

Abstract
The retina is considered to be the most metabolically active tissue in the body. However, the link between energy metabolism and retinal inflammation, as incited by microbial infection such as endophthalmitis remains unexplored. In this study, using a mouse model of Staphylococcus aureus (SA) endophthalmitis, we demonstrate that the activity (phosphorylation) of AMP-activated protein kinase alpha (AMPKα), a cellular energy sensor, and its endogenous substrate; acetyl-CoA carboxylase (ACC) is downregulated in the SA-infected retina. Intravitreal administration of an AMPK activator, 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) restored AMPKα and ACC phosphorylation. AICAR treatment reduced both the bacterial burden and intraocular inflammation in SA-infected eyes by inhibiting NF-κB and MAP kinases (p38 and JNK) signaling. The anti-inflammatory effects of AICAR were diminished in eyes pretreated with AMPK inhibitor, Compound C. The bioenergetics (Seahorse) analysis of SA-infected microglia and bone marrow-derived macrophages (BMDM) revealed an increase in glycolysis, which was reinstated by AICAR treatment. AICAR also reduced the expression of SA-induced glycolytic genes, including hexokinase 2 (HK2), and glucose transporter 1 (Glut1) in microglia, BMDM, and the mouse retina. Interestingly, AICAR treatment enhanced the bacterial phagocytic and intracellular killing activities of cultured microglia, macrophages, and neutrophils. Furthermore, AMPKα1 global knockout mice exhibited increased susceptibility towards SA endophthalmitis, as evidenced by increased inflammatory mediators and bacterial burden and reduced retinal function. Together, these findings provide the first evidence that AMPK activation promotes retinal innate defense in endophthalmitis by modulating energy metabolism, and that it can be targeted therapeutically to treat ocular infections.
Keywords: Staphylococcus aureus, inflammation, AMPK, bioenergetics, innate immunity
INTRODUCTION
Bacterial endophthalmitis remains a potentially blinding complication of ocular surgeries (Callegan et al., 2007). The incidence of bacterial endophthalmitis has risen steadily over the last two decades, predominantly due to the increased popularity of sutureless cataract surgery, small-gauge vitrectomy, and multiple intravitreal (IVT) injections for the treatment of age-related macular degeneration (AMD) and diabetic retinopathy (Hanscom, 1996, Campbell et al., 2010). Despite therapeutic and surgical interventions, most cases of endophthalmitis result in partial or complete loss of vision. One of the hallmarks of bacterial endophthalmitis is increased intraocular inflammation culminating to retinal tissue damage (Kumar et al., 2010). Since, the visual properties of the eyes are highly sensitive to inflammation-mediated damage, a rapid resolution of inflammation is critical in minimizing retinal damage in endophthalmitis (Kumar et al., 2013). Thus investigating the mechanisms underlying inflammation initiation and resolution may provide novel therapeutic targets for the prevention and/or treatment of bacterial endophthalmitis.
Considering the complexity of host-pathogen interactions (Park et al., 2013c), we recently performed transcriptome analysis of S. aureus-infected mouse retina (Rajamani et al., 2016) and observed that the major pathways impacted in endophthalmitis includes: metabolism, inflammatory/immune, antimicrobial, cell trafficking, and lipid biosynthesis. Among the metabolic pathways, AMPK signaling was the most significantly affected. Similar to transcriptome analysis, the metabolic profiling, commonly referred to as metabolomics, has also attracted significant interest in various diseases, as it holds great promise in formulating a systems-level interpretation of biological events at the cellular or tissue levels (Isobe et al., 2014, Suhre, 2014, Yin et al., 2015). Indeed, our recent metabolomics studies led to the identification of resolvin D1 (RvD1) as an endogenous metabolite possessing drug-like properties, whose therapeutic efficacy was tested in preclinical mouse models of multiple sclerosis (Poisson et al., 2015). Similarly, we performed the global metabolomics on Staphylococcus aureus (SA)–infected mouse retina and discovered that the pathways involved in energy metabolism were significantly altered, as evidenced by a decrease in the enzymes of the TCA cycle and oxidative phosphorylation (unpublished data). Since both transcriptome (Rajamani et al., 2016) and metabolomics analysis implicated metabolic regulation of innate responses in bacterial endophthalmitis, we decided to investigate the role of AMPK, a multi-substrate protein kinase which plays a central role in regulating various metabolic processes by switching off glycolysis, fatty acid synthesis, and gluconeogenesis, and switching on oxidative phosphorylation (Hardie et al., 1998, Hardie et al., 2006).
AMPK is a critical intermediate in the control of fundamental cellular processes such as growth, proliferation, and survival (Bijland et al., 2013, Dandapani et al., 2013). Additionally, AMPK orchestrates multiple signaling pathways controlling nutrient uptake and energy metabolism (Grahame Hardie, 2014). In response to energy-depletion signals (i.e. a higher AMP/ATP ratio), AMPK balances energy consumption by inhibiting energy/ATP-consuming pathways and activating the compensatory energy/ATP-producing processes. In addition to the traditional role of AMPK in energy metabolism, several studies have indicated a pivotal role of AMPK in immune regulation (Sag et al., 2008, Viollet et al., 2010, Lin et al., 2014). Using AMPK activators (5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside, AICAR) and inhibitors (Compound C) (Corton et al., 1995, Zhou et al., 2001), there is now ample evidence that AMPK1α, through its signaling network, suppress inflammation (Salt et al., 2012, Palomer et al., 2013). The activation of AMPK1α by AICAR has been found to decrease inflammation in acute and chronic colitis (Bai et al., 2010a), cystic fibrosis (Myerburg et al., 2010), and autoimmune encephalomyelitis (Nath et al., 2005), and the pro-inflammatory state following lung injury (Zhao et al., 2008). Similarly, the therapeutic potential of AICAR has been evaluated in some ocular diseases, including retinoblastoma (Theodoropoulou et al., 2010), diabetic retinopathy (Kubota et al., 2011), endotoxin-induced uveitis (Suzuki et al., 2011), and dry eye (Sung et al., 2015) experimental models. Despite these beneficial effects of AMPK activation in various inflammatory diseases, very few studies have investigated its role in infectious diseases, and studies related to ocular infection are almost nonexistent.
Since increased intraocular inflammation is linked to retinal tissue damage in endophthalmitis, we hypothesized that AICAR might exert protective effects on bacterial endophthalmitis via modulating the anti-inflammatory activities of AMPK signaling. Here, we demonstrate that AICAR exerted therapeutic effects and improved the outcome of S. aureus endophthalmitis in a murine model of the disease. The protective effects of AICAR were mediated, at least in part, by the downregulation of inflammatory mediators and enhanced bacterial phagocytosis. To our knowledge, this is the first report to show the role of AMPK activation in ocular infections, opening a new avenue for the treatment of infectious endophthalmitis.
RESULTS
AMPKα phosphorylation is reduced in S. aureus-infected mouse retina
Previous studies have shown that AMPK activation alleviates inflammation (O’Neill et al., 2013), while increased inflammatory cytokines promote the dephosphorylation, and hence inactivation, of AMPK activity (Giri et al., 2004, Nath et al., 2005, Nath et al., 2009a, Gauthier et al., 2011). To determine how AMPK activity is modulated in bacterial endophthalmitis, we assessed the phosphorylation state of AMPKα in S. aureus-infected mouse neuroretina. Western blot analysis revealed that although the total level of AMPK is increased in the infected vs. uninfected (PBS) retina, the active (phosphorylated) form of AMPK is decreased (Fig. 1A), as evidenced by reduced levels of phospho(p)- AMPKα at 24, 48, and 72h post-infection (Fig. 1B). Consistent with the downregulation of p-AMPKα, the phosphorylation of ACC, a substrate of AMPKα, was also decreased drastically (Fig. 1 A & B). As the phosphorylation of ACC is an important downstream target of AMPKα activation (Hardie et al., 2012), this data suggests that AMPK signaling is impaired in bacterial endophthalmitis.
Figure 1. Bacterial endophthalmitis reduced AMPK phosphorylation in the retina.
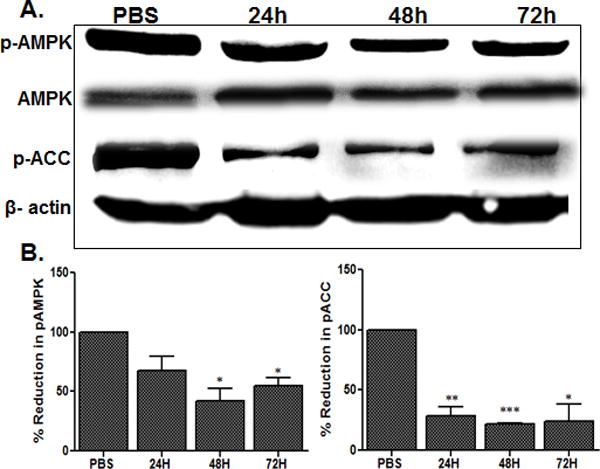
Endophthalmitis was induced by inoculation of S. aureus (RN6390, 5000 cfu) in the eyes of C57BL/6 mice (n = 6/time point). At the indicated time points (except PBS at 24h), the retinas were removed from the infected eyes, pooled (two retinas per sample), and sonicated to make retinal lysates. Following protein quantification, the retinal lysates were used for Western blot detection of total AMPK, phospho(p)-AMPK, and phospho(p)-ACC (A). Antibodies against total AMPK and β-actin were used to normalize protein loading. For quantification, the band intensities were measured using imageJ software and the percentage reduction of p-AMPK and p-ACC in infected samples were calculated with respect to the ratio of p-AMPK/AMPK and p-ACC/β-actin in PBS controls set at 100%, respectively. The data shown are representative of three independent experiments (B). Statistical analysis was performed by using one-way ANOVA with Dunnett’s post-hoc test. * p < 0.05, ** p < 0.001, *** p < 0.0001.
