

ABSTRACT
Aag from Bacillus subtilis has been implicated in in vitro removal of hypoxanthine and alkylated bases from DNA. The regulation of expression of aag in B. subtilis and the resistance to genotoxic agents and mutagenic properties of an Aag-deficient strain were studied here. A strain with a transcriptional aag-lacZ fusion expressed low levels of β-galactosidase during growth and early sporulation but exhibited increased transcription during late stages of this developmental process. Notably, aag-lacZ expression was higher inside the forespore than in the mother cell compartment, and this expression was abolished in a sigG-deficient background, suggesting a forespore-specific mechanism of aag transcription. Two additional findings supported this suggestion: (i) expression of an aag-yfp fusion was observed in the forespore, and (ii) in vivo mapping of the aag transcription start site revealed the existence of upstream regulatory sequences possessing homology to σG-dependent promoters. In comparison with the wild-type strain, disruption of aag significantly reduced survival of sporulating B. subtilis cells following nitrous acid or methyl methanesulfonate treatments, and the Rifr mutation frequency was significantly increased in an aag strain. These results suggest that Aag protects the genome of developing B. subtilis sporangia from the cytotoxic and genotoxic effects of base deamination and alkylation.
IMPORTANCE In this study, evidence is presented revealing that aag, encoding a DNA glycosylase implicated in processing of hypoxanthine and alkylated DNA bases, exhibits a forespore-specific pattern of gene expression during B. subtilis sporulation. Consistent with this spatiotemporal mode of expression, Aag was found to protect the sporulating cells of this microorganism from the noxious and mutagenic effects of base deamination and alkylation.
INTRODUCTION
The integrity of genomes of organisms is constantly compromised by intracellular and extracellular factors that have the potential to generate different base modifications, including, oxidations, alkylations, and deaminations (1). These types of nonbulky genetic insults are detected primarily by specific DNA glycosylases and eliminated through the base excision repair (BER) pathway (2). DNA deamination is a major type of spontaneous genetic damage with which cells must contend (3), and the spontaneous loss of the exocyclic amino groups in cytosine, guanine, and adenine yields the bases uracil, xanthine, and hypoxanthine (HX), respectively (4, 5). HX in DNA is potentially mutagenic, since it can pair not only with thymine but also with cytosine and therefore would result in AT-to-GC transitions after DNA replication (6). Organisms such as Escherichia coli and Saccharomyces cerevisiae employ the 3-methyladenine DNA glycosylases AlkA and MAG, respectively, to process HX and the modified bases 3-methyladenine, 7-methylguanine, and 7-methyladenine (7, 8). Other enzymes of mammalian origin, which are structurally unrelated to E. coli AlkA, include alkyl-adenine-DNA glycosylase (AAG), alkyl-N-purine-DNA glycosylase (ANPG), and N-methylpurine-DNA glycosylase (MPG) from human, mouse, and rat, respectively, and these also can excise alkylated and deaminated bases from DNA (7, 9–16). The physiological relevance of eliminating the base analog HX from DNA is evidenced by the mutator phenotype exhibited by bacteria and mammals deficient in these glycosylases (6, 17).
Deaminated bases can also be excised from DNA by endonuclease V (EndoV), an endonuclease that hydrolyzes the second phosphodiester bond located at the 3′ end of the modified base, and homologs of such enzymes have been described in bacteria, archaea, and eukaryotes (18–21).
A recent report revealed that Bacillus subtilis employs uracil DNA glycosylase (Ung) as well as YwqL, an EndoV homolog, to contend with the mutagenic effects of base deamination (22). However, HX can be processed by another repair protein, termed Aag; this alkyl adenine glycosylase is encoded in the genome of B. subtilis by aag (formerly yxlJ), and its product possesses functional and structural similarity with human AAG (23). In addition, a biochemical study indicated that the purified Aag protein preferentially eliminates HX from DNA over alkylated bases (23). In the genome of B. subtilis, aag is oriented adjacent to but in the opposite orientation from katX, a gene expressed only in the developing forespore compartment of sporulating cells and encoding a catalase that protects germinating spores against hydrogen peroxide (24). Despite studies reporting induction of aag as part of the general stress response (25, 26), inspection of the katX-aag intergenic region does not reveal DNA sequences with obvious homology to vegetative or sporulation promoters that may regulate aag expression. Therefore, the molecular mechanisms involved in regulating the expression of aag during the life cycle of B. subtilis were studied in this work. Our results demonstrated a forespore-specific mode of expression of aag. In addition, exposure of B. subtilis sporulating cells lacking Aag to deaminating and alkylating chemicals resulted in increased mutagenesis and more cell killing than for wild-type cells. We propose that aag is expressed during sporulation and that its protein product counteracts the mutagenic effects of hypoxanthine and alkylated bases that may potentially interfere with spore morphogenesis.
MATERIALS AND METHODS
Strain construction and culture conditions.
The strains of B. subtilis and plasmids used in this work are listed in Table 1 and were constructed using standard molecular biology techniques (27).
TABLE 1.
B. subtilis strains and plasmids used in this study
| Strain or plasmid | Genotype or descriptiona | Reference or sourceb |
|---|---|---|
| Strains | ||
| 168 | trpC2 (wild type) | Laboratory stock |
| WN118 | sigGΔ1 trpC2 | 30 |
| PERM791 | ywqL::lacZ Err | 22 |
| PERM1246 | aag::lacZ Err | pPERM1243→168 |
| PERM1286 | aag::yfp Cmr | pPERM1283→168 |
| PERM1322 | sigGΔ1 trpC2 Δaag::lacZ Cmr | pPERM1318→WN118 |
| PERM1374 | sigGΔ1 trpC2 Δaag::lacZ Cmr (pDG148 [Pspac-empty Kanr]) | pDG148→PERM1322 |
| PERM1375 | sigGΔ1 trpC2 Δaag::lacZ Cmr (pDG298 [pDG148::Pspac-sigG Kanr]) | pDG298→PERM1322 |
| PERM1385 | Δaag::lacZ Cmr ΔywqL::lacZ Err | pPERM1318→PERM791 |
| Plasmids | ||
| pDG148 | Shuttle IPTG-inducible Pspac vector; Ampr Kanr | Laboratory stock |
| pDG298 | pDG148 containing Pspac-sigG gene; Ampr Kanr | 30 |
| pMUTIN4 | Integrational lacZ fusion vector; Ampr Err | 28 |
| pMUTIN4cat | Integrational lacZ fusion vector; Cmr | 29 |
| pSG1187 | Integrational yfp fusion vector; Ampr Cmr | 31 |
| pPERM1243 | pMUTIN4 containing an internal region (360 bp) of aag; Err | This study |
| pPERM1283 | pSG1187 containing the RBS and ORF (606 bp) of aag; Ampr Cmr | This study |
| pPERM1318 | pMUTIN4cat containing an internal region (360 bp) of aag; Err | This study |
Selection markers: Amp, ampicillin; Cm, chloramphenicol; Er, erythromycin; Kan, kanamycin.
