

ABSTRACT
We recently discovered that exposure of enterohemorrhagic Escherichia coli (EHEC) to d-serine resulted in accumulation of this unusual amino acid, induction of the SOS regulon, and downregulation of the type III secretion system that is essential for efficient colonization of the host. Here, we have investigated the physiological relevance of this elevated SOS response, which is of particular interest given the presence of Stx toxin-carrying lysogenic prophages on the EHEC chromosome that are activated during the SOS response. We found that RecA elevation in response to d-serine, while being significant, was heterogeneous and not capable of activating stx expression or stx phage transduction to a nonlysogenic recipient. This “SOS-like response” was, however, capable of increasing the mutation frequency associated with low-level RecA activity, thus promoting genetic diversity. Furthermore, this response was entirely dependent on RecA and enhanced in the presence of a DNA-damaging agent, indicating a functional SOS response, but did not result in observable cleavage of the LexA repressor alone, indicating a controlled mechanism of induction. This work demonstrates that environmental factors not usually associated with DNA damage are capable of promoting an SOS-like response. We propose that this modulated induction of RecA allows EHEC to adapt to environmental insults such as d-serine while avoiding unwanted phage-induced lysis.
IMPORTANCE The SOS response is a global stress network that is triggered by the presence of DNA damage due to breakage or stalled replication forks. Activation of the SOS response can trigger the replication of lytic bacteriophages and promote genetic diversification through error-prone polymerases. We have demonstrated that the host-associated metabolite d-serine contributes to Escherichia coli niche specification and accumulates inside cells that cannot catabolize it. This results in a modulated activation of the SOS antirepressor RecA that is insufficient to trigger lytic bacteriophage but capable of increasing the SOS-associated mutation frequency. These findings describe how relevant signals not normally associated with DNA damage can hijack the SOS response, promoting diversity as E. coli strains adapt while avoiding unwanted phage lysis.
INTRODUCTION
The bacterial SOS response is a global network of DNA repair proteins that is triggered in the presence of DNA replication inhibition, DNA strand breaks, or DNA-damaging agents such as antibiotics, UV light, reactive oxygen species, and pressure (1, 2). The SOS regulon is under the control of the LexA repressor and consists of >40 genes in Escherichia coli (1, 3–5). Upon DNA damage, the LexA antirepressor RecA is activated by forming a filament structure with single-stranded DNA and promotes autocatalytic cleavage of LexA, thus relieving the repression of SOS-regulated genes (6–9). A second mechanism of SOS induction has also been described that involves the two-component sensor DpiAB, which activates the SOS network in response to β-lactam antibiotics (10). This induction is independent of DNA damage but instead is in response to replication inhibition by DpiA overproduction and results in transient halting of replication, allowing bacteria to survive otherwise lethal doses of antibiotics. Induction of the SOS response can also have further effects on the cell, including activation of the lytic life cycle of lysogenic bacteriophages (11, 12). Temperate lysogenic prophages are carried on bacterial chromosomes and are repressed against replication by the cI phage repressor, a structural homologue of LexA. During the SOS response, activated RecA can also cleave cI, forcing the phage out of latency and thus resulting in lysis of the host cell (13–15). Furthermore, bacteriophages also often carry virulence genes and therefore can act as shuttles of horizontal gene transfer once activated (16).
Enterohemorrhagic E. coli (EHEC) is a Gram-negative foodborne pathogen associated with severe diarrheal illness in humans worldwide (17). The Centers for Disease Control and Prevention estimates >260,000 EHEC infections each year in the United States alone, with the common O157:H7 serotype being responsible for ∼36% of these (www.cdc.gov). The natural host for EHEC is ruminants, particularly cattle, where the organism asymptomatically colonizes the recto-anal junction (18, 19), which acts as a reservoir for indirect transmission to the human host through contamination of meat, fresh produce, and water sources. Cattle feces have been reported to harbor as many as 106 CFU of EHEC/g, and the infectious dose of EHEC in humans can be extremely low (10 to 100 bacteria of the most common O157:H7 serotype) (20). Disease progression of EHEC infections can range from mild diarrhea to hemorrhagic colitis and, in extreme cases, the potentially life-threatening hemolytic-uremic syndrome.
EHEC pathogenesis is mediated by a number of virulence factors that are carried primarily by horizontally acquired genomic elements (21, 22). The locus of enterocyte effacement is a large pathogenicity island containing five polycistronic operons that are responsible for the formation of a type III secretion system (T3SS), which facilitates attachment to host cells and translocation of numerous effector proteins into the host cell cytosol (23–25). This complex process ultimately results in the modulation of diverse host signaling pathways, leading to rearrangement of the host cell actin cytoskeleton around the bacterial site of attachment, forming a raised pedestal upon which the EHEC cell resides that is known as an attaching and effacing lesion (26). The second major virulence factor carried by EHEC is the Shiga-like toxin (Stx), of which there are a number of variants that are carried by lambdoid bacteriophages (27). Stx is an AB5-type cytotoxin composed of the enzymatically active A subunit noncovalently linked to the B unit pentamer (28). The B subunit recognizes the glycolipid Gb3 receptor on Paneth cells of the intestinal epithelium, as well as kidney epithelial cells, leading to internalization of the active A subunit and cleavage of the 28S rRNA subunit, thus inhibiting protein synthesis and inducing apoptosis (19, 29). Stx is known to be the causative agent of hemolytic-uremic syndrome in infected patients (30).
Because the stx genes are carried by lambdoid bacteriophages, induction is triggered by the bacterial SOS response (13–15). For this reason, antibiotic treatment of EHEC infections is not recommended, as induction of stx-carrying phages would result in lysis of the host cell and release of the Stx toxin (27). We recently reported that d-serine, a host metabolite found in abundance at extraintestinal sites such as the urinary tract, was capable of independently repressing the T3SS and triggering transcription of the SOS regulon in EHEC O157:H7 (31). This d-serine-mediated “SOS response” is dependent on the intracellular accumulation of this amino acid by E. coli isolates that cannot catabolize it, such as EHEC, because of a lack of functional d-serine catabolic genes. In this work, we have explored the impact of the d-serine-induced SOS response on EHEC, in particular, the importance of this response for stx production. We found that levels of RecA reached by accumulation of d-serine resulted in a modulated induction of RecA that was lower than that of exposure to subinhibitory concentrations of mitomycin C (MMC). d-Serine did not result in activation of lytic stx phages but was found to induce SOS-mediated mutations caused by error-prone polymerase expression, thus resulting in genetic diversity and adaptation of exposed populations. These results demonstrate a novel method of SOS regulon induction and generation of genetic diversity in response to an environmentally relevant stress signal that can be encountered by EHEC.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are described in Table S1 in the supplemental material (32, 33). For liquid culture, single colonies from a pure culture on an LB agar plate were inoculated into 5 ml of LB broth and grown overnight at 37°C and 200 rpm. For growth and reporter assays, a 1/100 dilution of an overnight culture was used to inoculate prewarmed minimum essential medium (MEM)-HEPES (Sigma, St. Louis, MO; catalog no. m7278) and samples were cultured at 37°C with shaking at 200 rpm. Antibiotics were used where indicated (100 μg/ml ampicillin; 30 μg/ml chloramphenicol; 40 μg/ml kanamycin; 12 μg/ml tetracycline; 100 μg/ml rifampin). d-Amino acids and MMC were used at the concentrations indicated. All of the chemicals used were purchased from Sigma.
Generation of single gene deletions.