AICAR treatment reduced bacterial load and attenuated the inflammation in S. aureus-infected eyes
We next investigated the in vivo effects of AICAR, a pharmacological activator of AMPK. First, unexpectedly, we observed that intravitreal administration of AICAR following S. aureus infection, significantly reduced bacterial burden at the 24, 48, and 72h time points (Fig. 2A). Moreover, AICAR treatment was found to modulate inflammatory mediators as assessed by ELISA. As expected, S. aureus induced the production of IL-1β, TNF-α, and IL-6 in comparison with PBS-injected control eyes (Fig. 2B–D). The levels of all cytokines peaked at 24h, except for TNF-α (48h), followed by a time-dependent decline. In contrast, S. aureus-infected eyes treated with AICAR showed significant reduction in all inflammatory mediators at tested time points. Next we sought to determine whether the observed in vivo effects of AICAR treatment are due to increased AMPK activity, which is downregulated in endophthalmitis (Fig.1). Indeed, our data showed that AICAR treatment significantly restored the AMPKα phosphorylation/activity (Fig. 2E). A similar effect of AICAR treatment was observed in restoring the partial levels of p-ACC in S. aureus-infected retina (Fig. 2F).
Figure 2. Intravitreal injection of AICAR reduced bacterial load and inflammatory mediators in S. aureus-infected eyes.
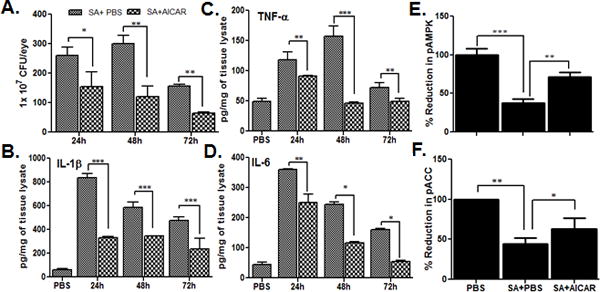
After 12h of S. aureus infection (5000 cfu/eye), the eyes of C57BL/6 mice (n=6) were treated with AICAR (30 μg/eye, delivered intravitreally in 2μl volume) and mice were sacrificed at 24, 48, and 72h post AICAR treatment. Control eyes received same volume of PBS (vehicle used to dissolve AICAR). Enucleated eyes were crushed in sterile PBS and used for bacterial enumeration (A), and the same lysate was used for quantitation of indicated inflammatory mediators using ELISA (B–D). In another experiment, pooled retinas from two eyes with or without AICAR, treatments (24h time point) were used for Western blot and quantification of p-AMPK (E), and p-ACC (F), as described in Fig. 1 legend. The data shown are mean ± SD of three independent experiments. Statistical analysis was performed using Student’s t-test by comparing S. aureus infected samples with or without AICAR treatment.* p < 0.05, ** p < 0.001, *** p < 0.0001.
Although, AICAR reinstated AMPK phosphorylation in the inflamed retina and reduced inflammatory cytokines, some studies suggest that AICAR can exert anti-inflammatory effects independent of AMPK activation (Lanner et al., 2012, Ying et al., 2013, Quan et al., 2015). To test this, eyes were pre-injected with Compound C, an AMPK inhibitor, followed by S. aureus challenge and AICAR treatment. We observed that the pre-treatment of eyes with Compound C reversed the anti-inflammatory effects of AICAR, as evidenced by higher or even exacerbated (TNF-α) levels of pro-inflammatory cytokines (Fig. 3). Together, these results indicate that the anti-inflammatory effects of AICAR in endophthalmitis are mediated through AMPK signalling.
Figure 3. The anti-inflammatory effects of AICAR in bacterial endophthalmitis are mediated via AMPK activation.
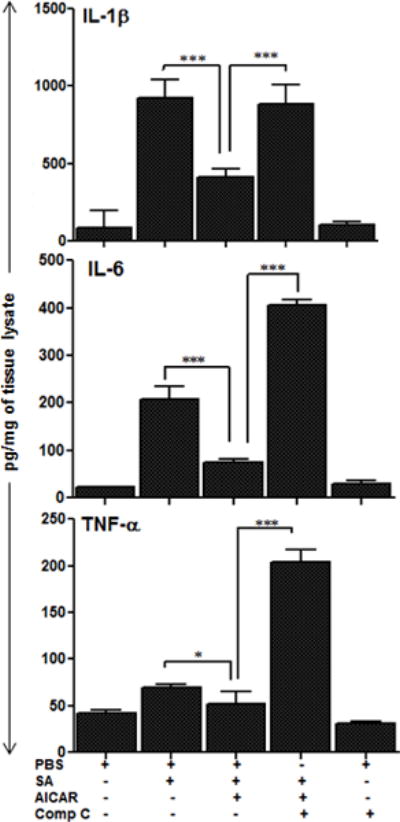
The eyes of C57BL/6 mice (n=6) were pretreated via intravitreal injection of AMPK inhibitor, Compound C (Comp C) or PBS (vehicle) control. After 12h of pretreatment, eyes of Comp C pretreated and two PBS groups were infected with S. aureus (5000 cfu/eye) and 12h post-bacterial challenge, AICAR was injected in Comp C treated and one PBS treated group. All mice were sacrificed after 24h of AICAR treatment, eyes were enucleated and whole eye lysates were subjected to TNF-α, IL-1β and IL-6 ELISA. Each data point represents mean ± SD, and the results are cumulative of two independent experiments. Statistical analysis was performed using one-way ANOVA with Bonferroni’s multiple-comparison test. * p < 0.05, *** p < 0.0001;
AICAR inhibits S. aureus-induced NF-kB and MAP Kinase signaling and inflammatory mediators production
The anti-inflammatory properties of AICAR-mediated AMPK activation have been attributed to its ability to inhibit NF-κB activation (Peairs et al., 2009, Katerelos et al., 2010, Salminen et al., 2011, Guo et al., 2014), a universal player in orchestrating inflammatory responses, including that of ocular infections (Kumar et al., 2004, Kumar et al., 2007, Kumar et al., 2012). To determine whether AICAR treatment influences NF-κB and other MAP Kinase signaling pathways in endophthalmitis, Western blot analysis was performed on retinal lysate of AICAR-treated and untreated eyes. As expected, S. aureus infection increased the activation of NF-κB, as evidenced by increased phosphorylation of IκB and corresponding decreases in total IκB levels. Similarly, S. aureus induced the activation of other MAP Kinases (i.e., phosphorylation of p38 and JNK) in the mouse retina, and these responses were significantly attenuated in eyes treated with AICAR (Fig. 4 A & B). A similar observation was made under in vitro conditions using microglial cells challenged with S. aureus (Fig. 4C & D).
Figure 4. AICAR treatment inhibited S. aureus-induced NF-kB, p38, and JNK signaling in the mouse retina and microglia cells.
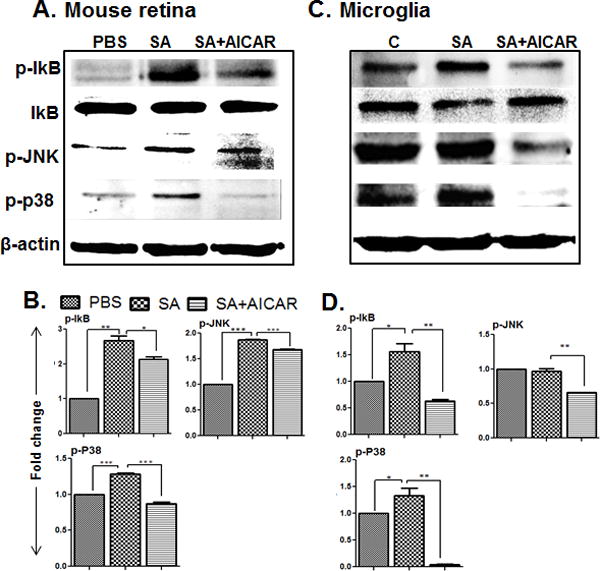
Retinal lysates from S. aureus-infected eyes with or without AICAR treatment (30 µg/eye, 24h) (described in Fig. 2 legend) were subjected to Western blot analysis with antibodies against phospho-IκB-α (p-IκB-α), phospho-p38 (p-p38), phospho-JNK (p-JNK), and IκB-α (IκB-α). Antibodies against non-phosphorylated p38 and JNK, and β-actin were used to normalize sample loading (A & B). BV2 microglia cells were plated in 6 well plate (1× 106 cells/well) and treated with AICAR (1mM) one hour before S. aureus infection for 8h. Cells supernatant was removed, and cells were used for Western blot analysis for the above indicated proteins (C & D). The results shown are representative of two independent experiments. The optical densities of the bands were quantified using imageJ and fold changes was calculated. Statistical analysis was performed by using One-Way ANOVA with Bonferroni’s multiple-comparison test. * p < 0.05, ** p < 0.001, *** p < 0.0001.
To further confirm the consequences of attenuated NF-κB signaling, the production of inflammatory cytokines was assessed in the residential (microglia) and infiltrating cells (macrophage and PMNs). As shown in figure 5, AICAR treatment significantly attenuated S. aureus-incited production of TNF-α, IL-6, and MIP-2 in BV2 microglia (Fig. 5A), macrophage (Fig. 5B), and PMNs (Fig. 5C). These findings provide the evidence that AICAR exerts inhibitory effects on the inflammatory response of retinal glial and infiltrated cells.
Figure 5. AICAR treatment diminished S. aureus-induced inflammatory mediators in microglia, macrophages and neutrophils.