X→Y indicates that strain Y was transformed with DNA from source X.
To obtain strains harboring a transcriptional aag-lacZ fusion, the integrative vector pMUTIN4 (28) or a chloramphenicol-resistant variant of this plasmid, pMUTIN4cat (29), was utilized. For recombinant plasmid construction, a 360-bp HindIII/BamHI internal fragment of aag (positions +121 to + 480 relative to the aag start codon) was amplified by PCR using chromosomal DNA of B. subtilis 168 and Vent DNA polymerase (New England BioLabs, Ipswich, MA). The oligonucleotide primers used for the amplification of the aag fragment were 5′-GCAAGCTTTATATTGTGGAAACAGAGG (forward) and 5′-GCGGATCCGTACCCGCTTTCGATGTAA (reverse), including the restriction sites (underlined) for cloning the amplified aag product between the HindIII/BamHI sites of pMUTIN4 or pMUTIN4cat. The resulting constructs, termed pPERM1243 (aag::lacZ, obtained in pMUTIN4) and pPERM1318 (aag::lacZ, obtained in pMUTIN4cat) were propagated in E. coli XL10-Gold cells (Stratagene, La Jolla, CA). Plasmid pPERM1243 was used to transform competent cells of B. subtilis 168, generating strain PERM1246 (aag-lacZ), and plasmid pPERM1318 was used to transform competent cells of B. subtilis WN118 (sigGΔ1 trpC2) (30), generating strain PERM1322 (sigGΔ1 trpC2 aag-lacZ). The correct integration of the constructions into the aag locus was corroborated by PCR using specific oligonucleotide primers (data not shown).
To investigate the subcellular localization of Aag during sporulation, a translational aag::yfp fusion was constructed by employing the integrative plasmid pSG1187 (31). In this construction, a 606-bp-long KpnI/ClaI fragment of aag (positions −18 to +588 relative to the aag start codon) including the entire open reading frame (ORF) and the ribosome binding site (RBS) of the aag gene was amplified by PCR using chromosomal DNA of B. subtilis 168. The oligonucleotide primers used for the amplification of this aag fragment were 5′-GCGGTACCAAAAGGAGAGGTCGATTCGTG (forward) and 5′-GCATCGATCGACACGTACCTGTTTCCCGT (reverse), including the restriction sites (underlined) for cloning the amplified products of aag between the KpnI/ClaI sites of pSG1187. The resulting construct, designated pPERM1283 (aag::yfp), was propagated in E. coli GM161 cells. This plasmid was used to transform competent cells of B. subtilis strain 168, generating strain PERM1286 (aag-yfp). The correct integration of constructions into the corresponding aag locus was verified by PCR using specific oligonucleotide primers (data not shown).
To complement the σG deficiency of B. subtilis strain PERM1322 bearing a chromosomal copy of the aag-lacZ fusion, plasmid pDG298 (30), carrying a copy of sigG under the control of the IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible Pspac promoter in plasmid pDG148, was transformed into competent cells of B. subtilis PERM1322, generating strain PERM1375 [sigGΔ1 trpC2 aag-lacZ(pDG298, pDG148::Pspac-sigG)]. As a control, the empty pDG148 vector (Pspac-empty) was also used to transform strain PERM1322, generating strain PERM1374 [sigGΔ1 trpC2 aag-lacZ(pDG148::Pspac-empty)]. The aag ywqL strain (PERM1385) was obtained by transformation of the ywqL strain (PERM791) (22) with pPERM1318.
Liquid cultures of B. subtilis were grown in Luria-Bertani (LB) medium, Difco antibiotic no. 3 (PAB) medium, or Difco sporulation medium (DSM) (32). When required, erythromycin (Er) (5 μg ml−1), chloramphenicol (Cm) (5 μg ml−1), kanamycin (Kn) (10 μg ml−1), or IPTG (1 mM) was added to media. E. coli cultures were grown in LB medium supplemented with ampicillin (Amp) (100 μg ml−1) or Cm (25 μg ml−1). Solid media were obtained by adding bacteriology agar (15 g liter−1) to the liquid media. For all experiments, strains were grown overnight in liquid LB medium at 37°C and then diluted 100-fold into appropriate medium. All liquid cultures were incubated at 37°C with vigorous aeration. Cultures on solid media were incubated at 37°C in the dark.
β-Galactosidase assay.
All B. subtilis strains harboring the aag-lacZ fusion were grown in either liquid PAB medium (to avoid sporulation) or liquid DSM (to promote sporulation) at 37°C. Cells from 1-ml samples were harvested by centrifugation during vegetative growth and throughout sporulation when appropriate. Cell pellets were immediately frozen and stored at −20°C prior to determination of β-galactosidase activity as previously described (33). The level of β-galactosidase during sporulation was determined as follows. Cells collected from the different sporulation stages were treated with lysozyme and centrifuged (14,500 × g, 2 min), and β-galactosidase in the supernatant was assayed using ortho-nitrophenyl-β-d-galactopyranoside (ONPG) as the substrate, and this value was assigned to the mother cell fraction. The pellet fraction (lysozyme-resistant forespores containing spore coats) from sporulation stages T4 (4 h after the onset of sporulation) to T8 was subjected to spore coat removal (33), washed five times with TS buffer (25 mM Tris-HCl [pH 7.4], 0.1 M NaCl), and subjected to lysozyme treatment, and the β-galactosidase assay was repeated to determine the β-galactosidase activity inside forespores. The β-galactosidase activity was expressed in Miller units (34). The endogenous ONPGase-specific activity expressed by the wild-type strain without a lacZ fusion during growth and sporulation was determined in parallel and subtracted from the data obtained for strains carrying the aag-lacZ fusion. This correction was at most ∼2 Miller units.
Microscopy.
B. subtilis containing the aag::yfp fusion was sporulated in DSM. FM4-64 (5 μg ml−1) was added to sporulating cells at stage T1 to stain membranes. At the appropriate times, samples of 0.5 ml of the culture were harvested by centrifugation (14,500 × g, 1 min) and resuspended in 50 μl of fresh DSM. Cells from 5 μl of this concentrated cell suspension were immobilized on an agarose spot (20 mg ml−1) placed on a microscopy slide and covered with a coverslip prior to microscopic analysis. Fluorescence microscopy was performed with a Zeiss Axioscope A1 microscope equipped with an AxioCam ICc1. Images were acquired and adjusted only for brightness and contrast with the AxioVision V 4.8.2 software. Exposure times were typically 2 s for Aag-yellow fluorescent protein (Aag-YFP) and 0.5 s for FM4-64. Samples of the wild-type parental strain were collected at the same stages of the sporulation process as described for the strain carrying the aag::yfp fusion and processed for microscopic analysis. No fluorescent background corresponding to the YFP channel was detected in the sporangia analyzed (results not shown).
Primer extension.