A nonpolar recA deletion mutant was generated by the lambda Red-mediated method (34). Briefly, the kanamycin resistance cassette from pKD4 was amplified by PCR with primers flanked by 50 bp directly homologous to flanking sequences immediately up- and downstream of the gene of interest (forward, CAACAGAACATATTGACTATCCGGTATTACCCGGCATGACAGGAGTAAAAGTGTAGGCTGGAGCTGCTTC; reverse, TAAAAAGCAAAAGGGCCGCAGATGCGACCCTTGTGTATCAAACAAGACGACATATGAATATCCTCCTTAG; sequences obtained from NCBI; GenBank accession no. for EDL933, NC_002655.2). This PCR product was purified by phenol-chloroform extraction and ethanol precipitated, DpnI treated, and transformed into electrocompetent TUV93-0 carrying pKD46 at a concentration of 1 μg/50 μl of cells. TUV93-0 carrying pKD46 was cultured in SOB medium at 30°C and induced with 10 mM arabinose prior to electroporation. After recovery for 2 h at 37°C in SOC medium, the reaction mixture was plated on LB agar containing 40 μg/ml kanamycin and grown at 37°C for 16 h. Phenotypically positive colonies were checked for positive gene knockouts by PCR with primers located up- and downstream of the gene of interest (forward, TGACAGGAGTAAAAATGGCT; reverse, ATTCTGTCATGGCATATCCT). Positive deletions were confirmed by isolation on kanamycin plates and subsequent sequencing. Removal of kanamycin resistance was performed by temperature selection with pCP20 (34).
In vitro GFP reporter fusion assays of recA and stx transcription.
To assay the promoter activities of individual genes, bacterial strains of interest were transformed with plasmids carrying fusions of specific promoter regions with green fluorescent protein (GFP) (precA-GFP or pstx2-GFP) (35). Transformed strains were used to inoculate 10 ml of MEM-HEPES for growth analysis and cultured at 37°C with shaking (200 rpm). Samples were taken at regular intervals for measurement of population cell density (optical density at 600 nm [OD600]) and fluorescence indicative of promoter activity. Population GFP expression was measured by transferring 200-μl aliquots to a black 96-well plate for GFP fluorescence measurement (excitation at 485 nm, emission at 550 nm) with a FLUOstar Optima Fluorescence Plate Reader (BMG Labtech, United Kingdom). Background fluorescence was accounted for by measuring fluorescence from cells carrying promoterless reporter plasmids and subtracting these values from corresponding data. GraphPad Prism 5.0 (GraphPad, San Diego, CA) was used to either plot scatter curves of GFP expression or generate a standard curve of OD600 versus fluorescence and obtain values at specific OD600s for quantification and direct comparison of samples. The data are presented as the mean value (±the standard error of the mean [SEM]) of at least three biological replicates.
qRT-PCR.
Quantitative reverse transcriptase PCR (qRT-PCR) was performed with KAPA SYBR FAST Universal master mix (KAPA Biosystems, Woburn, MA) and Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI). Total RNA was extracted with an RNeasy kit (Qiagen) in accordance with the manufacturer's specifications. qRT-PCR analysis was carried out in a one-step reaction, i.e., cDNA synthesis first, followed by qRT-PCR in accordance with the manufacturer's specifications. Individual reaction mixtures were prepared in technical triplicate, and each gene was analyzed in biological triplicate. The gapA housekeeping gene was used as a calibrator for the analysis. qRT-PCRs were carried out with the ECO real-time PCR system (Illumina, San Diego, CA), and data were analyzed by the 2−ΔΔCT method (36).
Immunoblot analysis of whole-cell lysate.
Levels of RecA and LexA production were determined by immunoblotting of whole-cell lysates with anti-RecA and anti-LexA antibodies (Abcam, Cambridge, United Kingdom). Samples were taken from growth cultures at specific time points, normalized by OD600, and lysed with BugBuster Protein Extraction buffer (Merck, New Jersey) before separation by SDS-PAGE and transfer to a nitrocellulose membrane with the Novex system (Invitrogen, Carlsbad, CA). Membranes were blocked with phosphate-buffered saline–Tween 20 (PBST) containing 5% skim milk powder. Anti-RecA and anti-LexA antibodies were used at 1/4,000 and 1/5,000, respectively, in PBST containing 1% skim milk powder to probe membranes. Anti-DnaK antibody was used at 1/10,000 to detect equal protein loading between lanes. Comparison of protein levels from immunoblot experiments was carried out with ImageJ. Experiments were performed in triplicate to confirm the results.
Fluorescence microscopy.
Strains of interest carrying the fluorescent recA-GFP reporter plasmid were cultured to an OD600 of 0.6 and mixed 1:1 with 4% paraformaldehyde at room temperature for 20 min. Twenty microliters of each sample was dotted onto a single well each of a multiwell microscope slide and allowed to dry. Slides were coverslip mounted with Dako fluorescent mounting fluid (Invitrogen). Slides were left to set and imaged on a Leica DM18 microscope (Leica Microsystems). Cells were imaged at ×100 magnification with oil immersion, and images were analyzed with Leica Application Suite X. To compare recA-GFP induction of the wild-type and d-serine-treated cultures directly, the exposure times were normalized between the samples compared. Cells fluorescing above the background level of wild-type cells were considered induced for recA expression. MMC-induced samples were adjusted for GFP exposure time accordingly, given the strong induction of the reporter. GFP fluorescence was measured at an excitation wavelength of 488 nm. Experiments were carried out in triplicate, with at least five random fields of views analyzed in each replicate.
Phage transduction assay.
To assay the production and transduction of phages in response to d-serine or MMC, we used E. coli strain JP10819, which contains ϕP27 of EHEC with the stx gene being replaced with a tetracycline resistance marker (37). JP10819 was cultured in LB at 37°C until an OD600 of ∼0.25 was reached. d-Serine or MMC was added at the concentrations indicated. These cultures were grown at 32°C with slow shaking (80 rpm) for 4 h and then left on the bench overnight at room temperature. Phage lysate was filtered through a 0.2-μm filter and serially diluted in phage buffer (100 mM NaCl, 0.5 M Tris [pH 7.8], 1 mM MgSO4, 4 mM CaCl2). A culture of MG1655 was grown in LB at 37°C until an OD600 of ∼1.4 was reached, at which point CaCl2 was added to a final concentration of 10 mM. A 100-μl volume of each serially diluted phage lysate was added to a 1-ml aliquot of MG1655 with CaCl2 before static incubation at 37°C for 30 min. A 3-ml volume of soft LB agar (0.75%) agar was added to these cultures and poured over LB agar plates (1.5%) containing 6 μg/ml tetracycline and 1.7 mM sodium citrate. Plates were incubated at 37°C overnight. Colonies were counted and multiplied by the appropriate dilution factor to calculate the relative transduction frequency. Each colony was interpreted as a single phage transduction event. GraphPad Prism 5.0 was used to generate figures and perform analysis. Mean data (±the SEM) were used to calculate statistical significance by way of a Student t test. Mutation assays were performed on three independent occasions.
Mutation frequency assay.
The SOS response-mediated mutation frequency was determined with a rifampin resistance assay as described previously (38). Overnight cultures of TUV93-0 were used to inoculate 5 ml of MEM-HEPES as described above. Cultures were treated with either 1 mM d-serine or 0.05 μg/ml of MMC after 3 h (OD of ∼0.3). Cultures were harvested by centrifugation after a further 2 h of incubation at late exponential phase (OD of ∼0.8) or 8 h (prolonged stationary phase). A sample of each culture was removed prior to centrifugation for serial dilution in PBS and determination of CFU counts on plain LB agar plates. The pelleted culture was resuspended in 200 μl of prewarmed PBS and plated on LB agar plates containing 100 μg/ml rifampin. Plates were incubated at 37°C for 24 h in darkness before being scored for the number spontaneous rifampin-resistant (Rifr) colonies per plate. The mutation frequency was defined as the number Rifr CFU over the total colony count per culture. GraphPad Prism 5.0 was used to generate figures and perform analysis. Mean data (±the SEM) were used to calculate statistical significance with a Student t test. Mutation assays were performed on at least five independent occasions.
RESULTS
Intracellular accumulation of d-serine increases RecA production in EHEC.
Previous work by our group identified d-serine as an important niche-specific signal that is involved in the regulation of the infection process by repressing the EHEC T3SS essential for colonization of the host (31). Transcriptomic analysis showed that exposure to millimolar concentrations of d-serine (similar to those found in the urinary tract) also resulted in upregulation of the SOS response regulon, and this was dependent on d-serine accumulation independently of T3SS repression. To verify the role of d-serine accumulation in SOS induction, we performed qRT-PCR analysis of total RNA extracted from EHEC TUV93-0 cells cultured in MEM-HEPES (conditions known to strongly induce T3SS expression) alone or supplemented with 1 mM d-serine. d-Serine accumulation resulted in a >5-fold increase (P ≤ 0.001) in recA transcription compared to that of the wild type (Fig. 1). As a control, EHEC was transformed with a plasmid expressing d-serine deaminase from UPEC CFT073 (pdsdA), allowing this strain to catabolize d-serine. Accordingly, the levels of recA were reduced to those of the wild type, indicating that d-serine accumulation is required for SOS induction.
