

ABSTRACT
The immune-modulating properties of certain bifidobacterial strains, such as Bifidobacterium longum subsp. longum 35624 (B. longum 35624), have been well described, although the strain-specific molecular characteristics associated with such immune-regulatory activity are not well defined. It has previously been demonstrated that B. longum 35624 produces a cell surface exopolysaccharide (sEPS), and in this study, we investigated the role played by this exopolysaccharide in influencing the host immune response. B. longum 35624 induced relatively low levels of cytokine secretion from human dendritic cells, whereas an isogenic exopolysaccharide-negative mutant derivative (termed sEPSneg) induced vastly more cytokines, including interleukin-17 (IL-17), and this response was reversed when exopolysaccharide production was restored in sEPSneg by genetic complementation. Administration of B. longum 35624 to mice of the T cell transfer colitis model prevented disease symptoms, whereas sEPSneg did not protect against the development of colitis, with associated enhanced recruitment of IL-17+ lymphocytes to the gut. Moreover, intranasal administration of sEPSneg also resulted in enhanced recruitment of IL-17+ lymphocytes to the murine lung. These data demonstrate that the particular exopolysaccharide produced by B. longum 35624 plays an essential role in dampening proinflammatory host responses to the strain and that loss of exopolysaccharide production results in the induction of local TH17 responses.
IMPORTANCE Particular gut commensals, such as B. longum 35624, are known to contribute positively to the development of mucosal immune cells, resulting in protection from inflammatory diseases. However, the molecular basis and mechanisms for these commensal-host interactions are poorly described. In this report, an exopolysaccharide was shown to be decisive in influencing the immune response to the bacterium. We generated an isogenic mutant unable to produce exopolysaccharide and observed that this mutation caused a dramatic change in the response of human immune cells in vitro. In addition, the use of mouse models confirmed that lack of exopolysaccharide production induces inflammatory responses to the bacterium. These results implicate the surface-associated exopolysaccharide of the B. longum 35624 cell envelope in the prevention of aberrant inflammatory responses.
INTRODUCTION
The gut microbiota contributes significantly to host health via multiple mechanisms, including the digestion of foods, competitive exclusion of pathogens, enhancement of epithelial cell differentiation, and promotion of mucosa-associated lymphoid tissue proliferation (1, 2). Furthermore, accumulating evidence suggests that the composition and metabolic activity of the gut microbiota has profound effects on proinflammatory activity and the induction of immune tolerance within mucosal tissue (3–5). Certain microbes induce regulatory responses, while others induce effector responses, resulting in the case of healthy individuals in a balanced homeostatic immunological state that protects against infection and controls aberrant, tissue-damaging inflammatory responses (6).
One bacterial strain that is known to induce tolerogenic responses within the gut is Bifidobacterium longum subsp. longum 35624 (7). Induction of T regulatory (Treg) cells by the B. longum 35624 strain in mice is associated with protection against colitis, arthritis, allergic responses, and pathogen-associated inflammation (8–12). Administration of this bacterium to humans increases Foxp3+ lymphocytes in peripheral blood, enhances interleukin-10 (IL-10) secretion ex vivo, and reduces the levels of circulating proinflammatory biomarkers in a wide range of patient groups (13, 14). A number of host mechanisms that contribute to the anti-inflammatory activity of this microbe have been described, including Toll-like receptor 2 (TLR-2) and dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) recognition and retinoic acid release by dendritic cells (13, 15–17). However, the bacterial-strain-specific structural and/or metabolic factors that contribute to these protective immune responses have as yet remained elusive.
A number of different exopolysaccharides (EPS) from gut microbes have been shown to induce immune-modulatory effects. Polysaccharide A (PSA) from Bacteroides fragilis mediates the conversion of naive CD4+ T cells into Foxp3+ Treg cells that produce IL-10 during commensal colonization. Functional Treg cells are also induced by PSA during intestinal inflammation, which requires TLR-2 signaling (18). Further studies have reported that PSA interacts directly with mouse plasmacytoid dendritic cells via TLR-2 and that PSA-exposed plasmacytoid dendritic cells express molecules involved in protection against colitis and stimulate CD4+ T cells to secrete IL-10 (19). An exopolysaccharide from Bacillus subtilis prevents gut inflammation stimulated by Citrobacter rodentium, which is dependent on TLR-4 and MyD88 signaling (20). Similarly, protection against C. rodentium infection by Bifidobacterium breve UCC2003 was dependent on the presence of its exopolysaccharide (21). Furthermore, it was described that an extracellular polymeric matrix, isolated from Faecalibacterium prausnitzii, displayed anti-inflammatory activity in the mouse dextran sodium sulfate colitis model (22).
