

Abstract
The KDM5C gene (also known as JARID1C and SMCX) is located on the X chromosome and encodes a ubiquitously expressed 1,560-aa protein, which plays an important role in lysine methylation (specifically reverses tri- and di-methylation of Lys4 of histone H3). Currently, thirteen missense mutations in KDM5C have been linked to X-linked mental retardation. However, the molecular mechanism of disease is currently unknown due to the experimental difficulties in expressing such large protein and the lack of experimental 3D structure. In this work, we utilize homology modeling, docking, and experimental data to predict 3D structures of KDM5C domains and their mutual arrangement. The resulting quaternary structure includes KDM5C JmjN, ARID, PHD1, JmjC, ZF domains, substrate histone peptide, enzymatic cofactors and DNA. The predicted quaternary structure was investigated with molecular dynamic simulation for its stability, and further analysis was carried out to identify features measured experimentally. The predicted structure of KDM5C was used to investigate the effects of disease-causing mutations and it was shown that the mutations alter domain stability and inter-domain interactions. The structural model reported in this work could prompt experimental investigations of KDM5C domain-domain interaction and exploration of undiscovered functionalities.
Keywords: KDM5C gene, quaternary structure, missense mutations, X-linked mental retardation, homology modeling, inter-domain interactions
Introduction
The epigenetic processes control transcription of genes and result in the widely different gene expression patterns in different tissues and organs (1, 2). The most frequently occurring histone modifications involve acetylation, phosphorylation, methylation, ubiquitination and crotonylation (3, 4). Lysine methylation is one of the most important histone modifications among them, and has a crucial role in heterochromatin formation, X-chromosome inactivation and transcriptional regulation. Histone lysine methylation occurs in histones H3 and H4, and this methylation results in lysine residue’s three different methylation states (mono-, di-, and tri-), which are associated with different nuclear features and transcriptional states(5).
The KDM5C gene (also known as JARID1C and SMCX) is located on the X chromosome and encodes a ubiquitously expressed 1,560-aa protein, which plays an important role in transcriptional regulation and chromatin remodeling (6). The KDM5C protein belongs to the JARID subfamily of JmjC-containing proteins. The function of JmjC domain is to specifically demethylate di- and trimethylated lysine 4 on histone 3 (7). The KDM5C acts as a transcriptional repressor and many mutations in the KDM5C gene has been shown to cause X-linked mental retardation (XLMR) and the syndromic Claes-Jensen-type disease (7–9). Most of disease-causing mutations are located on JmjC domain, ZF domain (C5HC2 zinc finger domain) and inter-domain regions. These mutations are expected to affect the protein stability and enzymatic activity (7, 10).
Overall KDM5C is a multi-functional protein, consisting structurally of several well-defined domains, including ARID, JmjN, JmjC, ZF, and two PHD zinc finger domains. Little experimental data is available about the 3D structure of the entire protein, individual domains and the interactions with its partners. The first experimental structure of one of the KDM5C domains is the solution structure of ARID domain, released in 2007 (11). In the past, we used it to generate a 3D model of the ARID-DNA complex (9). Very recently, the 3D structure of part of the human KDM5C protein (JmjN, JmjC and ZF domains) was released (12). The same work revealed that ARID and PHD1 domains contribute to the histone substrate recognition despite not being directly required for demethylase activity (12). However, there is still no experimental or theoretical 3D structure of KDM5C protein which includes JmjN, ARID, JmjC, C5HC2 zinc finger (ZF), and two PHD domains.
Several XLMR-associated mutations were shown to reduce KDM5C demethylase activity and binding to the H3K9me3 peptide (13). Specifically, ARID domain is a DNA-binding domain in which two missense mutations (A77T and D87G) were reported (8). The other domain, the JmjC domain, catalyzes demethylation of H3K4me3 to H3K4me1, and three missense mutations (D402Y, S451R and Y642L) are experimentally shown to reduce KDM5C demethylase activity (13). Several pathogenic mutations (A388P, R731W and Y751C) were reported in the PHD1 domain (a histone methyl-lysine binding motif) and ZF domain and were shown to reduce the demethylase activity as well (13). However, the molecular mechanism of the above-mentioned disease-causing mutations is mostly unknown. Perhaps this is due to the lack of experimental or theoretical 3D structure of KDM5C protein, which would allow for modeling the effects of mutations on domain stability and inter-domain interactions. The goal of this work is to fill this gap by developing a 3D structural model of KDM5C quaternary structure and using it to model the effects of disease-causing mutations on KDM5C stability, dynamics and inter-domain interactions. It is understood that such multi-functional and multi-domain proteins may adopt various domain arrangements in different functional states. Thus, the quaternary structure that is reported in this work represents one of several domain arrangements of KDM5C that the protein may adopt during its functional cycle.
Materials and Methods
Sequence alignment and homology modeling
The homologues of the KDM5C protein and their domains were retrieved from the Protein Data Bank database using the protein basic local alignment search tool (14), applying Position-Specific Iterated BLAST. The homologues with the highest sequence similarity were used to generate the homology model of KDM5C domains using the Swiss-Model webserver (15).
Protein docking and inter domain linker building
The KDM5C domains interactions were predicted with ZDOCK 3.0.2 (16), which searches all possible binding modes in the translational and rotational space between two proteins/domains and evaluates each pose using an energy-based scoring function. The linker regions were predicted with LOOPY (17), a computer algorithm to predict loop conformation provided the amino acid sequence.
Molecular Dynamics simulation
Molecular dynamics (MD) simulations were performed with the NAMD program, version 2.11b (18). The force field used in the simulation, including the substrate peptide and enzymatic cofactors, was Amber force field in the Amber tools 15 (19). The inpcrd and prmtop files were generated with Amber tools 15 (18). Other simulations, with only standard protein residues, were performed with CHARMM22 force field (20). A 10,000 steps minimization was performed for all simulations to relax plausible overlaps. Generalized Born implicit solvent (GBIS) was applied in the simulations and the time step was set to 2 fs. The temperature in the simulation was set to 298 K. The trajectory files were investigated using VMD 1.9.2 (21) with related plugins in order to analyze the RMSD, RMSF, and salt bridges.
Change of folding and binding free energies upon missense mutations
The binding free energy changes upon missense mutations (ΔΔΔG) were predicted with webservers including BeAtMuSiC (22), MutaBind (23) and SAAMBE (24, 25). The folding free energy changes upon missense mutations (ΔΔG) were evaluated with mCSM (26), SDM (27), DUET (28) and SAAFEC (29) servers.
Electrostatic Potential Calculation
The DelPhi program was used to perform the electrostatic potential calculations. The following parameters were applied in the calculation: scale = 2 grid/Å, percentage of protein filling of the cube = 70%, dielectric constant = 2 for the protein and 80 for the solvent, and water probe radius = 1.4 Å.
pKa shifts analysis
To investigate the possibility that some titratable residues may undergo protonation change upon formation of quaternary structure of KDM5C, we performed the pKa calculations with DelPhiPKa (30, 31), which is a surface-free Poisson-Boltzmann based approach to calculate the pKa values of protein ionizable residues, nucleotides of RNA and DNA. We first calculated the pKa’s of titratable residues for unbound ARID, PHD1 domains and KDM5C catalytic core and then repeated the calculations using quaternary KDM5C structure. The pKa shifts are calculated by subtracting the pKa of quaternary KDM5C structure and the pKa of individual domains (details are provided in SI).