The 5′ end of aag mRNA was mapped by primer extension of aag transcripts produced during wild-type B. subtilis sporulation or following induction of sigG expression during logarithmic growth of strain PERM1375 as follows. Samples of total RNA (40 μg), isolated as previously described (35), were hybridized with the 20-mer oligonucleotide 5′-TAGCCTGACGCTGTGCCTTC-3′, which was complementary to the aag mRNA from nucleotide +103 to +122 relative to the translational start codon of aag. The oligonucleotide primer was radiolabeled on its 5′ end with [γ-32P]ATP (PerkinElmer, Foster City, CA) and T4 polynucleotide kinase (New England BioLabs, Ipswich, MA), and the primer was extended with 40 units of Moloney murine leukemia virus (M-MLV) RNase H Minus reverse transcriptase (Promega, Madison, WI). The position of the extension product was determined by running a DNA sequencing reaction generated with the same 20-nucleotide (nt) primer and a 1-kb PCR product extending from bp −412 to +588 relative to the aag start codon using a Thermo Sequenase dye primer manual cycle sequencing kit (Affymetrix-USB, Cleveland, OH). The extension products and the sequencing DNA ladder were separated by electrophoresis through an 8 M urea–6% polyacrylamide DNA sequencing gel and analyzed using a personal molecular imager (Bio-Rad Laboratories, Hercules, CA) and Quantity-One software.
Determination of sporangial sensitivity to HNO2 and MMS.
The sensitivity of B. subtilis sporulating cells to HNO2 or methyl methanesulfonate (MMS) was determined from dose-response curves for the wild-type strain and the aag, ywqL, and aag ywqL strains. Cells of various strains were sporulated in liquid DSM, collected by centrifugation (5,000 × g, 10 min) at 4.5 h after the onset of sporulation (T4.5, with T0 the time when exponential growth of cultures stopped), washed twice with phosphate-buffered saline (PBS) (0.7% Na2HPO4, 0.3% KH2PO4, 0.4% NaCl [pH 7.5]), and adjusted to an optical density at 600 nm (OD600) of 1.0. Two-milliliter samples of cellular suspension were treated with different concentrations of either HNO2 (0, 2.5, 5, or 7.5 mM), prepared as was previously described (36), or MMS (0, 30, 60, or 90 mM) and incubated for 1 h at room temperature with shaking. After treatment, cells were washed again with PBS, and cell viability was determined by plating aliquots of serial dilutions on LB agar plates that were incubated overnight at 37°C and counting colonies.
Two-milliliter aliquots of T4.5 sporulating cells of all strains adjusted to an OD600 of 1.0 were treated for 1 h at room temperature with the 50% lethal dose (LD50) of HNO2 or MMS calculated for wild-type cells and washed twice with PBS, and 10-fold serial dilutions were spot plated on solid LB medium. The plates were incubated overnight at 37°C and then photographed in a Gel-Doc EZ imaging system (Bio-Rad Laboratories, Hercules, CA).
Determination of HNO2- or MMS-induced mutagenesis.
Cells of different strains were sporulated in liquid DSM and collected by centrifugation (5,000 × g, 10 min) at stage T4.5, and each culture was divided in half and washed with PBS. One of the cultures was untreated, and the other was treated for 1 h at room temperature with the LD50 of either HNO2 or MMS (calculated for each strain from the survival curves in Fig. 6A and 7A). Mutation frequencies were calculated by plating aliquots on six LB plates containing 10 μg ml−1 of rifampin (Rif) as well as plating aliquots of the appropriate dilution on LB plates to determine the total viable count. Rifr colonies were counted after 48 h of incubation at 37°C.
FIG 6.

Sensitivity of sporulating cells to HNO2. (A) Survival of B. subtilis wild-type (●), aag (◆), ywqL (▲), and aag ywqL (■) strains following HNO2 treatment. Cells from various strains were sporulated in liquid DSM and treated with different concentrations of HNO2 at 4.5 h after the onset of sporulation, and cell viability was determined as described in Materials and Methods. Results are expressed as averages ± SD from at least three independent experiments. (B) Resistance of B. subtilis wild-type (WT), aag, ywqL, and aag ywqL strains after exposure to HNO2. Cells from all strains were sporulated in liquid DSM, treated with the LD50 of HNO2 (calculated from the dose-response curve in panel A for the wild-type strain) at 4.5 h after the onset of sporulation, spot plated, grown, and photographed as described in Materials and Methods.
FIG 7.

Sensitivity of sporulating cells to MMS. (A) Killing of B. subtilis wild-type (●), aag (◆), ywqL (▲), and aag ywqL (■) strains by MMS. Cells from all strains were sporulated in liquid DSM and treated with different concentrations of MMS at 4.5 h after the onset of sporulation, and cell viability was determined as described in Materials and Methods. Results are expressed as averages ± SD from at least three independent experiments. (B) Resistance of B. subtilis wild-type (WT), aag, ywqL, and aag ywqL strains after exposure to MMS. Cells from all strains were sporulated in liquid DSM, treated with the LD50 of MMS (calculated from the dose-response curve in panel A for the wild-type strain) at 4.5 h after the onset of sporulation, spot plated, grown, and photographed as described in Materials and Methods.
Statistical analyses.
Differences between lethal doses obtained from survival curves for HNO2 and MMS treatments were calculated by performing one-way analysis of variance (ANOVA) followed by Tukey's post hoc analyses, with a P value of <0.05 considered significant. For HNO2- or MMS-induced mutagenesis, results were tested by the Mann-Whitney U test, and analyses were done using Minitab17 software.
RESULTS
Expression of aag is primarily during spore development.
To analyze the expression of aag during the life cycle of B. subtilis, a recombinant strain bearing a transcriptional in-frame aag-lacZ fusion (PERM1246) was inoculated in liquid PAB medium to promote vegetative growth. Under these conditions, aag-directed β-galactosidase activity was very low during exponential growth and stationary phase (Fig. 1A). This aag-lacZ fusion was also used to measure aag expression during the differentiation process of sporulation. Cells of strain PERM1246 were sporulated in liquid DSM, and samples harvested at various times during growth and sporulation were assayed for β-galactosidase. These assays showed low levels of expression of the reporter gene during exponential growth and early sporulation (Fig. 1B). Cell fractionation experiments with cell samples collected from sporulation stages T4 to T8 revealed that whereas β-galactosidase activity from the aag-lacZ fusion decreased in the mother cell compartment, the levels of this enzyme activity increased significantly in the forespore fraction (Fig. 1B). Together these results suggest that aag is expressed primarily during sporulation and in the forespore compartment of the sporulating cell.
FIG 1.