We recently described that the B. longum 35624 strain-specific EPS gene cluster, designated the EPS624 cluster, is responsible for the production of a cell surface-associated exopolysaccharide (sEPS) composed of a branched hexasaccharide repeating unit with two galactoses, two glucoses, galacturonic acid, and the unusual sugar 6-deoxytalose (23). The overall aim of the current study was to determine whether the exopolysaccharide produced by B. longum 35624 is related to the immunoregulatory effects of this microorganism. To address this aim, we investigated whether an isogenic derivative of B. longum 35624 that does not produce exopolysaccharide, designated sEPSneg, is able to exert immunological effects similar to those of its parent strain in vitro and in colitis and asthma mouse models.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are detailed in Table 1. Bifidobacteria were routinely cultured in either de Man Rogosa Sharpe medium (MRS; Oxoid Ltd., Basingstoke, United Kingdom) supplemented with 0.05% cysteine–HCl or reinforced clostridial medium (RCM; Oxoid Ltd.). Bifidobacterial cultures were incubated at 37°C under anaerobic conditions in a Don Whitley anaerobic chamber. Escherichia coli strains were cultured in lysogeny broth (LB; Oxoid Ltd.) at 37°C with agitation. Where appropriate, the growth medium contained chloramphenicol (Cm; 10 μg ml−1 for E. coli and 5 μg ml−1 for B. longum 35624), erythromycin (Em; 100 μg ml−1 for E. coli), tetracycline (Tet; 10 μg ml−1 for E. coli and 10 μg ml−1 for B. longum 35624), ampicillin (Amp; 100 μg ml−1 for E. coli), or kanamycin (Km; 50 μg ml−1 for E. coli). All antibiotics were obtained from Sigma-Aldrich (England). The commercially available B. longum 35624 culture was provided by Alimentary Health Limited (Cork, Ireland).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Reference or source |
|---|---|---|
| Strains | ||
| E. coli EC101 | Cloning host, repA negative, Kmr | 26 |
| E. coli XL1 Blue | Cloning host, Tetr | Stratagene |
| E. coli EC101/pNZ-M.1185 | E. coli EC101 harboring pNZ-M.1185 | This study |
| B. longum 35624 | Parent strain | Alimentary Health |
| sEPSneg | B. longum 35624 harboring insertion mutation in priming glycosyl transferase-encoding gene pgt624 | This study |
| sEPScomp | sEPSneg harboring pBC1.2_pgt624+BI0343 | This study |
| Plasmids | ||
| pNZEm | Gene expression vector, Emr | 55 |
| pORI19 | Emr, repA negative, ori+, cloning vector | 26 |
| pORI19tet(W)_pgt | pORI19 harboring internal fragment of BI0342 (pgt) and TetW gene | This study |
| pBC1.2 | E. coli-Bifidobacterium shuttle vector | 27 |
| pBC1.2_pgt624+BI0343 | pBC1.2 harboring the cotranscribed genes pgt624 and BI0343 under the control of their native promoter | This study |
| pNZ-M.1185 | BI1185 (M.1185, isoschizomer of M.EcoRII) cloned with its own promoter in pNZEM | This study |
DNA manipulations.
Chromosomal DNA was isolated from bifidobacteria as previously described (24). Preparation of plasmid DNA from E. coli or B. longum 35624 was achieved using the QIAprep spin plasmid miniprep kit (Qiagen GmBH, Hilden, Germany). For B. longum 35624, an initial lysis step was incorporated into the plasmid isolation procedure, and cells were resuspended in lysis buffer supplemented with lysozyme (30 mg ml−1) and incubated at 37°C for 30 min. Restriction enzymes and T4 DNA ligase were used according to the supplier's instructions (Roche Diagnostics, United Kingdom). The synthetic single-stranded oligonucleotide primers used in this study were obtained from Eurofins (Ebersberg, Germany) and are detailed in Table 2. Standard PCRs were performed using Taq PCR mastermix (Qiagen), while high-fidelity PCR was achieved using PfuII polymerase (Agilent, Santa Clara, CA). B. longum 35624 colony PCRs were performed according to standard procedures with the addition of an initial incubation step of 95°C for 5 min to perform cell lysis. PCR fragments were purified using the Qiagen PCR purification kit. Following electroporation of plasmid DNA into E. coli strain EC101, electrotransformation of B. longum 35624 or sEPSneg was performed essentially as described by Maze et al. (25) with the following modifications. An overnight culture of B. longum 35624 was subcultured twice (first using a 2% inoculum and then a 1% inoculum) in MRS medium supplemented with 0.05% cysteine–HCl and 0.2 M sucrose prior to inoculating (4%) modified Rogosa medium supplemented with 0.05% cysteine–HCl, 1% (wt/vol) glucose, and 0.2 M sucrose. Bacteria were grown until the optical density at 600 nm (OD600) had reached 0.3 to 0.4, after which cells were harvested by centrifugation (6,500 rpm for 10 min at 4°C) and washed twice using 1 mM ammonium citrate buffer (pH 6.0) supplemented with 0.5 M sucrose. An additional centrifugation step (9,800 × g for 10 min at 4°C) was included to concentrate the competent cells. For electroporation, 5 μl of plasmid DNA was mixed with 50 μl of competent cells, transferred into an electroporation cuvette with a 0.2-cm interelectrode distance, and pulsed at 2.5 kV, 25 μF, and 200 Ω using a Gene Pulser II electroporation system (Bio-Rad, Hercules, CA). For recovery, 800 μl of RCM supplemented with 0.05% l-cysteine hydrochloride was added to the bacteria and they were incubated anaerobically for 2.5 h at 37°C. Transformants were plated on reinforced clostridial agar (RCA) plates supplemented with appropriate concentrations of relevant antibiotics and incubated for 2 to 3 days anaerobically at 37°C. The correct orientation and integrity of all constructs was verified by PCR and subsequent DNA sequencing, which was performed at Eurofins (Ebersberg, Germany).
TABLE 2.
Oligonucleotide primers used in this study
| Purpose | Primer | Sequencea |
|---|---|---|
| Cloning of M.1185 in pNZEm | BI1185F_PstI | GACTGCAGGCCCACTAGGTAACCAAACG |
| BI1185R_SpeI | GCGCACTAGTCTAGAGCAAAGCCAGTATAG | |
| Cloning of internal 583-bp fragment of pgt in pORI19 | BI0342F_HindIII | GATAAGCTTGCGTCGGCAACTCAACTACC |
| BI0342R_XbaI | GATTCTAGACGTCGGCGTTCACTACCATC | |
| Cloning of pgt624 and BI0343 in pBC1.2 | BI0342FSalI | GACGTCGACACTCCACTCTCGCTGATCG |
| BI0343EcoRI | GGCGAATTCTAATCAACCAAGGGGGTCTG |
Restriction sites incorporated into oligonucleotide primer sequences are indicated in boldface.
Construction of sEPSneg.