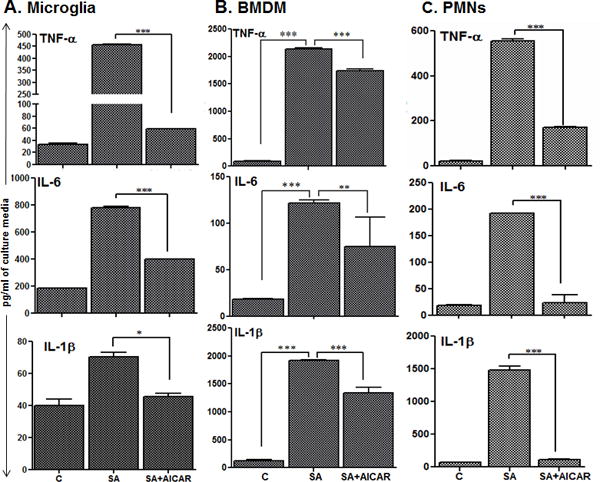
Cultured BV2 mouse microglia (A), bone marrow derived macrophages (B) and neutrophils (C) were plated in 6 well plate (1× 106 cells/well) (n = 6/condition), and treated with AICAR (1mM) one hour before S. aureus infection (MOI 10:1) for 8 h. Culture supernatant was used to quantify the expression of the indicated cytokines by using ELISA. Each data point represents the mean ± SD and the results are representative of at least two independent experiments. Statistical analysis was performed between SA alone and SA+AICAR. Statistical analysis was performed using one-way ANOVA with Bonferroni’s multiple-comparison test ** p < 0.001, *** P < 0.0001.
Bioenergetics analysis of S. aureus-challenged microglia and macrophage revealed glycolysis as the major source of energy
To determine, how the retina/retinal cells withdraw energy in response to microbial infection, currently not known, we performed the bioenergetics profile of S. aureus-infected BV2 microglia and BMDM. We utilized an extracellular flux analyzer (Seahorse Bioscience), which allows for the simultaneous assay of glycolysis intermediates in real-time by measuring extracellular acidification rate (ECAR). The Seahorse analysis of S. aureus-challenged BV2 microglia revealed increased ECAR, while AICAR treatment was found to reduce ECAR (Fig. 6A), indicating glycolysis as the major source of energy in bacterial infection. Although S. aureus alone in culture media (i.e. without in contact with cells) did not contribute significantly towards measured ECAR, to distinguish between the bacterial metabolism and the eukaryotic metabolism, the experiment was repeated using heat-killed S. aureus (HKSA). To this end, our data showed that similar to live S. aureus, HKSA challenge increased ECAR in BV2 cells and the presence of AICAR reduced ECAR levels (Fig. 6B). However, it should be noted that ECAR levels (absolute value) were much higher in live S. aureus versus HKSA infected cells. These findings were further confirmed by qPCR analysis, which revealed the increased expression of glycolytic enzymes, hexokinase 2 (HK2), and glucose transporter 1 (Glut1) in the S. aureus-infected BV2 cells and their downregulation by AICAR treatment (Fig. 6C). Similarly, AICAR treatment reduced the expression of HK2 and Glut1 in S. aureus-infected mouse retinas (Fig. 6D).
Figure 6. S. aureus-infected microglia and retinal tissue showed increased glycolysis and AICAR treatment inhibited this response.
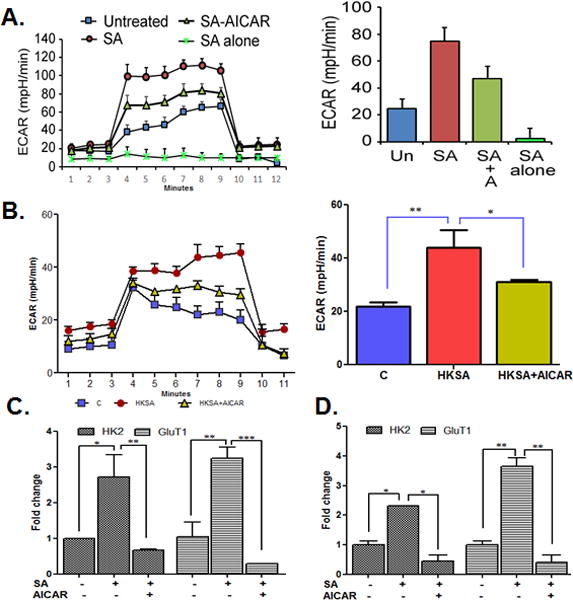
BV2 cells (3 × 104 cells/well) were plated in XF96V3-PS 96 cell culture plates (Seahorse Bioscience) and treated with AICAR (1mM) one hour before challenged with live S. aureus (MOI 10:1) (A) or HKSA (108 cfu) for 8h (B). The extracellular acidification rate (ECAR) was measured by the sequential addition of glucose, oligomycin and 2 deoxyglucose (2-DG). To examine the contribution of bacteria alone in the total response of the cells, live S. aureus was added in media only. In the another experiment, BV2 mouse microglia cells were plated in 6 well plate (1× 106 cells/well) and treated with AICAR (1mM) one hour before S. aureus infection for 8h. Cells were collected, RNA was extracted, cDNA was synthesized, and qRT-PCR was performed for glycolytic pathway genes HK2 and Glut1 and fold changes were calculated using housekeeping gene GAPDH (C). Eyes of WT (C57BL/6) (n = 5) were infected with S. aureus (5000 cfu/eye) followed by AICAR treatment (30 μg/eye) for 24h. Retinas were removed and pooled for RNA extraction. cDNA was prepared, and qPCR was performed for the glycolytic pathway genes HK2 and Glut1 and fold change was calculated using GAPDH (D). Each data point represents mean ± SD (n = 6/condition/experiment), and results are representative of at least 3 independent experiments. The results represent the mean ± SD of triplicates from three independent experiments. Statistical analysis was performed using one-way ANOVA with Bonferroni’s multiple-comparison test. * P < 0.05, ** P < 0.001, *** P < 0.0001. HKSA- heat killed S. aureus, HK2 - hexokinase II, GluT1- glucose transporter 1, mpH- milli-pH units.
To determine whether the bacterial-induced glycolytic response is cells specific, bioenergetics analysis was performed in mouse BMDM challenged with both live (Fig. 7A) and HKSA (Fig. 7B). BMDM exhibited similar trend as BV2 cells i.e. induction of ECAR and its downregulation by AICAR treatment including the reduced expression of HK2 and Glut1 (Fig. 7C). Collectively, this data indicates that AICAR reduces glycolysis, an energy-consuming pathway, in microglia and macrophages.
Figure 7. Bioenergetics analysis of macrophages challenged with S. aureus.
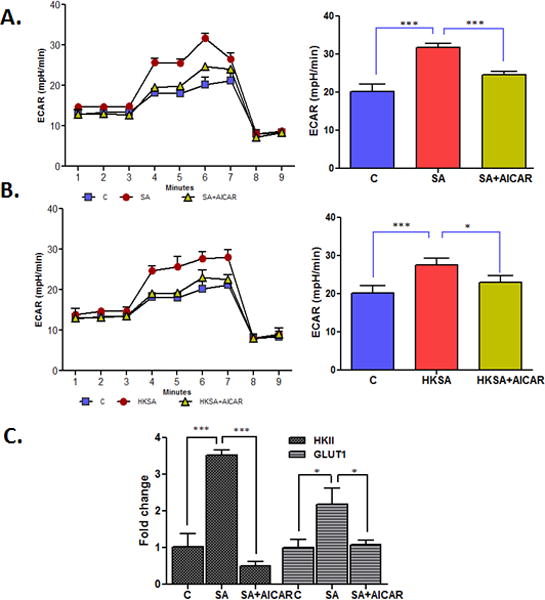
Mouse BMDM (5 × 104) were plated in XF96V3-PS cell culture plates and treated with AICAR (1mM) one hour before challenge with S. aureus (MOI 10:1) (A) and HKSA (108 cfu) (B) for 8h. The extracellular acidification rate (ECAR) was measured by the sequential addition of glucose, oligomycin and 2DG. In a separate experiment, macrophages were plated in 6 well plate (5× 106 cells/well) and treated with AICAR (1mM) one hour before S. aureus infection for 8h. Cells were collected, RNA was extracted, cDNA was synthesized, and qRT PCR was performed for the glycolytic pathway genes HK2 and Glut1 and fold change was calculated using GAPDH (C). Each data point represents mean ± SD (n = 6/condition/experiment), and results are representative of at least 3 independent experiments. Statistical analysis was performed using one-way ANOVA with Bonferroni’s multiple-comparison test. *P < 0.05, ***P < 0.0001. HK2 - hexokinase II, GluT1- glucose transporter 1.
AICAR treatment increases phagocytosis and intracellular killing of S. aureus in microglia, macrophages and neutrophils
Our in vivo studies showed that AICAR treatment reduces the bacterial burden (Fig. 2C), suggesting the potential role of AMPK activation in pathogen clearance in the eye. To determine the underlying mechanism (s) of this activity, we propose that AMPK signaling enhances the antibacterial properties of innate immune cells such as microglia, macrophages and neutrophils (Bae et al., 2011, Park et al., 2013a). To this end, our data showed that AICAR treatment enhances the phagocytic activity of microglia, as evidenced by the increased presence of CFSE-labelled S. aureus within the cells and their decreased presence following pretreatment with Compound C (Fig. 8A). The bacterial count assay revealed a significantly higher number of intracellular S. aureus after 2 h of incubation in AICAR-treated vs. control untreated microglia, indicating increased bacterial uptake or phagocytosis (Fig. 8B). However, the total number of bacteria inside and outside of the cells was found to be reduced following treatment with AICAR indicating the bacterial killing efficacy of AMPK activation, which was decreased by Compound C treatment (Fig. 8B). Similarly, AICAR treatment increased the phagocytic activity and bacterial killing ability of BMDM (Fig. 8C) and PMNs (Fig. 8D).