Expression of aag during B. subtilis growth and sporulation. (A) β-Galactosidase activity from an aag-lacZ fusion during vegetative growth. B. subtilis strain PERM1246 harboring an aag-lacZ fusion was grown in PAB medium and the OD600 measured (●). Cells were collected at various times, and the β-galactosidase activity of growing cells (△) was determined as described in Materials and Methods. Values for β-galactosidase specific activity are the averages of values from three independent experiments ± standard deviations (SD). (B) β-Galactosidase specific activity from an aag-lacZ fusion during sporulation. B. subtilis strain PERM1246 harboring an aag-lacZ fusion was induced to sporulate in DSM and the OD600 measured (●). Time zero of sporulation (T0) is defined as the time when log-phase cell growth stops. Cells were collected at various times, and β-galactosidase assays were conducted in both mother cell (△) and forespore (▲) fractions as described in Materials and Methods. Values for β-galactosidase specific activity are the averages of values from three independent experiments ± SD.
Expression of a chimeric Aag-YFP protein occurs mainly inside the forespore compartment.
To further confirm the forespore-specific transcription of aag, we analyzed the compartmental synthesis of a chimeric Aag-YFP protein during sporulation of a B. subtilis strain bearing a translational in-frame aag-yfp fusion in the aag locus (PERM1286). Samples of sporulating cells stained with FM4-64, a lipophilic dye that strongly adheres to the cell membrane and allows discernment of both compartments of sporangia (mother cell and forespore), were collected from stages T2, T4.5, T6, and T8 of sporulation. Each sample was examined by epifluorescence microscopy. The results revealed that the Aag-YFP protein was present in the forespore compartment but absent from the mother cell compartment (Fig. 2). These results further support the concept that aag is transcribed in the forespore compartment during late stages of sporulation.
FIG 2.

Localization of Aag-YFP during B. subtilis sporulation. B. subtilis strain PERM1286 harboring a translational in-frame aag-yfp fusion was grown and sporulated in DSM. At 2, 4.5, 6, and 8 h after the onset of sporulation, sporulating cells were analyzed by bright-field (BF) and fluorescence (YFP and FM4-64) microscopy as described in Materials and Methods. Overlain images of YFP and FM4-64 at each time point are depicted as merge. MC, mother cell; FS, forespore compartments. Scale bar, 5 μM.
Expression of aag is dependent on the forespore-specific sigma factor σG.
The forespore-specific pattern of expression exhibited by aag was reminiscent of a group of genes, the σG regulon, that are transcribed during late stages of sporulation exclusively in the forespore compartment by RNA polymerase with the forespore-specific sigma factor σG (37, 38). To investigate whether aag expression was indeed dependent on σG, the transcriptional aag-lacZ fusion was inserted into the aag locus of a B. subtilis ΔsigG strain (WN118) (30) by homologous recombination. The resulting strain (B. subtilis PERM1322) was sporulated in liquid DSM, and the aag-directed β-galactosidase activity was determined from various samples collected during growth and sporulation. Notably, expression of the aag-lacZ fusion was almost completely abolished inside the forespore compartment in the ΔsigG genetic background (Fig. 3A), strongly suggesting that aag transcription is σG dependent. To further test this suggestion, strain PERM1322 was transformed with the self-replicating plasmid pDG298 harboring a copy of sigG under the control of an IPTG-inducible Pspac promoter (30), giving strain PERM1375. As a control, strain PERM1322 was also transformed with plasmid pDG148 (empty vector), generating strain PERM1374. Both the PERM1374 and PERM1375 strains were grown in PAB medium to an OD600 of 0.5, and IPTG was added to 1 mM. As shown in Fig. 3B, the addition of IPTG induced the expression of the aag-lacZ fusion in the strain that contained the Pspac-sigG cassette but not in the control strain bearing the empty vector pDG148.
FIG 3.

σG dependence of aag-lacZ expression. (A) β-Galactosidase activity of aag-lacZ during sporulation of the ΔsigG strain. B. subtilis strain PERM1375 harboring an aag-lacZ fusion in a ΔsigG genetic background was sporulated in DSM and the OD600 measured (●). Cells were collected at various times, and β-galactosidase activity was assayed in mother cell (△) and forespore (▲) fractions as described in Materials and Methods. Values for β-galactosidase specific activity are the averages of values from three independent experiments ± SD. (B) β-Galactosidase activity from an aag-lacZ fusion induced by overexpression of sigG during vegetative growth. B. subtilis strains PERM1375 and PERM1374 harboring an aag-lacZ fusion in a ΔsigG genetic background containing either a Pspac::sigG gene fusion (●) or the empty vector (○) were grown in PAB medium and induced with IPTG (1 mM) when an OD600 of 0.5 was reached. The OD600s of cultures were measured (● and ○), cells were collected at various times, and β-galactosidase assays from cells that were (▲) or were not (△) complemented with sigG were conducted as described in Materials and Methods. Values for β-galactosidase specific activity are the averages of values from three independent experiments ± SD.
Mapping the 5′ ends of aag mRNA.
The genetic arrangement of the aag locus reveals that this gene is preceded by katX, encoding the major catalase in dormant spores and transcribed divergently with respect to aag (Fig. 4A) (24), and followed by yxzF, which codes for an uncharacterized protein. The aag and yxzF genes are separated by only 28 bp, and there is not an apparent transcriptional terminator in this intergenic region, suggesting that these two genes are in a bicistronic operon (Fig. 4A). As demonstrated in this work, the expression of aag was dependent on the forespore-specific sigma factor σG. Therefore, the likely promoter sequence recognized by σG to activate aag transcription was located by mapping the 5′ end of aag mRNA produced in vivo. Total RNA isolated from the wild-type strain during stage T5 of sporulation as well as from vegetative cells of PERM1375, 3 h after IPTG induction of sigG, was hybridized with a 19-nt-long radioactive primer complementary to aag mRNA from nt +103 to +122 relative to the aag translational start codon and subjected to primer extension analysis. This analysis revealed the synthesis of an extension product beginning 69 bp upstream of the aag translation start codon using mRNAs from either sporulating cells of the wild-type strain or vegetative cells of the strain overexpressing sigG (Fig. 4B, lanes 1 and 2).
FIG 4.