An internal 583-bp fragment of the pgt624 gene was amplified by PCR using B. longum 35624 chromosomal DNA as a template and the oligonucleotide primers BI0342F_HindIII and BI0342R_XbaI. The PCR product generated was ligated to pORI19, an Ori+ RepA− integration plasmid (26), using the unique HindIII and XbaI restriction sites that were incorporated into the primers for the pgt fragment-encompassing amplicon, and the plasmid introduced into E. coli EC101 by electroporation. Recombinant E. coli EC101 derivatives containing pORI19 constructs were selected on LB agar containing Em and supplemented with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (40 μg ml−1) and IPTG (isopropyl-β-D galactopyranoside) (1 mM). The expected genetic structure of the recombinant plasmid, designated pORI19-pgt, was confirmed by restriction mapping prior to subcloning of the Tet resistance antibiotic cassette, tet(W), from pAM5 (27) as a SacI fragment into the unique SacI site on pORI19-pgt. The expected structure of a single representative of the resulting plasmid, designated pORI19tet(W)_pgt, was confirmed by restriction analysis. The plasmid was introduced into E. coli EC101 harboring pNZ-M.1185 (this is a plasmid expressing a B. longum 35624-encoded methylase) by electroporation, and transformants were selected based on Em and Tet resistance. Methylation of the resulting plasmid complement of such transformants by M.1185 (isoschizomer of M.EcoRII) was confirmed by their observed resistance to EcoRII restriction. Plasmid preparations of methylated pORI19tet(W)_pgt were introduced by electroporation into B. longum 35624 with subsequent selection on RCA plates supplemented with Tet.
Construction of sEPScomp.
For the construction of plasmid pBC1.2_pgt624+BI0343, a DNA fragment encompassing pgt624 plus the gene with locus tag BI0343 located downstream and the presumed promoter region, was generated by PCR amplification from chromosomal DNA of B. longum 35624 using PfuII polymerase and primer combinations BI0342FSalI and BI0343EcoRI, where SalI or EcoRI restriction sites were incorporated at the 5′ ends of the forward primer and reverse primer, respectively. Amplicons were digested with SalI and EcoRI and ligated into similarly digested pBC1.2 prior to introduction into E. coli XL1 Blue by electroporation and subsequent selection of transformants on LB agar supplemented with Tet and Amp. The integrity of positive clones was confirmed by sequencing, and one selected clone, designated pBC1.2 pgt+BI0343, was introduced into sEPSneg by electroporation with subsequent selection of transformants on RCA supplemented with Tet and Cm. The resultant complemented sEPSneg strain, harboring pBC1.2_pgt+BI0343, was designated sEPScomp.
Electron microscopy.
After culture in MRS medium, bacteria were gently rinsed in PIPES (piperazine-N,N-bis-2-ethane sulfonic acid) buffer (0.1 M, pH 7.4) before being fixed in 2.5% glutaraldehyde in PIPES buffer for 5 min. The samples were rinsed twice (2 min each time) in PIPES buffer and postfixed with 1% osmium tetroxide in 0.1 M PIPES buffer (pH 6.8) for 60 min in the dark. The samples were rinsed three times in double-distilled water (2 min each wash) before being dehydrated through an ethanol series (50, 70, 96, and 100%) for 5 min each step. All fixation and washing steps were carried out at room temperature. Following dehydration, the samples were critically point dried in a Polaron E3100 critical point drier (Agar Scientific, Stansted, United Kingdom) and coated with 10 nm of gold/palladium (80/20) using a Baltec MED 020 unit (Baltec, Buchs, Liechtenstein). Bacterial preparations were examined using a Hitachi S-4700 scanning electron microscope (SEM) operated in secondary electron detection mode (3 kV, 40 μA), and images captured with Quartz PCI (Quartz Imaging Corporation, Vancouver, Canada).
Exopolysaccharide isolation.
The exopolysaccharide was isolated as previously described (23). Briefly, following harvesting of B. longum 35624 cells, which were grown on agar plates to minimize carryover of medium components, an exopolysaccharide solution was generated by agitating the cells in phosphate-buffered saline (PBS). The harvested exopolysaccharide solution was mixed with ethanol, and the exopolysaccharide aggregated at the center of the surface of the ethanol solution, which facilitated harvesting of the exopolysaccharide without the need for centrifugation. The exopolysaccharide aggregations were taken with a spatula, resuspended in water and dialyzed against water to remove contaminants and residual ethanol. The exopolysaccharide was applied 2 times on Bakerbond SPE C18 columns (Avantor, Deventer, The Netherlands) as indicated by the manufacturer using a HyperSep-96 vacuum manifold (Thermo Scientific, Waltham, MA). The flowthrough fraction was collected and filtered through 0.45-μm syringe filters. Quantification of total carbohydrate levels was performed as previously described (28) using a phenol-sulfuric acid method in microplate format. The absence of contaminating proteins was confirmed by measuring the total soluble protein content of the exopolysaccharide preparation using the bicinchoninic acid (BCA) protein quantification kit (Thermo Scientific) according to the manufacturer's instructions. Bovine serum albumin was used to generate standards. Lipopolysaccharide (LPS) contamination was monitored using the PyroGene recombinant factor C assay (Lonza, Bettlach, Switzerland).
In vitro immune assays.