Results and Discussion
The results section has three major components, namely a report of building 3D models of the corresponding KDM5C domains and arranging them into quaternary structure of KDM5C, and then validating and using the 3D structure of KDM5C protein to predict the effect of XLMD-linked mutations. Below we describe the results in sequential order.
(1) Modeling quaternary structure of KDM5C protein
Homology model of KDM5C PHD1 domain
As mentioned above, KDM5C is multi-domain protein consisting of well-defined domains as JmjN, ARID, PHD, JmjC and ZF domains. For most of them, JmjN, ARID, JmjC and ZF domains, there is experimental 3D structure available (Protein Data Bank (PDB) ID:2JRZ and 5FWJ)(11). However, the 3D structure of PHD1 domain is not available and must be modeled. For this purpose, we used homology modeling, since high homology templates do exist. Thus, the amino acid sequence of KDM5C was submitted to the PSI-Blast (14) and the search was performed against the sequences of proteins in PDB database (32). The best template for KDM5C PHD1 domain is the solution structure of another PHD domain within JARID family (PDB: 2E6R), with 84% sequence identity (33). Then, we used SWISS-MODEL server (15) to generate the homology model for KDM5C PHD1 domain.
Quaternary structure of KDM5C catalytic core
We began the modeling of quaternary structure of KDM5C protein by taking advantage of recently released experimental structure (PDB ID: 5FWJ), which includes JmjN, JmjC and ZF domains. The experimental structure revealed a previously known fact that JmjN interacts with JmjC domain and that this interaction is crucial for the KDM5C protein stability and catalytic function (12, 34). Furthermore, it is known that the ZF domain is required for catalytic activity of JARID proteins (35).
While it is known that JmjC domain demethylates di- and trimethylated lysine 4 on histone 3 (H3K4me2 and H3K4me3) along with the cofactors Ferrous ion (Fe2+) and alpha–ketoglutarate (2-oxoglutaric acid), the binding mode of histone peptide to JmjC domain is still unknown for the JARID family. Developing such a model is one of the goals of this work and we intend to generate it by taking the advantage of the experimental structures of other JmjC domain containing proteins. For this purpose, we first collected all existing experimental structures of JmjC domain bound to histone peptide. The experimental structures include the structure of KDM2A bound to H3K36me1 (PDB ID: 4QXH) (36), the structure of JMJD2B complexed with H3K9me3 (PDB ID: 4LXL) (37), the structure of human JMJD2D/KDM4D in complex with an H3K9me3 peptide (PDB ID:4HON) (38), the structure of KDM6B bound with H3K27me3 peptide (PDB ID: 4EZH) (39), the structure of KDM7A from C.elegans complexed with H3K4me3 peptide, H3K9me2 peptide and NOG (PDB ID: 3N9O) (40), and the complex structure of JMJD2A and trimethylated H3K36 peptide (PDB ID:2P5B) (41). The structural alignment was applied using the Chimera (42) for all collected structures. Figure 1A shows the results of structural alignment and it can be observed that the structures are highly conserved among different JmjC containing proteins. The positions of the histone peptides are also similar among different proteins, especially for the regions close to the bound Lys4 residue. Therefore, we took the average coordinates of these histone peptide backbones, which were manually assigned to the histone 3 peptide residues from 1 to 10. The side chain of each residue was generated using the most probable rotamer from the Dunbrack backbond-dependent rotamer library (43) as implemented in Chimera (42). Further, Lys4 and Lys9 in the peptide were modified to H3K4me3 and H3K9me3 using Avogadro (44), since in vivo they are methylated as they bind to JmjC and PHD1 domains. Finally, auto-optimization was performed for the peptide with Avogadro (44) to correct for bad contacts and improper bonds.
Figure 1.
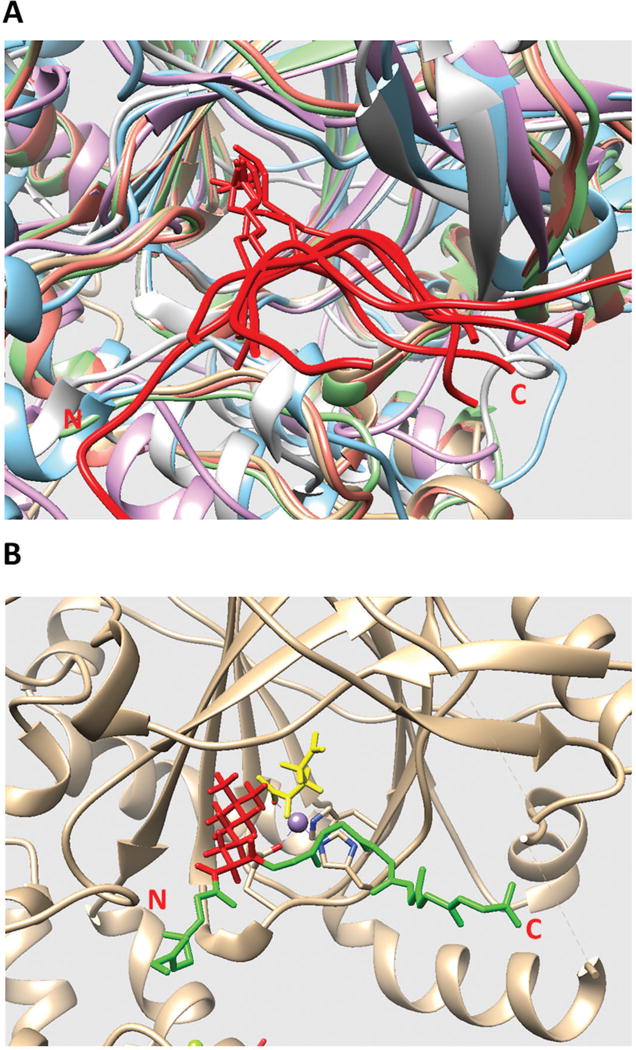
(A) Structural alignment of all existing JmjC domains bound to histone peptide. The histone peptides are marked in red. The JmjC domains are marked with other colors. The C and N terminals of the histone peptide are labeled as C and N, respectively. (B) The model of KDM5C catalytic core bound to histone peptide and enzymatic cofactors. The histone peptide backbone is shown in green and H3K4me3 is shown in red. Enzymatic cofactors Fe2+ ion and 2-oxoglutaric acid are marked with purple and yellow, respectively. The C and N terminals of the histone peptide are also labeled.
Since the template experimental structure contains Mn2+ ion and inhibitor MMK instead of enzymatic cofactors Fe2+ ion and 2-oxoglutaric acid, the enzymatic cofactors must be placed in the model to replace the template cofactors. This was done by superimposing template structure (structure of rice JMJ703 in complex with alpha-KG, PDB ID: 4IGO) onto the structure of KDM5C catalytic core and then replacing the template cofactors with enzymatic cofactors. The final model of KDM5C catalytic core bound with histone peptide and enzymatic cofactors is shown in Figure 1B.
