performed during the last 5 days (31–35 days post-injury) before planned death to estimate the learning function of hippocampus. The higher the percentage of time animals spend in the correct quadrant where the hidden platform is located in the water maze, the better their spatial learning function. The percentage of time spent by AcSDKP treated rats in the correct quadrant increased significantly between 33 and 35 days compared to the vehicle group (Fig. 2A, at Day 33, F2, 21 = 34.27, q = 8.729, p < 0.05; at Day 34, F2, 21 = 29.58, q = 7.092, p < 0.05; and at Day 35, F2, 21 = 36.94, q = 8.871, p < 0.05). These data indicate that AcSDKP improves memory/learning functional recovery after TBI.
Fig. 2.
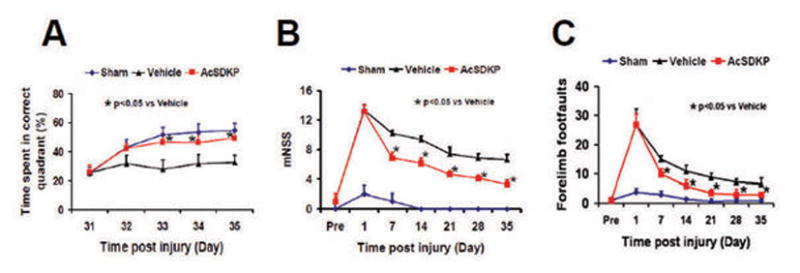
The effect of AcSDKP on spatial learning in the MWM test (A), sensorimotor function measured by mNSS (B), and right forelimb foot-fault test scores (C) in rats after TBI (8 rats per group). Data represent the mean ± SD.
AcSDKP Significantly Reduced Sensorimotor Deficits in Rats after TBI
The lower the score on the modified neurological severity score (mNSS) or footfault tests, the better the functional recovery. Spontaneous functional recovery occurred in the vehicle-treated group over the 35 days post injury. AcSDKP treatment significantly decreased mNSS score and footfault steps from Day 7 post injury compared to the vehicle group (Fig. 2B and 2C, for mNSS score, at Day 7, F2, 21 = 254.29, q = 19.886, p < 0.05; at Day 14, F2, 21 = 520.62, q = 16.174, p < 0.05; at Day 21, F2, 21 = 290.54, q = 13.298, p < 0.05; at Day 28, F2, 21 = 331.35, q = 15.109, p < 0.05; and at Day 35, F2, 21 = 245.87, q = 16.731, p < 0.05. For footfault test, at Day 7, F2, 21 = 114.28, q = 9.421, p < 0.05; at Day 14, F2, 21 = 46.68, q = 7.220, p < 0.05; at Day 21, F2, 21 = 91.57, q = 12.482, p < 0.05; at Day 28, F2, 21 = 39.87, q = 8.444, p < 0.05; and at Day 35, F2, 21 = 28.000, q = 6.792, p < 0.05). Together, these data indicate that AcSDKP treatment improves sensorimotor functional recovery after TBI.
AcSDKP Significantly Reduced Neuronal Cell Loss and Increased Neurogenesis in the Hippocampus in Rats after TBI
To investigate the effect of AcSDKP treatment on neuronal protection after TBI, anti-NeuN antibody was employed to identify neurons 35 days after injury. In the DG and CA3 regions of hippocampus, the NeuN (+) cell number in AcSDKP treatment group was significantly higher than that in the vehicle group (Fig. 3A–G, for DG, F2, 21 = 43.12, q = 9.708, p < 0.05; for CA3, F2, 21 = 56.12, q = 10.808, p < 0.05). In the DG, AcSDKP therapy significantly increased the number of BrdU+/NeuN+ cells (newly generated mature neurons) compared with the vehicle (Fig. 3H–K, F2, 21 = 120.50, q = 12.658, p < 0.05). These data in concert suggest that AcSDKP treatment promotes neuroprotection and neurogenesis in the DG in rats after TBI.
Fig. 3.
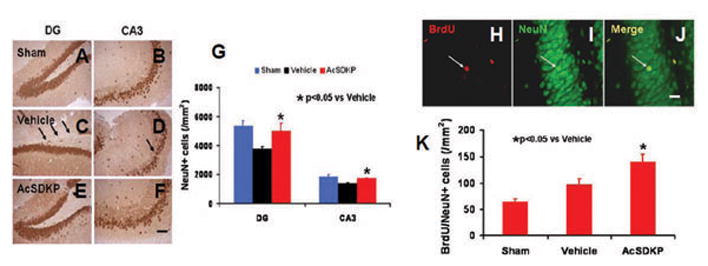
The effects of AcSDKP on the number of neuronal cells and BrdU/NeuN-positive newborn mature neurons in the dentate gyrus 35 days after TBI. Anti-NeuN staining was performed to identify neuronal cells in the dentate gyrus (A–F), Scale bar = 40 μm (A–F). Compared with the vehicle group (C, D), AcSDKP treatment (E, F) significantly increased NeuN-positive neuronal cells in the dentate gyrus 35 days after TBI. The bar graph (G) shows the number of NeuN-positive neuron cells. Compared to the vehicle group, AcSDKP treatment significantly increased newborn mature neurons identified with BrdU/NeuN double immunofluorescent staining in the dentate gyrus as indicated by white arrows (J), at 35 days postinjury. The bar graph (K) shows the number of BrdU/NeuN-positive newborn mature neurons. Scale bar = 20 μm (H–J). Data in the graphs represent mean ± SD. There were 8 rats per group.
AcSDKP Significantly Increased Angiogenesis in Rats after TBI
To identify newly generated - vasculature in the brain, BrdU/EBA double staining was performed 35 days after injury. BrdU+/EBA+ staining showed that AcSDKP treatment significantly increased newborn - endothelial cells compared to the vehicle group (Fig. 4, for cortex, F2, 21 = 244.00, q = 13.066, p < 0.05; for CC, F2, 21 = 333.23, q = 18.255, p < 0.05; for DG, F2, 21 = 244.95, q = 19.243, p < 0.05; for CA3, F2, 21 = 365.26, q = 11.593, p < 0.05; and for CA1, F2, 21 = 242.67, q = 13.064, p < 0.05), which suggests that AcSDKP promotes angiogenesis after TBI.
Fig. 4.
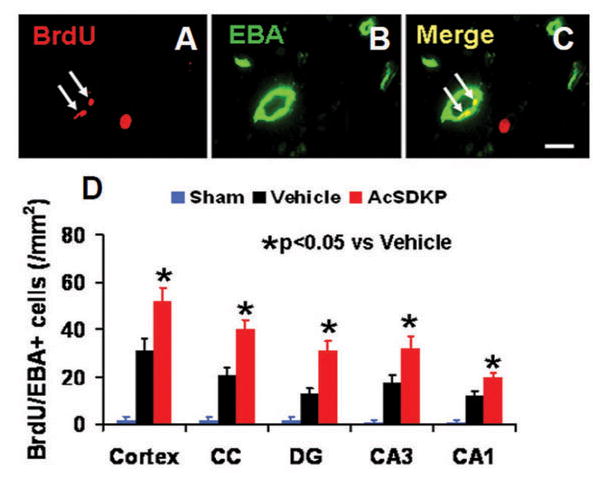
The effect of AcSDKP on the number of newly generated vessels in rats 35 days after TBI (8 rats per group). Double staining for BrdU (A, white arrows) and EBA (B, green signal) was performed to identify newly formed mature vessels (C, white arrows) in the brain at Day 35 after TBI in the lesion boundary zone and DG area. Scale bar = 20 μm (A–C). Data in the bar graphs (D) represent the mean ± SD.