FIG 1.
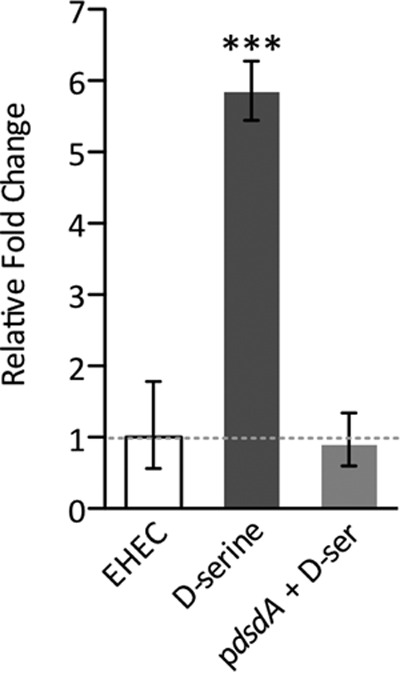
Induction of recA transcription upon intracellular d-serine accumulation. qRT-PCR analysis of relative recA transcription with mRNA from EHEC cultured to mid-exponential phase in MEM-HEPES alone and supplemented with 1 mM d-serine. As a control, recA levels from an EHEC strain transformed with pdsdA (encoding d-serine deaminase from UPEC) that does not accumulate d-serine were also analyzed. The broken line indicates the baseline wild-type recA level. ***, P ≤ 0.001 (calculated from three independent biological replicates). Error bars indicate standard deviations.
To demonstrate that elevation of RecA production was specific to d-serine, whole-cell lysates of EHEC TUV93-0 cultured in MEM-HEPES with or without supplementation with various d-amino acids at 1 mM were probed by immunoblotting with anti-RecA antibody. d-Serine resulted in a >6-fold increase in RecA production, whereas the variation in response to other d-amino acids was more modest, with little variation either above or below the wild-type level (see Fig. S1 in the supplemental material). As a control, whole-cell lysates were also probed with anti-DnaK antibody to demonstrate normalized total protein levels and equal loading of the lanes. This indicates that this SOS response is unique to d-serine, as was the repression of the EHEC T3SS (31).
d-Serine-induced RecA production does not activate lysogenic stx phages.
Given that d-serine accumulation induced the SOS response, we next addressed the physiological relevance of this. First, we investigated whether d-serine is capable of inducing the activation of lytic phages through the SOS response. This is an important question, as the stx genes of EHEC are carried by horizontally acquired lysogenic phages (21, 22). We profiled the growth of EHEC strain Sakai (stx mutant) in MEM-HEPES alone and with three concentrations of d-serine (0.02, 0.2, and 1 mM) or MMC (0.05, 0.5, and 2 μg/ml) added after 2 h at 37°C with shaking. Addition of d-serine at any of the concentrations tested had no effect on the growth rate of EHEC under these conditions (Fig. 2A). At 0.05 μg/ml, MMC also did not affect growth, and this was therefore considered a subinhibitory concentration of MMC under these conditions. At 2 μg/ml, MMC conversely resulted in a dramatic drop in OD, indicating phage-mediated lysis of the bacterial population and SOS response. At 0.5 μg/ml, MMC resulted in partial lysis of the population (Fig. 2B).
FIG 2.

d-Serine does not induce lytic bacteriophage and stx expression in EHEC. Growth of EHEC was profiled in MEM-HEPES supplemented with 1, 0.2, or 0.02 mM d-serine (A) and 2, 0.5, or 0.05 μg/ml MMC (B). The time point of SOS induction by d-serine or MMC is indicated by an arrow. A sharp drop in OD600 is indicative of phage-induced lysis. (C) stx-GFP reporter expression in response to d-serine and MMC was analyzed. Relative fluorescence units (RFU) were measured at various time points postinduction and compared to those of uninduced EHEC. **, P ≤ 0.01; ***, P ≤ 0.001 (calculated from three independent biological replicates). (D) Quantification of the number of phage transduction events in response to a d-serine- or MMC-induced SOS response in EHEC. Error bars represent the SEM calculated from at least three biological replicates. The concentrations of d-serine and MMC used are indicated individually in each panel.
Next, we addressed the effects of d-serine on stx expression directly. To facilitate this, we employed a reporter plasmid containing the stx2 promoter region fused to GFP. Transformed strains were cultured in MEM-HEPES for 2 h before induction with a range of d-serine or MMC concentrations. SOS induction by d-serine was insufficient to alter stx expression levels at all of the concentrations tested (Fig. 2C). Exposure to MMC at lytic concentrations resulted in a sharp increase in stx expression between 3 and 5 h postinduction, the point at which the cell population begins mass lysis due to the SOS response (Fig. 2B). This corresponds to an ∼7.5-fold increase in stx expression above the wild-type background at 2 μg/ml MMC (P ≤ 0.001). Accordingly, the partially lytic concentration of MMC (0.5 μg/ml) resulted in only a limited, albeit significant, increase in stx expression (P ≤ 0.001). No significant increase in stx transcription was observed at subinhibitory MMC concentrations.
At lytic concentrations of MMC, expression of the SOS response results in activation of dormant lysogenic phages and transduction of packaged phage particles to recipient cells via lysis of the donor cell. To assay the effect of d-serine or MMC on phage transduction, we used E. coli strain JP10819, a derivative of MG1655 containing a modified ϕP27 prophage (37). ϕP27 normally carries stx, but in this construct, it has been replaced with a tetracycline resistance marker (tetA), which is used to select for successful recipients of the activated phage. Lysates of JP10819 induced with either d-serine or MMC at various concentrations were used to transduce wild-type MG1655, resulting in tetracycline-resistant recipients. In agreement with the growth assays, d-serine at concentrations of up to 1 mM had no effect on the transduction of the ϕP27 phage from JP10819 (Fig. 2C). MMC induction resulted in an increased number of phage transductants at lytic concentrations, with the highest number of events occurring at 2 μg/ml MMC (P ≤ 0.001) and decreasing accordingly. Subinhibitory MMC concentrations resulted in only a modest increase in the phage titer over the uninduced wild-type level. Collectively, these data indicate that known d-serine levels encountered in the host cannot activate the transduction of lysogenic phages or the expression of stx, despite being able to induce the transcription of recA and the SOS regulon.
d-Serine-induced RecA production is sufficient to generate mutation-dependent genetic diversity at the population level.
Three DNA polymerases are induced as part of the SOS response in E. coli, high-fidelity PolB, error-prone DinB, and Pol V (UmuDC) (1). We previously demonstrated by transcriptome sequencing that multiple members of the SOS regulon were induced by d-serine accumulation in EHEC, including dinB, umuDC, and polB (31). The enzymes encoded by these genes are key members of the DNA repair process, but it is known that error-prone polymerase activity can lead to mutations in the population and that this can occur spontaneously in stationary phase or in response to antibiotic treatment, even at subinhibitory concentrations (38–40). We thus hypothesized that the SOS-like response induced by d-serine could be capable of promoting genetic diversity through this mechanism. Cultures of EHEC TUV93-0 in MEM-HEPES were induced with either 1 mM d-serine or sublytic MMC and allowed to grow for a further 2 or 8 h before being harvested by centrifugation. TUV93-0 has had the stx1- and stx2-carrying prophages deleted, and therefore it will not lyse upon SOS activation, allowing investigation of the d-serine- and MMC-induced SOS response over the full course of growth experiments. Incubation times of 2 and 8 h postinduction were chosen to represent the exponential and stationary phases on the basis of the growth experiments described in Fig. 2. Bacteria obtained in the exponential phase had no indication of the appearance of a higher Rifr frequency in response to d-serine than that of spontaneous mutation in the wild type alone (Fig. 3A). Treatment with MMC showed a higher count of Rifr colonies, but it was not significantly higher than the background at this stage. However, after prolonged growth into stationary phase, the frequency of Rifr colonies after d-serine exposure was significantly (P ≤ 0.05) higher than that of the wild type. Accordingly, prolonged exposure to MMC in stationary phase dramatically increased the Rifr mutation rate by >2 orders of magnitude over that of the wild type (P ≤ 0.05). As a control, we also analyzed the Rifr mutation frequency of TUV93-0 carrying plasmid pdsdA expressing d-serine deaminase to assess if d-serine accumulation contributed to the phenotype observed (Fig. 3B). Indeed, TUV93-0/pdsdA generated Rifr mutations with a frequency more similar to that of the wild type.
