

Abstract
Retinal dystrophies (RD) are a rare genetic disorder with high genetic heterogeneity. This study aimed at identifying disease-causing variants in fifteen consanguineous Tunisian families. Full ophthalmic examination was performed. Index patients were subjected to IROme analysis or whole exome sequencing followed by homozygosity mapping. All detected variations were confirmed by direct Sanger sequencing. Mutation analysis in our patients revealed two compound heterozygous mutations p.(R91W);(V172D) in RPE65, and five novel homozygous mutations: p.R765C in CNGB1, p.H337R in PDE6B, splice site variant c.1129-2A > G and c.678_681delGAAG in FAM161A and c.1133 + 3_1133 + 6delAAGT in CERKL. The latter mutation impacts pre-mRNA splicing of CERKL. The other changes detected were six previously reported mutations in CNGB3 (p.R203*), ABCA4 (p.W782*), NR2E3 (p.R311Q), RPE65 (p.H182Y), PROM1 (c.1354dupT) and EYS (c.5928-2A > G). Segregation analysis in each family showed that all affected individuals were homozygotes and unaffected individuals were either heterozygote carriers or homozygous wild type allele. These results confirm the involvement of a large number of genes in RD in the Tunisian population.
Retinal dystrophies (RD) are a heterogeneous group of diseases in which the photoreceptor and RPE cells of the retina degenerate, leading to partial or complete blindness and affecting approximately 1 in 2500–3500 individuals1. Classification of RD is generally based on whether the disease primarily affects cones or rods (predominantly affecting the macula or peripheral retina respectively) and whether it occurs alone (non-syndromic RD) or in conjunction with other systemic disorders, especially loss of hearing (syndromic RD).
RD phenotypes are variable in terms of onset, progression and severity. The disease may be mild and non progressive, such as in congenital stationary night blindness (CSNB), characterized by defective rod photoreceptors involved in night vision. Non progressive disorders may lead to severe visual impairment as well as achromatopsia (ACHM), stationary congenital cone dystrophies. Other disorders are progressive, leading to severe visual impairment such as in retinitis pigmentosa (RP), cone dystrophy (CD), cone-rod dystrophy (CRD)2 and Stargardt disease (STGD)3. In RP, rod photoreceptors are initially affected more severely than cones4. The most severe cases are Leber Congenital Amaurosis (LCA) and early-onset rod–cone dystrophies, in which infants suffer from complete blindness from birth or within the first years of life5. Cone dystrophies are characterized by progressive degeneration of cone photoreceptors with preservation of rod function6, whereas in CRD peripheral vision is also compromised, leading to early blindness. Stargardt disease is a juvenile macular degeneration characterized by central vision loss3.
This group of rare genetic disorders shows substantial clinical and genetic overlaps with high genetic heterogeneity involving more than 220 genes identified so far (https://sph.uth.edu/retnet/). Similar phenotypes may result from different gene mutations, and subtle differences in phenotypes may result from a similar mutation7.
The mutations in causative genes induce either in degeneration or dysfunction of retinal cells. Functionally, different gene products are involved in many cellular functions and fall into four categories: proteins directly involved in the phototransduction cascade, genes encoding proteins responsible for the structure and polarity of the photoreceptors, genes encoding proteins of the visual cycle, and regulatory genes (such as transcription and splicing factors)2.
All modes of mendelian inheritance have been described in RD, with autosomal recessive being the most prevalent2. In our study we focused on the Tunisian population, known to have a relatively high level of consanguineous marriages, leading to a relatively high frequency of autosomal recessive diseases. This study was designed to apply homozygosity mapping in 15 consanguineous Tunisian families segregating retinal degenerative disease and three families analyzed by IROme7, aiming at the identification of the genetic defects.
Materials and Methods
Subjects
The department B of Hedi Rais Institute of Ophthalmology in Tunisia recruited all subjects involved in the study over a 10-year period. 177 Tunisian families segregating RD were enrolled. In this pilot study, a subset of 61 individuals (32 affected, 29 unaffected) from 15 families with clear recessive transmission and two or more affected individuals was selected. The affected individuals comprised 17 males and 15 females, ranging in age from 4 to 72 years, with an onset of disease ranging from birth to 47 years. Demographic characteristics, age at onset, and personal and family history were recorded for all participants. Written informed consent was obtained from each study participant, analyses were done in accordance with local guidelines and regulations study was approved by the Local Ethics Committee of the Hedi Rais Institute.
All patients underwent a standard ophthalmological examination including determination of best corrected visual acuity using standard Snellen charts. Clinical examination was supplemented by fundus photography, color vision assessment using the Farnsworth-Munsell 100 hue color vision test (FM100, Munsell Color Company Inc., Baltimore, MD, USA) and analysis of dark adaptation. Goldmann kinetic perimetry (Carl Zeiss Meditec Inc., Dublin, CA, USA) using V-4e and I-4e targets, fluorescein angiography (Imagenet; Topcon Corporation, Tokyo, Japan) and optical coherence tomography (SD-OCT) (OCT 3D TOPCON 2000) was also performed. Electrophysiological investigation was carried out using the Métrovision vision monitor (Métrovision, Pérenchies, France) according to the International Society for Clinical Electrophysiology of Vision (ISCEV) protocol.