AcSDKP Significantly Increased the Number of Dendritic Spines in the Injured Brain Regions after TBI
Neuronal dendritic spines were detected following Golgi-Cox staining 35 days after injury. Compared to the vehicle group, AcSDKP treatment group showed a significantly increased spine number after TBI (Fig. 5, for cortex, F2, 9 = 19.13, q = 6.852, p < 0.05; for DG, F2, 9 = 12.80, q = 4.828, p < 0.05; for CA3, F2, 9 = 67.75, q = 7.778, p < 0.05; and for CA1, F2, 9 = 29.38, q = 7.575, p < 0.05), which suggests that AcSDKP has protective and/or neural plasticity effects on dendritic spines in TBI rats.
Fig. 5.
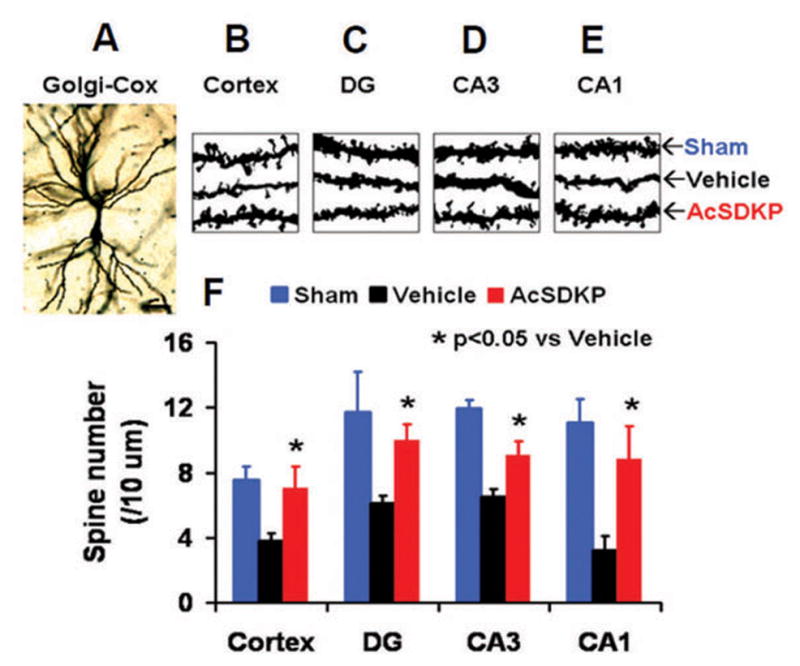
The effect of AcSDKP on the number of dendritic spines in the injured brain35 days after TBI. Golgi staining was used to identify neuronal dendritic spines in the brain (A–E). Compared to the vehicle group, AcSDKP treatment significantly increased the number of neuronal dendritic spines in the brain 35 days after TBI. The bar graph (F) shows the number of neuronal dendritic spines. There were 3–4 rats per group.
AcSDKP Significantly Increased Synaptophysin Expression in the Injured Brain after TBI
Synaptophysin was employed to measure the density of synapses 35 days after injury. In the injured rats treated with vehicle, the density of synapses was remarkably decreased. However, AcSDKP administration after injury significantly decreased the loss of synapses (Fig. 6, for cortex, F2, 21 = 58.77, q = 7.013, p < 0.05; for DG, F2, 21 = 45.57, q = 7.283, p < 0.05; for CA3, F2, 21 = 28.58, q = 10.688, p < 0.05; and for CA1, F2, 21 = 41.85, q = 7.126, p < 0.05), which suggests that AcSDKP reduces synapse loss and/or promotes synaptic plasticity after TBI.
Fig. 6.
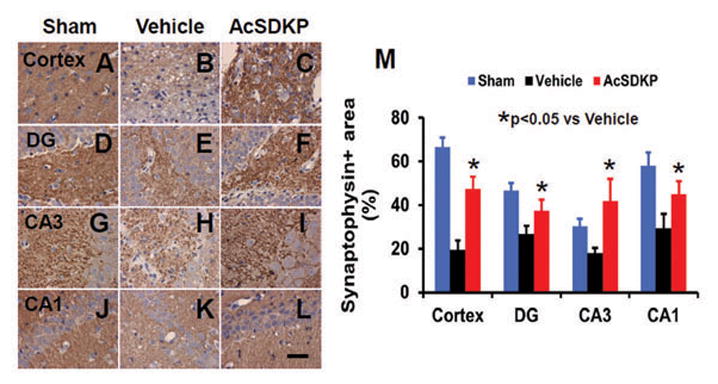
The effect of AcSDKP on synaptophysin expression 35 days after TBI. AcSDKP treatment (C, F, I, L) significantly increased synaptophysin expression in various brain regions 35 days after TBI compared to the saline group (B, E, H, K). DG = dentate gyrus. The bar graph (M) shows that synaptophysin-positive area. Scale bar = 20 μm. Data in graph represent mean ± SD. There were 8 rats per group.
AcSDKP Significantly Reduced the Number of CD68+Microglia/Macrophages in the Injured Brain Regions after TBI
To evaluate effects of AcSDKP on neuroinflammation, CD68 staining was used to detect microglia/microphages in TBI rats.87 Thirty-five day after injury, inflammation was notably increased, while AcSDKP treatment significantly decreased the CD68+ cells compared to vehicle group (Fig. 7, for cortex, F2, 21 = 600.39, q = 32.950, p < 0.05; for CC, F2, 21 = 259.80, q = 23.001, p < 0.05; for DG, F2, 21 = 658.24, q = 21.296, p < 0.05; for CA3, F2, 21 = 700.83, q = 29.112, p < 0.05; and for CA1, F2, 21 = 265.47, q = 28.610, p < 0.05), which indicates that AcSDKP significantly attenuates neuroinflammation after TBI.
Fig. 7.
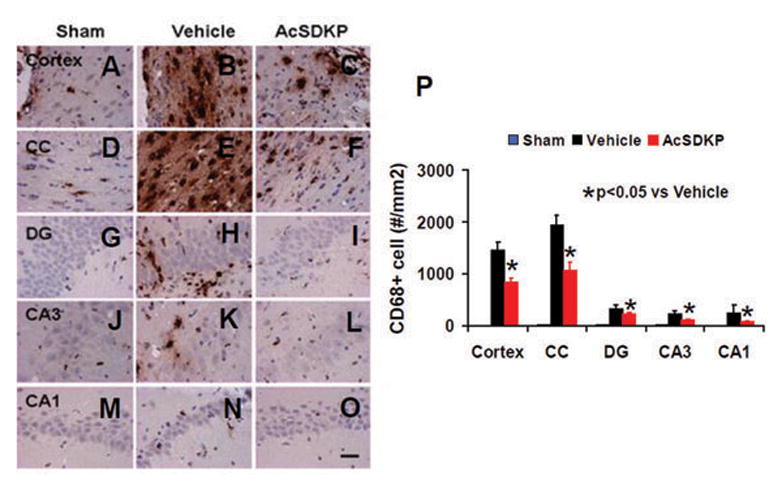
The effect of AcSDKP on microglia/macrophages in the injured brain 35 days after TBI. CD68 staining was performed to detect activation of microglia/macrophages 35 days after TBI. AcSDKP treatment (C, F, I, L, and O) significantly decreased CD68-positive cells in various brain regions 35 days after TBI compared to the saline group (B, E, H, K, and N). DG = dentate gyrus. The bar graph (P) shows the CD68-positive cells. Scale bar = 20 μm. Data in graph represent mean ± SD. There were 8 rats per group.