Figure 8. AICAR treatment increases the phagocytic activity of microglia, macrophages, and neutrophils.
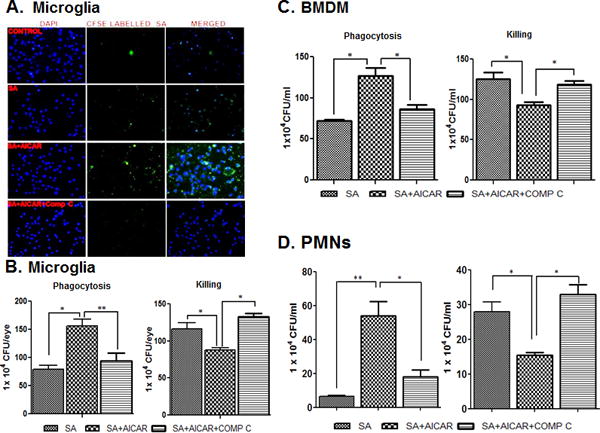
CFSE-labelled S. aureus was used to challenge the BV2 cells (5 × 105 cells/chamber) (n=4/condition) cultured in chamber slides under the indicated conditions (i.e., AICAR or Compound C treatment) for 2h. Stained cells were subjected to fluorescent imaging to visualize intracellular S. aureus (A). In another experiment, to assess phagocytic activity, cultured BV2 microglial cells) (B), bone-marrow derived macrophages (C) and neutrophils (D) were left untreated or treated with AICAR (1mM) and Comp C for one hour prior to S. aureus challenge (n=4/condition), as described in ‘Materials and Methods’. After 2 h of bacterial challenge, the cells were washed and kept in fresh medium containing gentamicin (200 μg/ml). After 2h of incubation, the cells were washed and lysed by using Triton-× (0.1%). The release of intracellular bacteria was quantitated via serial dilution and plate count (B, C & D). In the killing assay, cells were incubated with S. aureus for 2h, after which Triton × (0.1%) was added to the cells. Cells were then serially diluted and plated for counting (B, C & D). The data points represent the mean ± SD of three independent experiments. Statistical analysis was performed using a one-way ANOVA with Bonferroni’s multiple-comparison test. * p < 0.05, ** p < 0.001.
AMPKα1 KO mice exhibited increased bacterial burden and inflammatory mediators
Our previous results suggest that AICAR-mediated AMPK activation in the retina dampens pathogen-induced inflammatory responses, and induces a protective innate response, including enhanced phagocytosis. Thus, we hypothesized that the loss of AMPK will exacerbate inflammation and worsen the disease outcome. Our lab and others have reported that an infective dose of 5000 CFU/eye results in massive inflammation and retinal tissue damage (Engelbert et al., 2005, Whiston et al., 2008, Talreja et al., 2015), whereas eyes challenged with 500 CFU of S. aureus recover within 96h. Therefore, we propose that AMPK1α KO mice could be susceptible to endophthalmitis with a lower infective dose. Indeed, we observed that the bacterial burden was significantly higher in AMPKα1 KO vs. WT eyes at 24, 48, and 72h post-infection (Fig. 9A). Retinal function analysis, as determined by ERG, revealed a transient decline in the amplitude of a-and b-waves by 24 to 48h in WT eyes, which returned to ~70% by 72 h. In contrast, eyes of AMPKα1 KO mice exhibited a rapid decline in both waves by 24 h, and eventually a loss of measurable retinal function by 48h with slight recovery at 72h (Fig. 9B). Analysis of inflammatory mediators showed similar trends with significantly higher levels of IL-1β, TNF-α, and IL-6 in the eyes of AMPKα1 KO mice as compared to WT mice at all-time points (Fig. 9C). Collectively, these results show that the loss of AMPKα1signaling increases the severity of bacterial endophthalmitis.
Figure 9. Loss of AMPKα delays resolution of inflammation in S. aureus-infected eyes.
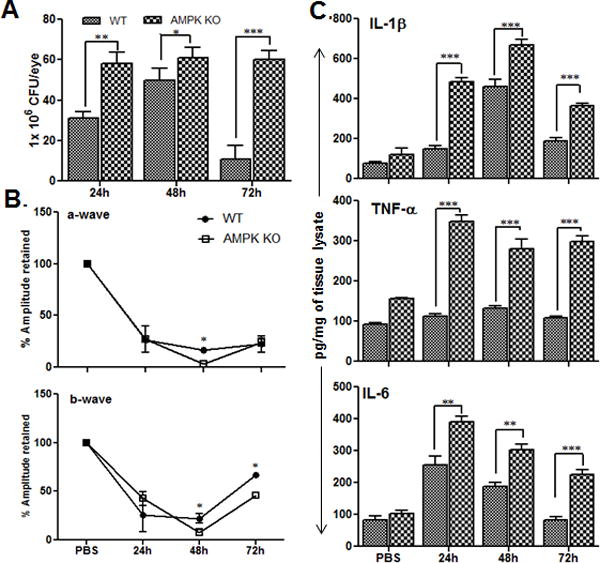
The eyes of WT (C57BL/6) (n = 6) and AMPKα KO mice (n=6) were infected with S. aureus (500 cfu/eye). At the indicated time points, infected eyes were subjected to retinal function testing by ERG (B), bacterial load estimation (A), and ELISA for pro-inflammatory cytokines expression for IL-1β, TNF-α, and IL-6 (C) as described in previous figure legends. The data presented as mean ± SD of three independent experiments. Statistical analysis was performed using Student’s t-test by comparing WT and AMPK KO mice. *p <0.05, **p <0.001, ***p <0.0001.
DISCUSSION
Bacterial endophthalmitis is an inflammatory disease of the eye and is, in part, mediated by the retinal innate response to infection (Singh et al., 2014b, Talreja et al., 2014, Parkunan et al., 2015). The current standard of care involves the intravitreal injection of antibiotics, which, while inhibiting bacterial growth, do little to suppress ongoing inflammation (Callegan et al., 2007, Pandey et al., 2013). Therefore, the identification of novel targets for the development of new anti-inflammatory therapeutics is needed, which may be used to treat endophthalmitis alone or in combination with antibiotics. In this study, we show AICAR to be a novel immunomodulatory agent with beneficial effects on the course of retinal infection. In a murine model of S. aureus endophthalmitis, treatment with AICAR after infection significantly attenuated intraocular inflammation, as well as reduced the bacterial burden in the eyes. Analysis of the mechanism(s) mediating AICAR’s anti-inflammatory effects revealed that it restored lost AMPK activity in the infected retina and inhibited NF-kB and MAP kinase signaling. Furthermore, AICAR treatment reduced the activity of the energy-consuming glycolytic pathway, and enhanced the bactericidal activity of retinal microglia and macrophages. Thus, AICAR may be used as an anti-inflammatory agent in the case of endophthalmitis and other ocular infections.
Although inflammation is necessary to defend the host against invading pathogens, the retina, being an immune-privileged tissue, is exquisitely sensitive to inflammation-mediated damage if inflammation persists (Shamsuddin et al., 2011, Kochan et al., 2012, Kumar et al., 2013, Kumar et al., 2015). We previously reported that in the retina, these early inflammatory responses are generated by retinal Müller glia (Kumar et al., 2012, Singh et al., 2014a) and microglia (Kumar et al., 2010, Kochan et al., 2012) via Toll-like receptor (TLR) signaling. This is followed by the profound recruitment of inflammatory cells, particularly myeloid cells, such as neutrophils and monocytes/macrophages (Ravindranath et al., 1995, Giese et al., 1998). As the ongoing inflammatory cell infiltration could dramatically shift retinal metabolism, causing tissue damage and visual cycle malfunction, we hypothesized that the altered activity of AMPK, a master regulator of cellular metabolism, may be involved in the pathogenesis of bacterial endophthalmitis. Indeed, our data show that the phosphorylation of AMPK is reduced in the infected retina at the early (24h) stages of infection and remained downregulated during the entire course of the infection, up to 96h (data not shown for 96h). This finding is consistent with the previous studies demonstrating the downregulation of phospho-AMPK under various inflammatory conditions (Viollet et al., 2010), including retinal inflammatory diseases (Hyttinen et al., 2011, Kubota et al., 2011, Kamoshita et al., 2014). In endophthalmitis, the increased production of predominantly pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 (Fig. 2) may have led to reduced AMPK activity, as shown in LPS-challenged neutrophils (Zhao et al., 2008) and macrophages (Jeong et al., 2009). Together, our findings suggest that AMPK may be a promising pharmacologic target for the treatment of bacterial endophthalmitis.
In an attempt to restore lost AMPK activity in the infected retina, we utilized a widely used AMPK activator, AICAR, and administered it to the mice via intraperitoneal and intravitreal routes. Interestingly, in contrast to LPS- (Kamoshita et al., 2014) or diabetes-induced retinal inflammation (Kubota et al., 2011) studies, we observed no significant difference in the level of intraocular inflammation in the treatment vs. control groups when AICAR was given intraperitoneally (data not shown). However, the intravitreal administration of AICAR following infection resulted in the activation (phosphorylation) of AMPK in the retina and drastically reduced S. aureus-induced inflammatory mediators. These data indicate that the localized delivery of anti-inflammatory agents may be more effective when treating acute retinal inflammation, whereas systemic administration may be used for chronic inflammation, as in the case of uveitis, diabetic retinopathy, or AMD. As intravitreal injection remains the primary route for the delivery of therapeutic modalities in retinal diseases (Peyman et al., 2011), including the administration of antibiotics in bacterial endophthalmitis (Callegan et al., 2007), our mode of AICAR delivery can be justified to regulatory bodies such as the US Food and Drug Administration (FDA). Moreover, the intraocular administration of AICAR may not have serious systemic effects on energy metabolism, as may be the case with the intraperitoneal route.