Modeling ARID domain’s interactions in quaternary structure of KDM5C
ARID domain is the DNA binding domain of KDM5C protein and a NMR structure is currently available (PDB ID: 2JRZ). Also, in our previous work (9), a model of ARID-DNA complex has been generated. The presence of DNA provides constrains of how ARID interacts with the rest of KDM5C domains. It should be noted that the ARID domain is the second domain of the KDM5C sequence (after the JmjN domain) and a link consisting of 23 amino acids connects these two domains. This provides further constrains of mutual orientation and positioning of ARID and JmjN domains. Since the quaternary structure of JmjN, JmjC and ZF domains is already available (see above), the next question is to predict ARID domain position and orientation with respect to the JmjN, JmjC and ZF domains.
Thus, we first applied the ZDOCK server (16) to search possible binding modes of the ARID domain to the quaternary structure of JmjN, JmjC and ZF domains (in parallel ClusPro (45) and GRAMM-X(46) were used as well, see SI for details). The ten best predictions were collected and analyzed. It should be reiterated that JmjN and ARID domains are connected via a linker of 23 amino acids. Thus, the first consideration that was made in the analysis of the ten best binding modes was to remove binding modes that result in ARID position and orientation that makes it impossible for ARID and JmjN to be connected by 23 amino acid linker. It should be pointed out that the linker region between JmjN and ARID domain is also included in the crystal structure (12) and it is simply wrapped around the JmjC domain. It is tempting to use the linker in the experimental structure of JmjN, JmjC and ZF domains (PDB ID 5FWJ) as a guide in positioning the ARID domain. However, it should be clarified that the linker adopts crystallographic conformation in absence of the ARID domain and thus its conformation in 5FWJ may be misleading. Due to this, we decided to delete the linker region from the experimental structure and rebuild 3D structure of the linker connecting ARID and JmjC domains using the LOOPY program (47). The results showed that the 23 amino acid linker is not long enough to connect JmjN and ARID domain in six out of ten binding modes. Thus, these six models were deleted. To compare the rest of the four binding modes, we computed the RMSD among them (Table 1). The RMSDs among four models is relatively small, ranging from 1.80 to 3.46Å. This indicates that these four binding modes are quite similar and that the binding interface is almost identical (Figure 2A).
Table 1.
The RMSD of various ARID binding modes (in Å). The last row shows the average RMSD calculated with respect with other three models.
| Model1 | Model2 | Model3 | Model4 | |
|---|---|---|---|---|
| Model 1 | 0 | 2.20 | 2.64 | 1.80 |
| Model 2 | 2.20 | 0 | 3.46 | 3.06 |
| Model 3 | 2.64 | 3.46 | 0 | 2.93 |
| Model 4 | 1.80 | 3.06 | 2.93 | 0 |
| average | 2.21 | 2.91 | 3.01 | 2.60 |
Figure 2.
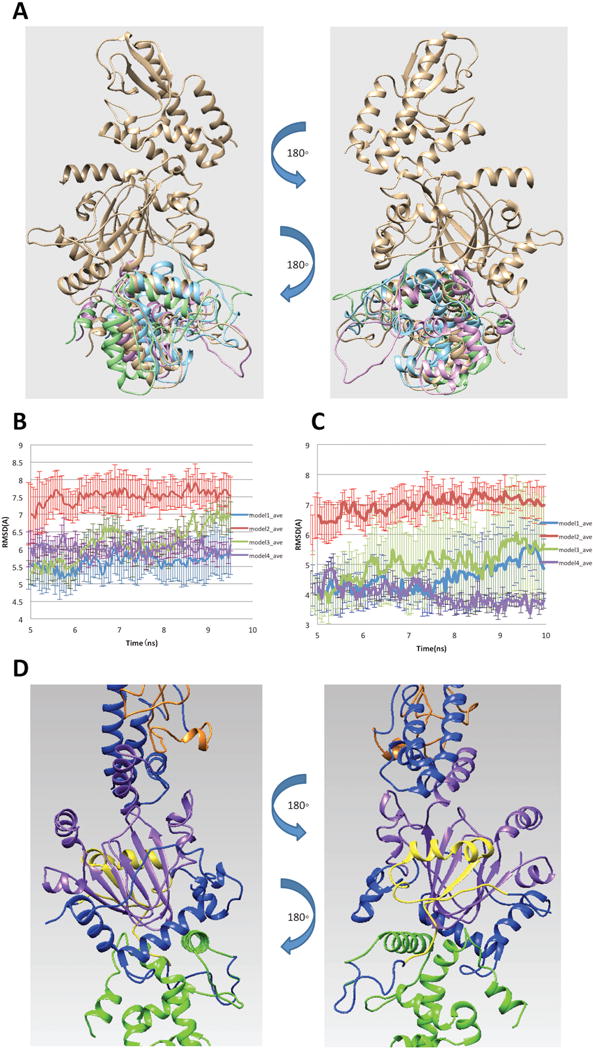
(A) Four possible binding modes of ARID domain onto 5FWJ structure after applying constraint of linker length. (B) RMSD of ARID domain and JmjC domain. (C) RMSD of the interfacial residues. (D) Finalized model of ARID bound to KDM5C catalytic core including JmjN, ARID, JmjC, ZF domains and the rest of inter domain regions marked with yellow, green, purple, orange and blue, respectively.
To further select the best model and test its stability, we run molecular dynamic (MD) simulations for each binding mode. Three 10ns parallel runs were performed for each model with CHARMM22 force field (20) in NAMD (18). Default charges of titratable residues were assigned, since the pKa analysis predicted no ionization changes in physiological pH (see SI for details). We first calculated RMSD of ARID and JmjC domains to observe the overall structural change along the simulation time (Figure 2B). The RMSD ranged from 5 to 7.5 Å and the overall structure became stable after 5ns for all models. To further study the binding mode stability, we also calculated the RMSD of the interfacial residues of each binding mode. We identified the interfacial residues by calculating the solvent accessible surface area (SASA) change of each residue in the complex and unbound domains. A residue is defined as an interfacial residue if the SASA change is not equal to 0. Since the identified interfacial residues were slightly different in each model due to the structure difference, we only selected the interfacial residues common in all models for our analysis and the RMSD results are shown in Figure 2C. The RMSD of interfacial residues ranges from 3.5 to 7 Å and reaches stable value after 5ns simulation time.
To finalize the model of ARID bound to KDM5C catalytic core, we took the twelve MD generated trajectories (Note that for each of the four binding modes we generated three MD trajectories) and selected 50% with lowest RMSD among them. The last 5ns of the trajectories were taken to calculate the averaged structure using VMD tcl script (21). The averaged structure was subjected to 10,000 steps of energy minimization to relax the structure. The finalized model of ARID bound to KDM5C catalytic core is shown in Figure 2D.
Modeling PHD1 domain’s interaction in the quaternary structure of KDM5C
The KDM5C PHD1 domain is close in sequence to the JmjC domain (the linker is 13 amino acids long) and it is expected to bind to tri-methylated H3K9 residue (H3K9me3) (7, 48). Indeed, a recent study indicated that the PHD1 domain is not required for the demethylase activity but does contribute to the recognition of the substrate peptide (7, 12). Currently, there is no available experimental structure of the KDM5C PHD1 domain bound to H3K9me3 and KDM5C catalytic core.