AcSDKP Decreased GFAP Expression in the Injured Brain after TBI
Astrocytes were activated post TBI as also shown in our previous studies.90,92 GFAP staining was performed to detect astrocytes 35 days after injury. There was a significantly decreased GFAP+ cell number in AcSDKP treatment group compared to vehicle group (Fig. 8, for cortex, F2, 21 = 195.31, q = 17.726, p < 0.05; for CC, F2, 21 = 386.08, q = 25.169, p < 0.05; for DG, F2, 21 = 181.07, q = 15.360, p < 0.05; for CA3, F2, 21 = 192.40, q = 14.777, p < 0.05; and for CA1, F2, 21 = 572.92, q = 26.673, p < 0.05), which suggests that AcSDKP reduces reactive astrogliosis after TBI.
Fig. 8.
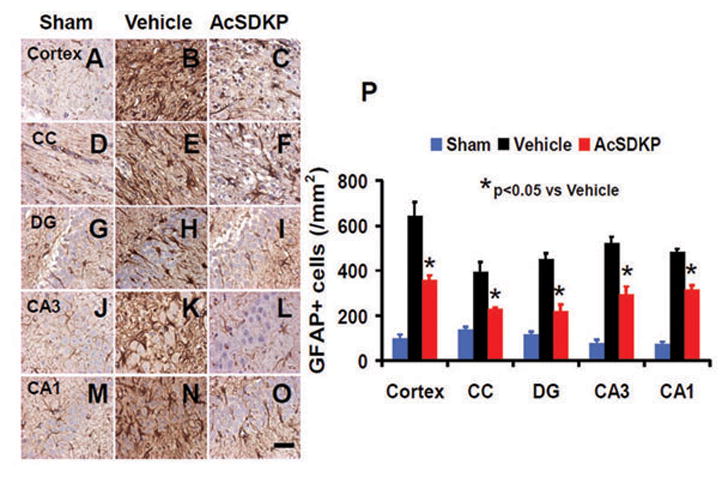
The effect of AcSDKP on astrocyte activation. GFAP staining was performed to detect activation of astrocytes 35 days after TBI. Some weak expression of GFAP was observed in brain regions of sham animals (A, D, G, J, and M). AcSDKP treatment (C, F, I, L, and O) significantly decreased GFAP-positive cells in various brain regions 35 days after TBI compared with the saline group where prominent astrogliosis exists (B, E, H, K, and N). CC = corpus callosum; DG = dentate gyrus. The data on GFAP-positive cells are shown in the bar graph (P). Scale bar = 20 μm. Data in graph represent mean ± SD. There were 8 rats per group.
AcSDKP Significantly Reduced Fibrin in the Injured Brain after TBI
To investigate the effects of AcSDKP on fibrin deposition, staining with fibrinogen/fibrin antibody was performed on brain sections one day after TBI. Compared to vehicle-treated rats, the percentage of fibrin+ area was significantly decreased after AcSDKP treatment in TBI rats (Fig. 9, for cortex, F2, 9 = 42.88, q = 8.908, p < 0.05; for CC, F2, 9 = 50.53, q = 12.312, p < 0.05; for DG, F2, 9 = 23.32, q = 7.978, p < 0.05; for CA3, F2, 9 = 87.16, q = 16.905, p < 0.05; and for CA1, F2, 9 = 14.36, q = 5.686, p < 0.05), which indicates that AcSDKP may have protective effect of vascular and blood-brain barrier (BBB) integrity after TBI.
Fig. 9.
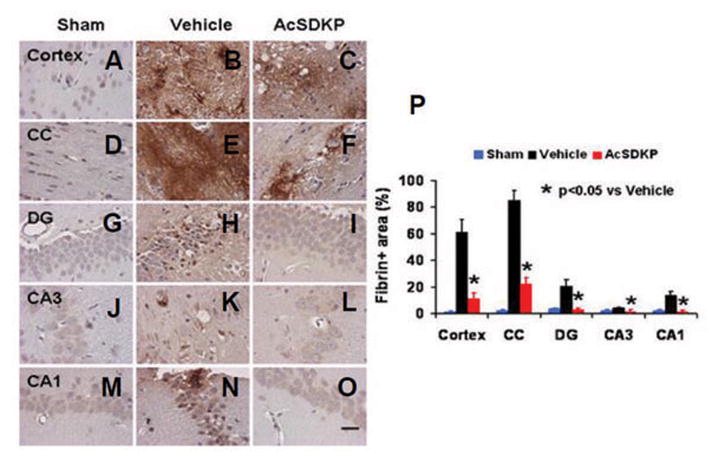
The effect of AcSDKP on brain fibrin accumulation one day after TBI. Fibrin was weakly expressed in brain regions of sham animals (A, D, G, J, and M). Compared with the vehicle group (B, E, H, K, and N), AcSDKP treatment (C, F, I, L, and O) significantly reduced fibrin accumulation in various brain regions 1 day after TBI. The data on fibrin positive areas are shown in the bar graph (P). CC = corpus callosum; DG = dentate gyrus. Scale bar = 20 μm. Data in the graph represent mean ± SD. There were 4 rats per group.
AcSDKP Significantly Reduced Expression of TGF-β1 and NFkB after TBI
Compared to the vehicle treatment, AcSDKP significantly reduced the expression of TGF-β1 and NFkB protein in the lesion boundary zone one day after TBI (Fig. 10, for TGF-β1, F2, 6 = 7.69, q = 5.075, p < 0.05; and for NFkB, F2, 6 = 9.21, q = 5.612, p < 0.05),which suggests that AcSDKP contributes to neuroprotection by suppressing the TGF-β1 and NFkB expression which modulates cerebral vascular patency and integrity, and neuroinflammation after brain injury.28
Fig. 10.
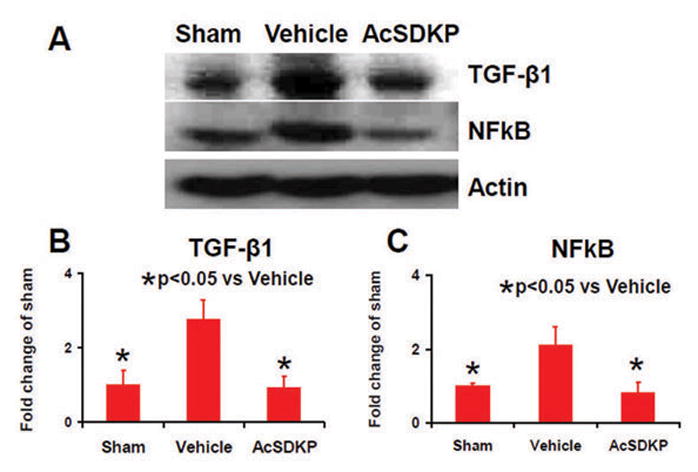
The effects of AcSDKP on the expression of TGF-β1 and NFkB one day after TBI. Protein levels of TGF-β1 and NFkB were measured by Western blot analysis in the injured cortical tissue (A). Bar graphs (B, C) show that AcSDKP treatment significantly reduced the expression of TGF-β1 and NFkB one day after TBI. There were 3 rats per group.