AICAR has been shown to attenuate the progression of disease in various in vivo models of inflammation, including a mouse lung injury model (Zhao et al., 2008), a murine colitis model (Bai et al., 2010b), a model of autoimmune encephalomyelitis (Prasad et al., 2006). Although the protective effects of AICAR have been attributed to its anti-inflammatory and anti-cancer properties, the underlying cellular signaling cascade is not fully understood. However, it is well known that AICAR’s effect on AMPK activity plays an important role. Therefore, research on the anti-inflammatory effects of AICAR-induced AMPK activation remains an important area of investigation (Salt et al., 2012, Scheen et al., 2015). However, only a few studies have explored the therapeutic potential of AICAR in ocular diseases. In our study, using the inhibitory Compound C, we demonstrate that the protective effects of AICAR in bacterial endophthalmitis are mediated through AMPKα. This finding was further supported by our data showing increased susceptibility and intraocular inflammation in AMPK1α KO mice. To investigate the molecular mechanisms underlying the anti-inflammatory effects of AMPK activation in endophthalmitis, we examined the ability of AICAR to attenuate the activation of NF-κB and other MAPKs (p38 and JNK), the key inflammatory signaling cascade. We found that AICAR significantly inhibited S. aureus-induced degradation of IκBα and other MAPKs in the mouse retina and also decreased the nuclear translocation of NF-κB in cultured microglial cells (Figure 10). Taken together, our study is the first to show the role of AICAR-mediated AMPK activation in innate defense against bacterial infection in the retina.
Figure 10. Effect of AICAR treatment in regulating the innate response of retinal residential (glial) and infiltrating cells in bacterial endophthalmitis.
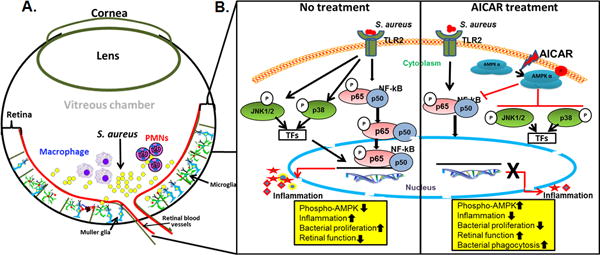
In response to retinal infection, such as bacterial (S. aureus) endophthalmitis, the retinal glial (microglial and Muller glia) recognize the pathogen in the vitreous chamber via Toll-like receptors (TLRs, e.g. TLR2). The activated glial cells secrete inflammatory mediators to recruit myeloid cells such as PMNs and macrophages to combat pathogen invasion (A). In the current study, we report that AICAR-mediated AMPK activation induces protective innate responses by affecting both glial and infiltrating innate immune cells. AICAR treatment was found to downregulate the inflammation, restored Phospho-AMPK levels, and promoted bacterial clearance via enhanced bacterial phagocytic activity. At molecular levels, AICAR inhibited bacterial-induced NF-KB and MAPK kinase signaling (B). The downward arrows indicate decrease whereas upward arrows indicate increase responses.
One of the most notable observations in our study was that AMPKα activation (by AICAR treatment) reduced bacterial burden in the eyes of wild-type mice. Similarly, the loss of AMPKα promoted bacterial growth in the eyes. As the clearance of pathogens and the removal of apoptotic cells is critical for the resolution of inflammation (Bae et al., 2010, Yang et al., 2010, Donnelly et al., 2012), we focused on evaluating the antibacterial properties of AICAR. Since AMPKα activation has been shown to enhance the phagocytic activity of microglia, macrophages and neutrophils, including the uptake of bacteria (Bae et al., 2011, Park et al., 2013a), we reasoned that AICAR would stimulate retinal microglia and may increase their phagocytic activity towards bacteria, as reported in our recent study (Kochan et al., 2012). Our data showed that AICAR pretreatment of microglia, macrophages, and neutrophils enhanced their ability to phagocytose and kill S. aureus, and that Compound C treatment reduced this effect. These findings indicate that AICAR-mediated AMPK activation augments the antibacterial properties of retinal innate immune cells and that this could be the underlying mechanism of increased pathogen clearance in AICAR-treated eyes. Although the BV-2 cell line has been extensively used to represent brain microglial cells (Stansley et al., 2012), a recent study by Butovsky et al while investigating TGF-β–dependent molecular and functional signature of microglia revealed that BV-2 cells express a macrophage-like signature rather an adult microglial signature (Butovsky et al., 2014). Our rationale to use BV-2 cells was based on our prior study (Kochan et al., 2012), where we demonstrate that both BV2 cells and the primary retinal microglia respond similarly to S. aureus challenge. However, in view of this studies, further investigation involving depletion of retinal microglial using genetic (Wang et al., 2016) or pharmacological (Kataoka et al., 2011, Ferrer-Martin et al., 2015) models, is needed to determine the role of AMPK-signaling in this cell type.
In the retina, the glial cells constitute the frontline cells of innate immunity. Upon sensing the invading pathogens via pattern recognition receptors (e.g., TLRs), these cells shift their phenotype from a quiescent/resting state to a highly activated state. This response is accompanied by the activation of multiple intracellular signaling cascades, resulting in the production of mediators with antimicrobial, chemotactic, and inflammatory functions. As expected, developing and maintaining this activated phenotype is considerably energy demanding, as it requires that nutrients such as glucose, fatty acids, and amino acids be degraded into various metabolic intermediates, thereby providing the cell with a constant supply of energy (ATP) (energy surplus, low AMP/ATP ratio) (Fox et al., 2005). Therefore, there is a growing interest in investigating the interplay between cellular metabolic pathways and immune response signaling. Several studies have demonstrated that cells of the myeloid lineage (i.e., PMNs, monocytes/macrophages, and dendritic cells) derive their energy almost exclusively from glycolysis, whereas the lymphocytes (T and B cells) predominantly use oxidative phosphorylation (Fox et al., 2005, Kramer et al., 2014). In this study, bioenergetics analysis of microglia and macrophages (both myeloid-derived cells) in response to S. aureus challenge revealed increased ECAR, suggesting glycolysis as the source of energy in activated retinal microglia and infiltrating macrophages. However, we observed some discrepancy in ECAR measurements, for example, BV2 cells infected with live S. aureus exhibited higher (2 fold) ECAR levels compared to those challenged with HKSA. As our recent study (Kumar et al., 2015) demonstrate that live S. aureus evoked higher inflammatory response than HKSA, we reasoned that higher ECAR values could be due to 1) increased virulence of the live bacteria inducing more lactate secretion by host cells and/or 2) higher basal ECAR levels of BV2 cells (data not shown). Similarly, we could not explain the observed differences in ECAR levels of BV2 versus BMDM other than BV-2 being an immortalized and BMDM as primary cells. Regardless, AICAR treatment was found to inhibit the expression of major glycolytic enzymes and reduce ECAR, indicating the potential role of AICAR-mediated AMPK1α activation in switching energy metabolism from glycolysis to oxidative phosphorylation. Considering the fact that TLR signaling plays an important role in the pathogenesis of bacterial endophthalmitis and that TLR activation has been implicated in the metabolic reprogramming of DCs and macrophages (Krawczyk et al., 2010, Tannahill et al., 2013, Cortese et al., 2014), further studies are warranted to establish this link in our model. Similarly, several other important topics that need to be addressed include: 1) How the metabolic shift occurs in the inflammatory vs. the resolution phase of the disease and 2) AMPK activation has been shown to switch the phenotype of monocytes/macrophages from M1 (pro-inflammatory) to M2 (anti-inflammatory) (Mounier et al., 2013, Li et al., 2015) and this macrophage phenotype switching and its role has not been studied in the pathogenesis of bacterial endophthalmitis.
In summary, our study describes an essential role of AMPK in regulating the retinal innate response to microbial infection by modulating both residential (glial) and infiltrating cells such as neutrophils and macrophages (Fig. 10). Further studies using bone-marrow chimeric mouse or myeloid specific ablation of AMPK are warranted to dissect the in vivo role of AMPK-signaling in glial versus infiltrating innate cells in bacterial endophthalmitis. Considering the safe use of AICAR in clinical trials (Cuthbertson et al., 2007, Boon et al., 2008), our study implies that AICAR may have a therapeutic potential not only in the treatment of human endophthalmitis, but in other infectious diseases too. Moreover, based on these findings, we propose that the modulation of energy metabolism of retinal residential or infiltrated myeloid cells via AMPK signaling might represent a novel therapeutic strategy in mitigating endophthalmitis-associated vision loss.
EXPERIMENTAL PROCEDURES
Animals
Female C57BL/6 (aged ~8 weeks) mice obtained from the Jackson Laboratory were maintained in the DLAR facility of the Kresge Eye Institute. AMPKα1 knockout (KO) mice (B6/129 background) (Nath et al., 2009b) were kindly provided by Dr. Shailendra Giri, Department of Neurology, Henry Ford Hospital, Detroit, MI. Animals were provided free access to water and standard laboratory chow and maintained in a 12h light/dark cycle at 22°C temperature. All procedures were conducted in compliance with the ARVO statement for the Use of Animals in Ophthalmic and Vision Research, and were approved by the Institutional Animal Care and Use Committee (IACUC).