Above we described the modeling of quaternary structure of KDM5C catalytic core bound to histone peptide and ARID domain. Since a homology model of KDM5C PHD1 domain was already generated (see above), we applied the ZDOCK server (16) to predict the binding modes of PHD1 domain and the model of KDM5C catalytic core bound to histone peptide (ClusPro(45) and GRAMM-X(46) were used in parallel as well, see SI for details). The Z-dock server predicted the ten most plausible binding modes. We evaluated them by applying constraint of the linker length between PHD1 and JmjC domain. Since PHD1 and JmjC domains are connected with a 13-residue-long liner (we built a 3D structure of the linker connecting the PHD1 and JmjC domains using the LOOPY program (47)), we only selected the binding mode in which a stretch of 13 amino acids is able to connect the two domains. This resulted in three possible binding modes (Figure 3A). The PHD1 domain in all binding models is close to the substrate peptide but adopts different orientations.
Figure 3.
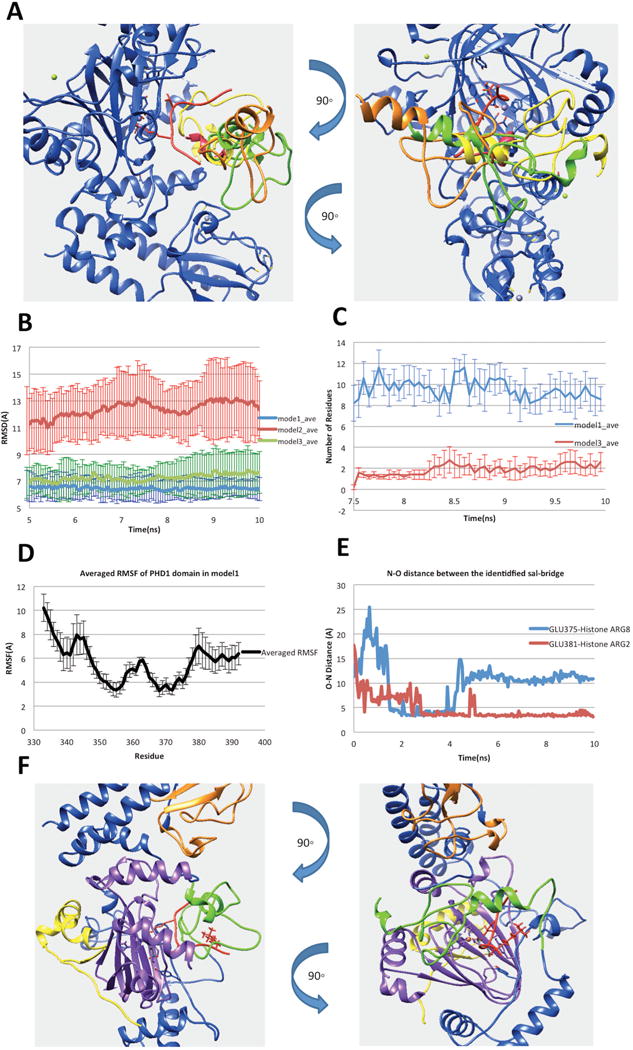
(A) Three possible binding modes after applying the constraint of linker length. (B) RMSDs of the complex of PHD1and JmjC domains. (C) Number of residues in PHD1 domain, which have any atom within 6Å of H3K9me3 in the last 2.5ns simulation time. (D) The N–O distance of identified salt bridges (Glu375-H3R8 and Glu381-H3R2) between PHD1 and substrate peptide. (E) Averaged RMSF of PHD1 domain residues calculated from model1 MD simulations. (F) Finalized model of PHD1 domain bound to KDM5C catalytic core including JmjN, PHD1, JmjC, ZF domains, inter domain region and histone peptide marked with yellow, green, purple, orange, blue and red, respectively
To further test the binding stability in the rest of the three binding modes, we performed MD simulations. Since the substrate peptide is very flexible and tends to move away from the JmjC catalytic core, the position of H3K4me3 residue was fixed to make the substrate peptide stay in the catalytic core during the simulation time. The enzymatic cofactors, Fe2+ ion and 2-oxoglutaric acid, were also fixed in the simulation to prevent them from moving away from the JmjC catalytic core. Five 10ns parallel runs were performed for each model using AMBER ff14SB force field (49) in NAMD (18). As mentioned above, default charges were used for titratable groups since no protonation changes were predicted by the pKa analysis (see SI for details). The averaged RMSDs calculated for the complex JmjC domain, substrate peptide and the PHD1 domain are shown in the Figure 3B. The RMSD calculated for model1 and model3 is around 7 Å and the PHD1 domain stays bound with the JmjC domain during the simulation time. At the same time, model2 shows much larger RMSD values. It was observed that the PHD1 domain moves away from the JmjC domain and substrate peptide. Therefore, model2 was removed from our protocol, while model1 and model3 were subjected to further considerations.
To further select the best binding mode among mode1 and model3, we took advantage of experimental data that indicates that PHD1 domain binds substrate peptide at H3K9me3. Therefore, we identified all residues in PHD1 domain, which have any atom within 6 Å distance from H3K9me3 in the last 2.5ns simulation time (Figure 3C). Comparing with model1, the analysis of the trajectories indicated that the PHD1 domain in model3 is farther away from the H3K9me3. Due to this, we selected model1 for further investigations. In particular, we paid attention to plausible stabilizing charge-charge interactions between PHD1 domain and peptides. Since the methylation of Lys does not neutralize the charge, we searched for acidic residue in PHD1 domain within a distance of 6 Å from H3K9me3. Thus, we identified two acidic residues: Glu360 and Glu375. Further N–O distance analysis did not indicate stable salt bridges between H3K9me3 and acidic residues in PHD1 domain. However, two other salt bridges within PHD1, Glu381–Histone ARG2 (H3R2) and PHD1 Glu375–Histone Arg8 (H3R8), were identified (shown in Figure 3D). It should be mentioned that the structural segment (the linker) connecting ARID domain and PHD1 domain was not included in the model due to lack of homology template and thus PHD1 domain may appear to be more flexible in the simulations than it actually is and cause us to observe less salt-bridges. Noting the consistence with experimental data, we took model1 as the most plausible binding mode (the structure is shown in Figure 3F).
Quaternary structure of KDM5C JmjN, ARID, PHD1, JmjC and ZF domains bound with DNA, substrate histone peptide and enzymatic cofactors
In our previous work, we generated the 3D structure of ARID-DNA complex (9). Here, we took advantage of our previous work and included the DNA in the KDM5C quaternary structure model discussed above. Combining all the models described above, we finally generated the quaternary model of KDM5C protein – including JmjN, ARID, PHD1, JmjC, ZF domains, substrate histone peptide, enzymatic cofactors and DNA (Figure 4).
Figure 4.