Discussion
The principal findings of the present study are as follows, AcSDKP treatment 1 hour after TBI, significantly: 1) improved cognitive and sensorimotor functional recovery compared to the vehicle treatment; 2) reduced lesion volume, hippocampal neuronal cell loss, fibrin deposit and the number of activated microglia/macrophages and reactive astrogliosis in the injured brain; 3) increased the number of newborn vessels and mature neurons as well as synaptophysin expression and the number of dendritic spines in the injured brain; 4) reduced protein expression of TGF-β1 and NFkB in the injured brain. These findings suggest that continuous infusion of AcSDKP initiated 1 hour post injury for 3 days not only provides neuroprotection but also promotes neurovascular remodeling in TBI rats. These dual effects may maximize functional recovery in TBI rats, implying that AcSDKP is a multifunctional agent and has promising therapeutic potential for treatment of TBI.
Increasing evidence suggests that neuroinflammation occurs in both acute and chronic stages of TBI,1,31,73 which may impact the pathophysiology and treatment of TBI. Neuroinflammation is present after injury and persists for a long period of time,72 which may provide a wide therapeutic window for anti-inflammatory intervention after TBI. After TBI, the rapid inflammatory response is followed by neuronal injury and BBB rupture.51,69 Within minutes, activated microglial cells are detected and resemble peripheral macrophages by proinflammatory cytokines and chemokines.10 In our study, CD68, a marker for microglia/macrophages,32 was used to evaluate the neuroinflammation after TBI. Compared to vehicle-treated rats, the number of CD68 positive microglia/macrophages was dramatically decreased in the AcSDKP-treated rats, suggesting infusion of AcSDKP initiated 1 hour post injury and continued for 3 days effectively suppresses activation of microglia/macrophages after TBI and reduces neuroinflammation.
Astrocytes secrete inflammatory mediators which play an important role in secondary injury after TBI.22 GFAP, is a sensitive marker for activated astrocytes.22 Our study showed that few astrocytes expressed GFAP in normal brain tissue, while a robust increase of GFAP was expressed in astrocytes after TBI, a finding consistent with our prior study9. AcSDKP administration significantly reduced numbers of GFAP reactive astrocytes. A previous clinical study indicates that TBI patients who died within first 24 hours have a significantly higher GFAP concentration in the blood at 6, 12, and 24 hours after TBI, moreover, a high level of GFAP in blood was associated with poor outcome at 1–6 months.19
From our study, newly generated vessels identified with BrdU/EBA (+) staining were detected in the lesion boundary zone and DG of the rats after TBI, indicating that TBI induces angiogenesis, which is consist with previous studies.50,82,85 AcSDKP further enhances angiogenesis in these regions. AcSDKP promotes formation of capillary-like structure in vitro by activating the proliferation and migration of endothelial cells and increases capillary density in rat heart with myocardial infarction.43,78 Magnetic resonance imaging (MRI) indices including cerebral blood volume (CBV), cerebral blood flow (CBF), blood-to-brain transfer constant (Ki) were used to monitor development of angiogenesis after TBI noninvasively, which found that new vessels were permeable at the early phase of angiogenesis and became less permeable as they mature.39,40 On the Ki map, angiogenic areas became apparent 3 to 4 weeks after TBI.39 Furthermore, in the ipsilateral DG regions after TBI, elevated CBV was observed starting from day 1 to 2 weeks after injury, which suggests that newly generated vessels by TBI-induced angiogenesis are functional. These newborn vessels may contribute to functional recovery after TBI, since angiogenesis is coupled with and may drive neurogenesis in the DG.75,82
Neurogenesis occurs in the subventricular zone of the lateral ventricle and the subgranular zone of the dentate gyrus in mammals during adulthood and the pathology of different neurological disorders.24,74,93 Neurogenesis is stimulated by TBI in rodents and humans.60,94 Newly generated neuroblasts in the subventricular zone can migrate to the injured area and become mature neurons after TBI.11,17,26 AcSDKP not only significantly reduced neuronal cell loss in the hippocampus, but also increased the number of newly born neurons and dendritic spines, which may contribute to the memory recovery after TBI. Additionally, cortical lesion volume of brain was significantly reduced with AcSDKP treatment initiated 1 hour after TBI, which is consistent with our previous study showing that Tβ4 administered 6 hours after TBI significantly reduced lesion volume.86 Further studies to investigate the effects of delayed, e.g. 6 hour AcSDKP treatment post TBI, time points that are more realistic for pre-clinical and clinical trials, are now warranted. The protection of cortex by AcSDKP may contribute to improving sensorimotor functional recovery (reduced mNSS and footfault number). Synaptophysin is a well-established marker for a presynaptic vesicle membrane protein.80 The elevated expression of synaptophysin in our study with AcSDKP treatment is consistent with previous studies,23 suggesting that increased synaptophysin level in the rat brain is related to improved spatial memory. This is also in agreement with our Golgi-Cox staining showing that AcSDKP treatment increased the number of dendritic spines. Based on our current study, AcSDKP amplifies angiogenesis, neurogenesis and synaptogenesis in the hippocampus, which may contribute to cognitive functional recovery after TBI. However, increased neurogenesis is not always good for brain function. Experimental status epilepticus can also cause a marked increase in adult neurogenesis.61 It has been speculated that seizure-induced aberrant neurogenesis contributes to deficits in hippocampal learning and memory that are associated with status epilepticus.61 However, the present study showed an enhancement in spatial memory, suggesting that AcSDKP-induced newborn neurons are functionally integrated into or enhanced neuronal circuitry. Thus, an AcSDKP-induced increase in neurogenesis may be favorable for spatial learning after TBI. Our present data are in agreement with a recent study demonstrating that intrahippocampal infusion of AcSDKP facilitated the generation of new neurons in the hippocampus and enhanced spatial memory in normal mice.36 In our current study, the level of AcSDKP was not determined in the plasma and the brain after TBI. AcSDKP can pass the BBB in normal rats and stroke rats,88 indicating that AcSDKP is a promising potential treatment for neural injuries.
Fibrinogen is known for its role as the protein component of blood clots and is normally excluded from the brain parenchyma via the BBB.42,63,65 BBB damage occurs after TBI.76 One of the earliest events after TBI is leakage of blood components into brain parenchyma in areas that correlate with the formation of reactive astrocytes.58,66 The soluble blood protein fibrinogen is converted to insoluble fibrin by the action of thrombin and is deposited in the nervous system promptly following vascular damage or BBB disruption.2 Fibrinogen plays a causative role in nervous system disease as a regulator of inflammation,2,54 remyelination,3 and neurodegeneration.18,64 Fibrinogen regulates TGF-β1-mediated signal transduction within central nervous system (CNS) tissues after vascular damage and induces reactive astrogliosis and deposition of chondroitin sulfate proteoglycans (CSPGs).65 Mice genetically or pharmacologically depleted of fibrinogen show a dramatic reduction in TGF-β1 and reduced astrogliosis and neurocan deposition after injury.65 In primary astrocyte cultures, fibrinogen is a potent inducer of secretion of proteoglycans, and conditioned medium of fibrinogen-treated astrocytes inhibits neurite outgrowth.65 Our data indicate that fibrin accumulates in the injured brain regions after TBI, and AcSDKP decreases the accumulation and improves functional recovery, suggesting that fibrin plays a critical role in pathophysiology contributing to functional deficits after TBI.