Microglia cell culture
In vitro experiments were performed using a BV2 mouse microglia cell line, as described in our earlier studies (Kumar et al., 2010, Kochan et al., 2012) and were provided by V. Bocchini (University of Perugia, Italy). Cells were cultured in a humidified 5% CO2 incubator at 37°C in low-glucose DMEM containing 5% FBS and a penicillin-streptomycin cocktail (Invitrogen, Carlsbad, CA, USA). Before treatment, media was replaced with antibiotic-free and serum-free DMEM for 18 h (growth factor starvation). At the indicated time points (shown in figures), conditioned media was assayed ELISA for cytokine/chemokine production measurement, and cells were processed either for protein or RNA extraction, as described in following sections.
Generation of bone marrow derived macrophage (BMDM) and neutrophil
BMDM and neutrophils (PMNs) were isolated as described previously (Swamydas et al., 2013). Briefly, mice were euthanized and bone marrow cells were flushed from femurs and tibias with RPMI media containing 10% FBS and 0.2 mM EDTA. For RBC lysis, a hypotonic solution of 0.2% NaCl was added for 20 sec., followed by 1.6% NaCl. After centrifugation at 1400 rpm for 7 min., the cells were rinsed with RPMI media as shown above. For macrophage isolation, cells were resuspended in RPMI supplemented with 10% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin, counted, and cultured at 37°C in 5% CO2. For neutrophil extraction, cells were mixed in 1 ml PBS and placed over the histopaque gradient of 1119 and 1077 followed by centrifugation at 25°C for 30 min. without interruption. The layer between histopaques 1119 and 1077 was collected and washed two times with RPMI media at 1400 rpm for 7 min and cultured as described for macrophages.
Induction of endophthalmitis and AICAR treatment
During our experiments, the mice were anesthetized by intraperitoneal injection of ketamin/xylazine (ketamin, 100–125 mg/kg; xylazine, 10–12.5 mg/kg). Endophthalmitis was induced by intravitreal injections of S. aureus (strain RN 6390) using a 32-G needle attached to a 10 μl glass syringe (Hamilton, Reno, USA) under a dissecting microscope. In the treatment group, AICAR (Toronto Research Chemicals, Ontario, Canada) was injected intravitreally (30 μg/eye) 12h after S. aureus infection. In the control group, eyes received same volume (2 μl) of PBS (vehicle) injection. In all experiments, only one eye (left) was used to induce endophthalmitis and/or AICAR treatment whereas the right eyes served as uninfected control in the same animal. The detailed experimental procedure is described in our previous studies (Kumar et al., 2010, Kochan et al., 2012, Kumar et al., 2015, Talreja et al., 2015). Retinal function was determined by scotopic electroretinogram (ERG), as described in our previous study (Kumar et al., 2010, Kumar et al., 2015).
Immunoblotting
For Western blot analysis, neuroretina (without RPE and choroid) from eyes were pooled (two retinas per sample) and processed in the RIPA lysis buffer. Protein concentration was measured using a protein estimation kit (Thermoscientific, MA) according to the manufacturer’s instructions. Following incubation, BV2 microglia cells were also lysed using radioimmunoprecipitation (RIPA) lysis buffer (Sigma Aldrich, St. Louis, USA). The denatured proteins were resolved by 8% or 12% SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane (0.45 μm) (Bio-Rad, Harcules, CA) followed by incubation with anti-phospho (Thr 172)-AMPKα, anti-phospho acetyl-CoA carboxylase (Ser-79) (ACC), anti-AMPKα (Cell signaling Technology, Beverly, MA), anti-phospho IkB (Cell signaling Technology, Boston, MA), anti-phospho JNK (Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho p38 (Cell signaling Technology, Boston, MA), and anti-IkB (Santa Cruz Biotechnology, Santa Cruz, CA) with a dilution of 1:1000, and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) with a dilution of 1:4000. The secondary antibodies used in the study were goat anti-rabbit-IgG-HRP (Bio-Rad, Harcules, CA) and rabbit anti-mouse-IgG-HRP (Bio-Rad, Harcules, CA). To visualize the protein bands, the membranes were exposed to Supersignal West Femto chemiluminescent substrate (Thermo scientific, Rockford, IL). For semi-quantitative analysis, immunodetected protein band intensities were measured using ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, http://rsb.info.nih.gov/ij/, 1997–2009).
Bacterial load determination and Enzyme-linked immunosorbent assay (ELISA)
Following infection, at the indicated time points, the mice were euthanized, their eyes were enucleated, and whole-eye lysates were made in 250 μl of PBS using tissue lyser. A small portion (50 μl) of the lysate was used to enumerate bacterial load by serial plate dilution. The rest of the lysate was used for the protein determination and ELISA assay to measure in vivo levels of cytokines and chemokines. For in vitro studies, culture supernatant of S. aureus-challenged cells (BV2, PMNs and macrophage) was used for ELISA detection of TNF-α, IL-1β, IL-6 (BD biosciences, San Diego, CA, USA), and MIP2 and KC (R & D systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. All of the values are expressed as mean ± standard deviations (SD).
Bioenergetics analysis
Seahorse Bioscience XFe 96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA) was used to monitor extracellular acidification rate (ECAR) in BV2 microglia cells and BMDM. Briefly, BV2 cells (3 × 104 cells/well) and BMDM (5 × 104 cells/well) were seeded to in a XFe 96-well cell culture microplate (seahorse Bioscience) in 200 μl of DMEM and incubated overnight (14–16h) at 37°C in 5% CO2. After treatment of the cells (as described in figures 6 & 7 legends), growth medium was removed, the wells were washed two times, and 175 μl of bicarbonate free glycolysis stress test media with glutamine (pre-warmed at 37°C) was added to each well. The cells were then incubated at 37°C for 1h. ECAR was measured by adding glucose (10 μM), oligomycin (2 μM) and 2-deoxyglucose (2-DG) (100 μM) to the indicated final concentrations using the included ports on the XFe 96 cartridges. The data obtained were normalized with cell number counts, as described previously (Tebbe et al., 2014).
Real-time (RT) PCR
Retinas were removed and pooled (two retinas per sample) for RNA isolation in TRIzol solution (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. RNA was reversed transcribed to cDNA using a cDNA synthesis kit (Superscript, Invitrogen Carlsbad, CA, USA). Quantitative assessment of gene expression was carried out by Sybergreen based real time PCR using specific primers on a StepOne Plus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The data was analyzed using the 2−ΔΔCt method.
Fluorescent labeling of S. aureus and phagocytosis
For intracellular visualization, S. aureus (RN 6390) was labelled with CFDA/SE using the method described by Vander Top E. et. al. (Vander Top et al., 2006). For the phagocytosis assay, BV2 microglia cells (106 cells/well), macrophage and neutrophils (1× 106 cells/well) were grown in small (60 mm) Petri dishes containing DMEM medium (10% FBS and 1% penicillin/streptomycin). The next day, the cells were infected with S. aureus (MOI 10:1) in each well and incubated for two hours. The cells were then washed and treated with gentamicin (200 μg/ml) for another two hours to kill all extracellular and/or adherent bacteria. To confirm the absence of extracellular bacteria, the supernatant was cultured on Tryptic Soy Agar (TSA) plates. The cells were rinsed three times with PBS and lysed with 0.1% Triton X-100. The lysed cells were scraped, collected, and centrifuged at 5,000 × g for five minutes. The cell pellet was washed with PBS and centrifuged at 5,000 × g twice for five minutes. The pellets were then re-suspended in 1 ml of sterile PBS, and serial ten-fold dilutions were prepared. Afterwards, the dilutions were plated on TSA plates and incubated overnight at 37°C. The following day, the colonies present on the TSA plates were counted, and counts were expressed in CFU/ml. To examine the killing activity of AICAR, cells were challenged with S. aureus for 2h with or without AICAR/Compound C treatment. After 2h, cells were lysed with 0.1% Triton X-100 and serially diluted in PBS followed by plating on the TSA plates for bacterial enumeration.
Statistical analysis
Graphpad Prism software was used for the data analysis and to plot the graphs. All plots represent the mean value ± SD. Statistical significance was assessed using the Student’s t-test and one-way ANOVA.
Acknowledgments
This study is supported by grants from the National Institute of Health and the Research to Prevent Blindness (RPB) Williams and Mary Greve special scholar award.
The authors would like to thank Pawn K. Singh, Ph.D. (Kresge Eye Institute, Wayne State University) for his technical help in performing intravitreal injections. We also acknowledge Jaspreet Singh, Ph.D. and Hamid Suhail, Ph.D. (Department of Neurology, Henry Ford Hospital) for their assistance in performing bioenergetics analysis using seahorse.
Grant Support: NIH-EY019888 and Research to Prevent Blindness.
Footnotes
Author’s Contributions-A.K., A.K, and S.G., conceived the project and designed the experiments; A.K. performed the experiments; A.K., A.K, and S.G. analyzed the data; A.K., A.K, and S.G. contributed reagents/materials/analysis tools; A.K., A.K, and S.G. wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Declaration-Authors have no financial conflict of interest.