FIG 3.
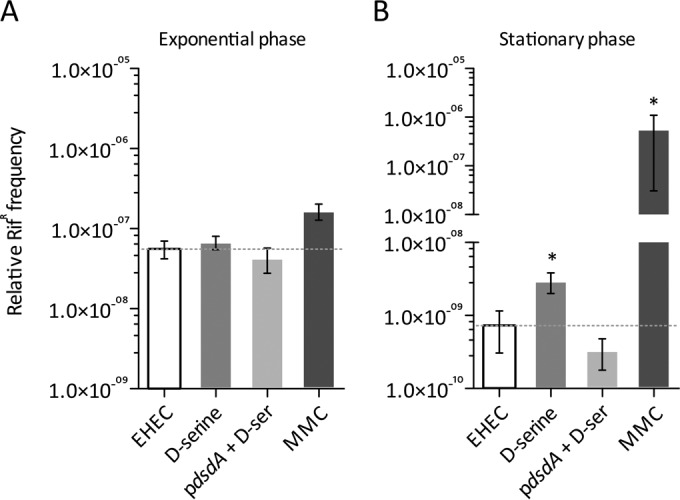
Increased SOS-like mutation frequency after d-serine accumulation. The emergence of resistance to rifampin (Rifr) in EHEC after SOS induction by 1 mM d-serine and 0.05 μg/ml MMC was assayed. EHEC cell cultures were harvested and plated on rifampin-containing LB plates in exponential phase at 2 h postinduction (A) and in stationary phase at 8 h postinduction (B). The dotted line indicates the background level of spontaneous Rifr emergence in uninduced EHEC cells, to which all other conditions were compared. The Rifr frequency after each incubation is relative to the number of CFU present in each culture. As a control, EHEC/pdsdA, which does not accumulate d-serine, was also assayed for the emergence of Rifr. Error bars represent the SEM calculated from at least five biological replicates. *, P ≤ 0.05 (compared to the wild-type control).
We previously proposed that EHEC was adapted to tolerate d-serine accumulation in order to promote survival within stressful environments. Conversely, a d-serine deaminase (ΔdsdA) mutant uropathogenic E. coli (UPEC) strain displayed decreased fitness under the same conditions (41). We thus postulated that E. coli strains normally capable of catabolizing d-serine would be susceptible to d-serine-mediated stress in the absence of dsdA. Transformation of both UPEC and an isogenic ΔdsdA mutant with precA-GFP and induction of SOS demonstrated that UPEC ΔdsdA indeed activated an SOS response associated with d-serine accumulation, a phenotype not observed in wild-type UPEC (see Fig. S2A in the supplemental material). Interestingly, this response was more than twice as pronounced as that of the EHEC background. Exposure of both UPEC and a ΔdsdA mutant to MMC resulted in indistinguishable SOS response levels. These results were mirrored in Rifr mutation assays, which indicated that a UPEC ΔdsdA background is also more susceptible to SOS-associated mutation, whereas a UPEC wild-type background is not (see Fig. S2B and C). These data indicate that d-serine accumulation activates a low level of RecA induction capable of inducing genetic diversity, although the extent of the frequency of mutation varies in different E. coli pathotypes.
d-Serine induces a modulated level of RecA production.
Given that d-serine was capable of causing an increase in the mutation frequency normally associated with SOS, we next addressed the extent of recA induction under these various conditions in order to gain mechanistic insights into the phenomenon. Using EHEC/precA-GFP, we compared d-serine- and MMC-induced transcription of recA. At 1 mM, d-serine significantly (∼3-fold; P ≤ 0.05) increased recA transcription at the mid to late exponential phase (OD600 of 0.8), in agreement with our previous findings. This induction, however, was markedly lower than the transcription of recA in response to even subinhibitory concentrations of MMC (Fig. 4A). At 0.05 μg/ml, MMC significantly increased recA (∼13-fold; P ≤ 0.001) severalfold above that of the wild type at the mid-to-late exponential phase. The effect of a lytic concentration of MMC (2 μg/ml) was even more pronounced, activating recA transcription to around double that of the lower concentration (∼28-fold; P ≤ 0.001). Whole-cell lysate samples were taken from growth assays at 0, 2, and 5 h postinduction with either d-serine or MMC and probed for RecA levels by immunoblotting. At time zero, the levels of RecA in all of the samples were comparable. By 2 h postinduction, an increase in RecA was observed at higher d-serine concentrations (see Fig. S3 in the supplemental material). In agreement with transcriptional reporter data, RecA production by MMC was much more pronounced over all of the concentrations tested. At 5 h postinduction, RecA levels in response to d-serine did not continue to increase; however, prolonged MMC exposure resulted in a large increase in RecA production at all of the concentrations tested (see Fig. S2). Levels of DnaK were also probed at each time point to control for protein loading effects.
FIG 4.

Modulated expression of recA induced by d-serine accumulation. (A) recA-GFP reporter expression was analyzed in response to d-serine and MMC. Relative fluorescence units (RFU) were measured at various stages of growth postinduction and compared to those of uninduced EHEC at all of the time points tested. The concentrations of d-serine or MMC used are indicated individually in each panel. Error bars represent the SEM. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (calculated from three independent biological replicates). (B) Fluorescence microscopy analysis of EHEC/precA-GFP cell populations left untreated or treated with 1 mM d-serine or 2.0 μg/ml MMC. Bacterial cells were imaged by phase-contrast microscopy, and recA induction was determined by detection of GFP-expressing cells. The merged channel indicates the level of recA-GFP induction per cell. Representative images are displayed for each condition, and data are summarized below as the percentage of the population expressing enhanced recA-GFP activity (green bars, cells induced for recA-GFP expression; gray bars, cells uninduced for recA-GFP expression). Experiments were performed in triplicate, and the results are shown as the mean ± the SEM. WT, wild type. (C) Immunoblot analysis of RecA, LexA, and DnaK levels in whole-cell lysates of EHEC cultured in MEM-HEPES with or without SOS induction. An EHEC ΔrecA mutant was used as a negative control for SOS induction. The concentrations of d-serine and MMC used to induce the SOS response are indicated above the immunoblots. DnaK levels were used to assess equal protein loading of samples. Experiments were performed at least in triplicate to confirm the results.
The SOS response is known to be a stochastic process, and within a population, a small percentage of the cells in a living culture can be spontaneously induced (42, 43). To determine if the low-level SOS response is indeed representative of the population or rather a sporadic occurrence in a small number of cells, we visualized EHEC/precA-GFP cells by fluorescence microscopy after treatment with both d-serine and MMC. d-Serine treatment resulted in heterogeneous induction of recA throughout the population, with >35% of the population observably increasing recA-GFP expression. This heterogeneity may explain how such a modest population-wide recA induction can affect the mutation frequency if the response is actually stronger but in a subpopulation of the cells. In contrast, a genuine SOS response induced by MMC results in nearly 100% of the population expressing recA-GFP to a much greater extent than with d-serine (Fig. 4B). Furthermore, MMC-treated cells were morphologically distinct, forming filamentous structures likely associated with inhibition of cell division, a phenomenon not observed in d-serine-treated cultures. These results collectively indicate that while d-serine is capable of inducing a significant increase in the transcription of the SOS regulon and the production of RecA, the dynamics and consequence of this response are quite separate from those of the classical SOS response to a DNA-damaging agent.