NFkB can be activated by inflammatory mediators7 and its activation results in the induction of inflammatory responsive genes in TBI.89 Activation of NFkB is present in astrocytes after injury and induces astrocyte swelling/brain edema after TBI.33 Correspondingly, NFkB inhibition significantly reduced trauma-induced astrocyte swelling.33 In our study, the NFkB level was obviously decreased to nearly normal level in the AcSDKP treatment group 1 day after TBI compared to vehicle-treated injured rats. This is in agreement with our previous study demonstrating that inactivation of the TGF-β1/NFkB signaling by AcSDKP may contribute to its neuroprotection in stroke rats.88
TGF-β1 has multiple functions in various organs, regulating the growth, differentiation and survival of different cell types.21 The role of TGF-β1 is complex. TGF-β1 has neuroprotective effects,21 but also participates in neuroinflammation and the scarring response in the rat brain.44 A recent study indicates that blood protein fibrinogen, after leaking into the CNS after BBB disruption, served as an early signal for the induction of glial scar formation via the TGF-β1 signaling pathway.65 Our data suggest that brain TGF-β1 expression was increased after TBI. More importantly, our data show that AcSDKP treatment significantly reduced fibrin accumulation, TGF-β1 expression, neuroinflammation, and astrogliosis. These data suggest that TGF-β1 plays a critical role in brain damage after TBI and AcSDKP can attenuate these detrimental effects mediated by TGF-β1. Together, our findings strongly suggest that: 1) the fibrin-mediated TGF-β1 signaling pathway contributes to brain damage; and 2) AcSDKP provides neuroprotection and promotes neurovascular remodeling likely by inhibiting TGF-β1 signaling. These neuroprotective and neurorestorative effects of AcSDKP, in concert, may contribute to improved functional recovery after TBI.
Our data indicate that continuous infusion of AcSDKP initiated 1 hour post injury for 3 days not only provides neuroprotection but also promotes neurovascular remodeling in TBI rats. These dual effects may maximize functional recovery in TBI rats. We are aware that in the vast majority of preclinical research, the treatment compounds are administered early and, frequently, even prior to the TBI.47 The administration of a compound early by pre-hospital care personnel may be problematic because practical time constraints associated with injury and patient management, and of the difficulty in obtaining informed consent.48 Our previous study suggests that delayed Tβ4 treatment initiated 24 hours post injury significantly improves histological and functional outcomes in rats with TBI.84 Thus, investigation whether AcSDKP delivered in the subacute (e.g. at or >24 hours postinjury) period after TBI improves functional outcome is warranted. Treatments targeting neurovascular remodeling at a greatly extended treatment window if successful can be made available for all brain injury.
Several characteristics of AcSDKP indicate that this tetrapeptide is a promising neurovascular protective and remodeling agent for TBI, and therefore merits further preclinical development and evaluation. Firstly, while the BBB poses significant challenges to the intraparenchymal delivery of neuroprotective agents, AcSDKP with a low molecular weight of 488 Daltons can readily cross the BBB in the intact and injured brain, as demonstrated in our previous study.88 Secondly, as a naturally occurring peptide, the pharmacokinetics and metabolism of AcSDKP have been well established and there is no apparent toxicity in rodents.5,35,67,81 More importantly, with the identification of multiple molecular mechanisms and mediators on the pathogenesis of TBI, the multi-targeted effects of AcSDKP on the neurovascular unit could achieve optimized therapeutic efficacy. AcSDKP is cleared almost exclusively by angiotensin I-converting enzyme (ACE).4 An alternative approach to increase AcSDKP levels in plasma is the use of ACE inhibitors.4 Some centrally acting ACE inhibitors may slow disease progression in patients with Alzheimer’s disease.53 However, ACE inhibitors have multiple other effects and exacerbate the histological damage and motor deficits in a rat model of diffuse TBI induced by impact acceleration, likely by regulating substance P-mediated neuronal injury.29 Our data imply that administration of AcSDKP may be a novel treatment of TBI.
In conclusion, in the present study, we demonstrate that early (1 hour post injury) administration of AcSDKP provides neuroprotection (reduced neuronal loss and cortical lesion size, fibrin accumulation), promotes neurovascular remodeling (increased angiogenesis, neurogenesis and dendritic spines), reduces neuroinflammation and astrogliosis, as well as improves functional recovery in rats after TBI, which may be mediated, in part, by AcSDKP’s inhibition of TGF-β1/NFkB signal pathway. Thus, AcSDKP is a multifunctional neuroprotective, neurorestorative and anti-inflammatory agent for treatment of TBI. Further investigation of the optimal dose and therapeutic window of AcSDKP treatment for TBI and the associated underlying mechanisms of action are warranted.
Acknowledgments
Sources of financial support: National Institutes of Health Grant RO1 NS 079612 (Zheng Gang Zhang)
Abbreviations
- AcSDKP
N-acetyl-seryl-aspartyl-lysyl-proline
- ACE
Angiotensin I-converting enzyme
- APC
antigen presenting cell
- BrdU
5′-bromo-2′-deoxyuridine
- BBB
blood-brain barrier
- CDC
Centers for Disease Control and Prevention
- DG
dentate gyrus
- ED
emergency department
- GFAP
glial fibrillary acidic protein
- MCAO
middle cerebral artery occlusion
- mNSS
Modified Neurological Severity Score Test
- MWM
Morris water maze
- NeuN
neuronal nuclei
- NFkB
nuclear factor-kappa B
- NO
nitric oxide
- TBI
traumatic brain injury
- Tβ4
thymosin β4
- TGF-β1
Transforming growth factor β1
Footnotes
The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.
Disclosure
This work was partially supported by National Institutes of Health Grant RO1 NS 079612 (Zheng Gang Zhang).
Author contributions to the study and manuscript preparation include the following: Conception and design: Xiong, Chopp, L Zhang, ZG Zhang, Mahmood. Acquisition of data: Y Zhang, Meng, Xiong, L Zhang. Analysis and interpretation of data: all authors. Drafting the article: Y Zhang, Xiong, Chopp. Critically revising the article: all authors. Reviewed submitted version of manuscript: all authors. Approved the final version of the manuscript on behalf of all authors: Xiong. Statistical analysis: Xiong, Y Zhang. Administrative/technical/material support: Xiong, Chopp, L Zhang, ZG Zhang, Mahmood. Study supervision: Xiong, Chopp.