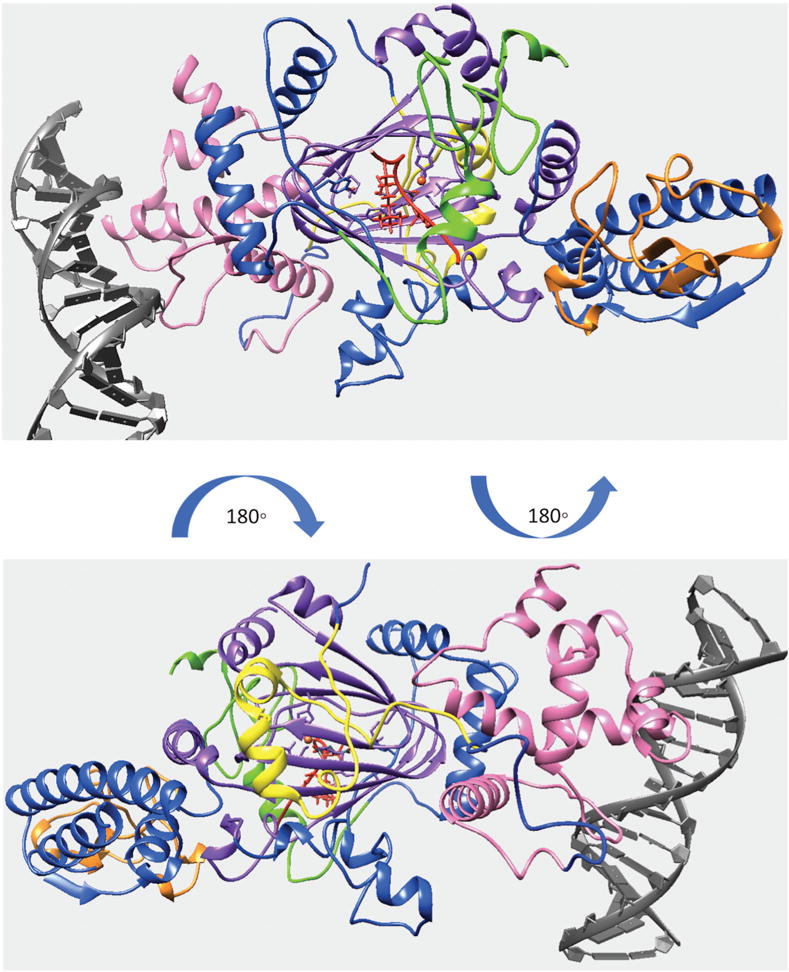
Finalized model of quaternary structure for the KDM5C including JmjN, ARID, PHD1, JmjC and ZF domains bound to DNA, substrate histone peptide and enzymatic cofactors. KDM5C JmjN, ARID, PHD1, JmjC, ZF domains, inter domain region, substrate histone peptide, enzymatic cofactors and DNA are marked with yellow, pink, green, purple, orange, blue, red, orange, red, and gray, respectively.
(2) Validation of KDM5C quaternary structure model
In this paragraph we will investigate domain spatial arrangement within the quaternary structure of KDM5C model using biophysical considerations (charge complementarity, inter-domain salt bridges, binding motif preservation, MD simulations and experimental observations) and predictions of interfacial patches. With our work focusing on predicting spatial positions of ARID and PHD1 domains within KDM5C quaternary structure, we will continue to focus on validating ARID and PHD1 interactions with the rest of the KDM5C domains.
ARID domain
The binding interface is mostly made of ARID domain’s N-terminal and C-terminal regions, including Helix1 and Helix6 (Figure 5A). Our previous evolutionary conservation study showed that the N-terminal of the KDM5C ARID domain is the most highly conserved region, indicating its essential function for KDM5C protein (9) and thus implicating its involvement in some important interactions. There exists an interface of the ARID domain in which the domain binds the DNA. In the model, it is Loop1 and a helix-turn-helix DNA binding motif formed by Helix4, Loop2, and Helix5, which is typical for DNA binding motifs. In the previous study (9), we demonstrated that DNA binding interface is positively charged, which is expected for interface binding to negatively charged DNA. Similarly, we calculated electrostatic potential of ARID domain and KDM5C JmjC and JmjN domain using Delphi (50). The binding interface of JmjC domain is highly negatively charged while the binding interface of ARID domain is slightly positively charged (Figure 5B). This indicates electrostatic complementarity and further validates the model. Furthermore, we submitted the sequence to cons-PPISP webserver (51, 52), a consensus neural network method for predicting protein-protein interaction sites. The webserver predicts the residues which form binding sites for another protein. Here, we separately submitted ARID, PHD and JmjC domains to the server to predict the interaction sites on each domain. The results are shown in Figure 6D,E. The predicted interaction sites in ARID domain are mostly in the N terminal helix, which is highly consistent with our model (Figure 6D). We also perfomed MD simulations on the model of ARID bound with KDM5C catalytic core (see above). The RMSD analysis of the interfacial residues indicated that the binding is stable (Figure 2C). Lastly we investigated plausible salt bridges formed at the interface of ARID and PHD1 domains, because salt bridges are frequently observed at transient domain-domain interfaces (53, 54). Since we had previously performed 5 parallel runs, we only selected the salt-bridges observed in no less than two runs. The list of identified interfacial salt bridges is shown in Table 2. It can be seen that ARID interface is rich of interfacial salt bridges, indirectly indicating that the interface is correctly predicted.
Figure 5.
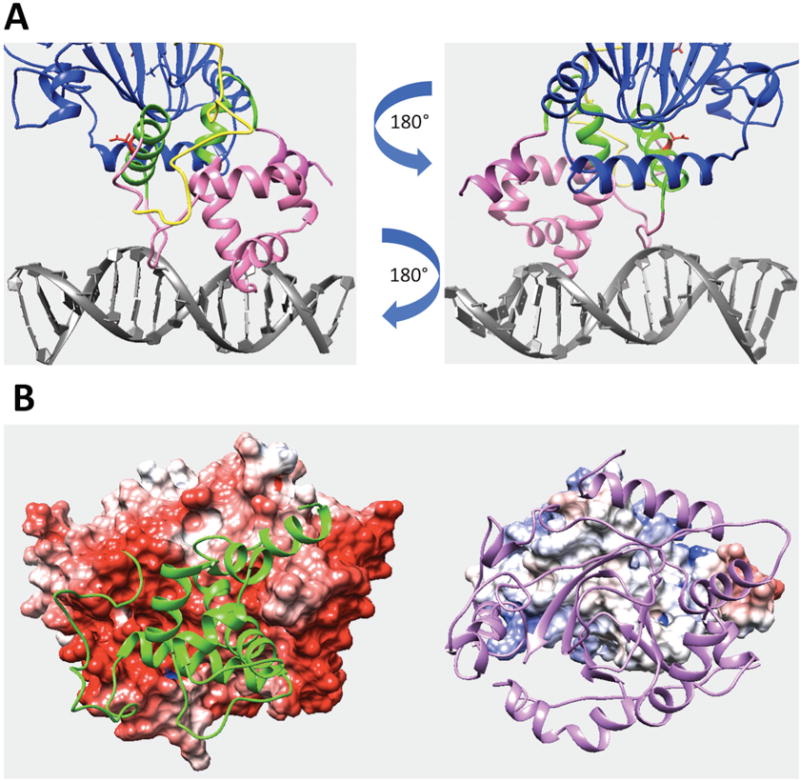
(A) Predicted interfacial region of ARID domain involved in domain interactions in the KDM5C quaternary structure. The interfacial region of ARID domain is marked with green while the rest of ARID domain is marked with pink. JmjN domain and DNA are marked with yellow and gray. Other regions, including JmjC and ZF domains, are marked with blue. The reside Asp87 side chain is shown with red. (B) The electrostatic potential map of ARID domain and KDM5C catalytic core. The electrostatic potential map of KDM5C catalytic core (including JmjN, JmjC and ZF domains) is shown on the left (the structure of ARID domain is marked with green). The electrostatic potential map of ARID domain is shown on the right (the structure of JmjN and JmjC domains are marked with purple).