Human blood was purchased from the Swiss blood bank (Blutspendezentrum, Basel, Switzerland), which obtains the blood following appropriate screening and with the consent of volunteers. Blood samples were anonymized and coded prior to leaving the blood bank. Research procedures on human blood were performed in accordance with Swiss law (ethical approval number KEK Nr. 19/08). All experiments with human blood-derived cells were conducted under biosafety level 2 conditions. Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors using density gradient centrifugation. Human monocyte-derived dendritic cells (MDDCs) were differentiated with 1,000 IU ml−1 granulocyte-macrophage colony-stimulating factor (GM-CSF; Peprotech, London, United Kingdom) and 1,000 IU ml−1 IL-4 (Novartis, Basel, Switzerland) from purified CD14+ cells using magnetically activated cell sorting (MACS) separation (Miltenyi Biotec, Bergisch Gladbach, Germany). Bacterial strains for in vitro assays were cultured in MRS medium supplemented with 0.05% l-cysteine for 48 h under anaerobic conditions at 37°C. Cells were harvested and washed once with sterile PBS by centrifugation at 6,500 rpm for 10 min. Bacterial cell numbers were determined by microscopy using a Petroff-Hausser chamber, and bacteria were diluted as appropriate in PBS for incubation with human cells. PBMCs and MDDCs were stimulated for 24 h at 37°C, 5% CO2 with bacterial strains at a concentration of 50 bacterial cells to 1 PBMC or MDDC. Human and bacterial cell cocultures were performed in complete RPMI 1640 (cRPMI) medium (Sigma, Buchs, Switzerland) supplemented with 10% fetal bovine serum (Sigma), penicillin (100 U ml−1), and streptomycin (0.1 mg ml−1) (Sigma). Purified exopolysaccharide from B. longum 35624 was also added (final concentration, 100 μg/ml) to PBMC cultures (in duplicate) stimulated with sEPSneg. Cytokine concentrations were measured using the Bio-Plex multiplex system (Bio-Rad). For human dendritic cell staining, the following antibodies were used: phycoerythrin (PE)-Cy7 anti-human CD274 antibody (programmed death ligand-1 [PD-L1]), allophycocyanin (APC) anti-human CD273 antibody (PD-L2), and Pacific blue anti-human CD11c antibody (eBioscience, Vienna, Austria). The THP-1-Blue NF-κB monocyte cell line (Invivogen, San Diego, CA) was maintained and subcultured in cRPMI medium (Sigma) in the presence of 200 μg/ml Zeocin (Invivogen). For the coculture experiment with bacteria, 105 cells/well were seeded in a 96-well plate in a total volume of 200 μl/well of cRPMI medium. The cells were stimulated over a range of different bacterial concentrations for 24 h, and activation of the NF-κB/AP-1 pathway was evaluated with the Quanti-Blue assay according to the manufacturer's instructions. In addition, MDDCs were stimulated with different bacterial strains, and NF-κB phosphorylation measured over a time course. MDDCs were lysed using the Bio-Plex Pro cell signaling reagent kit (Bio-Rad), and cell lysates were stored at −80°C until analysis. Protein concentrations were determined using Bio-Rad's DC protein assay, and equal amounts of protein were used to measure NF-κB p65 (Ser536) in the Bio-Plex Pro magnetic cell signaling assay (Bio-Rad). Results are expressed as mean fluorescence intensity (MFI).
T cell transfer colitis model.
C.B-17 severe combined immunodeficient (SCID) and BALB/c female mice (8 to 12 weeks of age) were obtained from Charles River (Sulzfeld, Germany) and maintained under specific-pathogen-free conditions. The animals were housed at the AO Research Institute, Davos, Switzerland, in individually ventilated cages for the duration of the study, and all experimental procedures were carried out in accordance with Swiss law (permit number 2013_32). Colitogenic CD4+ CD25− CD45RBhi cells were isolated from BALB/c donor mouse spleens using the MACS Miltenyi system (depletion of CD4+ CD25+ cells, followed by positive selection of CD45RB fluorescein isothiocyanate [FITC]-labeled cells). At day 0, colitis was induced by intraperitoneal transfer of 4 × 105 cells per C.B-17 SCID mouse (8 mice per group). Bacterial cells were prepared as described above and counted using microscopy (Petroff-Hausser chamber) prior to dilution in sterile PBS. An amount of 1 × 109 B. longum 35624 cells or of one of its isogenic derivatives was administered to each mouse by intragastric gavage (total volume of 200 μl). Bacteria were gavaged from the beginning of the study (day 0) and continued to be gavaged every second day until animals were euthanized at the end of the study. Sixteen days after study initiation, disease severity scores were recorded, while animal weights were monitored every day. Disease severity scores included the condition of feces (1, wet; 2, diarrhea; and 3, bloody diarrhea or rectal prolapse), activity (1, isolated, abnormal position; 2, huddled, hypoactive or hyperactive; and 3, unconscious), coordination of movement (1, slightly uncoordinated; 2, very uncoordinated; and 3, paralysis), and fur quality (1, reduced grooming; 2, disheveled; and 3, hair loss). Gut transit was determined by quantifying fecal B. longum 35624 levels by PCR. B. longum 35624-specific primers were designed using Primer3 software (http://simgene.com/Primer3). The primers, designated 2420t (forward, CAG TGG GGT GCG ACT ACA, and reverse, GCG CGA ACC AGA AGA TGT), generated a 494-bp amplicon. Bacterial DNA from fecal samples was extracted using the QIAamp DNA stool minikit (Qiagen). DNA was quantified using NanoDrop (Thermo Scientific), and 100 ng of total DNA was assayed using SYBR green PCR master mix (Bio-Rad). The thermal cycling conditions consisted of an initial denaturation step of 15 min at 95°C, followed by 30 cycles of denaturation at 94°C for 45 s, annealing for 45 s at 56°C, and extension at 72°C for 45 s. B. longum 35624 DNA concentrations were quantified using the absolute quantitation protocol of the ABI 7900 fast real-time PCR system (Applied Biosystems). In a parallel experiment, healthy BALB/c mice (6 mice per group) were gavaged with B. longum 35624 or its isogenic derivative for 3 weeks as described above for the colitis study.
Following euthanasia on day 26, mesenteric lymph nodes were isolated in order to obtain single-cell suspensions. Lymph nodes were mechanically disrupted using a syringe plunger to grind the nodes on a nylon cell strainer (70 μm). The strainer was washed with PBS, and the single-cell suspensions were centrifuged at 300 × g for 10 min. Cell pellets were resuspended in 1 ml of cRPMI medium (Sigma), and cells were counted using a Scepter cell counter (Millipore, Billerica, MA). Cells were diluted to a final density of 1 × 106 cells ml−1 in cRPMI, and cells were dispensed in propylene tubes for fluorescence-activated cell sorting (FACS) staining. Following removal of epithelial cells and collagenase VIII/DNase I (Roche) digestion of the tissue, lamina propria mononuclear cells were isolated as described previously (29). At the end of the process, cells were counted using the Scepter cell counter (Millipore) and diluted to a final concentration of 1 × 106 cells ml−1 in cRPMI in propylene FACS staining tubes.