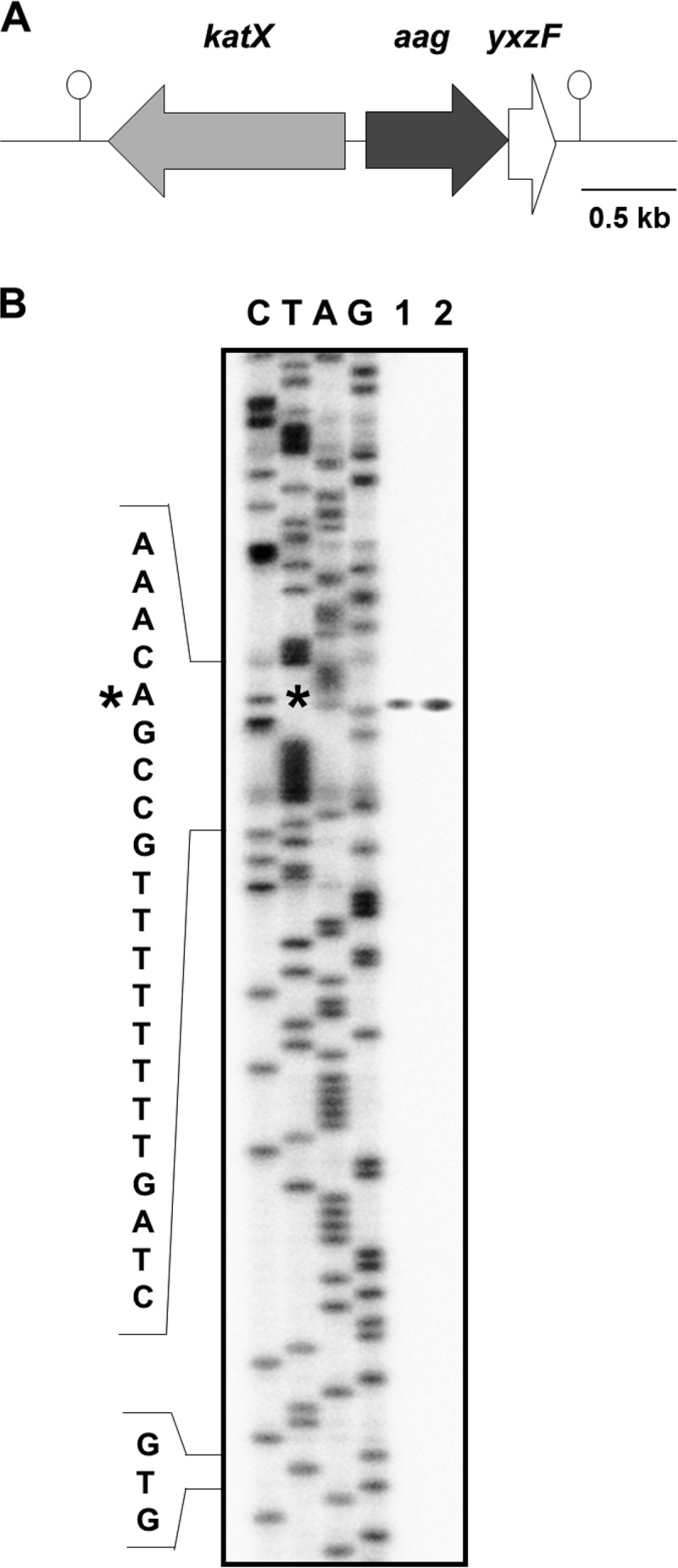
Genetic map of the aag region and mapping of the aag promoter. (A) The B. subtilis chromosomal region around aag. The katX gene is divergently transcribed upstream of aag, and aag and yxzF may comprise a bicistronic operon. Hairpin structures denote putative transcriptional terminators. The drawing is to scale. (B) Primer extension analysis to identify the 5′ end of aag mRNA. Total RNA was isolated from T6 sporulating cells of B. subtilis strain 168 (wild type) grown in DSM (lane 1) or vegetative cells of strain PERM1375 grown in PAB medium, induced with IPTG, and harvested 3 h after IPTG addition (lane 2). Primer extension was performed as described in Materials and Methods. The asterisk indicates the position of the primer extension product relative to the DNA sequencing ladder (lanes C, T, A, and G). The position of the mapped 5′ end of aag mRNA is denoted with an asterisk. “GTG” indicates the position of the aag translation start codon.
Having found that an adenine located 69 nt upstream of the first codon of the aag ORF functions as the transcription start for the synthesis of aag mRNAs, we inspected the sequences preceding this site to identify possible σG promoter sequences (39). Notably, regions located at −10 and −35 with respect to the transcription start site, and separated by 18 nt, exhibited a good level of homology with the consensus −10 and −35 regions of σG-dependent promoters (Fig. 5).
FIG 5.

Comparison of the consensus σG (39) promoter sequence with the putative promoter sequences (Paag) upstream of the aag ORF. Perfect matches are in bold. The position of the aag transcription start site is denoted with an asterisk. Abbreviations: H, A or C; R, A or G; and X, A or T.
Aag and YwqL protect sporulating B. subtilis cells from cytotoxic and genotoxic effects of HNO2.
The forespore-specific expression exhibited by aag prompted us to investigate the DNA repair functions of Aag during B. subtilis spore development. To this end, sporangia from wild-type and aag strains collected at T4.5 of sporulation were challenged with increasing doses of nitrous acid (HNO2), an agent that promotes deamination of adenine to HX (40). Notably, the Aag-deficient sporangia were only slightly more susceptible to the genotoxic effects of HNO2 than sporangia from the parental wild-type strain (Fig. 6), although the LD90 values for HNO2 were significantly different between wild-type and aag sporangia (Table 2). The relatively small differences seen in the latter experiment suggested the existence of additional pathways involved in counteracting HNO2-induced base deamination in these sporulating cells. In agreement with this notion, disruption of ywqL, encoding an endonuclease capable of processing a wider range of deaminated bases (22; V. M. Ayala-Garcia and M. Pedraza-Reyes, unpublished data), generated sporangia that were significantly more sensitive to HNO2 than those of the Aag-deficient strain (Fig. 6; Table 2). Importantly, disruption of aag did not significantly increase the sensitivity of the ywqL sporangia to the genotoxic effects of HNO2 (Fig. 6; Table 2). Together, these results suggest that YwqL rather than Aag is most important in eliminating HX and probably other deaminated bases from DNA during B. subtilis sporulation.
TABLE 2.
LD50 and LD90 values for wild-type and mutant sporulating cells treated with HNO2 and MMS
| Strain | HNO2 |
MMS |
||
|---|---|---|---|---|
| LD50 (mM)a | LD90 (mM) | LD50 (mM) | LD90 (mM) | |
| WT | 3.47 ± 0.11 a | 7.49 ± 0.06 a | 38.86 ± 0.30 a | 71.20 ± 0.57 a |
| PERM1246 (aag) | 2.92 ± 0.05 a | 6.82 ± 0.09 b | 15.16 ± 0.17 b | 32.69 ± 0.37 b |
| PERM791 (ywqL) | 2.17 ± 0.09 b | 5.14 ± 0.10 c | 37.93 ± 0.61 a | 69.49 ± 0.67 a |
| PERM1385 (aag ywqL) | 1.90 ± 0.06 b | 4.80 ± 0.08 c | 14.94 ± 0.31 b | 32.47 ± 0.40 b |
Values are means ± standard errors of the means (SEM). Letters indicate statistically significantly different values between strains as determined by one-way analysis of variance (ANOVA) followed by Tukey's post hoc tests (P < 0.05).
Aag protects sporulating B. subtilis cells from cytotoxic and genotoxic effects of MMS.
In addition to processing HX, Aag also can remove 3-mA and 3-mG from DNA (23). Therefore, we compared the abilities of wild-type and mutant sporangia lacking Aag and/or YwqL to survive treatment with the alkylating agent MMS. Interestingly, the absence of Aag but not YwqL sensitized sporulating cells to the genotoxic effects of MMS, and this effect did not increase in cells deficient for both repair proteins (Fig. 7; Table 2). Taken together, these results suggest that Aag is primarily involved in eliminating alkylated DNA bases during spore development.