Homozygosity mapping and mutation analysis
Genomic DNA of participating individuals was isolated from peripheral blood using a standard salting out procedure. One index patient of 12 families was selected for whole exome sequencing (WES) analysis and unaffected family members were collected for co-segregation analysis. Three index patients were analyzed with IROme8.
Exome capturing was performed with the Roche Nimble-Gen version 2 (44.1–megabase pair) at Otogenetics Corporation (Norcross, Georgia) and sequencing was done on an Illumina HiSeq2000 at a mean coverage×31. Sequence reads were aligned to the human genome reference sequence (build hg19), and variants were identified and annotated using the Nextgene software package v.2.3.5. (Softgenetics, State College, PA). Homozygosity was evaluated from SNPs obtained by WES. In families with CERKL mutations, SNPs: rs4667591, rs1050354, rs4894140, rs1143676, rs3754929, rs1400130, rs12476147, rs17228441, rs1800255, rs1225090 were selected and analyzed by Sanger sequencing in the index patients of families F12-F15.
Assessment of the Pathogenicity of Candidate Variants
We applied various bioinformatic tools to filter variants against NCBI dbSNP to facilitate the identification of disease-causing mutations. The Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org) data set was used as a reference data set of population-based allele frequencies9. Novel DNA variations were compared with data from public databases (HGMD10 and 1000 genomes11). Although most causative mutations associated with RD are rare (MAF < 0.01). The putative pathogenicity of the novel missense variants reported in this study was evaluated using two in silico tools: Polymorphism Phenotyping v2 (PolyPhen-2)12 and Sorting Intolerant from Tolerant (SIFT)13. The PolyPhen-2 score ranges from 0.0 (tolerated) to 1.0 (deleterious). Variants with scores of 0.0 are predicted to be benign. Values of 1.0 or close to 1.0 are more confidently predicted to be deleterious. PolyPhen-2 and SIFT scores use the same range, 0.0 to 1.0, but with opposite meanings. A variant with a SIFT score of 1.0 is predicted to be benign. In addition, we determined with PolyPhen-2 a measure of evolutionary nucleotide conservation.
Mutation analysis
After all filtering steps, Sanger sequencing was used to verify each predicted disease-causing variants remaining in the autozygous regions of interest and to perform cosegregation analyses. PCR was realized in a total reaction mixture of 20 μl, containing 20 ng of genomic DNA, 10 pmol of each primer (Eurogentec, Liège, Belgium) and 10 μl of FastStart PCR Master Mix (Roche, Basel, Switzerland). Three amplification steps (30 s each), with the annealing temperature ranging from 58 °C to 60 °C, were performed. Primer sequences were designed using the PRIMER3 software (http://frodo.wi.mit.edu/) and PCR conditions are provided in Table 1. The purified PCR products were then directly sequenced on an ABI 3100XL DNA automated sequencer (Applied Biosystems, Foster city, CA), using the Big Dye Terminator Labeling Kit version 1 (Life Technologies, Zug, Switzerland). Cosegregation analysis was performed in all families.
Table 1. Sequences of primers and PCR condition.
| Genes | Exon | Forward | Reverse | Hybridation temp. (°C) |
|---|---|---|---|---|
| CNGB3 | 5 | GTGAGAACATGCGGTGTTTG | CAAAGATGGGCAAATGATCC | 58 |
| CNGB1 | 23 | AGAGACTCCGCCTCTCACTC | GGGGCAGACACGAAGATG | 58 |
| ABCA4 | 15 | AGCACATGGAGTGTGCGTAG | TGCCCCTGTACATTTTAGCC | 58 |
| NR2E3 | 6 | TCTGAGCCTCTGGCTGATGTCA | AGAAGGGAGTCCAGCCTCAC | 60 |
| RPE65 | 4 | GACTTGATGAGGACACATAG | CAGTCCAGTAATTTTCAAGC | 60 |
| RPE65 | 6 | TTCAAGGGGTAGTGATGACC | GCACAAAATGCTATTCTGAC | 60 |
| RPE65 | 11 | CTTAGGAGCCAAGACTTAAG | GAGGAAACTCAAATGCTACG | 60 |
| PROM1 | 12 | ctccagccttagtccagcag | gtcccatcacagcaggatct | 58 |
| EYS | 29 | AATCTGCTTCTGGCTTTGTTT | GCCCCACTAGCCAGAAAATA | 58 |
| PDE6B | 7 | CCTGCACACAGACATCCAGT | TGGCAGAGACAAGGAGAAGC | 58 |
| FAM161A | 3 | TGGTCACATACAACTGAAAGTATAACA | GCTTCTGTTCCTCCCTTGCT | 60 |
| CERKL | 8 | GGGAACTGAAGAGAAAGTGAGG | GGCTGAGTGAGTAGTTGTTTGC | 58 |