OVA respiratory allergy model.
Female BALB/c mice (8 to 12 weeks of age) were obtained from Charles River and were maintained under specific-pathogen-free conditions at the AO Research Institute, Davos, Switzerland, in individually ventilated cages for the duration of the study. All experimental procedures were carried out in accordance with Swiss law (permit number 2013_20). Eight mice per group were used in this model. Three intraperitoneal immunizations with 50 μg of ovalbumin (OVA, grade V > 98%; Sigma) emulsified in Imject alum adjuvant (Life Technologies, Carlsbad, CA) were performed on days 0, 14, and 21, followed by OVA aerosol challenges on days 26, 27, and 28. On days 19, 25, and 27, mice received B. longum 35624 or sEPSneg intranasally (∼1 × 109 bacteria per dose in a total volume of 50 μl of PBS). Bacterial cells were prepared as described above. Control animals received three intraperitoneal injections with alum adjuvant (without OVA) on days 0, 14, and 21, followed by OVA aerosol challenges on days 26, 27, and 28. Control animals also received 50 μl of PBS intranasally on days 19, 25, and 27. All mice were sacrificed at day 29 for isolation of lung tissue and flow cytometric staining. Lung-derived single-cell suspensions were obtained using a combination of enzymatic digestion (lung dissociation kit, Miltenyi) and mechanical dissociation with a gentleMACS dissociator (Miltenyi) according to the manufacturer's protocol. Lung cells were plated at 1 × 106 cells/ml in complete RPMI (Sigma) and stimulated ex vivo with 50 μg/ml OVA, grade VI (Sigma) or with 500 ng/ml LPS (Sigma) for 48 h, and cytokine secretion quantified by using the Bio-Plex multiplex system (Bio-Rad).
Flow cytometry.
All flow cytometry analyses were performed on the Gallios flow cytometer (Beckman Coulter, La Brea, CA). Mesenteric lymph node or lung single-cell suspensions were stimulated with phorbol myristate acetate (PMA) and ionomycin at 50 ng ml−1 and 500 ng ml−1, respectively, for 4 h in the presence of brefeldin A (eBioscience). The viability dye eFluor780 (eBioscience) and the following surface-staining antibodies were used: PE-Cy7 anti-mouse CD3 antibody and Pacific blue anti-mouse CD4 antibody (BioLegend, San Diego, CA). Cells were stained for intracellular cytokines using PE anti-mouse IL-10 antibody, Alexa Fluor 488 anti-mouse/rat IL-17A antibody, and peridinin chlorophyll protein (PerCP)-Cy5.5 anti-mouse interferon (IFN-γ) antibody after fixation and permeabilization (intracellular fixation and permeabilization buffer set; eBioscience). Lamina propria cells were in addition stained for the gut-homing molecule CCR9 using Alexa Fluor 647 anti-mouse CD199 antibody (CCR9) from BioLegend.
Statistical analysis.
Unless otherwise indicated, data are presented as box-and-whisker plots with the median values and maximum/minimum values illustrated. In experiments with technical replicates, the mean was calculated from the technical replicates for each donor and only the mean value was used for the statistical analysis. The Mann-Whitney U test was used for the nonparametric statistical analysis of differences between two groups. For analysis of more than two groups, statistical significance was determined using the Kruskal-Wallis test and Dunn's multiple-comparison test. A two-way analysis of variance (ANOVA) was used to compare groups over time. A P value of less than 0.05 was considered statistically significant.
RESULTS
Generation of an isogenic exopolysaccharide-negative derivative of B. longum 35624, designated sEPSneg, by insertion mutagenesis.
In order to determine the role, if any, of the exopolysaccharide in the reported immunomodulatory activities of the B. longum 35624 strain, we set out to generate an isogenic derivative of this strain that was unable to produce this cell surface-associated glycan exopolymer. For this purpose, we employed a mutagenesis strategy that was based on previously described methods (30). The particular mutagenesis strategy employed for B. longum 35624 involved the heterologous expression of a B. longum 35624-encoded DNA methylase in E. coli strain EC101 so as to methylate any plasmid DNA in the latter cloning host. When such methylated plasmid DNA is subsequently introduced into B. longum 35624, it will be protected from digestion by the native restriction-modification systems encoded by the latter strain and will therefore allow homology-guided, site-directed mutagenesis as has been described previously (30). Employing this strategy, we created an insertion mutation in the first gene of the EPS624 cluster, i.e., pgt624, encoding the predicted priming glycosyl transferase (pGT), resulting in an isogenic derivative of B. longum 35624 that was designated sEPSneg.
In order to assess whether the sEPSneg mutant had, as would be expected, lost its ability to produce exopolysaccharide, we performed electron microscopy analysis, which indeed revealed that the “stringy”-appearing sEPS layer present on the parent strain B. longum 35624 is absent on sEPSneg, thus confirming its exopolysaccharide-deficient phenotype (Fig. 1). Furthermore, and in contrast to the parent strain B. longum 35624, the sEPSneg strain exhibits a so-called dropping phenotype when grown in liquid medium (i.e., the sEPSneg strain was found to sediment during growth in liquid medium, but the B. longum 35624 strain remained in suspension). A similar phenotype was observed for exopolysaccharide-negative variants of B. breve UCC2003 and Bifidobacterium animalis subsp. lactis (21, 31), thereby substantiating the loss of exopolysaccharide production. To ensure that the observed phenotype is directly linked to the inactivation of pgt624, the adjacent and cotranscribed genes pgt624 and BI0343 were cloned together with the presumed promoter sequence in plasmid pBC1.2, after which the resultant construct was introduced into the sEPSneg strain. The resulting strain, designated sEPScomp, was shown to produce exopolysaccharide (Fig. 1). A similar complementation approach was described previously (32, 33). The sEPSneg and sEPScomp mutants grew more slowly than the parent strain B. longum 35624, likely due to the presence of antibiotics in their culture medium, but by 38 h of culture, all bacteria were at similar numbers and had reached stationary phase (see Fig. S1 in the supplemental material).