Aag and YwqL counteract the mutagenic effects of HNO2 and MMS.
We next investigated whether Aag and/or YwqL was involved in preventing the mutagenic effects promoted by HNO2 and MMS during spore development. To this end, sporulating B. subtilis wild-type, aag, ywqL, and aag ywqL cells at T4.5 were treated or not with the LD50 of HNO2 or MMS, and the mutation frequency for colonies resistant to rifampin (Rifr) was determined for each strain. With respect to wild-type sporulating cells, the lack of Aag or YwqL alone did not increase the spontaneous Rifr mutation frequency. However, this parameter increased significantly when both aag and ywqL were absent (Fig. 8). HNO2 treatment increased mutagenesis in sporangia of all the strains tested, but the effect was maximal in strains that were deficient for either Aag or YwqL and did not increase in aag ywqL sporangia (Fig. 8A). In contrast, MMS had a large mutagenic effect on sporulating cells of the aag and aag ywqL strains but not those of the ywqL strain (Fig. 8B). Overall, Aag and YwqL both seem to participate in counteracting the mutagenic effects promoted by factors that damage the DNA of sporulating cells through deamination and alkylation of bases.
FIG 8.

Frequencies of mutation to Rifr. Cells of B. subtilis wild-type, aag, ywqL, and aag ywqL strains were sporulated in liquid DSM and treated with the LD50 for each strain of either HNO2 (A) or MMS (B) at 4.5 h after the onset of sporulation. The levels of Rifr cells with (+) or without (−) treatment exposure were determined. Each bar represents the mean of data collected from three independent experiments, and error bars represent the standard error of the mean (SEM). *, P < 0.05; **, P < 0.01 (by the Mann-Whitney U test).
DISCUSSION
The soil bacterium B. subtilis may be exposed to a plethora of environmental pollutants, including reactive nitrogen species (RNS) generated by oxidation or reduction of nitric oxide (NO·). RNS resulting from microbial denitrification (41) include nitrogen dioxide (NO2·), peroxynitrite (ONOO−), and peroxynitrous acid (ONOOH) (42), all of which are potent triggers of DNA base deamination, generating, among others, the base analog HX (40). In vegetative cells of B. subtilis, HX may potentially be removed from DNA by the endonuclease YwqL (22). However, this organism also possesses aag, encoding a DNA glycosylase that operates on HX as well as alkylated adenine derivatives (23).
In contrast to alkA, B. subtilis aag does not seem to be under the control of the Ada response (43); however, aag expression is regulated in concert with the general stress σB regulon (25, 26). Furthermore, inducible levels of expression of this gene were detected during spore germination/outgrowth (44). Our analysis using a B. subtilis strain carrying a transcriptional aag-lacZ fusion revealed that aag transcription is very low during exponential growth and early stages of sporulation but increases significantly during late stages of this developmental process. Of note, genes encoding a number of DNA repair proteins, including splB, ywjD, mfd, uvrA, and recA, exhibit similar expression patterns (45–48). In addition, results from one transcriptomic study did report aag expression associated with sporulation stages T7 and T8 (49). In the current work, results from fractionation of sporulating cells of the aag-lacZ strain and microscopic analysis of sporangia carrying an aag-yfp gene fusion provided strong evidence that transcription of aag takes place mainly in the forespore compartment of B. subtilis sporangia. Furthermore, genetic evidence showing that expression of aag-lacZ was eliminated in a σG-deficient strain and restored by in trans expression of σG strongly supports the notion that aag is expressed in the forespore compartment under the control of RNA polymerase with σG. Based on an in silico analysis, a previous report proposed that common regulatory elements may direct the expression of the katX and aag genes (23). However, primer extension analysis detected specific regulatory regions directing aag that are different from those described for katX (24). Indeed, the localization of the 5′ end of aag mRNA allowed the identification of DNA sequences centered at −10 and −35 relative to the transcription start site that exhibited a reasonable level of homology to −10 and −35 sequences of σG-dependent promoters (39). Based on these results, we postulate that aag is an additional member of the σG regulon. Of note, katX was also reported to be part of the σB regulon (25, 26), and previous work also demonstrated that σF directs katX expression inside the forespore compartment during B. subtilis sporulation (24).
In agreement with the forespore-specific expression of aag, B. subtilis aag sporangia exhibited slight but significantly increased susceptibility to HNO2; however, sporangia deficient for the endonuclease YwqL were more severely sensitized to this genotoxic chemical. These results are in agreement with the ability of YwqL to process the full range of deaminated bases generated by nitrous acid (V. M. Ayala-García and M. Pedraza-Reyes, unpublished data), while Aag acts specifically on HX (23). Interestingly, these two DNA repair proteins played equivalent roles in counteracting the mutagenic effects of HNO2, since compared to the wild-type strain, the aag and ywqL strains exhibited higher but almost identical levels of HNO2-induced mutagenesis. Therefore, Aag and YwqL may work together to protect sporulating cells of B. subtilis from the genotoxic, cytotoxic, and mutagenic effects of base deamination.
In a marked contrast, aag but not ywqL sporangia exhibited increased sensitivity to the alkylating agent MMS. This alkylating agent also induced mutagenesis in the aag sporangia but not in ywqL sporangia, confirming that YwqL does not act on alkylated DNA. Interestingly, although B. subtilis possesses additional pathways to process alkylated bases, including AlkA and the products of the adaA-adaB operon (50–53), our results showed that the absence of Aag alone generated sporangia that were more susceptible to the cytotoxic and mutagenic effects of MMS. However, the following observations may explain the latter, somewhat anomalous results: (i) MMS mainly induces the formation of the nonbulky DNA lesions 3-mA, 3-mG, and 7-mG (54, 55), while (ii) the products of the adaA-adaB operon and AlkA are more important in removing bulky alkylated lesions generated by N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) and N-propyl-N′-nitro-N-nitrosoguanidine (PNNG) (43).
Finally, as shown in this work, the synthesis of a chimeric Aag-Yfp protein took place inside the forespore compartment, strongly suggesting that Aag is packaged in the dormant spores putatively to eliminate alkylated DNA during spore germination/outgrowth. Indeed, B. subtilis spores are not resistant to the genotoxic effects of alkylating agents like MMS (56). Therefore, experiments are under way to explore the contribution of Aag in ensuring the successful return to life of B. subtilis spores.
ACKNOWLEDGMENTS
V.M.A.-G. and L.I.V.-G. were supported by a scholarship from the Consejo Nacional de Ciencia y Tecnología (CONACYT).
We are grateful for the excellent technical assistance of Rocío del C. Barajas-Ornelas and Fernando H. Ramírez-Guadiana.