Figure 6.
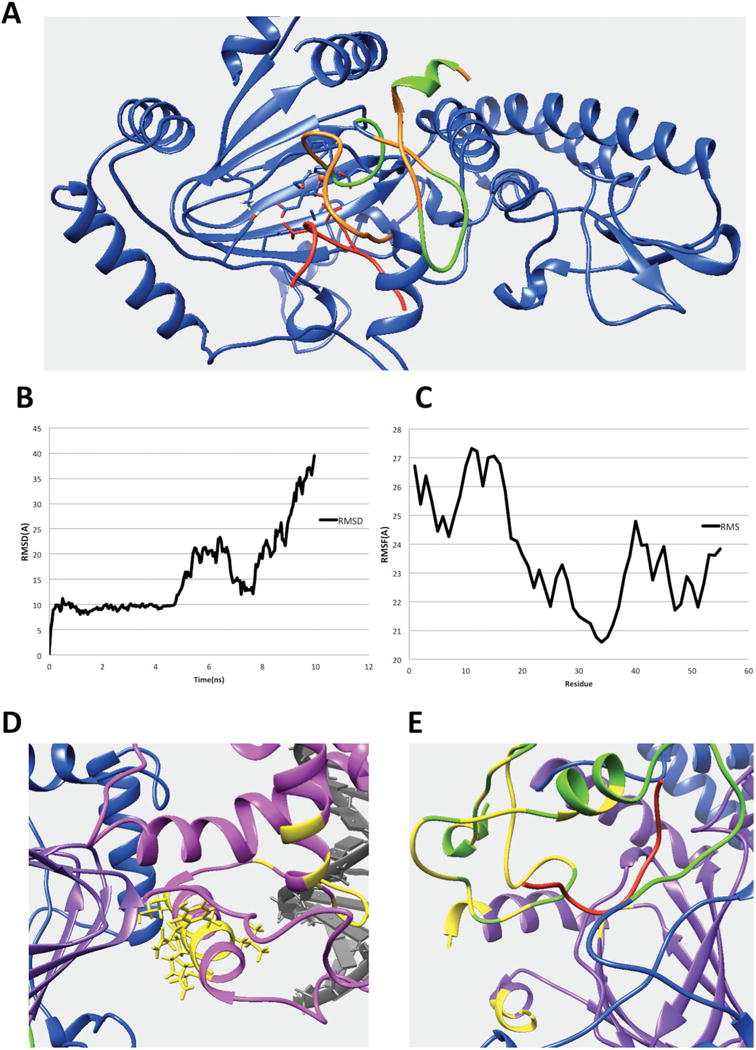
(A) The interfacial residues of PHD1 domain involved in the interaction in the KDM5C quaternary structure. The interfacial residues of PHD1 domain are marked with green while the rest of the PHD1 domain are marked with orange. The substrate histone peptide is marked with red while the rest of the region including JmjC and ZF domain are marked with blue. (B) The RMSD results for the KDM5B PHD1 domain bound to substrate peptide complex. (C) The RMSF results for the KDM5B PHD1 domain. (D) The predicted interfacial residues in ARID domain. The ARID domain, JmjC domain, DNA and inter-domain region are marked with pink, purple, gray, and blue, respectively. The predicted interfacial residues in ARID domain are colored with yellow. (E) The predicted interfacial residues in PHD and JmjC domains. The PHD domain, JmjC domain, histone substrate and inter-domain regions are marked with green, purple, red, and blue, respectively. Predicted interfacial residues in PHD and JmjC domains are colored with yellow.
Table 2.
Lists of identified salt bridges involved in interfacial ARID and PHD1 domains interactions.
| Salt bridge involved in the domain interactions | |
|---|---|
| ARID | Arg80-Glu465, Arg80-Glu467, Arg80-Gu468, Asp87-Lys459, Asp87-Arg460 Lys91-Glu465, Lys91-Glu466, Glu94-Arg390, Glu94-Lys459, Arg159-Glu399, Arg159-Asp402, Arg159-Glu419, Arg159-Glu422 |
| PHD1 | Asp334-Lys551, Glu334-Lys550, Glu335-Lys551, Glu335-Arg635, Asp336-Arg637, Asp337-Lys550, Asp347-Arg637, Asp347-Lys711 |
PHD1 domain
The predicted binding interface between PHD1 and JmjC domains is shown in Figure 6A and marked with green. It includes a short helix and two loops. The KDM5C PHD1 domain binds to H3K9me3 and reduction of its binding decreases enzyme activity (7, 48). This mechanism is still unknown. However, a recent study indicated that PHD1 domain contributes to the histone substrate recognition, despite being not directly required for demethylase activity (12). In our model, the PHD1 domain simultaneously binds to peptide and JmjC domain. Here we investigated whether PHD1 domain binds with the substrate histone peptide alone, or whether it also interacts with JmjC domain to help further stabilize its binding to substrate peptide. For this purpose, we carried out MD stimulations and monitored RMSDs (Figure 3). It is shown that the PHD1 domain is highly flexible - the salt bridge formed between the substrate peptide and the PHD1 domain were not very stable during the simulations as well (Figure 3D,E). To better understand this finding, we carried out the same type of MD simulations of closely related complexes with experimentally available 3D structures. Thus, we collected the experimental structure of the KDM5B PHD1 finger (having 61% sequence identity with KDM5C PHD1 domain) in complex with H3K4me0 (55) and ran 10ns simulation to study the RMSD of PHD1 domain and histone peptide. The results showed that the peptide moves away from PHD1 domain around 5ns (Figure 6B). The overall PHD1 domain is very flexible as well (Figure 6C). Thus, the simulations indicate that the binding between the substrate histone peptide and PHD domain alone is not stable. This is perhaps due to the high flexibility of both PHD domain and substrate peptide. This advocates that the binding of histone peptide is supported by PHD1 interactions with JmjC catalytic core. Indeed, very recent study of domain arrangement in KDM5B protein by small-angle X-ray scattering (SAXS) and rigid-body modeling (12) provided an evidence of the interaction between PHD1 domain and JmjC catalytic core. The model derived from SAXS indicates that the PHD1 domain is in close contact with the JmjC catalytic core and suggested cooperation between PHD1 and the catalytic core in KDM5 enzymes (12). Considering both these experimental results and our model (7, 12), we speculate that the PHD1 domain interacts with the JmjC domain to help stabilize the substrate peptide and itself, and further to position the H3K4 in the JmjC catalytic core. Furthermore, as we did for the ARID domain, we submitted the sequence to cons-PPISP webserver (51, 52). The cons-PPISP predicted interaction sites in PHD domain are located in the binding interface in our model (Figure 6E). Lastly, we identified interfacial salt bridges at the PHD1 interface (Table 2).