References
- 1.Acosta SA, Tajiri N, Shinozuka K, Ishikawa H, Sanberg PR, Sanchez-Ramos J, et al. Combination therapy of human umbilical cord blood cells and granulocyte colony stimulating factor reduces histopathological and motor impairments in an experimental model of chronic traumatic brain injury. PLoS One. 2014;9:e90953. doi: 10.1371/journal.pone.0090953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams RA, Schachtrup C, Davalos D, Tsigelny I, Akassoglou K. Fibrinogen signal transduction as a mediator and therapeutic target in inflammation: lessons from multiple sclerosis. Curr Med Chem. 2007;14:2925–2936. doi: 10.2174/092986707782360015. [DOI] [PubMed] [Google Scholar]
- 3.Akassoglou K, Yu WM, Akpinar P, Strickland S. Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. Neuron. 2002;33:861–875. doi: 10.1016/s0896-6273(02)00617-7. [DOI] [PubMed] [Google Scholar]
- 4.Azizi M, Ezan E, Nicolet L, Grognet JM, Menard J. High plasma level of N-acetyl-seryl-aspartyl-lysyl-proline: a new marker of chronic angiotensin-converting enzyme inhibition. Hypertension. 1997;30:1015–1019. doi: 10.1161/01.hyp.30.5.1015. [DOI] [PubMed] [Google Scholar]
- 5.Azizi M, Ezan E, Reny JL, Wdzieczak-Bakala J, Gerineau V, Menard J. Renal and metabolic clearance of N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP) during angiotensin-converting enzyme inhibition in humans. Hypertension. 1999;33:879–886. doi: 10.1161/01.hyp.33.3.879. [DOI] [PubMed] [Google Scholar]
- 6.Azizi M, Rousseau A, Ezan E, Guyene TT, Michelet S, Grognet JM, et al. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J Clin Invest. 1996;97:839–844. doi: 10.1172/JCI118484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 8.Baskin YK, Dietrich WD, Green EJ. Two effective behavioral tasks for evaluating sensorimotor dysfunction following traumatic brain injury in mice. J Neurosci Methods. 2003;129:87–93. doi: 10.1016/s0165-0270(03)00212-7. [DOI] [PubMed] [Google Scholar]
- 9.Beschorner R, Dietz K, Schauer N, Mittelbronn M, Schluesener HJ, Trautmann K, et al. Expression of EAAT1 reflects a possible neuroprotective function of reactive astrocytes and activated microglia following human traumatic brain injury. Histol Histopathol. 2007;22:515–526. doi: 10.14670/HH-22.515. [DOI] [PubMed] [Google Scholar]
- 10.Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience. 2012;225:65–75. doi: 10.1016/j.neuroscience.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capilla-Gonzalez V, Lavell E, Quinones-Hinojosa A, Guerrero-Cazares H. Regulation of subventricular zone-derived cells migration in the adult brain. Adv Exp Med Biol. 2015;853:1–21. doi: 10.1007/978-3-319-16537-0_1. [DOI] [PubMed] [Google Scholar]
- 12.Castoldi G, di Gioia CR, Bombardi C, Perego C, Perego L, Mancini M, et al. Prevention of myocardial fibrosis by N-acetyl-seryl-aspartyl-lysyl-proline in diabetic rats. Clin Sci (Lond) 2009;118:211–220. doi: 10.1042/cs20090234. [DOI] [PubMed] [Google Scholar]
- 13.Castoldi G, di Gioia CR, Bombardi C, Preziuso C, Leopizzi M, Maestroni S, et al. Renal antifibrotic effect of N-acetyl-seryl-aspartyl-lysyl-proline in diabetic rats. Am J Nephrol. 2013;37:65–73. doi: 10.1159/000346116. [DOI] [PubMed] [Google Scholar]
- 14.Cavasin MA. Therapeutic potential of thymosin-beta4 and its derivative N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) in cardiac healing after infarction. Am J Cardiovasc Drugs. 2006;6:305–311. doi: 10.2165/00129784-200606050-00003. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32:2682–2688. doi: 10.1161/hs1101.098367. [DOI] [PubMed] [Google Scholar]
- 16.Choi SH, Woodlee MT, Hong JJ, Schallert T. A simple modification of the water maze test to enhance daily detection of spatial memory in rats and mice. J Neurosci Methods. 2006;156:182–193. doi: 10.1016/j.jneumeth.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Christie KJ, Turnley AM. Regulation of endogenous neural stem/progenitor cells for neural repair-factors that promote neurogenesis and gliogenesis in the normal and damaged brain. Front Cell Neurosci. 2012;6:70. doi: 10.3389/fncel.2012.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cortes-Canteli M, Zamolodchikov D, Ahn HJ, Strickland S, Norris EH. Fibrinogen and altered hemostasis in Alzheimer’s disease. J Alzheimers Dis. 2012;32:599–608. doi: 10.3233/JAD-2012-120820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Battista AP, Buonora JE, Rhind SG, Hutchison MG, Baker AJ, Rizoli SB, et al. Blood Biomarkers in Moderate-To-Severe Traumatic Brain Injury: Potential Utility of a Multi-Marker Approach in Characterizing Outcome. Front Neurol. 2015;6:110. doi: 10.3389/fneur.2015.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 21.Dobolyi A, Vincze C, Pal G, Lovas G. The neuroprotective functions of transforming growth factor beta proteins. Int J Mol Sci. 2012;13:8219–8258. doi: 10.3390/ijms13078219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- 23.Frick KM, Fernandez SM. Enrichment enhances spatial memory and increases synaptophysin levels in aged female mice. Neurobiol Aging. 2003;24:615–626. doi: 10.1016/s0197-4580(02)00138-0. [DOI] [PubMed] [Google Scholar]
- 24.Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22:612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaudron S, Grillon C, Thierry J, Riches A, Wierenga PK, Wdzieczak-Bakala J. In vitro effect of acetyl-N-Ser-Asp-Lys-Pro (AcSDKP) analogs resistant to angiotensin I-converting enzyme on hematopoietic stem cell and progenitor cell proliferation. Stem Cells. 1999;17:100–106. doi: 10.1002/stem.170100. [DOI] [PubMed] [Google Scholar]
- 26.Ghashghaei HT, Lai C, Anton ES. Neuronal migration in the adult brain: are we there yet? Nat Rev Neurosci. 2007;8:141–151. doi: 10.1038/nrn2074. [DOI] [PubMed] [Google Scholar]
- 27.Grillon C, Rieger K, Bakala J, Schott D, Morgat JL, Hannappel E, et al. Involvement of thymosin beta 4 and endoproteinase Asp-N in the biosynthesis of the tetrapeptide AcSerAspLysPro a regulator of the hematopoietic system. FEBS Lett. 1990;274:30–34. doi: 10.1016/0014-5793(90)81322-f. [DOI] [PubMed] [Google Scholar]
- 28.Harari OA, Liao JK. NF-kappaB and innate immunity in ischemic stroke. Ann N Y Acad Sci. 2010;1207:32–40. doi: 10.1111/j.1749-6632.2010.05735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harford-Wright E, Thornton E, Vink R. Angiotensin-converting enzyme (ACE) inhibitors exacerbate histological damage and motor deficits after experimental traumatic brain injury. Neurosci Lett. 2010;481:26–29. doi: 10.1016/j.neulet.2010.06.044. [DOI] [PubMed] [Google Scholar]
- 30.Hayes RL, Jenkins LW, Lyeth BG. Neurotransmitter-mediated mechanisms of traumatic brain injury: acetylcholine and excitatory amino acids. J Neurotrauma. 1992;9(Suppl 1):S173–187. [PubMed] [Google Scholar]
- 31.Hernandez-Ontiveros DG, Tajiri N, Acosta S, Giunta B, Tan J, Borlongan CV. Microglia activation as a biomarker for traumatic brain injury. Front Neurol. 2013;4:30. doi: 10.3389/fneur.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holness CL, Simmons DL. Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood. 1993;81:1607–1613. [PubMed] [Google Scholar]