The finding that d-serine could activate the transcription of the SOS network but failed to induce stx expression or phage lysis suggested that this was an indirect consequence of d-serine accumulation in the cell and perhaps not a genuine SOS response to a DNA-damaging event. Indeed, prolonged exposure to MMC resulted in the continued elevation of the SOS network (see Fig. S3 in the supplemental material). The lack of effects on stx expression, as well as its heterogeneous nature, suggests that d-serine exposure does not have DNA-damaging effects. d-Serine was originally described as being bacteriostatic to susceptible strains by interfering with l-serine and pantothenic acid biosynthetic pathways (44). However, growth inhibition by d-serine is dependent on the culture conditions used and the transcriptional response described here is independent of growth defects. This raises the important question of whether this observed SOS response is indeed genuine. To begin to examine this, we investigated the dependency of this SOS activation on RecA. We generated a TUV93-0 recA (ΔrecA) deletion mutant and analyzed the precA-GFP reporter activity in this genetic background, with the hypothesis being that if d-serine activated the SOS response by an indirect mechanism, recA promoter activity may still be observable. RecA was absolutely required for SOS induction by both d-serine and, as expected, MMC, confirming the dependency of these responses on RecA (see Fig. S4 in the supplemental material).
We next investigated whether d-serine accumulation leads to cleavage of the LexA repressor protein during the SOS response. Immunoblotting for LexA following exposure to 1 mM d-serine resulted in no observable decrease in the quantity of LexA protein detectable in whole-cell lysates (Fig. 4C). In contrast, subinhibitory (0.05 μg/ml) MMC exposure resulted in a >2-fold reduction in the quantity of LexA detectable in whole-cell lysates. Accordingly, a lytic concentration of MMC (2 μg/ml) completely abolished detectable levels of LexA in a RecA-dependent manner (Fig. 4C). The lack of detectable LexA cleavage suggests two scenarios, i.e., (i) that the modesty of the d-serine-induced SOS response renders LexA degradation undetectable and (ii) that accumulation of d-serine activates RecA production by an undetermined mechanism and not a genuine SOS response.
SOS induction in a RecA-dependent manner suggests that the canonical mechanism of induction but no observable LexA cleavage was seen in response to d-serine accumulation. To explore this further, we supplemented a sublytic concentration of MMC with high concentrations of d-serine in an attempt to increase recA or stx transcription above that achieved with MMC alone. Addition of either 1 or 5 mM d-serine to 0.05 μg/ml MMC increased recA transcription by 1.3- or 1.4-fold, respectively, over that achieved by exposure to 0.05 μg/ml MMC but was not as extreme as that of a lytic 2.0-μg/ml MMC concentration (∼1.6-fold) (Fig. 5A). More strikingly, stx transcription was increased approximately 1.8- and 1.9-fold (P ≤ 0.01), respectively, in d-serine-supplemented assays but again was still not high enough to mirror the level of activation achieved by the lytic concentration of MMC (4.6-fold above the subinhibitory MMC concentration; P ≤ 0.001) (Fig. 5B). In accordance with these data, immunoblotting for LexA in whole-cell lysate from the combined treatment of MMC and d-serine resulted in enhanced degradation of detectable LexA (∼2-fold) compared with MMC alone. However, as shown in Fig. 4, this result was obtainable only by treatment with a DNA-damaging agent (Fig. 5C). These results suggest that d-serine is capable of activating the transcription of the SOS regulon in an unusual but genuinely functional manner. However, the lack of DNA-damaging events in response to d-serine indicates a possible unique mechanism of transcriptional activation and a controlled increase in functional RecA within the cell.
FIG 5.
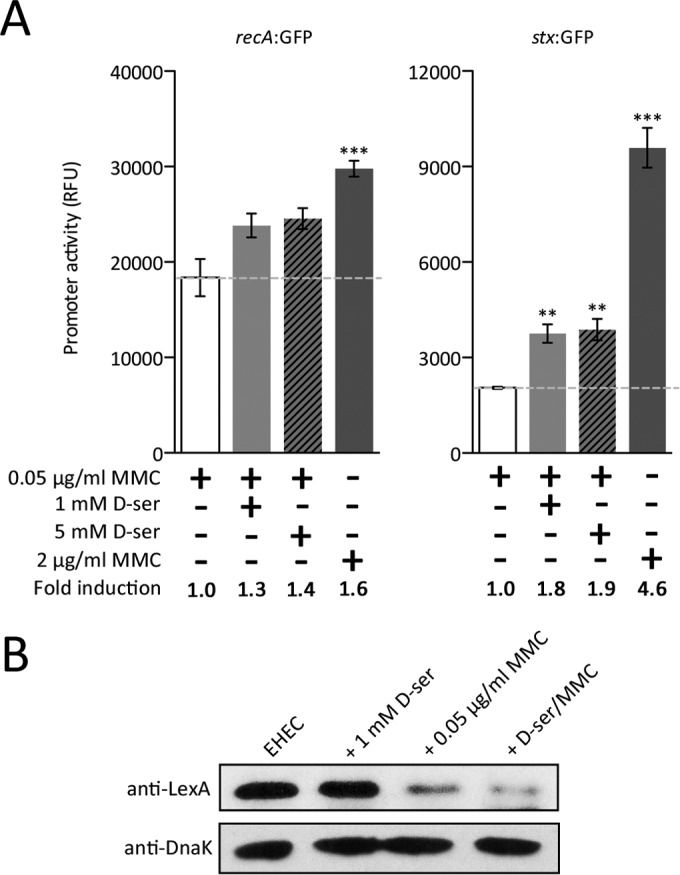
d-Serine (d-ser) accumulation induces a functional SOS-like response in EHEC. (A) The levels of recA-GFP (left) and stx-GFP (right) reporter expression were analyzed in cell cultures treated with 0.05 or 2.0 μg/ml MMC. The levels of reporter activity in response to a combination of a low MMC concentration supplemented with either 1 or 5 mM d-serine were also analyzed and compared to that of a low MMC concentration. Relative fluorescence units (RFU) were measured at 3 h postinduction (OD600, ∼0.8). The concentrations and combinations of d-serine and MMC used are indicated at the bottom, where a plus sign means present and a minus sign means absent. The broken line indicates the level of recA or stx expression in response to a low subinhibitory MMC concentration. Error bars represent the SEM. **, P ≤ 0.01; ***, P ≤ 0.001 (calculated from three independent biological replicates). The enhanced SOS response is also represented as the fold change in recA or stx expression indicated at the bottom. (B) Immunoblot analysis of LexA and DnaK levels in whole-cell lysates of EHEC induced for an SOS response with d-serine, MMC, or both. The concentration of d-serine or MMC used to induce the SOS response is indicated above the immunoblots. DnaK levels were used to assess equal protein loading of samples. Experiments were performed at least in triplicate to confirm the results.
DISCUSSION
The bacterial SOS response is a well-studied reaction to stresses that ultimately result in damage to DNA or halting of cellular replication. The system is ubiquitous in the bacterial kingdom and plays a fundamental role in the successful life cycle of all bacteria (1). Alternative mechanisms of SOS activation are less well understood, and specifically what role they play in the absence of direct DNA damage is a less-well-explored area. The finding that d-serine accumulation results in upregulation of the SOS regulon in EHEC was indicative of a stress that the bacteria must respond to in some way. However, the mechanism of SOS induction by d-serine was obscure, considering that the negative effects of this amino acid are known to be at the level of metabolism (44). Here we demonstrate that d-serine accumulation induces a functional but mechanistically distinct SOS response in EHEC at a tolerated low level capable of promoting genetic diversification.
Our results collectively indicate that the SOS regulon is transcriptionally activated in response to d-serine and results in increased production of RecA associated with such an event. This level of induction, however, is more modest than that observed after MMC exposure, even at a subinhibitory concentration. In contrast, MMC treatment resulted in both an increased SOS response and phage activation. Levels of stx activation in response to a subinhibitory MMC concentration, however, were not increased, likely representing the extent of DNA damage from this treatment. The fact that high concentrations of d-serine could induce the “SOS response” but not phage activation suggests either a level of induction too low to observe such side effects or an alternative mechanism that is independent of direct DNA damage. Cleavage of the SOS repressor LexA was not detectable after d-serine induction, suggesting a very controlled response and maintenance of normal LexA levels. Indeed, transcriptomic analysis of the response to d-serine has previously shown that the members of the SOS regulon upregulated include the cell division inhibitor sulA and the RecA destabilizers recX and dinI, as well as lexA itself (31, 45). This suggests that the response may be modulated in some way to maintain normal and controlled cellular replication. Importantly, the data have shown that d-serine exposure can actually enhance the SOS response and stx activation, as well as LexA cleavage, when used in combination with MMC, thus providing key evidence that this is in fact a functional SOS-like response but is activated in an unusual manner that is not specific to a DNA-damaging agent.