FIG 1.
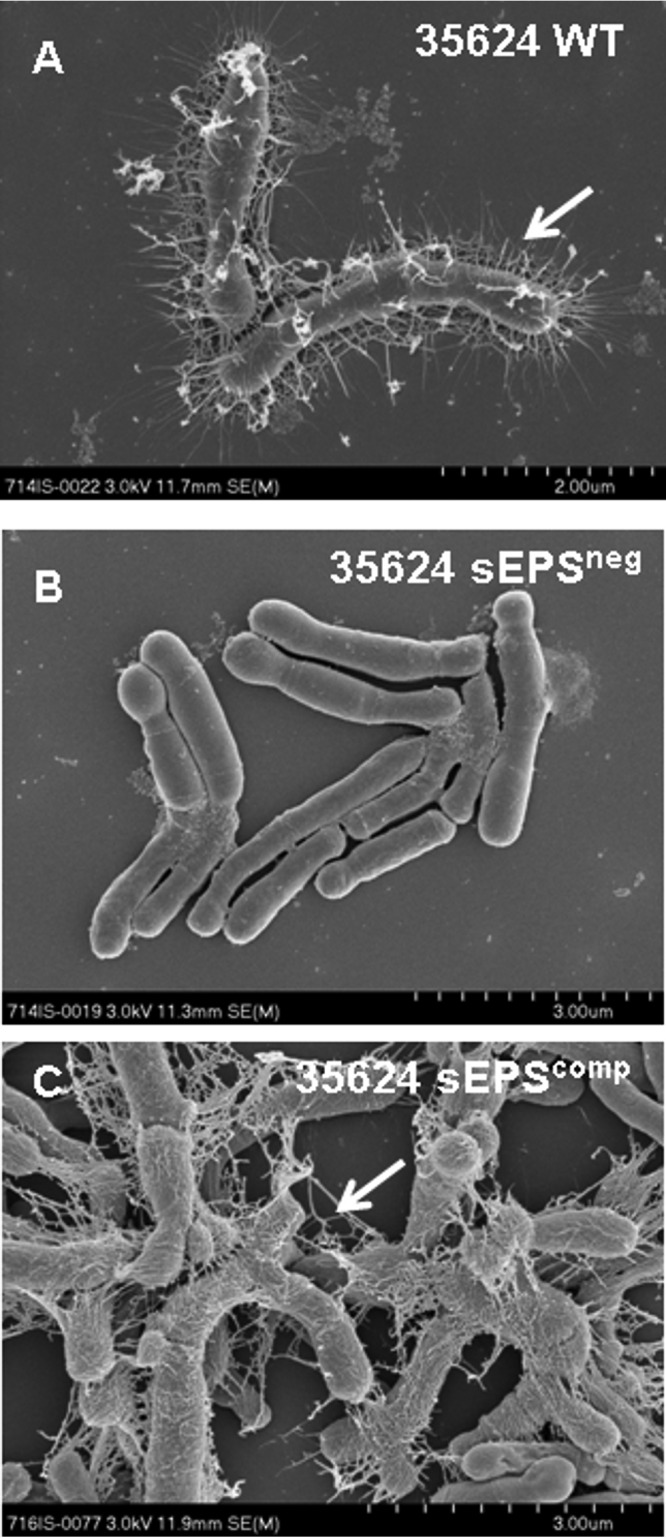
Electron microscopy of B. longum 35624. Representative scanning electron microscopy (SEM) images for the B. longum 35624 parent strain (A) and its isogenic derivatives sEPSneg (B) and sEPScomp (C). Arrows indicate the stringy-appearing layer of extracellular polysaccharide visible on the B. longum 35624 parent strain and the sEPScomp strain. Scale bars are indicated at the bottom right of each panel.
In vitro responses to B. longum 35624, sEPSneg, and sEPScomp.
The wild-type strain B. longum 35624 and its derivatives sEPSneg and sEPScomp were coincubated with human PBMCs for 24 h, followed by analysis of cytokine secretion in cell-free supernatants. Compared with B. longum 35624, the sEPSneg strain was shown to induce higher levels of IL-12p70, IFN-γ, and IL-17 secretion but comparable levels of IL-10 (Fig. 2A). The sEPScomp strain induced cytokine secretion at levels similar to their induction by B. longum 35624, confirming that enhanced proinflammatory cytokine secretion is specifically associated with the lack of exopolysaccharide production. The addition of isolated exopolysaccharide to the cocultures significantly reduced IL-12p70 and IFN-γ secretion in response to the sEPSneg strain but did not alter the IL-17 or IL-10 response to the sEPSneg strain (Fig. 2B).
FIG 2.

PBMC cytokine response to bacterial strains. (A) PBMCs from 6 healthy donors were stimulated with B. longum 35624 or its isogenic derivatives sEPSneg or sEPScomp (50 bacteria/1 PBMC) for 24 h, and cytokine secretion into the culture supernatant was quantified. Data are presented as box-and-whisker plots with the median values and maximum/minimum values illustrated. Statistical significance was determined using the Kruskal-Wallis test and Dunn's multiple-comparison test (*, P < 0.05). (B) Effect of adding isolated exopolysaccharide on sEPSneg strain-induced PBMC secretion of IL-12p70, IFN-gamma, IL-17, and IL-10. Each line connects the data for PBMC from the same donor. The Mann-Whitney U test was used for the statistical analysis (*, P < 0.05 versus the sEPSneg strain alone). Error bars show standard deviations.