- 33.Jayakumar AR, Tong XY, Ruiz-Cordero R, Bregy A, Bethea JR, Bramlett HM, et al. Activation of NF-kappaB mediates astrocyte swelling and brain edema in traumatic brain injury. J Neurotrauma. 2014;31:1249–1257. doi: 10.1089/neu.2013.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Junot C, Menard J, Gonzales MF, Michaud A, Corvol P, Ezan E. In vivo assessment of captopril selectivity of angiotensin I-converting enzyme inhibition: differential inhibition of acetyl-ser-asp-lys-pro and angiotensin I hydrolysis. J Pharmacol Exp Ther. 1999;289:1257–1261. [PubMed] [Google Scholar]
- 35.Kanasaki K, Nagai T, Nitta K, Kitada M, Koya D. N-acetyl-seryl-aspartyl-lysyl-proline: a valuable endogenous anti-fibrotic peptide for combating kidney fibrosis in diabetes. Front Pharmacol. 2014;5:70. doi: 10.3389/fphar.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim DH, Moon EY, Yi JH, Lee HE, Park SJ, Ryu YK, et al. Peptide fragment of thymosin beta4 increases hippocampal neurogenesis and facilitates spatial memory. Neuroscience. 2015;310:51–62. doi: 10.1016/j.neuroscience.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 37.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- 38.Lenfant M, Wdzieczak-Bakala J, Guittet E, Prome JC, Sotty D, Frindel E. Inhibitor of hematopoietic pluripotent stem cell proliferation: purification and determination of its structure. Proc Natl Acad Sci U S A. 1989;86:779–782. doi: 10.1073/pnas.86.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L, Chopp M, Ding GL, Qu CS, Li QJ, Lu M, et al. MRI measurement of angiogenesis and the therapeutic effect of acute marrow stromal cell administration on traumatic brain injury. J Cereb Blood Flow Metab. 2012;32:2023–2032. doi: 10.1038/jcbfm.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li L, Jiang Q, Qu CS, Ding GL, Li QJ, Wang SY, et al. Transplantation of marrow stromal cells restores cerebral blood flow and reduces cerebral atrophy in rats with traumatic brain injury: in vivo MRI study. J Neurotrauma. 2011;28:535–545. doi: 10.1089/neu.2010.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linz W, Scholkens BA, Han YF. Beneficial effects of the converting enzyme inhibitor, ramipril, in ischemic rat hearts. J Cardiovasc Pharmacol. 1986;8(Suppl 10):S91–99. doi: 10.1097/00005344-198600101-00017. [DOI] [PubMed] [Google Scholar]
- 42.Liu HM, Sturner WQ. Extravasation of plasma proteins in brain trauma. Forensic Sci Int. 1988;38:285–295. doi: 10.1016/0379-0738(88)90174-0. [DOI] [PubMed] [Google Scholar]
- 43.Liu JM, Lawrence F, Kovacevic M, Bignon J, Papadimitriou E, Lallemand JY, et al. The tetrapeptide AcSDKP, an inhibitor of primitive hematopoietic cell proliferation, induces angiogenesis in vitro and in vivo. Blood. 2003;101:3014–3020. doi: 10.1182/blood-2002-07-2315. [DOI] [PubMed] [Google Scholar]
- 44.Logan A, Berry M, Gonzalez AM, Frautschy SA, Sporn MB, Baird A. Effects of transforming growth factor beta 1 on scar production in the injured central nervous system of the rat. Eur J Neurosci. 1994;6:355–363. doi: 10.1111/j.1460-9568.1994.tb00278.x. [DOI] [PubMed] [Google Scholar]
- 45.Lu D, Mahmood A, Qu C, Hong X, Kaplan D, Chopp M. Collagen scaffolds populated with human marrow stromal cells reduce lesion volume and improve functional outcome after traumatic brain injury. Neurosurgery. 2007;61:596–602. doi: 10.1227/01.NEU.0000290908.38438.B2. discussion 602–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahmood A, Lu D, Qu C, Goussev A, Chopp M. Treatment of traumatic brain injury with a combination therapy of marrow stromal cells and atorvastatin in rats. Neurosurgery. 2007;60:546–553. doi: 10.1227/01.NEU.0000255346.25959.99. discussion 553–544. [DOI] [PubMed] [Google Scholar]
- 47.Marklund N, Hillered L. Animal modelling of traumatic brain injury in preclinical drug development: where do we go from here? Br J Pharmacol. 2011;164:1207–1229. doi: 10.1111/j.1476-5381.2010.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menon DK. Unique challenges in clinical trials in traumatic brain injury. Crit Care Med. 2009;37:S129–135. doi: 10.1097/CCM.0b013e3181921225. [DOI] [PubMed] [Google Scholar]
- 49.Monpezat JP, Frindel E. Further studies on the biological activities of the CFU-S inhibitory tetrapeptide AcSDKP. I. The precise point of the cell cycle sensitive to AcSDKP. Studies on the effect of AcSDKP on GM-CFC and on the possible involvement of T-lymphocytes in AcSDKP response. Exp Hematol. 1989;17:1077–1080. [PubMed] [Google Scholar]
- 50.Morgan R, Kreipke CW, Roberts G, Bagchi M, Rafols JA. Neovascularization following traumatic brain injury: possible evidence for both angiogenesis and vasculogenesis. Neurol Res. 2007;29:375–381. doi: 10.1179/016164107X204693. [DOI] [PubMed] [Google Scholar]
- 51.Namas R, Ghuma A, Hermus L, Zamora R, Okonkwo DO, Billiar TR, et al. The acute inflammatory response in trauma / hemorrhage and traumatic brain injury: current state and emerging prospects. Libyan J Med. 2009;4:97–103. doi: 10.4176/090325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, et al. Clinical trials in head injury. J Neurotrauma. 2002;19:503–557. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Caoimh R, Healy L, Gao Y, Svendrovski A, Kerins DM, Eustace J, et al. Effects of centrally acting angiotensin converting enzyme inhibitors on functional decline in patients with Alzheimer’s disease. J Alzheimers Dis. 2014;40:595–603. doi: 10.3233/JAD-131694. [DOI] [PubMed] [Google Scholar]
- 54.Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J Exp Med. 2007;204:1999–2008. doi: 10.1084/jem.20070304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 2. Sydney; Orlando: Academic Press; 1986. [Google Scholar]
- 56.Peng H, Carretero OA, Raij L, Yang F, Kapke A, Rhaleb NE. Antifibrotic effects of N-acetyl-seryl-aspartyl-Lysyl-proline on the heart and kidney in aldosterone-salt hypertensive rats. Hypertension. 2001;37:794–800. doi: 10.1161/01.hyp.37.2.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Porritt MJ, Chen M, Rewell SS, Dean RG, Burrell LM, Howells DW. ACE inhibition reduces infarction in normotensive but not hypertensive rats: correlation with cortical ACE activity. J Cereb Blood Flow Metab. 2010;30:1520–1526. doi: 10.1038/jcbfm.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Preston E, Webster J, Small D. Characteristics of sustained blood-brain barrier opening and tissue injury in a model for focal trauma in the rat. J Neurotrauma. 2001;18:83–92. doi: 10.1089/089771501750055794. [DOI] [PubMed] [Google Scholar]
- 59.Prevention CfDCa: REPORT TO CONGRESS Traumatic Brain Injury In the United States:Epidemiology and Rehabilitation. National Center for Injury Prevention and Control; Division of Unintentional Injury Prevention; Atlanta, GA: 2014. [Google Scholar]
- 60.Richardson RM, Sun D, Bullock MR. Neurogenesis after traumatic brain injury. Neurosurg Clin N Am. 2007;18:169–181. xi. doi: 10.1016/j.nec.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 61.Rotheneichner P, Marschallinger J, Couillard-Despres S, Aigner L. Neurogenesis and neuronal regeneration in status epilepticus. Epilepsia. 2013;54(Suppl 6):40–42. doi: 10.1111/epi.12274. [DOI] [PubMed] [Google Scholar]
- 62.Rousseau A, Michaud A, Chauvet MT, Lenfant M, Corvol P. The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J Biol Chem. 1995;270:3656–3661. doi: 10.1074/jbc.270.8.3656. [DOI] [PubMed] [Google Scholar]