(3) Effects of disease-associated mutations on KDM5C domain’s stability and interactions
Currently there are eleven amino acid mutations in KDM5C protein known to be causing XLMD. They were investigated to predict their effects on the KDM5C domain’s stability and interactions. First we predict the effect on folding free energy (domain stability) using various webservers (Table 3). The results indicate that most of the disease-associated mutations substantially decrease domain stability, particularly mutations A77T, D87G, F642L, E698K and Y751C. Furthermore, mutations D87G and D402Y are located at the binding interface of the ARID and JmjC domains and their effect on domain-domain interactions was predicted using several webservers (Table 4). It can be seen that the mutation D87G significantly decreases the binding affinity while D402Y slightly increases it. Asp87 is in the binding interface (shown in Figure 5A) and participates in two salt bridges (Asp87-Lys459, Asp87-Arg460) across the binding interface. Replacing negatively charged D87 with small, uncharged residue will alter the salt bridges and reduce the ARID-JmjC binding. The other mutation, D402, involves negatively charged residue in which the wild type KDM5C is predicted to form a salt bridge with R159 of JmjC domain. Preplacing it with neutral residue is expected to alter the salt bridge and to decrease binding affinity. However, the mutation is predicted to slightly increase binding affinity of ARID domain to JmjC domain. Perhaps this is due to the fact that R159 is involved in another salt bridge (Table 1) and the D402-R159 salt bridge does not contribute to inter-domain interactions. However, this bridge may be essential for forming the wild type binding pose. Overall, all investigated mutations are predicted to alter the wild type domain stability and inter-domain interactions.
Table 3.
Folding free energy change upon mutations (kcal/mol). Positive value indicates that the mutation decreases domain stability. The most contradictory predictions are underlined and not used. The averaged prediction is shown in the last column.
| mCSM | SDM | DUET | SAAFEC | Average | |
|---|---|---|---|---|---|
| A77T | 0.88 | 2.48 | 0.83 | 1.44 | 1.41 |
| D87G | 0.84 | 1.9 | 0.86 | 5.01 | 2.16 |
| A388P | 0.44 | 2.12 | 0.57 | −0.13 | 0.75 |
| D402Y | 1.03 | −0.8 | 1.04 | −7.93 | 0.42 |
| S451R | 0.52 | 0.25 | 0.43 | −3.74 | 0.40 |
| V504M | 0.16 | 0.88 | 0.16 | −0.14 | 0.27 |
| F642L | 1.43 | 1.05 | 1.56 | 1.71 | 1.44 |
| E698K | 0.22 | 3.27 | 0.29 | 4.16 | 1.99 |
| L731F | 1.5 | 0.31 | 1.69 | 0.01 | 0.88 |
| R750W | −0.56 | −1.74 | 0.76 | 1.01 | 0.40 |
| Y751C | 1.66 | −1.76 | 1.57 | 1.44 | 1.56 |
Table 4.
Binding free energy change upon mutations (kcal/mol). Positive value indicates that the mutation decreases binding affinity. The most contradictory predictions are underlined and not used. The averaged prediction is shown in the last column.
| BeAtMuSiC | SAAMBE | MutaBind | Average | |
|---|---|---|---|---|
| D87G | 1.65 | −0.54 | 0.42 | 1.04 |
| D402Y | −0.22 | −0.66 | 0.18 | −0.23 |
Conclusion
The KDM5 protein is of significant interest for the biomedical community due to its relevance to X-linked mental retardation (56) and importance in oncological drug development (57). KDM5C protein is a multi-functional protein of 1560-aa length and 3D structure of the entire protein is currently unavailable. Only the 3D structure of ARID domain, and very recently of catalytic core, is available (12). Here we fill this gap by reporting a 3D model of KDM5C protein quaternary structure, including JmjN, ARID, PHD1, JmjC and ZF domains bound to DNA, substrate histone peptide and enzymatic cofactors. The model was used to infer the effect of disease-causing mutations of domain stability and domain-domain interactions. From the model, it was demonstrated that the mutations significantly alter wild type domain stability and inter-domain interactions. This suggests that domain stability and domain spatial arrangement with KDM5C protein are essential for its wild type function.
Supplementary Material
Acknowledgments
The work was supported by a grant from NIH grant number R01GM093937. The 3 D model of KDM5C protein, in PDB format, is available for download from compbio.clemson.edu/KDM5C.pdb.
Footnotes
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330(6004):612–6. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 3.Huang C, Xu M, Zhu B. Epigenetic inheritance mediated by histone lysine methylation: maintaining transcriptional states without the precise restoration of marks? Philos Trans R Soc Lond B Biol Sci. 2013;368(1609):20110332. doi: 10.1098/rstb.2011.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–49. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 6.Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447(7144):601–5. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- 7.Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128(6):1077–88. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 8.Goncalves TF, Goncalves AP, Fintelman Rodrigues N, dos Santos JM, Pimentel MM, Santos-Reboucas CB. KDM5C mutational screening among males with intellectual disability suggestive of X-Linked inheritance and review of the literature. Eur J Med Genet. 2014;57(4):138–44. doi: 10.1016/j.ejmg.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 9.Peng Y, Suryadi J, Yang Y, Kucukkal TG, Cao W, Alexov E. Mutations in the KDM5C ARID Domain and Their Plausible Association with Syndromic Claes-Jensen-Type Disease. Int J Mol Sci. 2015;16(11):27270–87. doi: 10.3390/ijms161126022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brookes E, Laurent B, Ounap K, Carroll R, Moeschler JB, Field M, Schwartz CE, Gecz J, Shi Y. Mutations in the intellectual disability gene KDM5C reduce protein stability and demethylase activity. Hum Mol Genet. 2015;24(10):2861–72. doi: 10.1093/hmg/ddv046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koehler C, Bishop S, Dowler EF, Schmieder P, Diehl A, Oschkinat H, Ball LJ. Backbone and sidechain 1H, 13C and 15N resonance assignments of the Bright/ARID domain from the human JARID1C (SMCX) protein. Biomol NMR Assign. 2008;2(1):9–11. doi: 10.1007/s12104-007-9071-7. [DOI] [PubMed] [Google Scholar]
- 12.Johansson C, Velupillai S, Tumber A, Szykowska A, Hookway ES, Nowak RP, Strain-Damerell C, Gileadi C, Philpott M, Burgess-Brown N, Wu N, Kopec J, Nuzzi A, Steuber H, Egner U, Badock V, Munro S, LaThangue NB, Westaway S, Brown J, Athanasou N, Prinjha R, Brennan PE, Oppermann U. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat Chem Biol. 2016 doi: 10.1038/nchembio.2087. [DOI] [PubMed] [Google Scholar]