The toxicity of d-serine to bacteria is well known. E. coli isolates that carry an intact d-serine tolerance locus (dsdCXA) are capable of internalizing d-serine, regulating its uptake, and catabolizing it as a nutrient source into nontoxic pyruvate and ammonia (44, 46–49). The mechanism of toxicity for cells that do not carry functional dsdCXA has been reported to be bacteriostatic inhibition of cell growth due to interference with l-serine and pantothenate metabolic pathways (44). Several E. coli isolates carry a conserved truncation in dsdCXA that renders them susceptible to d-serine toxicity (48, 49). We previously reported that carriage of the genes encoding the T3SS was widespread in isolates with mutated dsdCXA, suggesting an incompatibility between EHEC virulence and d-serine tolerance (31). Indeed, d-serine represses the T3SS and therefore host cell attachment in EHEC, but d-serine is not found in abundance at sites normally colonized by EHEC. d-Serine is conversely very abundant at extraintestinal sites (such as the urinary tract) and is used by pathogens in this niche as a positive fitness trait and a regulator of virulence (50, 51). This led us to propose that EHEC (and similar pathotypes) has evolved to lose d-serine tolerance, thus limiting the possibility that it disseminates to an environment rich in d-serine, where its primary colonization factor would be redundant (31). More recently, we discovered a novel d-serine uptake system in E. coli. This system is highly conserved in all E. coli strains but is regulated differently in unique genetic backgrounds. EHEC counterintuitively uses this system to increase d-serine uptake in its presence but is capable of tolerating d-serine accumulation without actively breaking down this toxic metabolite. However, despite this apparent tolerance, expression of the SOS regulon persists, suggesting a mechanism of transcriptionally responding to this signal in order to quickly adapt to changing d-serine concentrations that may be encountered (31, 41). Furthermore, SOS activation can lead to an increased mutational frequency and, in turn, increase bacterial fitness after exposure to even subinhibitory concentrations of antibiotics, for example (38, 39, 52). Indeed, one study reported that ciprofloxacin treatment of E. coli could increase the mutation frequency by a factor of 104 over the spontaneous rate (53). In this work, we found that d-serine accumulation could actually enhance the mutation rate of the population through RecA production and that this was dependent on d-serine accumulation in the cell. Interestingly, d-serine accumulation in a UPEC ΔdsdA background is a much less tolerated event and exemplifies a fitness defect under these conditions (41). In the UPEC ΔdsdA mutant, d-serine accumulation also results in a higher recA induction than that of EHEC, suggesting that this stress may account for the apparent adaptation to d-serine tolerance in EHEC. Recent studies have suggested that antibiotic-mediated mutations can not only increase resistance to antibiotics but also generally enhance the fitness of the affected bacterial cells (39, 52). It was also demonstrated that after rapid adaptation, SOS expression is actually decreased, limiting further unwanted mutagenic capability (52). This may explain why recA induction by d-serine is weaker in adapted EHEC than in a UPEC ΔdsdA mutant that is more susceptible to toxic d-serine and is perhaps how EHEC has evolved to tolerate d-serine exposure and the associated induction of a stress response. We propose that this is to allow EHEC to continue monitoring and sampling its environment within the host for inhibitory d-serine by a mechanism that is unique to DNA damage or even spontaneous prophage induction and subsequently would result in a modulated transcriptional response to this stress.
Factors that may activate stx expression are of particular importance when studying EHEC biology and potential intervention strategies. There are currently no recommended antibiotic treatments for Shiga-toxigenic E. coli infections, given the known repercussions of such procedures. Recently, there has been much interest in how niche-specific metabolites found in the host can regulate colonization by EHEC (54). This pathogen has evolved intricate mechanisms to specifically localize to the colonic epithelial surface by responding to numerous physiologically relevant signals such as host- and microbiota-derived nutrient sources, hormone-like signals, and short-chain fatty acids (55–58). Furthermore, an understanding of these mechanisms has led to the possibility of using such metabolites and small molecules as treatments themselves to reduce colonization (59–61). However, for an approach like this to work, it is important to understand fully the “off-target” effects of said treatment, such as induction of SOS-related genes. Xu et al. have shown that spontaneous stx activation results in the lysis of a small portion of the EHEC population, but this, in turn, promotes colonization by priming the epithelial surface for attachment of the nonlysed cells (62). Indeed, stx prophage induction has been associated with enhanced colonization of EHEC in vivo (63, 64). Furthermore, the T3SS of enteropathogenic E. coli has been shown to be under direct control of the SOS response (65). However, despite the fact that these studies link prophage induction and the SOS response with colonization by the T3SS, each case requires prophage activation or DNA damage. In contrast, this study highlights that d-serine does not result in phage expression, suggesting a different mechanism and role for this response.
How does d-serine accumulation induce the SOS regulon? Cases of unusual mechanisms leading to LexA derepression have been reported. In Bacillus subtilis, a transcription factor, ComK, is capable of activating recA transcription without displacing LexA itself (66). A second study has described an unusual mechanism of LexA displacement in which the nucleoid-associated protein HU affects DNA topology close to the site of LexA binding, thus resulting in dissociation from its binding site (67). It has also been demonstrated experimentally that mutation of interaction sites between RecA and LexA can interrupt the cleavage of LexA while still allowing induction of RecA in response to DNA damage (68). The concept of LexA displacement is certainly intriguing, considering the lack of observable LexA cleavage in response to d-serine; however, the dependency of this response on RecA seems to suggest a more common route of activation that is possibly masked by the subtlety of the response. One could hypothesize that the bacteriostatic effects of d-serine in minimal medium could indirectly lead to SOS induction by halting cellular replication. Indeed, the DpiAB mechanism results in transient inhibition of replication via activation of the cell division inhibitor SulA in an SOS-dependent manner (10). However, we have observed growth tolerance of EHEC irrespective of sulA upregulation, suggesting that there are downstream factors that counteract the effects of increased SulA expression. What is most intriguing is that this tolerance appears to be unique to EHEC, given that a UPEC ΔdsdA mutant is unable to tolerate d-serine accumulation and shows arrested growth under the same conditions (41). These results suggest EHEC-specific regulation that counteracts recA induction by d-serine.
Rapid adaptation to the environment is paramount to survival within a complex host environment where incoming threats are immediate but also long-term niche adaptation promotes a competitive advantage. EHEC isolates, while attracted specifically to the distal colon, have the capacity to at least come into contact with the urinary tract and, in some rare cases, have even been associated with urinary tract infections (69). Our data suggest, however, that in these cases, high concentrations of d-serine would not be a driver of stx expression and that other unknown factors contribute to this rare phenomenon. This ability to survive in an unusual environment may be a result of rapid adaptation to factors such as d-serine, but the uncommon nature of T3SS-mediated colonization of this niche may be due to the regulatory effects of d-serine and not its properties as a toxic carbon source (31). Taken together, these studies collectively suggest that d-serine may be a contributing factor in the evolution of E. coli niche specificity, depending on the individual's genotypic and pathogenic properties.
Supplementary Material
ACKNOWLEDGMENTS
We thank Alejandro Huerte Uribe for generating the EHEC ΔrecA mutant and Nicky O'Boyle for assistance with the microscopy. We are very grateful to David Gally for providing the Sakai stx strain and stx2-GFP reporter plasmid, Rodney Welch for providing the CFT073 wild-type and ΔdsdA mutant strains, and Jose Penades for the JP10819 strain generously supplied as gifts for use in this study.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00548-16.