REFERENCES
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. 2006. DNA repair and mutagenesis, 2nd ed American Society for Microbiology, Washington, DC. [Google Scholar]
- 2.Dalhus B, Laerdahl JK, Backe PH, Bjørås M. 2009. DNA base repair—recognition and initiation of catalysis. FEMS Microbiol Rev 33:1044–1078. doi: 10.1111/j.1574-6976.2009.00188.x. [DOI] [PubMed] [Google Scholar]
- 3.Yonekura SI, Nakamura N, Yonei S, Zhang-Akiyama QM. 2009. Generation, biological consequences and repair mechanisms of cytosine deamination in DNA. J Radiat Res 50:19–26. doi: 10.1269/jrr.08080. [DOI] [PubMed] [Google Scholar]
- 4.Lindahl T. 1993. Instability and decay of the primary structure of DNA. Nature 362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 5.Barnes DE, Lindahl T. 2004. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet 38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 6.Hill-Perkins M, Jones MD, Karran P. 1986. Site-specific mutagenesis in vivo by single methylated or deaminated purine bases. Mutat Res 162:153–163. doi: 10.1016/0027-5107(86)90081-3. [DOI] [PubMed] [Google Scholar]
- 7.Saparbaev M, Laval J. 1994. Excision of hypoxanthine from DNA containing dIMP residues by the Escherichia coli, yeast, rat, and human alkylpurine DNA glycosylases. Proc Natl Acad Sci U S A 91:5873–5877. doi: 10.1073/pnas.91.13.5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao B, O'Brien PJ. 2011. Kinetic mechanism for the excision of hypoxanthine by Escherichia coli AlkA and evidence for binding to DNA ends. Biochemistry 50:4350–4359. doi: 10.1021/bi200232c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakravarti D, Ibeanu GC, Tano K, Mitra S. 1991. Cloning and expression in Escherichia coli of a human cDNA encoding the DNA repair protein N-methylpurine-DNA glycosylase. J Biol Chem 266:15710–15715. [PubMed] [Google Scholar]
- 10.O'Connor TR, Laval J. 1991. Human cDNA expressing a functional DNA glycosylase excising 3-methyladenine and 7-methylguanine. Biochem Biophys Res Commun 176:1170–1177. doi: 10.1016/0006-291X(91)90408-Y. [DOI] [PubMed] [Google Scholar]
- 11.Samson L, Derfler B, Boosalis M, Call K. 1991. Cloning and characterization of a 3-methyladenine DNA glycosylase cDNA from human cells whose gene maps to chromosome 16. Proc Natl Acad Sci U S A 88:9127–9131. doi: 10.1073/pnas.88.20.9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Connor TR. 1993. Purification and characterization of human 3-methyladenine-DNA glycosylase. Nucleic Acids Res 21:5561–5569. doi: 10.1093/nar/21.24.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dosanjh MK, Roy R, Mitra S, Singer B. 1994. 1, N6-ethenoadenine is preferred over 3-methyladenine as substrate by a cloned human N-methylpurine-DNA glycosylase (3-methyladenine-DNA glycosylase). Biochemistry 33:1624–1628. doi: 10.1021/bi00173a002. [DOI] [PubMed] [Google Scholar]
- 14.Saparbaev M, Kleibl K, Laval J. 1995. Escherichia coli, Saccharomyces cerevisiae, rat and human 3-methyladenine DNA glycosylases repair 1, N6-ethenoadenine when present in DNA. Nucleic Acids Res 23:3750–3755. doi: 10.1093/nar/23.18.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T. 2000. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci U S A 97:13573–13578. doi: 10.1073/pnas.97.25.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Brien PJ, Ellenberger T. 2004. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J Biol Chem 279:9750–9757. doi: 10.1074/jbc.M312232200. [DOI] [PubMed] [Google Scholar]
- 17.Kamiya H, Miura H, Kato H, Nishimura S, Ohtsuka E. 1992. Induction of mutation of a synthetic c-Ha-ras gene containing hypoxanthine. Cancer Res 52:1836–1839. [PubMed] [Google Scholar]
- 18.Gates FT, Linn S. 1977. Endonuclease V of Escherichia coli. J Biol Chem 252:1647–1653. [PubMed] [Google Scholar]
- 19.Yao M, Hatahet Z, Melamede RJ, Kow YW. 1994. Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3′endonuclease, from Escherichia coli. J Biol Chem 269:16260–16268. [PubMed] [Google Scholar]
- 20.He B, Qing H, Kow YW. 2000. Deoxyxanthosine in DNA is repaired by Escherichia coli endonuclease V. Mutat Res 459:109–114. doi: 10.1016/S0921-8777(99)00063-4. [DOI] [PubMed] [Google Scholar]
- 21.Cao W. 2013. Endonuclease V: an unusual enzyme for repair of DNA deamination. Cell Mol Life Sci 70:3145–3156. doi: 10.1007/s00018-012-1222-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lopez-Olmos K, Hernández MP, Contreras-Garduño JA, Robleto EA, Setlow P, Yasbin RE, Pedraza-Reyes M. 2012. Roles of endonuclease V, uracil-DNA glycosylase, and mismatch repair in Bacillus subtilis DNA base-deamination-induced mutagenesis. J Bacteriol 194:243–252. doi: 10.1128/JB.06082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aamodt RM, Falnes PØ, Johansen RF, Seeberg E, Bjørås M. 2004. The Bacillus subtilis counterpart of the mammalian 3-methyladenine DNA glycosylase has hypoxanthine and 1,N6-ethenoadenine as preferred substrates. J Biol Chem 279:13601–13606. doi: 10.1074/jbc.M314277200. [DOI] [PubMed] [Google Scholar]
- 24.Bagyan I, Casillas-Martinez L, Setlow P. 1998. The katX gene, which codes for the catalase in spores of Bacillus subtilis, is a forespore-specific gene controlled by σF, and KatX is essential for hydrogen peroxide resistance of the germinating spore. J Bacteriol 180:2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Höper D, Völker U, Hecker M. 2005. Comprehensive characterization of the contribution of individual SigB-dependent general stress genes to stress resistance of Bacillus subtilis. J Bacteriol 187:2810–2826. doi: 10.1128/JB.187.8.2810-2826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reder A, Höper D, Gerth U, Hecker M. 2012. Contributions of individual σB-dependent general stress genes to oxidative stress resistance of Bacillus subtilis. J Bacteriol 194:3601–3610. doi: 10.1128/JB.00528-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 28.Vagner V, Dervyn E, Ehrlich SD. 1998. A vector for systematic gene inactivation in Bacillus subtilis. Microbiology 144:3097–3104. doi: 10.1099/00221287-144-11-3097. [DOI] [PubMed] [Google Scholar]
- 29.Barajas-Ornelas RC, Ramirez-Guadiana FH, Juarez-Godinez R, Ayala-Garcia VM, Robleto EA, Yasbin RE, Pedraza-Reyes M. 2014. Error-prone processing of apurinic/apyrimidinic (AP) sites by PolX underlies a novel mechanism that promotes adaptive mutagenesis in Bacillus subtilis. J Bacteriol 196:3012–3022. doi: 10.1128/JB.01681-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun DX, Stragier P, Setlow P. 1989. Identification of a new sigma-factor involved in compartmentalized gene expression during sporulation of Bacillus subtilis. Genes Dev 3:141–149. doi: 10.1101/gad.3.2.141. [DOI] [PubMed] [Google Scholar]