- 13.Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-Linked Mental Retardation GeneSMCX/JARID1C Defines a Family of Histone H3 Lysine 4 Demethylases. Cell. 2007;128(6):1077–88. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 14.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 15.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–8. doi: 10.1093/nar/gku340. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30(12):1771–3. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins. 2012;80(7):1715–35. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. Journal of computational chemistry. 2005;26(16):1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuster-Bockler B, Bateman A. Protein interactions in human genetic diseases. Genome biology. 2008;9(1):R9. doi: 10.1186/gb-2008-9-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 21.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. Journal of Molecular Graphics. 1996;14(1):33–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 22.Dehouck Y, Kwasigroch JM, Rooman M, Gilis D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013;41:W333–9. doi: 10.1093/nar/gkt450. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M, Simonetti FL, Goncearenco A, Panchenko AR. MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res. 2016;44(W1):W494–501. doi: 10.1093/nar/gkw374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petukh M, Dai L, Alexov E. SAAMBE: Webserver to Predict the Charge of Binding Free Energy Caused by Amino Acids Mutations. Int J Mol Sci. 2016;17(4) doi: 10.3390/ijms17040547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petukh M, Li M, Alexov E. Predicting Binding Free Energy Change Caused by Point Mutations with Knowledge-Modified MM/PBSA Method. PLoS Comput Biol. 2015;11(7):e1004276. doi: 10.1371/journal.pcbi.1004276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pires DE, Ascher DB, Blundell TL. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014;30(3):335–42. doi: 10.1093/bioinformatics/btt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Worth CL, Preissner R, Blundell TL. SDM–a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–22. doi: 10.1093/nar/gkr363. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pires DE, Ascher DB, Blundell TL. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014;42:W314–9. doi: 10.1093/nar/gku411. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Getov I, Petukh M, Alexov E. SAAFEC: Predicting the Effect of Single Point Mutations on Protein Folding Free Energy Using a Knowledge-Modified MM/PBSA Approach. Int J Mol Sci. 2016;17(4) doi: 10.3390/ijms17040512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Li L, Alexov E. pKa predictions for proteins, RNAs, and DNAs with the Gaussian dielectric function using DelPhi pKa. Proteins. 2015;83(12):2186–97. doi: 10.1002/prot.24935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Zhang M, Alexov E. DelPhiPKa web server: predicting pKa of proteins, RNAs and DNAs. Bioinformatics. 2016;32(4):614–5. doi: 10.1093/bioinformatics/btv607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berman HM. The Protein Data Bank. Nucleic Acids Research. 2000;28(1):235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kadirvel S, He F, Muto Y, Inoue M, Kigawa T, Shirouzu M, Terada T, Yokoyama S. RIKEN Structural Genomics/Proteomics Initiative. Solution structure of the PHD domain in SmcY protein [Google Scholar]
- 34.Huang F, Chandrasekharan MB, Chen YC, Bhaskara S, Hiebert SW, Sun ZW. The JmjN domain of Jhd2 is important for its protein stability, and the plant homeodomain (PHD) finger mediates its chromatin association independent of H3K4 methylation. J Biol Chem. 2010;285(32):24548–61. doi: 10.1074/jbc.M110.117333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, Taylor-Papadimitriou J, Tempst P, Zhang Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell. 2007;25(6):801–12. doi: 10.1016/j.molcel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Cheng Z, Cheung P, Kuo AJ, Yukl ET, Wilmot CM, Gozani O, Patel DJ. A molecular threading mechanism underlies Jumonji lysine demethylase KDM2A regulation of methylated H3K36. Genes Dev. 2014;28(16):1758–71. doi: 10.1101/gad.246561.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang W-C, Chu C-H, Chen C-C. Crystal structure of JMJD2B complexed with pyridine-2,4-dicarboxylic acid and H3K9me3 [Google Scholar]
- 38.Krishnan S, Trievel RC. Structural and functional analysis of JMJD2D reveals molecular basis for site-specific demethylation among JMJD2 demethylases. Structure. 2013;21(1):98–108. doi: 10.1016/j.str.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 39.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, Mosley J, Ramirez-Molina C, Rowland P, Schofield CJ, Sheppard RJ, Smith JE, Swales C, Tanner R, Thomas P, Tumber A, Drewes G, Oppermann U, Patel DJ, Lee K, Wilson DM. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488(7411):404–8. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Y, Hu L, Wang P, Hou H, Lin Y, Liu Y, Li Z, Gong R, Feng X, Zhou L, Zhang W, Dong Y, Yang H, Lin H, Wang Y, Chen CD, Xu Y. Structural insights into a dual-specificity histone demethylase ceKDM7A from Caenorhabditis elegans. Cell Res. 2010;20(8):886–98. doi: 10.1038/cr.2010.86. [DOI] [PubMed] [Google Scholar]
- 41.Chen Z, Zang J, Kappler J, Hong X, Crawford F, Wang Q, Lan F, Jiang C, Whetstine J, Dai S, Hansen K, Shi Y, Zhang G. Structural basis of the recognition of a methylated histone tail by JMJD2A. Proc Natl Acad Sci U S A. 2007;104(26):10818–23. doi: 10.1073/pnas.0704525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 43.Dunbrack RL. Rotamer Libraries in the 21st Century. Current Opinion in Structural Biology. 2002;12(4):431–40. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- 44.Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012;4(1):17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32:W96–9. doi: 10.1093/nar/gkh354. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tovchigrechko A, Vakser IA. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006;34:W310–4. doi: 10.1093/nar/gkl206. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiang Z, Soto CS, Honig B. Evaluating conformational free energies: the colony energy and its application to the problem of loop prediction. Proc Natl Acad Sci U S A. 2002;99(11):7432–7. doi: 10.1073/pnas.102179699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, Lacoste N, Cayrou C, Davrazou F, Saha A, Cairns BR, Ayer DE, Kutateladze TG, Shi Y, Cote J, Chua KF, Gozani O. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442(7098):96–9. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput. 2015;11(8):3696–713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li L, Li C, Sarkar S, Zhang J, Witham S, Zhang Z, Wang L, Smith N, Petukh M, Alexov E. DelPhi: a comprehensive suite for DelPhi software and associated resources. BMC Biophys. 2012;5:9. doi: 10.1186/2046-1682-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen H, Zhou HX. Prediction of interface residues in protein-protein complexes by a consensus neural network method: test against NMR data. Proteins. 2005;61(1):21–35. doi: 10.1002/prot.20514. [DOI] [PubMed] [Google Scholar]
- 52.Deng L, Guan J, Dong Q, Zhou S. Prediction of protein-protein interaction sites using an ensemble method. BMC Bioinformatics. 2009;10:426. doi: 10.1186/1471-2105-10-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bairagya HR, Mukhopadhyay BP, Bera AK. Role of salt bridge dynamics in inter domain recognition of human IMPDH isoforms: an insight to inhibitor topology for isoform-II. J Biomol Struct Dyn. 2011;29(3):441–62. doi: 10.1080/07391102.2011.10507397. [DOI] [PubMed] [Google Scholar]
- 54.Gc JB, Johnson KA, Husby ML, Frick CT, Gerstman BS, Stahelin RV, Chapagain PP. Interdomain salt-bridges in the Ebola virus protein VP40 and their role in domain association and plasma membrane localization. Protein Sci. 2016 doi: 10.1002/pro.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Yang H, Guo X, Rong N, Song Y, Xu Y, Lan W, Zhang X, Liu M, Xu Y, Cao C. The PHD1 finger of KDM5B recognizes unmodified H3K4 during the demethylation of histone H3K4me2/3 by KDM5B. Protein & cell. 2014;5(11):837–50. doi: 10.1007/s13238-014-0078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, Janecke AR, Tariverdian G, Chelly J, Fryns JP, Van Esch H, Kleefstra T, Hamel B, Moraine C, Gecz J, Turner G, Reinhardt R, Kalscheuer VM, Ropers HH, Lenzner S. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 2005;76(2):227–36. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rasmussen PB, Staller P. The KDM5 family of histone demethylases as targets in oncology drug discovery. Epigenomics. 2014;6(3):277–86. doi: 10.2217/epi.14.14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.