REFERENCES
- 1.Baharoglu Z, Mazel D. 2014. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev 38:1126–1145. doi: 10.1111/1574-6976.12077. [DOI] [PubMed] [Google Scholar]
- 2.Michel B. 2005. After 30 years of study, the bacterial SOS response still surprises us. PLoS Biol 3:e255. doi: 10.1371/journal.pbio.0030255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernández De Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R. 2000. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol 35:1560–1572. [DOI] [PubMed] [Google Scholar]
- 4.Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. 2001. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158:41–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wade JT, Reppas NB, Church GM, Struhl K. 2005. Genomic analysis of LexA binding reveals the permissive nature of the Escherichia coli genome and identifies unconventional target sites. Genes Dev 19:2619–2630. doi: 10.1101/gad.1355605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phizicky EM, Roberts JW. 1981. Induction of SOS functions: regulation of proteolytic activity of E. coil RecA protein by interaction with DNA and nucleoside triphosphate. Cell 25:259–267. doi: 10.1016/0092-8674(81)90251-8. [DOI] [PubMed] [Google Scholar]
- 7.Little JW. 1984. Autodigestion of lexA and phage lambda repressors. Proc Natl Acad Sci U S A 81:1375–1379. doi: 10.1073/pnas.81.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo Y, Pfuetzner RA, Mosimann S, Paetzel M, Frey EA, Cherney M, Kim B, Little JW, Strynadka NCJ. 2001. Crystal structure of LexA: a conformational switch for regulation of self-cleavage. Cell 106:585–594. doi: 10.1016/S0092-8674(01)00479-2. [DOI] [PubMed] [Google Scholar]
- 9.Kovačič L, Paulič N, Leonardi A, Hodnik V, Anderluh G, Podlesek Z, Žgur-Bertok D, Križaj I, Butala M. 2013. Structural insight into LexA-RecA* interaction. Nucleic Acids Res 41:9901–9910. doi: 10.1093/nar/gkt744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller C, Thomsen LE, Gaggero C, Mosseri R, Ingmer H, Cohen SN. 2004. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science 305:1629–1631. doi: 10.1126/science.1101630. [DOI] [PubMed] [Google Scholar]
- 11.Nanda AM, Thormann K, Frunzke J. 2015. Impact of spontaneous prophage induction on the fitness of bacterial populations and host-microbe interactions. J Bacteriol 197:410–419. doi: 10.1128/JB.02230-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, McDaniel AD, Wolf LE, Keusch GT, Waldor MK, Acheson DW. 2000. Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. J Infect Dis 181:664–670. doi: 10.1086/315239. [DOI] [PubMed] [Google Scholar]
- 13.Ptashne M. 1992. A genetic switch, 3rd ed, phage lambda revisited. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 14.Mühldorfer I, Hacker J, Keusch GT, Acheson DW, Tschäpe H, Kane AV, Ritter A, Olschläger T, Donohue-Rolfe A. 1996. Regulation of the Shiga-like toxin II operon in Escherichia coli. Infect Immun 64:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plunkett G, Rose DJ, Durfee TJ, Blattner FR. 1999. Sequence of Shiga toxin 2 phage 933W from Escherichia coli O157:H7: Shiga toxin as a phage late-gene product. J Bacteriol 181:1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 17.Croxen MA, Law RJ, Scholz R, Keeney KM, Wlodarska M, Finlay BB. 2013. Recent advances in understanding enteric pathogenic Escherichia coli. Clin Microbiol Rev 26:822–880. doi: 10.1128/CMR.00022-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naylor SW, Low JC, Besser TE, Mahajan A, Gunn GJ, Pearce MC, McKendrick IJ, Smith DGE, Gally DL. 2003. Lymphoid follicle-dense mucosa at the terminal rectum is the principal site of colonization of enterohemorrhagic Escherichia coli O157:H7 in the bovine host. Infect Immun 71:1505–1512. doi: 10.1128/IAI.71.3.1505-1512.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pruimboom-Brees IM, Morgan TW, Ackermann MR, Nystrom ED, Samuel JE, Cornick NA, Moon HW. 2000. Cattle lack vascular receptors for Escherichia coli O157:H7 Shiga toxins. Proc Natl Acad Sci U S A 97:10325–10329. doi: 10.1073/pnas.190329997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chase-Topping ME, McKendrick IJ, Pearce MC, MacDonald P, Matthews L, Halliday J, Allison L, Fenlon D, Low JC, Gunn G, Woolhouse MEJ. 2007. Risk factors for the presence of high-level shedders of Escherichia coli O157 on Scottish farms. J Clin Microbiol 45:1594–1603. doi: 10.1128/JCM.01690-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perna NT, Plunkett G, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Pósfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409:529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi T, Makino K, Ohnishi M, Kurokawa K, Ishii K, Yokoyama K, Han CG, Ohtsubo E, Nakayama K, Murata T, Tanaka M, Tobe T, Iida T, Takami H, Honda T, Sasakawa C, Ogasawara N, Yasunaga T, Kuhara S, Shiba T, Hattori M, Shinagawa H. 2001. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res 8:11–22. doi: 10.1093/dnares/8.1.11. [DOI] [PubMed] [Google Scholar]
- 23.McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci U S A 92:1664–1668. doi: 10.1073/pnas.92.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elliott SJ, Wainwright LA, Timothy K, Jarvis KG, Deng Y, Lai L, Mcnamara BP, Michael S, Kaper JB. 1998. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia. Mol Microbiol 28:1–4. [DOI] [PubMed] [Google Scholar]
- 25.Wong ARC, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, Frankel G, Hartland EL. 2011. Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol Microbiol 80:1420–1438. doi: 10.1111/j.1365-2958.2011.07661.x. [DOI] [PubMed] [Google Scholar]
- 26.Croxen MA, Finlay BB. 2010. Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol 8:26–38. doi: 10.1038/nrmicro2265. [DOI] [PubMed] [Google Scholar]
- 27.Schüller S. 2011. Shiga toxin interaction with human intestinal epithelium. Toxins (Basel) 3:626–639. doi: 10.3390/toxins3060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fraser ME, Fujinaga M, Cherney MM, Melton-Celsa AR, Twiddy EM, O'Brien AD, James MNG. 2004. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J Biol Chem 279:27511–27517. doi: 10.1074/jbc.M401939200. [DOI] [PubMed] [Google Scholar]
- 29.Lindberg AA, Brown JE, Strömberg N, Westling-Ryd M, Schultz JE, Karlsson KA. 1987. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J Biol Chem 262:1779–1785. [PubMed] [Google Scholar]
- 30.Tarr PI, Gordon CA, Chandler WL. 2005. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 31.Connolly JPR, Goldstone RJ, Burgess K, Cogdell RJ, Beatson SA, Vollmer W, Smith DGE, Roe AJ. 2015. The host metabolite d-serine contributes to bacterial niche specificity through gene selection. ISME J 9:1039–1051. doi: 10.1038/ismej.2014.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campellone KG, Giese A, Tipper DJ, Leong JM. 2002. A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol Microbiol 43:1227–1241. doi: 10.1046/j.1365-2958.2002.02817.x. [DOI] [PubMed] [Google Scholar]