- 31.Feucht A, Lewis PJ. 2001. Improved plasmid vectors for the production of multiple fluorescent protein fusions in Bacillus subtilis. Gene 264:289–297. doi: 10.1016/S0378-1119(01)00338-9. [DOI] [PubMed] [Google Scholar]
- 32.Schaeffer P, Millet J, Aubert JP. 1965. Catabolic repression of bacterial sporulation. Proc Natl Acad Sci U S A 54:704–711. doi: 10.1073/pnas.54.3.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicholson WL, Setlow P. 1990. Sporulation, germination and outgrowth, p 391–450. In Harwood CR, Cutting SM (ed), Molecular biological methods for Bacillus. John Wiley & Sons, Sussex, England. [Google Scholar]
- 34.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 35.Mason JM, Fajardo-Cavazos P, Setlow P. 1988. Levels of mRNAs which code for small, acid-soluble spore proteins and their LacZ gene fusions in sporulating cells of Bacillus subtilis. Nucleic Acids Res 16:6567–6583. doi: 10.1093/nar/16.14.6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tennen R, Setlow B, Davis KL, Loshon CA, Setlow P. 2000. Mechanisms of killing of spores of Bacillus subtilis by iodine, glutaraldehyde and nitrous acid. J Appl Microbiol 89:330–338. doi: 10.1046/j.1365-2672.2000.01114.x. [DOI] [PubMed] [Google Scholar]
- 37.Steil L, Serrano M, Henriques AO, Völker U. 2005. Genome-wide analysis of temporally regulated and compartment-specific gene expression in sporulating cells of Bacillus subtilis. Microbiology 151:399–420. doi: 10.1099/mic.0.27493-0. [DOI] [PubMed] [Google Scholar]
- 38.Wang ST, Setlow B, Conlon EM, Lyond JL, Imamura D, Sato T, Setlow P, Losick R, Eichenberger P. 2006. The forespore line of gene expression in Bacillus subtilis. J Mol Biol 358:16–37. doi: 10.1016/j.jmb.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 39.Haldenwang WG. 1995. The sigma factors of Bacillus subtilis. Microbiol Rev 59:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shapiro R, Pohl SH. 1968. The reaction of ribonucleosides with nitrous acid. Side products and kinetics. Biochemistry 7:448–455. [DOI] [PubMed] [Google Scholar]
- 41.Choi PS, Naal Z, Moore C, Casado-Rivera E, Abruna HD, Helmann JD, Shapleigh JP. 2006. Assessing the impact of denitrifier-produced nitric oxide on other bacteria. Appl Environ Microbiol 72:2200–2205. doi: 10.1128/AEM.72.3.2200-2205.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang FC. 2004. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol 2:820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 43.Morohoshi F, Hayashi K, Munkata N. 1993. Bacillus subtilis alkA gene encoding inducible 3-methyladenine DNA glycosylase is adjacent to the ada operon. J Bacteriol 175:6010–6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keijser BJ, Ter Beek A, Rauwerda H, Schuren F, Montijn R, van der Spek H, Brul S. 2007. Analysis of temporal gene expression during Bacillus subtilis spore germination and outgrowth. J Bacteriol 189:3624–3634. doi: 10.1128/JB.01736-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pedraza-Reyes M, Gutiérrez-Corona F, Nicholson WL. 1994. Temporal regulation and forespore-specific expression of the spore photoproduct lyase gene by sigma-G RNA polymerase during Bacillus subtilis sporulation. J Bacteriol 176:3983–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramírez-Guadiana FH, Barraza-Salas M, Ramírez-Ramírez N, Ortiz-Cortés M, Setlow P, Pedraza-Reyes M. 2012. Alternative excision repair of ultraviolet B- and C-induced DNA damage in dormant and developing spores of Bacillus subtilis. J Bacteriol 194:6096–6104. doi: 10.1128/JB.01340-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramírez-Guadiana FH, Barajas-Ornelas RC, Ayala-García VM, Yasbin RE, Robleto E, Pedraza-Reyes M. 2013. Transcriptional coupling of DNA repair in sporulating Bacillus subtilis cells. Mol Microbiol 90:1088–1099. doi: 10.1111/mmi.12417. [DOI] [PubMed] [Google Scholar]
- 48.Ramírez-Guadiana FH, Barajas-Ornelas RC, Corona-Bautista SU, Setlow P, Pedraza-Reyes M. 2016. The RecA-dependent SOS response is active and required for processing of DNA damage during Bacillus subtilis sporulation. PLoS One 11:e0150348. doi: 10.1371/journal.pone.0150348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicolas P, Mäder U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S, Becher D, Bisicchia P, Botella E, Delumeau O, Doherty G, Denham E, Fogg M, Fromion V, Goelzer A, Hansen A, Härtig E, Harwood C, Homuth G, Jarmer H, Jules M, Klipp E, Le Chat L, Lecointe F, Lewis P, Liebermeister W, March A, Mars R, Nannapaneni P, Noone D, Pohl S, Rinn B, Rügheimer F, Sappa P, Samson F, Schaffer M, Schwikowski B, Steil L, Stülke J, Wiegert T, Devine K, Wilkinson A, van Dijl J, Hecker M, Völker U, Bessières P, Noirot P. 2012. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335:1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- 50.Morohoshi F, Munakata N. 1983. Adaptive response to simple alkylating agents in Bacillus subtilis cells. Mutat Res 110:23–37. doi: 10.1016/0027-5107(83)90015-5. [DOI] [Google Scholar]
- 51.Morohoshi F, Munakata N. 1985. Bacillus subtilis mutants deficient in the adaptive response to simple alkylating agents. J Bacteriol 161:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morohoshi F, Munakata N. 1995. Diverse capacities for the adaptive response to DNA alkylation in Bacillus species and strains. Mutat Res 337:97–110. doi: 10.1016/0921-8777(95)00013-A. [DOI] [PubMed] [Google Scholar]
- 53.Morohoshi F, Hayashi K, Munakata N. 1990. Bacillus subtilis ada operon encodes two DNA alkyltransferases. Nucleic Acids Res 18:5473–5480. doi: 10.1093/nar/18.18.5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singer B, Grunberger D. 1983. Molecular biology of mutagens and carcinogens, p 45–96. Plenum Press, New York, NY. [Google Scholar]
- 55.Mielecki D, Wrzesiński M, Grzesiuk E. 2015. Inducible repair of alkylated DNA in microorganisms. Mutat Res 763:294–305. doi: 10.1016/j.mrrev.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 56.Setlow B, Tautvydas KJ, Setlow P. 1998. Small, acid-soluble spore proteins of the α/β type do not protect the DNA in Bacillus subtilis spores against base alkylation. Appl Environ Microbiol 64:1958–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]