References
- Bae HB, Tadie JM, Jiang S, Park DW, Bell CP, Thompson LC, et al. Vitronectin inhibits efferocytosis through interactions with apoptotic cells as well as with macrophages. J Immunol. 2010;190:2273–2281. doi: 10.4049/jimmunol.1200625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae HB, Zmijewski JW, Deshane JS, Tadie JM, Chaplin DD, Takashima S, Abraham E. AMP-activated protein kinase enhances the phagocytic ability of macrophages and neutrophils. FASEB J. 2011;25:4358–4368. doi: 10.1096/fj.11-190587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai A, Ma AG, Yong M, Weiss CR, Ma Y, Guan Q, et al. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem Pharmacol. 2010a;80:1708–1717. doi: 10.1016/j.bcp.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Bai A, Yong M, Ma AG, Ma Y, Weiss CR, Guan Q, et al. Novel anti-inflammatory action of 5-aminoimidazole-4-carboxamide ribonucleoside with protective effect in dextran sulfate sodium-induced acute and chronic colitis. J Pharmacol Exp Ther. 2010b;333:717–725. doi: 10.1124/jpet.109.164954. [DOI] [PubMed] [Google Scholar]
- Bijland S, Mancini SJ, Salt IP. Role of AMP-activated protein kinase in adipose tissue metabolism and inflammation. Clin Sci (Lond) 2013;124:491–507. doi: 10.1042/CS20120536. [DOI] [PubMed] [Google Scholar]
- Boon H, Bosselaar M, Praet SF, Blaak EE, Saris WH, Wagenmakers AJ, et al. Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia. 2008;51:1893–1900. doi: 10.1007/s00125-008-1108-7. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-[beta]-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callegan MC, Gilmore MS, Gregory M, Ramadan RT, Wiskur BJ, Moyer AL, et al. Bacterial endophthalmitis: therapeutic challenges and host-pathogen interactions. Prog Retin Eye Res. 2007;26:189–203. doi: 10.1016/j.preteyeres.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RJ, Bronskill SE, Bell CM, Paterson JM, Whitehead M, Gill SS. Rapid expansion of intravitreal drug injection procedures, 2000 to 2008: a population-based analysis. Arch Ophthalmol. 2010;128:359–362. doi: 10.1001/archophthalmol.2010.19. [DOI] [PubMed] [Google Scholar]
- Cortese M, Sinclair C, Pulendran B. Translating glycolytic metabolism to innate immunity in dendritic cells. Cell Metab. 2014;19:737–739. doi: 10.1016/j.cmet.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Cuthbertson DJ, Babraj JA, Mustard KJW, Towler MC, Green KA, Wackerhage H, et al. 5-Aminoimidazole-4-Carboxamide 1-β-d-Ribofuranoside Acutely Stimulates Skeletal Muscle 2-Deoxyglucose Uptake in Healthy Men. Diabetes. 2007;56:2078–2084. doi: 10.2337/db06-1716. [DOI] [PubMed] [Google Scholar]
- Dandapani M, Hardie DG. AMPK: opposing the metabolic changes in both tumour cells and inflammatory cells? Biochem Soc Trans. 2013;41:687–693. doi: 10.1042/BST20120351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly LE, Barnes PJ. Defective phagocytosis in airways disease. Chest. 2012;141:1055–1062. doi: 10.1378/chest.11-2348. [DOI] [PubMed] [Google Scholar]
- Engelbert M, Gilmore MS. Fas ligand but not complement is critical for control of experimental Staphylococcus aureus Endophthalmitis. Invest Ophthalmol Vis Sci. 2005;46:2479–2486. doi: 10.1167/iovs.04-1139. [DOI] [PubMed] [Google Scholar]
- Ferrer-Martin RM, Martin-Oliva D, Sierra-Martin A, Carrasco MC, Martin-Estebane M, Calvente R, et al. Microglial Activation Promotes Cell Survival in Organotypic Cultures of Postnatal Mouse Retinal Explants. PLoS One. 2015;10:e0135238. doi: 10.1371/journal.pone.0135238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–852. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- Gauthier MS, O’Brien EL, Bigornia S, Mott M, Cacicedo JM, Xu XJ, et al. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun. 2011;404:382–387. doi: 10.1016/j.bbrc.2010.11.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese MJ, Sumner HL, Berliner JA, Mondino BJ. Cytokine expression in a rat model of Staphylococcus aureus endophthalmitis. Invest Ophthalmol Vis Sci. 1998;39:2785–2790. [PubMed] [Google Scholar]
- Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci. 2004;24:479–487. doi: 10.1523/JNEUROSCI.4288-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahame Hardie D. AMP-activated protein kinase: a key regulator of energy balance with many roles in human disease. J Intern Med. 2014;276:543–559. doi: 10.1111/joim.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Zhang Y, Hong K, Luo F, Gu Q, Lu N, Bai A. AMPK inhibition blocks ROS-NFkappaB signaling and attenuates endotoxemia-induced liver injury. PLoS One. 2014;9:e86881. doi: 10.1371/journal.pone.0086881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanscom T. The Endophthalmitis Vitrectomy Study. Arch Ophthalmol. 1996;114:1029–1030. doi: 10.1001/archopht.1996.01100140233036. author reply 1028–1029. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase–development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature reviews. Molecular cell biology. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyttinen JM, Petrovski G, Salminen A, Kaarniranta K. 5′-Adenosine monophosphate-activated protein kinase–mammalian target of rapamycin axis as therapeutic target for age-related macular degeneration. Rejuvenation Res. 2011;14:651–660. doi: 10.1089/rej.2011.1220. [DOI] [PubMed] [Google Scholar]
- Isobe Y, Arita M. Identification of novel omega-3 fatty acid-derived bioactive metabolites based on a targeted lipidomics approach. J Clin Biochem Nutr. 2014;55:79–84. doi: 10.3164/jcbn.14-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong HW, Hsu KC, Lee JW, Ham M, Huh JY, Shin HJ, et al. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. 2009:E955–E964. doi: 10.1152/ajpendo.90599.2008. [DOI] [PubMed] [Google Scholar]
- Kamoshita M, Ozawa Y, Kubota S, Miyake S, Tsuda C, Nagai N, et al. AMPK-NF-kappaB axis in the photoreceptor disorder during retinal inflammation. PLoS One. 2014;9:e103013. doi: 10.1371/journal.pone.0103013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K, Nishiguchi KM, Kaneko H, van Rooijen N, Kachi S, Terasaki H. The roles of vitreal macrophages and circulating leukocytes in retinal neovascularization. Invest Ophthalmol Vis Sci. 2011;52:1431–1438. doi: 10.1167/iovs.10-5798. [DOI] [PubMed] [Google Scholar]
- Katerelos M, Mudge SJ, Stapleton D, Auwardt RB, Fraser SA, Chen CG, et al. 5-aminoimidazole-4-carboxamide ribonucleoside and AMP-activated protein kinase inhibit signalling through NF-kappaB. Immunol Cell Biol. 2010;88:754–760. doi: 10.1038/icb.2010.44. [DOI] [PubMed] [Google Scholar]
- Kochan T, Singla A, Tosi J, Kumar A. Toll-Like Receptor 2 Ligand Pretreatment Attenuates Retinal Microglial Inflammatory Response but Enhances Phagocytic Activity toward Staphylococcus aureus. Infect Immun. 2012;80:2076–2088. doi: 10.1128/IAI.00149-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PA, Ravi S, Chacko B, Johnson MS, Darley-Usmar VM. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic biomarkers. Redox Biology. 2014;2:206–210. doi: 10.1016/j.redox.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota S, Ozawa Y, Kurihara T, Sasaki M, Yuki K, Miyake S, et al. Roles of AMP-activated protein kinase in diabetes-induced retinal inflammation. Invest Ophthalmol Vis Sci. 2011;52:9142–9148. doi: 10.1167/iovs.11-8041. [DOI] [PubMed] [Google Scholar]
- Kumar A, Kumar A. Role of Staphylococcus aureus Virulence Factors in Inducing Inflammation and Vascular Permeability in a Mouse Model of Bacterial Endophthalmitis. PLoS One. 2015;10:e0128423. doi: 10.1371/journal.pone.0128423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Pandey RK, Miller LJ, Singh PK, Kanwar M. Muller glia in retinal innate immunity: a perspective on their roles in endophthalmitis. Crit Rev Immunol. 2013;33:119–135. doi: 10.1615/critrevimmunol.2013006618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Shamsuddin N. Retinal Muller glia initiate innate response to infectious stimuli via toll–like receptor signaling. PLoS One. 2012;7:e29830. doi: 10.1371/journal.pone.0029830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Singh CN, Glybina IV, Mahmoud TH, Yu FS. Toll-like receptor 2 ligand-induced protection against bacterial endophthalmitis. J Infect Dis. 2010;201:255–263. doi: 10.1086/649589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Yin J, Zhang J, Yu FS. Modulation of corneal epithelial innate immune response to pseudomonas infection by flagellin pretreatment. Invest Ophthalmol Vis Sci. 2007;48:4664–4670. doi: 10.1167/iovs.07-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Zhang J, Yu FS. Innate immune response of corneal epithelial cells to Staphylococcus aureus infection: role of peptidoglycan in stimulating proinflammatory cytokine secretion. Invest Ophthalmol Vis Sci. 2004;45:3513–3522. doi: 10.1167/iovs.04-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Dagnino-Acosta A, Ainbinder A, Cheng Q, Joshi AD, et al. AICAR prevents heat-induced sudden death in RyR1 mutant mice independent of AMPK activation. Nat Med. 2012;18:244–251. doi: 10.1038/nm.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Ding XY, Xiang DM, Xu J, Huang XL, Hou FF, Zhou QG. Enhanced M1 and Impaired M2 Macrophage Polarization and Reduced Mitochondrial Biogenesis via Inhibition of AMP Kinase in Chronic Kidney Disease. Cell Physiol Biochem. 2015;36:358–372. doi: 10.1159/000430106. [DOI] [PubMed] [Google Scholar]
- Lin JT, Chen HM, Chiu CH, Liang YJ. AMP-activated protein kinase activators in diabetic ulcers: from animal studies to Phase II drugs under investigation. Expert Opin Investig Drugs. 2014;23:1253–1265. doi: 10.1517/13543784.2014.922951. [DOI] [PubMed] [Google Scholar]
- Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, et al. AMPKalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013;18:251–264. doi: 10.1016/j.cmet.2013.06.017. [DOI] [PubMed] [Google Scholar]
- Myerburg MM, King JD, Jr, Oyster NM, Fitch AC, Magill A, Baty CJ, et al. AMPK agonists ameliorate sodium and fluid transport and inflammation in cystic fibrosis airway epithelial cells. Am J Respir Cell Mol Biol. 2010;42:676–684. doi: 10.1165/rcmb.2009-0147OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:566–574. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- Nath N, Khan M, Paintlia MK, Singh I, Hoda MN, Giri S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol. 2009a;182:8005–8014. doi: 10.4049/jimmunol.0803563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath N, Khan M, Rattan R, Mangalam A, Makkar RS, Meester Cd, et al. Loss of AMPK exacerbates experimental autoimmune encephalomyelitis disease severity. Biochemical and Biophysical Research Communications. 2009b;386:16–20. doi: 10.1016/j.bbrc.2009.05.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- Palomer X, Salvado L, Barroso E, Vazquez-Carrera M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int J Cardiol. 2013;168:3160–3172. doi: 10.1016/j.ijcard.2013.07.150. [DOI] [PubMed] [Google Scholar]
- Pandey RK, Yu FS, Kumar A. Targeting toll-like receptor signaling as a novel approach to prevent ocular infectious diseases. Indian J Med Res. 2013;138:609–619. [PMC free article] [PubMed] [Google Scholar]
- Park DW, Jiang S, Tadie JM, Stigler WS, Gao Y, Deshane J, et al. Activation of AMPK enhances neutrophil chemotaxis and bacterial killing. Molecular medicine. 2013a;19:387–398. doi: 10.2119/molmed.2013.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PJ, Shukla D. Role of heparan sulfate in ocular diseases. Experimental Eye Research. 2013c;110:1–9. doi: 10.1016/j.exer.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkunan SM, Randall CB, Coburn PS, Astley RA, Staats RL, Callegan MC. Unexpected Roles for TLR4 and TRIF in Gram-Positive Intraocular Infection. Infect Immun. 2015 doi: 10.1128/IAI.00502-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peairs A, Radjavi A, Davis S, Li L, Ahmed A, Giri S, Reilly CM. Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clin Exp Immunol. 2009;156:542–551. doi: 10.1111/j.1365-2249.2009.03924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyman GA, Hosseini K. Combination Therapies in Ophthalmology: Implications for Intravitreal Delivery. Journal of Ophthalmic & Vision Research. 2011;6:36–46. [PMC free article] [PubMed] [Google Scholar]
- Poisson LM, Suhail H, Singh J, Datta I, Denic A, Labuzek K, et al. Untargeted Plasma Metabolomics Identifies Endogenous Metabolite with Drug-like Properties in Chronic Animal Model of Multiple Sclerosis. Journal of Biological Chemistry. 2015;290:30697–30712. doi: 10.1074/jbc.M115.679068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R, Giri S, Nath N, Singh I, Singh AK. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside attenuates experimental autoimmune encephalomyelitis via modulation of endothelial-monocyte interaction. J Neurosci Res. 2006;84:614–625. doi: 10.1002/jnr.20953. [DOI] [PubMed] [Google Scholar]
- Quan H, Kim JM, Lee HJ, Lee SH, Choi JI, Bae HB. AICAR Enhances the Phagocytic Ability of Macrophages towards Apoptotic Cells through P38 Mitogen Activated Protein Kinase Activation Independent of AMP-Activated Protein Kinase. PLoS ONE. 2015;10:e0127885. doi: 10.1371/journal.pone.0127885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamani D, Singh PK, Rottmann BG, Singh N, Bhasin MK, Kumar A. Temporal retinal transcriptome and systems biology analysis identifies key pathways and hub genes in Staphylococcus aureus endophthalmitis. Scientific Reports. 2016;6:21502. doi: 10.1038/srep21502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranath RM, Mondino BJ, Adamu SA, Pitchekian-Halabi H, Hasan SA, Glasgow BJ. Immunopathologic features of Staphylococcus aureus endophthalmitis in the rat. Invest Ophthalmol Vis Sci. 1995;36:2482–2491. [PubMed] [Google Scholar]
- Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 2011;89:667–676. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt IP, Palmer TM. Exploiting the anti-inflammatory effects of AMP-activated protein kinase activation. Expert Opin Investig Drugs. 2012;21:1155–1167. doi: 10.1517/13543784.2012.696609. [DOI] [PubMed] [Google Scholar]
- Scheen AJ, Esser N, Paquot N. Antidiabetic agents: Potential anti-inflammatory activity beyond glucose control. Diabetes Metab. 2015;41:183–194. doi: 10.1016/j.diabet.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Shamsuddin N, Kumar A. TLR2 mediates the innate response of retinal Muller glia to Staphylococcus aureus. J Immunol. 2011;186:7089–7097. doi: 10.4049/jimmunol.1100565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Shiha M, Kumar A. Antibacterial responses of retinal Muller glia: production of antimicrobial peptides, oxidative burst and phagocytosis. Journal of Neuroinflammation. 2014a;11:33. doi: 10.1186/1742-2094-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PK, Donovan DM, Kumar A. Intravitreal Injection of the Chimeric Phage Endolysin Ply187 Protects Mice from Staphylococcus aureus Endophthalmitis. Antimicrobial Agents and Chemotherapy. 2014b;58:4621–4629. doi: 10.1128/AAC.00126-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansley B, Post J, Hensley K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J Neuroinflammation. 2012;9:115. doi: 10.1186/1742-2094-9-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K. Metabolic profiling in diabetes. J Endocrinol. 2014;221:R75–85. doi: 10.1530/JOE-14-0024. [DOI] [PubMed] [Google Scholar]
- Sung MS, Li Z, Cui L, Choi JS, Choi W, Park MJ, et al. Effect of Topical 5-Aminoimidazole-4-carboxamide-1-β-d-Ribofuranoside in a Mouse Model of Experimental Dry EyeAICAR and Dry Eye. Investigative Ophthalmology & Visual Science. 2015;56:3149–3158. doi: 10.1167/iovs.14-16153. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Manola A, Murakami Y, Morizane Y, Takeuchi K, Kayama M, et al. Inhibitory effect of aminoimidazole carboxamide ribonucleotide (AICAR) on endotoxin-induced uveitis in rats. Invest Ophthalmol Vis Sci. 2011;52:6565–6571. doi: 10.1167/iovs.11-7331. [DOI] [PubMed] [Google Scholar]
- Swamydas M, Lionakis MS. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp. 2013:e50586. doi: 10.3791/50586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talreja D, Kaye KS, Yu FS, Walia SK, Kumar A. Pathogenicity of ocular isolates of Acinetobacter baumannii in a mouse model of bacterial endophthalmitis. Invest Ophthalmol Vis Sci. 2014;55:2392–2402. doi: 10.1167/iovs.13-13401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talreja D, Singh PK, Kumar A. In vivo role of TLR2 and MyD88 signaling in eliciting innate immune responses in Staphylococcal endophthalmitis. Invest Ophthalmol Vis Sci. 2015 doi: 10.1167/iovs.14-16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbe C, Chhina J, Dar SA, Sarigiannis K, Giri S, Munkarah AR, Rattan R. Metformin limits the adipocyte tumor-promoting effect on ovarian cancer. Oncotarget. 2014;5:4746–4764. doi: 10.18632/oncotarget.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoropoulou S, Kolovou PE, Morizane Y, Kayama M, Nicolaou F, Miller JW, et al. Retinoblastoma cells are inhibited by aminoimidazole carboxamide ribonucleotide (AICAR) partially through activation of AMP-dependent kinase. FASEB J. 2010;24:2620–2630. doi: 10.1096/fj.09-152546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Top E, Perry G, Gentry-Nielsen M. A novel flow cytometric assay for measurement of In Vivo pulmonary neutrophil phagocytosis. BMC Microbiology. 2006;6:61. doi: 10.1186/1471-2180-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, et al. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol. 2010;45:276–295. doi: 10.3109/10409238.2010.488215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao L, Zhang J, Fariss RN, Ma W, Kretschmer F, et al. Requirement for Microglia for the Maintenance of Synaptic Function and Integrity in the Mature Retina. The Journal of Neuroscience. 2016;36:2827–2842. doi: 10.1523/JNEUROSCI.3575-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiston EA, Sugi N, Kamradt MC, Sack C, Heimer SR, Engelbert M, et al. alphaB-crystallin protects retinal tissue during Staphylococcus aureus-induced endophthalmitis. Infect Immun. 2008;76:1781–1790. doi: 10.1128/IAI.01285-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Friggeri A, Banerjee S, Bdeir K, Cines DB, Liu G, Abraham E. Urokinase-type plasminogen activator inhibits efferocytosis of neutrophils. Am J Respir Crit Care Med. 2010;182:1516–1523. doi: 10.1164/rccm.201003-0452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P, Lehmann R, Xu G. Effects of pre-analytical processes on blood samples used in metabolomics studies. Anal Bioanal Chem. 2015;407:4879–4892. doi: 10.1007/s00216-015-8565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Wang Z, Zhang Y, Yang TY, Ding ZH, Liu SY, et al. Capsaicin induces apoptosis in human osteosarcoma cells through AMPK-dependent and AMPK-independent signaling pathways. Mol Cell Biochem. 2013;384:229–237. doi: 10.1007/s11010-013-1802-8. [DOI] [PubMed] [Google Scholar]
- Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, Tsuruta Y, Abraham E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L497–504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]