- 33.Anfora AT, Welch RA. 2006. DsdX is the second d-serine transporter in uropathogenic Escherichia coli clinical isolate CFT073. J Bacteriol 188:6622–6628. doi: 10.1128/JB.00634-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaslaver A, Bren A, Ronen M, Itzkovitz S, Kikoin I, Shavit S, Liebermeister W, Surette MG, Alon U. 2006. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat Methods 3:623–628. doi: 10.1038/nmeth895. [DOI] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Quiles-Puchalt N, Carpena N, Alonso JC, Novick RP, Marina A, Penadés JR. 2014. Staphylococcal pathogenicity island DNA packaging system involving cos-site packaging and phage-encoded HNH endonucleases. Proc Natl Acad Sci U S A 111:6016–6021. doi: 10.1073/pnas.1320538111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baharoglu Z, Mazel D. 2011. Vibrio cholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: a route towards multiresistance. Antimicrob Agents Chemother 55:2438–2441. doi: 10.1128/AAC.01549-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long H, Miller SF, Strauss C, Zhao C, Cheng L, Ye Z, Griffin K, Te R, Lee H, Chen C-C, Lynch M. 2016. Antibiotic treatment enhances the genome-wide mutation rate of target cells. Proc Natl Acad Sci U S A 113:E2498–2505. doi: 10.1073/pnas.1601208113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yeiser B, Pepper ED, Goodman MF, Finkel SE. 2002. SOS-induced DNA polymerases enhance long-term survival and evolutionary fitness. Proc Natl Acad Sci U S A 99:8737–8741. doi: 10.1073/pnas.092269199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Connolly JPR, Gabrielsen M, Goldstone RJ, Grinter R, Wang D, Cogdell RJ, Walker D, Smith DGE, Roe AJ. 2016. A highly conserved bacterial d-serine uptake system links host metabolism and virulence. PLoS Pathog 12:e1005359. doi: 10.1371/journal.ppat.1005359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pennington JM, Rosenberg SM. 2007. Spontaneous DNA breakage in single living Escherichia coli cells. Nat Genet 39:797–802. doi: 10.1038/ng2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimoni Y, Altuvia S, Margalit H, Biham O. 2009. Stochastic analysis of the SOS response in Escherichia coli. PLoS One 4:e5363. doi: 10.1371/journal.pone.0005363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cosloy SD, McFall E. 1973. Metabolism of d-serine in Escherichia coli K-12: mechanism of growth inhibition. J Bacteriol 114:685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lusetti SL, Drees JC, Stohl EA, Seifert HS, Cox MM. 2004. The DinI and RecX proteins are competing modulators of RecA function. J Biol Chem 279:55073–55079. doi: 10.1074/jbc.M410371200. [DOI] [PubMed] [Google Scholar]
- 46.McFall E. 1975. Escherichia coli K-12 mutant forming a temperature-sensitive d-serine deaminase. J Bacteriol 121:1074–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heincz MC, McFall E. 1978. Role of the DsdC activator in regulation of d-serine deaminase synthesis. J Bacteriol 136:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jahreis K, Bentler L, Bockmann J, Meyer A, Siepelmeyer J, Lengeler JW. 2002. Adaptation of sucrose metabolism in the adaptation of sucrose metabolism in the Escherichia coli wild-type strain EC3132. J Bacteriol 184:5307–5316. doi: 10.1128/JB.184.19.5307-5316.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moritz RL, Welch RA. 2006. The Escherichia coli argW-dsdCXA genetic island is highly variable, and E. coli K1 strains commonly possess two copies of dsdCXA. J Clin Microbiol 44:4038–4048. doi: 10.1128/JCM.01172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roesch PL, Redford P, Batchelet S, Moritz RL, Pellett S, Haugen BJ, Blattner FR, Welch RA. 2003. Uropathogenic Escherichia coli use d-serine deaminase to modulate infection of the murine urinary tract. Mol Microbiol 49:55–67. doi: 10.1046/j.1365-2958.2003.03543.x. [DOI] [PubMed] [Google Scholar]
- 51.Anfora AT, Haugen BJ, Roesch P, Redford P, Welch RA. 2007. Roles of serine accumulation and catabolism in the colonization of the murine urinary tract by Escherichia coli CFT073. Infect Immun 75:5298–5304. doi: 10.1128/IAI.00652-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Torres-Barceló C, Kojadinovic M, Moxon R, MacLean RC. 2015. The SOS response increases bacterial fitness, but not evolvability, under a sublethal dose of antibiotic. Proc Biol Sci 282:20150885. doi: 10.1098/rspb.2015.0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cirz RT, Chin JK, Andes DR, De Crécy-Lagard V, Craig WA, Romesberg FE. 2005. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol 3:e176. doi: 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Connolly JPR, Finlay BB, Roe AJ. 2015. From ingestion to colonization: the influence of the host environment on regulation of the LEE encoded type III secretion system in enterohaemorrhagic Escherichia coli. Front Microbiol 6:568. doi: 10.3389/fmicb.2015.00568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sperandio V, Torres AG, Jarvis B, Nataro JP, Kaper JB. 2003. Bacteria-host communication: the language of hormones. Proc Natl Acad Sci U S A 100:8951–8956. doi: 10.1073/pnas.1537100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pacheco AR, Curtis MM, Ritchie JM, Munera D, Waldor MK, Moreira CG, Sperandio V. 2012. Fucose sensing regulates bacterial intestinal colonization. Nature 492:113–117. doi: 10.1038/nature11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kendall MM, Gruber CC, Parker CT, Sperandio V. 2012. Ethanolamine controls expression of genes encoding components involved in interkingdom signaling and virulence in enterohemorrhagic Escherichia coli O157:H7. mBio 3:e00050-12. doi: 10.1128/mBio.00050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakanishi N, Tashiro K, Kuhara S, Hayashi T, Sugimoto N, Tobe T. 2009. Regulation of virulence by butyrate sensing in enterohaemorrhagic Escherichia coli. Microbiology 155:521–530. doi: 10.1099/mic.0.023499-0. [DOI] [PubMed] [Google Scholar]
- 59.Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, Morita H, Hattori M, Ohno H. 2011. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469:543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- 60.Fukuda S, Toh H, Taylor TD, Ohno H, Hattori M. 2012. Acetate-producing bifidobacteria protect the host from enteropathogenic infection via carbohydrate transporters. Gut Microbes 3:449–454. doi: 10.4161/gmic.21214. [DOI] [PubMed] [Google Scholar]
- 61.Yang B, Feng L, Wang F, Wang L. 2015. Enterohemorrhagic Escherichia coli senses low biotin status in the large intestine for colonization and infection. Nat Commun 6:6592. doi: 10.1038/ncomms7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu X, McAteer SP, Tree JJ, Shaw DJ, Wolfson EBK, Beatson SA, Roe AJ, Allison LJ, Chase-Topping ME, Mahajan A, Tozzoli R, Woolhouse MEJ, Morabito S, Gally DL. 2012. Lysogeny with Shiga toxin 2-encoding bacteriophages represses type III secretion in enterohemorrhagic Escherichia coli. PLoS Pathog 8:e1002672. doi: 10.1371/journal.ppat.1002672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson CM, Sinclair JF, Smith MJ, O'Brien AD. 2006. Shiga toxin of enterohemorrhagic Escherichia coli type O157:H7 promotes intestinal colonization. Proc Natl Acad Sci U S A 103:9667–9672. doi: 10.1073/pnas.0602359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tyler JS, Beeri K, Reynolds JL, Alteri CJ, Skinner KG, Friedman JH, Eaton KA, Friedman DI. 2013. Prophage induction is enhanced and required for renal disease and lethality in an EHEC mouse model. PLoS Pathog 9:e1003236. doi: 10.1371/journal.ppat.1003236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mellies JL, Haack KR, Galligan DC. 2007. SOS regulation of the type III secretion system of enteropathogenic Escherichia coli. J Bacteriol 189:2863–2872. doi: 10.1128/JB.01859-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hamoen LW, Haijema B, Bijlsma JJ, Venema G, Lovett CM. 2001. The Bacillus subtilis competence transcription factor, ComK, overrides LexA-imposed transcriptional inhibition without physically displacing LexA. J Biol Chem 276:42901–42907. doi: 10.1074/jbc.M104407200. [DOI] [PubMed] [Google Scholar]
- 67.Preobrajenskaya O, Boullard A, Boubrik F, Schnarr M, Rouvière-Yaniv J. 1994. The protein HU can displace the LexA repressor from its DNA-binding sites. Mol Microbiol 13:459–467. doi: 10.1111/j.1365-2958.1994.tb00440.x. [DOI] [PubMed] [Google Scholar]
- 68.Adikesavan AK, Katsonis P, Marciano DC, Lua R, Herman C, Lichtarge O. 2011. Separation of recombination and SOS response in Escherichia coli RecA suggests LexA interaction sites. PLoS Genet 7:e1002244. doi: 10.1371/journal.pgen.1002244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Toval F, Schiller R, Meisen I, Putze J, Kouzel IU, Zhang W, Karch H, Bielaszewska M, Mormann M, Müthing J, Dobrindt U. 2014. Characterization of urinary tract infection-associated Shiga toxin-producing Escherichia coli. Infect Immun 82:4631–4642. doi: 10.1128/IAI.01701-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.