Similarly to PBMCs, human MDDCs were coincubated with B. longum 35624 or sEPSneg strains, and cytokine secretion was measured after a 24-h exposure. Secreted IL-17, IL-6, and tumor necrosis factor alpha (TNF-α) levels, but not IL-10, were all shown to be significantly higher for the sEPSneg-stimulated MDDCs than for the B. longum 35624 -stimulated MDDCs (Fig. 3A). In contrast, no differences were found in the B. longum 35624 or sEPSneg-induced expression of the MDDC inhibitory molecules programmed death ligand-1 (PD-L1) and PD-L2, which bind to PD-1 on activated lymphocytes and play an important role in downregulating the immune system (Fig. 3B).
FIG 3.

MDDC response to bacterial strains. MDDCs were generated from 4 healthy donors and were stimulated with B. longum 35624 or its isogenic derivative sEPSneg (50 bacteria/1 MDDC) for 24 h. (A and B) Cytokine secretion into the culture supernatant (A) and cell surface expression of the inhibitory molecules PD-L1 and PD-L2 (B) were quantified. Data are presented as box-and-whisker plots with the median values and maximum/minimum values illustrated. The Mann-Whitney U test was used for the statistical analysis (*, P < 0.05 B. longum 35624 versus the sEPSneg strain). (C) NF-κB activation in THP-1 cells following exposure to increasing concentrations of B. longum 35624 or its isogenic derivative sEPSneg (n = 4 experimental replicates). (D) Activation of NF-κB in MDDCs exposed to B. longum 35624 or its isogenic derivative sEPSneg (n = 3, 50 bacteria/1 MDDC). Statistical significance was determined using two-way ANOVA (*, P < 0.05 B. longum 35624 versus the sEPSneg strain). Error bars show standard deviations.
Activation of the transcription factor NF-κB is critical for the induction of inflammatory genes, including cytokines. Thus, we measured NF-κB activation in the monocyte cell line THP-1, containing a SEAP reporter for NF-κB and AP-1 activation. The sEPSneg strain was shown to induce higher levels of NF-κB/AP-1 activation than B. longum 35624 in THP-1 cells (Fig. 3C). To confirm this result, NF-κB phosphorylation was measured in MDDCs over time, following exposure to bacteria. Both B. longum 35624 and sEPSneg induced similar levels of NF-κB phosphorylation at early time points (Fig. 3D). However, sustained high levels of phosphorylated NF-κB were observed at later time points for the sEPSneg-stimulated MDDCs, which were not observed for B. longum 35624-stimulated cells.
Taken together, these results suggest a role for this exopolysaccharide in preventing in vitro inflammatory responses to B. longum 35624.
The sEPSneg strain does not protect against colitis development.
Colitis was induced in SCID mice by adoptively transferring CD4+ CD25− CD45RBhi lymphocytes. Mice were administered B. longum 35624, sEPSneg, or sEPScomp daily by oral gavage. As previously described, B. longum 35624 treatment prevented weight loss and disease symptoms in this model (34). However, mice treated with the sEPSneg strain exhibited significant weight loss and severe disease symptoms, while restoration of EPS production in the sEPScomp strain promoted a response similar to that of mice treated with B. longum 35624 (Fig. 4A). Following euthanasia, the colon/body weight ratio was significantly higher in animals administered the sEPSneg strain, while macroscopically, the colons of these mice appeared severely inflamed with visible necrotic regions, which was not observed when animals had been administered B. longum 35624 (Fig. 4B). Within the mesenteric lymph nodes, there were significantly more IL-17+ lymphocytes in animals administered sEPSneg, with a trend toward increased numbers of IFN-γ+ lymphocytes which was not statistically significant (Fig. 4C). No significant difference in IL-10+ lymphocytes was observed. No difference in the gastrointestinal transit of B. longum 35624 or the sEPSneg derivative was observed (see Fig. S2 in the supplemental material).
FIG 4.

sEPSneg is not protective in a T cell transfer colitis model. Following receipt of CD4+ CD25− CD45RBhi T cells, C.B-17 SCID mice were orally administered B. longum 35624 (n = 8), sEPScomp (n = 8), or sEPSneg (n = 8). (A) Weight loss and disease activity were monitored over time. Statistical significance was determined using two-way ANOVA (*, P < 0.05). (B) Following euthanasia, the colon/body weight ratio was determined. A representative picture of colons from B. longum 35624- and sEPSneg-treated animals is provided. (C) The percentages of IL-17+, IFN-γ+, and IL-10+ lymphocytes from mesenteric lymph nodes are illustrated (n = 8 mice per group). Data are presented as box-and-whisker plots with the median values and maximum/minimum values illustrated. Statistical significance was determined using the Kruskal-Wallis test and Dunn's multiple-comparison test (*, P < 0.05 for sEPSneg strain versus the other strains). Error bars show standard deviations.
The administration of the sEPSneg strain to healthy immunocompetent animals did not result in gastrointestinal inflammation, indicating that the sEPSneg mutant did not induce colitis in healthy animals. However, the administration of sEPSneg did provoke a trend to increased percentages of IL-17+ and IFN-γ+ lymphocytes, associated with an increase in CCR9+ T cells, within the lamina propria of healthy animals, although these differences did not reach statistical significance (see Fig. S3 in the supplemental material). These data suggest that an inflamed microenvironment, such as that present in the SCID model, is required for sEPSneg to exert its TH17-enhancing effects.
sEPSneg exacerbates IL-17 responses within the lung.