| LRP2 | 69 | TTTTTGCTCCCCATCTTCTG | AATTCAGCAGGTGGGAGTTG | 58 |
| PPIG | 14 | CATTTCCCCAATTCTTTTTCTT | GATTGTGCGCGTCTATCCTT | 58 |
| CWC22 | 10 | GCTCCTGGAAAGACCAACAG | GCCTCTTTCCAACTAACATGC | 58 |
| ITGA4 | 24 | AAAACCAGGTTGCTTTTTGC | ACCATGCTTGTTCCTTCCAC | 58 |
| PDE1A | 7 | ACAGGGTGCACTGAAAGTCC | TTCTGGGCAGAGACAGATTG | 58 |
| NCKAP1 | 11 | AGCAACATAAGAAAGGCAGGA | GAAGAATGCAGGGAGGTACG | 58 |
| ZNF804A | 4 | CCACTGTTGCTGAAGATCCA | TTTGATCTGCTAATGGGACTGA | 58 |
| FSIP2 | 12 | ATCCCTGATTGTGGCAATTC | AAGCAGACTGTTCAAGGGCTA | 58 |
| COL3A1 | 30 | TAGTTCCCACCCAGCTGTTC | CAGCAGCACCCTGAAAATAA | 58 |
| ANKAR | 16 | TGTCAATATAGCATACCCCTTTG | TTTTTGGCTTATTTCTCCAAGA | 58 |
Splicing variant analysis
A minigene analysis was done to predict the putative impact of the deletion c.1133 + 3_1133 + 6delAAGT occurring in a splice site. The mutated region was amplified by PCR from DNA of patient VI-1, VI-2 in F13 and one healthy control using specific (forward: 5′-TCC AAA GGA TCC GGG AAC TGA AGA GAA AGT GAG G-3′ and reverse: 5′-TCC AAA GAA TTC GGC TGA GTG AGT AGT TGT TTG C-3′). The resulting PCR products were subsequently cloned into the pBK-CMV vector using the T4 DNA Ligase according to manufacturer’s protocol (Rapid DNA Ligation Kit). Plasmids were analyzed by direct Sanger sequencing and then transfected into HEK293 cells. Total RNA was extracted with TRIzol (Invitrogen product name), retrotranscribed with the AffinityScript RT (AffinityScript QPCR cDNA Synthesis Kit – Agilent), and the resulting cDNA was PCR-amplified using the previously described primers but without the restriction enzyme sites (forward: 5′-GGG AAC TGA AGA GAA AGT GAG G-3′) and (reverse: 5′-GGC TGA GTG AGT AGT TGT TTG C-3′). The amplified products were separated by electrophoresis on a 2% agarose gel and were subsequently analyzed by Sanger sequencing.
Results
Clinical features
As summarized in Table 2, patient phenotypes included RP and early-onset retinal degeneration (EORD) and STGD (Fig. 1). The clinical presentation of patients in families F2, F4, F5, F7, F8, F10 and F11 showed typical hallmarks of RP symptoms for which affected individuals initially experienced night blindness with progressive visual loss and hemeralopia in the majority of cases. Specifically, affected individual IV-5 (aged 40 years) of F2 family reported poor visual acuity. Funduscopy revealed typical RP changes with macular atrophy in F10 affected members suffering from photophobia; onset of symptoms was reported to occur during the first decade of life, with typical RP changes and normal macula. Fundus examination of patient III-10 (aged 36 years) of F10 revealed normal macular pattern with typical RP, with no nystagmus or hemeralopia. In F11, the index patient V-1 presented typical RP with atrophy of the retina and macular edema.
Table 2. Clinical features of the patients examined.
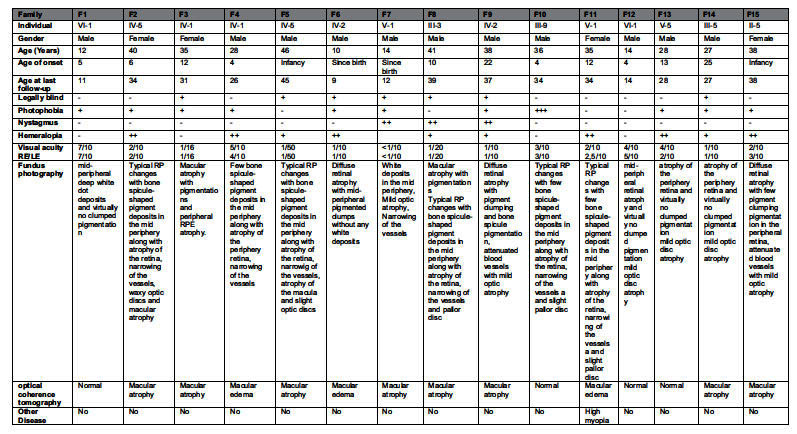
+ and − symbols indicate presence/absence, as well as degree of a given feature (+ mild, ++ moderate, +++ severe). F: family; RE: right eye; LE: Left eye.
Figure 1. Clinical features of patients.

Fundus photographs of (A) left eye of index patient (F8) with PROM1 mutation; (B) right eye of IV-4 patient (F3) with ABCA4 mutation; (C) left eye of index patient (F5) with RPE65 mutation; (D) right eye of the index case (F2) with CNGB1 mutation; (E,F) left eye of subject IV-1 in family F4 with NR2E3 mutation; (G) right eye of subject VI-1 (F1) with CNGB3 mutation; (H) right eye of IV-1patient (F6) with RPE65 mutation; (I) left eye of index case IV-2 (F9) with EYS mutation, (J) left eye of subject III-9 (F10) with PDE6B mutation, (K) left eye of the proband V-1 (F7) with RPE65 mutation, (L) right eye of subject V-1 (F11) with deletion in FAM161A.
In addition, clinical data of F9, F12, F13, F14 and F15 (Fig. 2) showed typical hallmarks of RP symptoms for which affected individuals initially experienced night blindness with progressive visual loss and hemeralopia in the majority of cases. Fundoscopy revealed mild optic disc and retinal atrophy without pigmentation. Macula was normal in young patients and showed atrophic alteration in advanced stages.