- 63.Ryu JK, Davalos D, Akassoglou K. Fibrinogen signal transduction in the nervous system. J Thromb Haemost. 2009;7(Suppl 1):151–154. doi: 10.1111/j.1538-7836.2009.03438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schachtrup C, Lu P, Jones LL, Lee JK, Lu J, Sachs BD, et al. Fibrinogen inhibits neurite outgrowth via beta 3 integrin-mediated phosphorylation of the EGF receptor. Proc Natl Acad Sci U S A. 2007;104:11814–11819. doi: 10.1073/pnas.0704045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, et al. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J Neurosci. 2010;30:5843–5854. doi: 10.1523/JNEUROSCI.0137-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH. Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur J Neurosci. 1999;11:3648–3658. doi: 10.1046/j.1460-9568.1999.00792.x. [DOI] [PubMed] [Google Scholar]
- 67.Shibuya K, Kanasaki K, Isono M, Sato H, Omata M, Sugimoto T, et al. N-acetyl-seryl-aspartyl-lysyl-proline prevents renal insufficiency and mesangial matrix expansion in diabetic db/db mice. Diabetes. 2005;54:838–845. doi: 10.2337/diabetes.54.3.838. [DOI] [PubMed] [Google Scholar]
- 68.Skolnick BE, Maas AI, Narayan RK, van der Hoop RG, MacAllister T, Ward JD, et al. A clinical trial of progesterone for severe traumatic brain injury. N Engl J Med. 2014;371:2467–2476. doi: 10.1056/NEJMoa1411090. [DOI] [PubMed] [Google Scholar]
- 69.Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI, et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1997;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- 70.Stier CT, Jr, Benter IF, Ahmad S, Zuo HL, Selig N, Roethel S, et al. Enalapril prevents stroke and kidney dysfunction in salt-loaded stroke-prone spontaneously hypertensive rats. Hypertension. 1989;13:115–121. doi: 10.1161/01.hyp.13.2.115. [DOI] [PubMed] [Google Scholar]
- 71.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 72.Szmydynger-Chodobska J, Gandy JR, Varone A, Shan R, Chodobski A. Synergistic interactions between cytokines and AVP at the blood-CSF barrier result in increased chemokine production and augmented influx of leukocytes after brain injury. PLoS One. 2013;8:e79328. doi: 10.1371/journal.pone.0079328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tajiri N, Acosta SA, Shahaduzzaman M, Ishikawa H, Shinozuka K, Pabon M, et al. Intravenous transplants of human adipose-derived stem cell protect the brain from traumatic brain injury-induced neurodegeneration and motor and cognitive impairments: cell graft biodistribution and soluble factors in young and aged rats. J Neurosci. 2014;34:313–326. doi: 10.1523/JNEUROSCI.2425-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taupin P. The therapeutic potential of adult neural stem cells. Curr Opin Mol Ther. 2006;8:225–231. [PubMed] [Google Scholar]
- 75.Teng H, Zhang ZG, Wang L, Zhang RL, Zhang L, Morris D, et al. Coupling of angiogenesis and neurogenesis in cultured endothelial cells and neural progenitor cells after stroke. J Cereb Blood Flow Metab. 2008;28:764–771. doi: 10.1038/sj.jcbfm.9600573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thal SC, Neuhaus W. The blood-brain barrier as a target in traumatic brain injury treatment. Arch Med Res. 2014;45:698–710. doi: 10.1016/j.arcmed.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 77.Vink R, Nimmo AJ. Multifunctional drugs for head injury. Neurotherapeutics. 2009;6: 28–42. doi: 10.1016/j.nurt.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang D, Carretero OA, Yang XY, Rhaleb NE, Liu YH, Liao TD, et al. N-acetyl-seryl-aspartyl-lysyl-proline stimulates angiogenesis in vitro and in vivo. Am J Physiol Heart Circ Physiol. 2004;287:H2099–2105. doi: 10.1152/ajpheart.00592.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Werner C, Hoffman WE, Kochs E, Rabito SF, Miletich DJ. Captopril improves neurologic outcome from incomplete cerebral ischemia in rats. Stroke. 1991;22:910–914. doi: 10.1161/01.str.22.7.910. [DOI] [PubMed] [Google Scholar]
- 80.Wiedenmann B, Franke WW. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell. 1985;41:1017–1028. doi: 10.1016/s0092-8674(85)80082-9. [DOI] [PubMed] [Google Scholar]
- 81.Worou ME, Liao TD, D’Ambrosio M, Nakagawa P, Janic B, Peterson EL, et al. Renal protective effect of N-acetyl-seryl-aspartyl-lysyl-proline in dahl salt-sensitive rats. Hypertension. 2015;66:816–822. doi: 10.1161/HYPERTENSIONAHA.115.05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs. 2010;11:298–308. [PMC free article] [PubMed] [Google Scholar]
- 83.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiong Y, Mahmood A, Meng Y, Zhang Y, Zhang ZG, Morris DC, et al. Treatment of traumatic brain injury with thymosin beta(4) in rats. J Neurosurg. 2011;114:102–115. doi: 10.3171/2010.4.JNS10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xiong Y, Zhang Y, Mahmood A, Meng Y, Qu C, Chopp M. Erythropoietin mediates neurobehavioral recovery and neurovascular remodeling following traumatic brain injury in rats by increasing expression of vascular endothelial growth factor. Transl Stroke Res. 2011;2:619–632. doi: 10.1007/s12975-011-0120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xiong Y, Zhang Y, Mahmood A, Meng Y, Zhang ZG, Morris DC, et al. Neuroprotective and neurorestorative effects of thymosin beta4 treatment initiated 6 hours after traumatic brain injury in rats. J Neurosurg. 2012;116:1081–1092. doi: 10.3171/2012.1.JNS111729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang J, Zhang ZG, Morris D, Li Y, Roberts C, Elias SB, et al. Neurological functional recovery after thymosin beta4 treatment in mice with experimental auto encephalomyelitis. Neuroscience. 2009;164:1887–1893. doi: 10.1016/j.neuroscience.2009.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang L, Chopp M, Teng H, Ding G, Jiang Q, Yang XP, et al. Combination treatment with N-acetyl-seryl-aspartyl-lysyl-proline and tissue plasminogen activator provides potent neuroprotection in rats after stroke. Stroke. 2014;45:1108–1114. doi: 10.1161/STROKEAHA.113.004399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang R, Liu Y, Yan K, Chen L, Chen XR, Li P, et al. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J Neuroinflammation. 2013;10:106. doi: 10.1186/1742-2094-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang Y, Chopp M, Meng Y, Katakowski M, Xin H, Mahmood A, et al. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J Neurosurg. 2015;122:856–867. doi: 10.3171/2014.11.JNS14770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y, Chopp M, Meng Y, Zhang ZG, Doppler E, Mahmood A, et al. Improvement in functional recovery with administration of Cerebrolysin after experimental closed head injury. J Neurosurg. 2013;118:1343–1355. doi: 10.3171/2013.3.JNS122061. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y, Chopp M, Meng Y, Zhang ZG, Doppler E, Winter S, et al. Cerebrolysin improves cognitive performance in rats after mild traumatic brain injury. J Neurosurg. 2015;122:843–855. doi: 10.3171/2014.11.JNS14271. [DOI] [PubMed] [Google Scholar]
- 93.Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- 94.Zheng W, ZhuGe Q, Zhong M, Chen G, Shao B, Wang H, et al. Neurogenesis in adult human brain after traumatic brain injury. J Neurotrauma. 2013;30:1872–1880. doi: 10.1089/neu.2010.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]