Figure 2. Spectrum of fundus findings among different families (F12, F13, F14, F15) with mutation in CERKL.

Intermediate phenotypes between LCA and RP were observed in two families. In F1 and F6 the affected individuals had poor visual acuity at the ages of 10–12 years respectively, with a clinical picture suggesting EORD. Two retinal patterns were identified: pattern 1 presented mid-peripheral deep white dot deposits and virtually no clumped pigmentation (F1), whereas pattern 2 showed mid-peripheral pigmented clumps with no white deposits (F6). Family F3 presented a very advanced form of STGD. Night blindness was the first symptom, followed by intense photophobia. All had useful vision until the second decade of life. Fundus examination revealed atrophy of the macula, pallor of the optic disk and bone spicule-shaped pigment deposits in the periphery. SD-OCT show diffuse atrophy with Epiretinal membrane. Only family F3 presented a Stargardt phenotype. All cases presented non-syndromic phenotype (no associated extraocular abnormalities).
Mutation analysis
Following the clinical diagnosis of retinal dystrophy, WES or IROme was performed to screen for all known genes and to look for novel candidate genes. Assuming identity-by-descent in these consanguineous families, we identified and examined autozygous regions when they spanned more than 5 Mb of genomic DNA. After all filtering steps, a few strong candidate genes remained in the autozygous regions of interest. Known RD genes were selected first for direct sequencing.
Mutation analysis in our patients revealed seven novel homozygous mutations. The first mutation found was a missense substitution, c.2293C > T, in exon 23 of CNGB1 (NM_001297.4), resulting in p.R765C (Fig. 3A) detected in family F2. It is considered to be probably damaging with a score of 1.0 by polyphen-2 and R765 is conserved from mammals to Danio rerio (Zebrafish). The second variant was the substitution of A to G at nucleotide position 1010 in exon 7 of the PDE6B (NM_001145291.1) and resulted in the substitution of histidine (H) by arginine (R) at codon 337 (p.H337R) (Fig. 3D) in family F10. This variant is predicted to be benign with polyphen-2 (Table 3). This amino acid is also conserved from mammals to Danio rerio. These two mutations were amongst those included in the evaluation of IROme8.
Figure 3. Chromatograms of the six novel mutations in CNGB1, RPE65, PDE6B, FAM161A and CERKL.

For each mutation, wild-type (wt) sequence is depicted with the homozygous and heterozygous sequences. (A) c.2293C > T; p.R765C (CNGB1), (B) c.1129-2A > G (RPE65), (C) c.[325C > T];[569T > A] (RPE65), (D) c.1010A > G; p.H337R (PDE6B), (E) c.678_681delGAAG (FAM161A), and (F) c.1133 + 3_1133 + 6delAAGT(CERKL). Arrows indicate the position of the mutated nucleotide.
Table 3. Homozygous regions and mutations identified in this study.
| Family ID | Disease | Genotyping Method | Size of homozygous region, in Mb | Chromosome | Gene | DNA mutation | Predicted protein variant | Coverage | Reference sequence | Previously reported | SIFT | Polyphen |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | Eord | WES | 30.4 | 8q21.3 | CNGB3 | c.[607C > T];[607C > T] | p.(R203*);(R203*) | 26 | NM_019098.4 | [21] | — | — |
| F2 | RP | IROme | — | 16q21 | CNGB1 | c.[2293C > T];[2293C > T] | p.(R765C); (R765C) | — | NM_001297.4 | This study and [8] | 0 | 1,00 |
| F3 | Stargardt | WES | 5.8 | 1p22.1 | ABCA4 | c.[2345G > A];[2345G > A] | p.(W782*); (W782*) | 5 | NM_000350.2 | [28] | — | — |
| F4 | RP | WES | 12.2 | 15q23 | NR2E3 | c.[932G > A];[932G > A] | p.(R311Q); (R311Q) | 24 | NM_014249.3 | [35] | 0.10 | 0.627 |
| F5 | RP | WES | 54.1 | 1p31.3-p31.2 | RPE65 | c.[544C > T];[544C > T] | p.(H182Y); (H182Y) | 36 | NM_000329.2 | [30] | 0 | 0.872 |
| F6 | Eord | WES | 18.2 | 1p31.3-p31.2 | RPE65 | c.[1129-2A > G];[1129-2A > G] | — | 30 | NM_000329.2 | This study | — | — |
| F7 | RP | IROme | — | 1p31.3-p31.2 | RPE65 | c.[325C > T];[569T > A] | p.(R91W); (V172D) | — | NM_000329.2 | This study | 0.01/0.01 | 0.997/0.721 |
| F8 | RP | WES | 9 | 4p15.32 | PROM1 | c.[1354dupT];[1354dupT] | p.(Y452fs*12); (Y452fs*12) | 12 | NM_001145848.1 | [42] | — | — |
| F9 | LCA | WES | 58 | 6q12 | EYS | c.[5928-2A > G];[5928-2A > G] | — | 24 | NM_001292009.1 | [23] | — | — |
| F10 | RP | IROme | — | 4p16.3 | PDE6B | c.[1010A > G];[1010A > G] | p.(H337R); (H337R) | — | NM_001145291.1 | This study and [8] | 1.00 | 0.046 |
| F11 | RP | WES | 13 | 2p15 | FAM161A | c.[678_681delGAAG]; [678_681delGAAG] | p.(K227Nfs*17); (K227Nfs*17) | 94 | NM_001201543 | This study | — | — |
| F12 | RP | WES | 8 | 2q31.3 | CERKL | c.[1133 + 3-1133 + 6delAAGT]; [1133 + 3-1133 + 6delAAGT] | — | 21 | NM_201548.4 | This study | — | — |
| F13 | RP | — | — | 2q31.3 | CERKL | c.[1133 + 3-1133 + 6delAAGT]; [1133 + 3-1133 + 6delAAGT] | — | — | NM_201548.4 | This study | — | — |
| F14 | RP | — | — | 2q31.3 | CERKL | c.[1133 + 3-1133 + 6delAAGT]; [1133 + 3-1133 + 6delAAGT] | — | — | NM_201548.4 | This study | — | — |
| F15 | RP | — | — | 2q31.3 | CERKL | c.[1133 + 3-1133 + 6delAAGT]; [1133 + 3-1133 + 6delAAGT] | — | — | NM_201548.4 | This study | — | — |
Two new mutations were found in RPE65. The first was in the splice site of RPE65 at position c.1129-2 and changed the canonical splice site second nucleotide to G in family F6 (Fig. 3B). The second mutation was identified in family F7 and was a new compound heterozygous mutations c.[325C > T];[569T > A] resulting in p.(R91W);(V172D) (Fig. 3C). The V172D mutation was not described in ExAC Browser9, 1000 genomes11, and HGMD10 databases; this mutation is predicted to be possibly damaging with a score of 0.721. In family F11 we identified several genomic homozygous regions, one of which included FAM161A. Sequence analysis and validation by Sanger sequencing showed a new homozygous deletion of four nucleotides GAAG in exon 3 (c.678_681delGAAG) (Fig. 3E) resulting in a frameshift and a termination codon at position p.K227Nfs*17. To our knowledge, this mutation has not previously been reported in the literature or as variation in databases such as 1000 genomes11 and ExAC. The last mutation was a 4-bp deletion, c.1133 + 3_1133 + 6delAAGT, located in the donor splice site of intron 8 of CERKL in family F12 (Fig. 3F). This new deletion was present at a homozygous state in the two affected sons and was heterozygous in the parents. It was not present in the unaffected sister. Furthermore, this deletion affects the invariant acceptor site of exon 8, which has not previously been linked to this particular RP phenotype. Minigene analysis confirmed that this variant has a strong impact on the normal splicing pattern of CERKL. Transfection of HEK 293 cells with minigene constructs supporting this change and its wild-type counterpart revealed that c.1133 + 3_1133 + 6delAAGT did affect the canonical splicing of exon 8 by knocking down its natural 3′ splice site (Fig. 4).
Figure 4. RT-PCR products with primers in exon 8 and 9.

(A) Schematic representation of normally spliced mRNA, (B) Aberrantly spliced mRNA. In the gel electrophoresis we can observe absence of splicing in the mutated patient (column M). In the WT sequence (column WT), splicing was not complete. *Non-spliced fragment, **spliced fragment.
As the fundus of affected F12 family members showed atrophy of the peripheral retina with no clumped pigmentation, we looked for this specific phenotype in families with simplex cases or where consanguinity was not evident. Fourteen additional Tunisian families were screened for this specific mutation. This allowed us to identify the homozygous c.1133 + 3_1133 + 6delAAGT deletion in three seemingly unrelated families (F13, F14, F15), but originating from the same geographical area. Genotype analysis of these three families showed a common homozygous region of 5.7 cM. This is indicative of an unknown consanguinity of these families (Table 4).
Table 4. Common homozygous genomic region.
| rs. | Distance position | F12 (VI-1) | F13 (V-5) | F14 (III-5) | F15 (II-5) |
|---|---|---|---|---|---|
| rs4667591 | −12.4 M | A/C | C/C | A/C | A/C |
| rs1050354 | −11.9 M | T/A | A/A | T/A | T/A |
| rs4894140 | −1.5 M | C/C | C/C | C/C | C/C |
| rs1143676 | −0.18 M | A/A | A/A | A/A | A/A |
| CERKL | 0 | — | — | — | — |
| rs3754929 | +0.67 M | T/T | T/T | T/T | T/T |
| rs1400130 | +1.43 M | G/G | G/G | G/G | G/G |
| rs12476147 | +3.3 M | T/T | T/T | T/T | T/T |
| rs17228441 | +4.2 M | C/C | C/C | C/C | C/C |
| rs1800255 | +7.4 M | G/A | G/A | G/A | G/A |
| rs1225090 | +8.1 M | G/T | T/T | G/T | G/T |
The mutations identified in the other families have already been reported in the literature. These changes were two nonsense substitutions: p.R203* in CNGB3 (NM_019098.4) in family F1 and p.W782* in ABCA4 (NM_000350.2) in family F3; two missense mutations: p.R311Q in NR2E3 (NM_014249.3) in F4 and p.H182Y in RPE65 (NM_000329.2) in F5, one frameshift mutation: c.1354dupT in PROM1 (NM_001145848.1) in F8, and one splice site mutation: c.5928-2A > G in EYS (NM_001292009.1) in F9. A summary of these results is presented in Table 3. Cosegregation analysis in each family showed that all affected individuals were homozygotes and unaffected individuals were either heterozygous carriers or homozygous for the wild type allele (Fig. 5).
Figure 5. Overview of the pedigree structure of the families and segregation analysis of disease causing variants in the RD cohort.

Arrows point to the probands. Affected individuals are indicated with filled symbols (blue), suspected (grey), whereas unaffected relatives are indicated by open symbols. +: wild type allele; −: mutation. All mutations are homozygous except for family F7.Genes with new mutations are in bold.
Discussion
Molecular diagnosis of RD is a challenging task given the high genetic heterogeneity of this group of diseases. Aiming to identify the molecular origin of RD in our Tunisian population, we performed molecular analysis of index cases from Tunisian families and revealed a great genetic variety among this population. Ten different genes were involved in 15 families. Previous studies in the Tunisian population have shown a high genetic heterogeneity in RP patients14 as well as in patients with Usher syndrome15. This could be explained by the degree of consanguinity observed in various parts of the country. The genes identified play different roles in the retina. Most genes mutated in this sub-cohort are a part of three major processes in rod photoreceptors and RPE. The first three genes, PDE6B, CNGB1 and CNGB3, are implicated in the phototransduction cascade: PDE6B, encoding the beta-subunit of rod cGMP-phosphodiesterase (cGMP-PDE), is a key enzyme of the retinal rod phototransduction cascade. PDE6B was reported by several groups as one of the causative gene associated with arRP, including previous studies of consanguineous Tunisian families16,17,18,19. Mutations in CNGB1 encoding the beta-subunit of the rod cGMP-gated channel explain about 4% of autosomal recessive non-syndromic RP4. We discovered a new p.R765C homozygous missense substitution in exon 23 of CNGB1 in F2. This mutation occurred at an evolutionarily conserved position from mammals to zebrafish and is predicted to damage the protein. So far, only eight point mutations were described with convincing evidence. Four of them were non-sense. Our result concurred with that of Bariel et al., who found mutation in CNGB1 in a consanguineous French family affected with severe autosomal recessive RP (arRP)20. CNGB3 encodes the β-subunit of the cone photoreceptor cGMP-gated channel, essential for phototransduction in all three classes of cones21. Mutations in this gene are a common cause of achromatopsia14. Missense mutations in CNGB3 have previously been reported in two individuals with cone–rod dystrophy and in a single individual with a progressive cone dystrophy phenotype22. Although the p.R203* mutation was previously associated with achromatopsia21, in family F1 it induced a more severe phenotype that was classified as EORD. In the past, CNGB3 was mainly screened in patients with achromatopsia in which it represents a common cause. With current high-throughput sequencing, most approaches analyze a large set of genes. This non-biased approach will most likely widen the genotype-phenotype correlation of most diseases23,24,25.
We also observed four families with homozygous mutations in the retinoid cycle genes. In ABCA4, a member of the ABC transporter superfamily associated with STGD disease26, CRD, RP27,28 as well as age-related macular degeneration29, a p.W782* was identified in family 3 with STGD phenotype. In RPE65, a vitamin A trans-cis isomerase, a p.H182Y was observed in family 5. This mutation was first reported by Morimura et al. as a compound heterozygous mutations in a single family with two boys affected with LCA30. Interestingly, this amino acid has also been mutated into Asparagine and Arginine as reported in Hanein et al. and Jacobson et al., respectively31,32. The p.H182N and p.H182R were associated with LCA31,32. Unfortunately, there are not enough families with mutations at H182 to establish a strong genotype-phenotype correlation for this position. Mutation analysis in the region of homozygosity in family F6 revealed a new mutation c.1129-2A > G that affects the consensus splice site of exon 11 of RPE65 and segregates with EORD. A compound heterozygous mutation p.(R91W);(V172D) was discovered in one family (F7). To date, a number of different RPE65 mutations have been reported in patients with retinal dystrophies classified as LCA, autosomal recessive EORD or arRP30,33,34. However, in our cohort we found three different RPE65 mutations in three different families that correlated with typical RP phenotype despite the early age of onset. Morimura et al., reported a family in which a child with LCA phenotype is the offspring of two parents with RP23. A recent Chinese study, including 88 candidate genes in 179 families with RD, confirmed that the average age at onset was 5 years in patients carrying mutation in RPE65 gene24. In patients from family F4 harboring the p.R311Q, the most prevalent mutation in the nuclear receptor subfamily 2, group E, member 3 (NR2E3), we observed typical RP with macular atrophy, as already reported35. Nevertheless, Gerber et al. have shown that the p.R311Q mutation could cause late-onset arRP in Crypto-Jewish population36. NR2E3 recessive mutations are usually associated with enhanced S-cone syndrome, an autosomal recessive retinopathy in which patients have increased sensitivity to the perception of blue light while the dominant p.G56R mutation is associated with severe RP21,22. A homozygous splice alteration in EYS (c.5928-2A > G) was reported in family F823. EYS is likely to play a role in the modeling of retinal architecture37. In previous studies, various mutations including point mutations, splice-site mutations and gross rearrangements were identified in patients with arRP23,38,39,40 and cone rod dystrophies41. The phenotype observed in family F8 resembles the RP phenotype reported by Barragan et al.38 in the Spanish population. The advanced age of onset of the disease could be correlated with our molecular result, as Huang et al. found a relationship between the advanced average age at onset and EYS mutation24. The mutation in EYS appears to be a frequent cause of arRP (15.9%) in the Spanish population28. Unfortunately, the prevalence of EYS mutations remains to be established in Tunisian population. Our preliminary analysis of the mutation load in the Tunisian population does not allow us to draw a similar conclusion. The fifteen families described here represent a selection that underwent whole-exome sequencing and thus do not represent a correct estimation of the yield of this approach.
In addition, a previously described variant (c.1354dupT) in PROM1, which encodes a pentaspan transmembrane glycoprotein that maintains the structural integrity of retina, was observed in patient III:1 from family F7 affected with RP42. Mutations in PROM1 are usually associated with RP or CRD, which is the case in this family43.
We also report the identification of a new small deletion in exon 3 of FAM161A, a ciliary gene previously associated with RP44. This mutation causes a shift in the reading frame resulting in a premature termination codon at position p.K227Nfs*17, presumably inhibiting protein production due to the action of the nonsense mediated mRNA decay. Most currently known mutations44,45 cluster in exon3 of FAM161A, which is by far the largest coding exon of this gene. FAM161A is member of the growing list of ciliary proteins implicated in human diseases. Phenotypes of ciliopathies are quite diverse and may involve impairment of multiple organs or functions such as kidney, brain, bones, obesity and vision46. However, in the patients investigated in this study we did not observe any other obvious features or anamnestic history of extraocular symptoms typically related to ciliopathies. The phenotype was typical of RP.
Finally, it is of note that the analysis of four families (F13, F14, F15 and F16) originating from the same Tunisian region revealed a new homozygous deletion c.1133 + 3_1133 + 6delAAGT in the Ceramide kinase Like (CERKL) gene in association with RP without pigmentation. Mutations in CERKL have been described in RP (RP26) together with significant macular involvement during the early stages of the disease47 and cone-rod dystrophy which progresses to an RP-like phenotype in advanced stage48. However, the exact function of CERKL remains unknown. Genotype analysis of these four families showed a common disease-associated haplotype and supports the hypothesis of a common ancestor in this area.
Despite intensive research and studies, it is still very difficult to screen for specific genes based on clinical observation and severity of disease. Molecular diagnosis of RD is a challenging task given the important genetic heterogeneity of this group of diseases.
Additional Information
How to cite this article: Habibi, I. et al. Identifying mutations in Tunisian families with retinal dystrophy. Sci. Rep. 6, 37455; doi: 10.1038/srep37455 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
We thank the family members for their invaluable participation and cooperation. We acknowledge the help provided by Dr. Leila Largueche in this study and the colleagues who referred patients to us. We thank Susan E Houghton for editing the manuscript, Celine Agosti and Cedric Schoepfer for technical assistance. This work was supported by the Institute for Research in Ophthalmology (IRO) and a grant from the Swiss government (Swiss Government Excellence Scholarships for Foreign Scholars and Artists).
The authors declare no competing financial interests.
Author Contributions D.F.S. and H.I. identified the mutations; A.C., Y.F., F.K. and L.E.M. referred patients and clinical data; D.F.S. and H.I. wrote the paper; D.F.S., A.C., Y.F. and H.I. prepared the figures; H.I. and N.A.P conducted the minigene experiments; D.F.S. and L.E.M designed the experiments. All authors reviewed and approved the manuscript.
05/04/2017
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has not been fixed in the paper.
References
- Holt R. et al. Identification of rod-and cone-specific expression signatures to identify candidate genes for retinal disease. Exp Eye Res 132, 161–173 (2015). [DOI] [PubMed] [Google Scholar]
- Berger W., Kloeckener-Gruissem B. & Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retina Eye Res 29, 335–375 (2010). [DOI] [PubMed] [Google Scholar]
- Weleber R. G. Stargardt’s macular dystrophy. Arch Ophthalmol 112, 752–754 (1994). [DOI] [PubMed] [Google Scholar]
- Hartong D. T., Berson E. L. & Dryja T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006). [DOI] [PubMed] [Google Scholar]
- den Hollander A. I., Roepman R., Koenekoop R. K. & Cremers F. P. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retina Eye Res 27, 391–419 (2008). [DOI] [PubMed] [Google Scholar]
- Simunovic M. P. & Moore A. The cone dystrophies. Eye 12, 553–565 (1998). [DOI] [PubMed] [Google Scholar]
- Wright A. F., Chakarova C. F., El-Aziz M. M. A. & Bhattacharya S. S. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nature Rev Genet 11, 273–284 (2010). [DOI] [PubMed] [Google Scholar]
- Schorderet D. F., Iouranova A., Favez T., Tiab L. & Escher P. IROme, a new high-throughput molecular tool for the diagnosis of inherited retinal dystrophies. Biomed Res Int 2012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper D. N., Ball E. V. & Krawczak M. The human gene mutation database. Nucleic Acids Res 26, 285–287 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium G. P. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I. A. et al. A method and server for predicting damaging missense mutations. Nature methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P. C. & Henikoff S. Predicting deleterious amino acid substitutions. Genome Res 11, 863–874 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebil A. et al. Genotype-phenotype correlation in ten Tunisian families with non-syndromic retinitis pigmentosa. J Fr Ophtalmol 39, 277–286 (2016). [DOI] [PubMed] [Google Scholar]
- Riahi Z. et al. Whole exome sequencing identifies mutations in Usher syndrome genes in profoundly deaf Tunisian patients. PLoS One 10, e0120584 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin M. E., Ehrhart T. L., Berson E. L. & Dryja T. P. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. P Natl Acad Sci USA 92, 3249–3253 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hmani-Aifa M. et al. Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family. Eur J Hum Genet 17, 474–482 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocquet B. et al. Homozygosity mapping in autosomal recessive retinitis pigmentosa families detects novel mutations. Mol Vis 19, 2487 (2013). [PMC free article] [PubMed] [Google Scholar]
- Tsang S. H. et al. A novel mutation and phenotypes in phosphodiesterase 6 deficiency. Am J Ophthalmol 146, 780–788. e781 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareil C. et al. Segregation of a mutation in CNGB1 encoding the β-subunit of the rod cGMP-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum Genet 108, 328–334 (2001). [DOI] [PubMed] [Google Scholar]
- Kohl S. et al. Mutations in the CNGB3 gene encoding the β-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet 9, 2107–2116 (2000). [DOI] [PubMed] [Google Scholar]
- Wissinger B. et al. CNGA3 mutations in hereditary cone photoreceptor disorders. P Natl Acad Sci USA 69, 722–737 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-del Pozo M. et al. Mutation screening of multiple genes in Spanish patients with autosomal recessive retinitis pigmentosa by targeted resequencing. PLoS One 6, e27894–e27894 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.-F. et al. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet Med 17, 271–278 (2014). [DOI] [PubMed] [Google Scholar]
- Littink K. W. et al. Homozygosity mapping in patients with cone–rod dystrophy: novel mutations and clinical characterizations. Invest Ophthalmol Vis Sci 51, 5943–5951 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets R. et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 15, 236–246 (1997). [DOI] [PubMed] [Google Scholar]
- Cremers F. P. et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Gen 7, 355–362 (1998). [DOI] [PubMed] [Google Scholar]
- Downes S. M. et al. Detection rate of pathogenic mutations in ABCA4 using direct sequencing: clinical and research implications. Arch Ophthalmol 130, 1486–1490 (2012). [DOI] [PubMed] [Google Scholar]
- Allikmets R. et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 277, 1805–1807 (1997). [DOI] [PubMed] [Google Scholar]
- Morimura H. et al. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci USA 95, 3088–3093 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson S. G. et al. Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc Natl Acad Sci USA 102, 6177–6182 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanein S. et al. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype–phenotype correlations as a strategy for molecular diagnosis. Hum Mutat 23, 306–317 (2004). [DOI] [PubMed] [Google Scholar]
- Marlhens F. et al. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat Genet 17, 139–141 (1997). [DOI] [PubMed] [Google Scholar]
- Gu S.-m. et al. Mutations in RPE65 cause autosomal recessive childhood–onset severe retinal dystrophy. Nat Genet 17, 194–197 (1997). [DOI] [PubMed] [Google Scholar]
- Hayashi T. et al. Novel NR2E3 mutations (R104Q, R334G) associated with a mild form of enhanced S-cone syndrome demonstrate compound heterozygosity. Ophthalmology 112, 2115. e2111-2115. e2110 (2005). [DOI] [PubMed] [Google Scholar]
- Gerber S. et al. The photoreceptor cell-specific nuclear receptor gene (PNR) accounts for retinitis pigmentosa in the Crypto-Jews from Portugal (Marranos), survivors from the Spanish Inquisition. Hum Genet 107, 276–284 (2000). [DOI] [PubMed] [Google Scholar]
- Zelhof A. C., Hardy R. W., Becker A. & Zuker C. S. Transforming the architecture of compound eyes. Nature 443, 696–699 (2006). [DOI] [PubMed] [Google Scholar]
- Barragán I. et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum Genet 31, E1772 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Aziz M. M. A. et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet 40, 1285–1287 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin R. W. et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am J Hum Genet 83, 594–603 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audo I. et al. EYS is a major gene for rod‐cone dystrophies in France. Hum Mutat 31, E1406–E1435 (2010). [DOI] [PubMed] [Google Scholar]
- Pras E. et al. Cone-rod dystrophy and a frameshift mutation in the PROM1 gene. Mol Vis 15, 1709 (2009). [PMC free article] [PubMed] [Google Scholar]
- Permanyer J. et al. Autosomal recessive retinitis pigmentosa with early macular affectation caused by premature truncation in PROM1. Invest Ophthalmol Vis Sci 51, 2656 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmann T. et al. Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa. Am J Hum Genet 87, 376–381 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandah-Rozenfeld D. et al. Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa. Am J Hum Genet 87, 382–391 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockel A. et al. Retinal dystrophy in Bardet–Biedl syndrome and related syndromic ciliopathies. Prog Retin Eye Res 30, 258–274 (2011). [DOI] [PubMed] [Google Scholar]
- Ali M. et al. A missense mutation (p. R106S) in the nuclear localization signal sequence of CERKL causes autosomal recessive retinal degeneration. Mol Vis 14, 1960–1964 (2008). [PMC free article] [PubMed] [Google Scholar]
- Aleman T. S. et al. CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Invest Ophthalmol Vis Sci 50, 5944–5954 (2009). [DOI] [PubMed] [Google Scholar]