In order to further assess the ability of sEPSneg to promote IL-17 responses in vivo, we utilized the ovalbumin (OVA) sensitization and respiratory challenge model, as we and others previously evidenced potent TH17 responses within the lungs of challenged animals (35). Either B. longum 35624 or its isogenic derivative sEPSneg was administered intranasally to examine the influence of these strains on IL-17 responses within the lung. OVA sensitization and challenge resulted in an increased percentage of IL-17+ lymphocytes within lung tissue compared to the level in control animals (Fig. 5A). Exposure to the B. longum 35624 strain did not influence the percentage of IL-17+ lymphocytes within the lung; however, exposure to sEPSneg significantly increased the percentage of IL-17+ lymphocytes (Fig. 5A). Single-cell suspensions were generated from the lungs of all animals challenged as indicated above, and these lung cells were restimulated ex vivo with OVA or LPS to assess IL-17 secretion. OVA-sensitized and challenged animals displayed increased ex vivo secretion of IL-17 in response to both OVA and LPS stimulation compared to the levels in nonsensitized animals, suggesting that innate TLR-4 responses to LPS and allergen-specific lymphocyte responses to OVA are increased in the inflamed lungs of allergic animals (Fig. 5A). The in vivo exposure to B. longum 35624 did not alter the ex vivo secretion of IL-17 by lung cells stimulated with either LPS or OVA. However, if animals had been exposed to sEPSneg in vivo previously, the isolated lung cells secreted significantly more IL-17 ex vivo in response to both TLR-4 stimulation with LPS and allergen restimulation with OVA (Fig. 5A). Thus, the ex vivo secretion of IL-17 and the percentage of IL-17+ cells within lung tissue correlate with the highest levels for both assay systems being observed for sEPSneg-treated animals.
FIG 5.

sEPSneg promotes TH17 responses in the lung. Nonsensitized animals received an OVA aerosol challenge and were administered PBS intranasally (Control, n = 8). Sensitized animals received an OVA aerosol challenge and were administered PBS (OVA, n = 8), B. longum 35624 (OVA & 35624, n = 8), or sEPSneg (OVA & sEPSneg, n = 8) intranasally. (A) Percentages of IL-17+ CD3+ CD4+ T lymphocytes isolated from lung tissue and secretion levels of IL-17 from isolated lung cells restimulated ex vivo with OVA or LPS. (B and C) IFN-γ+ (B) and IL-10+ (C) CD3+ CD4+ T lymphocytes and ex vivo IFN-γ and IL-10 secretion were quantified using identical methods. Data are presented as box-and-whisker plots with the median values and maximum/minimum values illustrated. Statistical significance was determined using the Kruskal-Wallis test and Dunn's multiple-comparison test (*, P < 0.05 compared to the OVA group). Error bars show standard deviations.
OVA sensitization and challenge resulted in an increased percentage of IFN-γ+ lymphocytes within lung tissue compared to the level in control animals (Fig. 5A). Exposure to the B. longum 35624 strain prevented the increase in the percentage of IFN-γ+ lymphocytes within the lung, which was not observed following exposure to the sEPSneg strain (Fig. 5B). Restimulation of lung single-cell suspensions ex vivo with OVA or LPS did not result in significant levels of IFN-γ being secreted, and no statistically significant differences were observed between the groups (Fig. 5B).
OVA sensitization and challenge resulted in an increased percentage of IL-10+ lymphocytes within lung tissue, and exposure to B. longum 35624 or the sEPSneg strains further increased the percentage of IL-10+ lymphocytes within the lung (Fig. 5C). Restimulation ex vivo was also associated with increased secretion of IL-10 following in vivo exposure to B. longum 35624 or the sEPSneg strains (Fig. 5C).
These findings suggest that the absence of the exopolysaccharide on B. longum 35624 promotes TH17 responses in the inflamed lung, similar to the effects described above for the inflamed gut.
DISCUSSION
In order to avoid immune-mediated destruction of mucosal tissues, the host can activate regulatory mechanisms that can block proinflammatory responses to commensal microbes present on mucosal surfaces. Bifidobacteria comprise a significant proportion of the gut microbiota, and many strains are currently used as probiotics. However, the precise mechanisms by which such bifidobacteria interact with host immune cells are not fully understood. In this report, we describe that the presence of a cell surface-associated exopolysaccharide produced by B. longum 35624 modulates cytokine secretion and NF-κB activation in vitro, while in murine models, exposure to a B. longum 35624 derivative that is unable to synthesize exopolysaccharide promotes TH17 responses within both the gut and the lung.
Bifidobacterial cell surface-associated exopolysaccharides have previously been proposed to (i) mediate some of the health-promoting benefits of these bacteria, (ii) contribute to bifidobacterial tolerance of the harsh conditions within the gut, and (iii) influence the composition of the gut microbiome through their use as a fermentable substrate by other microbes (21, 36–38). In general, bacterial exopolysaccharides consist of repeating mono- or oligosaccharide subunits connected by various glycosidic linkages that are structurally very diverse and may contribute to strain-specific traits due to the expected structural and, therefore, functional diversity of such molecules. Of note, pathogen-associated exopolysaccharides have long been known to be critical in host-microbe interactions, where they facilitate adherence and colonization within the human host, with additional immunomodulatory effects (39, 40). Exopolysaccharides can also mediate the beneficial immune effects associated with certain commensal microbes. As already mentioned, a strong modulator of intestinal immune responses is PSA from B. fragilis, which is well described to influence lymphocyte polarization and which suppresses IL-17 production by intestinal immune cells (41–43). In line with data presented here, exopolysaccharide gene knockout mutants of Lactobacillus casei Shirota induced significantly more proinflammatory cytokine secretion from a mouse macrophage cell line than did wild-type cells (44). In addition, the cytokine response of PBMCs to two isogenic strains of B. animalis subsp. lactis that differ only in their exopolysaccharide-producing phenotype suggest that the mucoid strain could have greater anti-inflammatory activity (31). The data presented here are in agreement with these previous reports and further support the concept that exopolysaccharides from bifidobacteria may elicit immune-modulatory activities.
Interestingly, the induction of PD-L1 and PD-L2 on dendritic cells was similar for the wild-type B. longum 35624 strain and its isogenic derivative, sEPSneg. Similarly, the induction of IL-10 was not negatively impacted by the loss of exopolysaccharide from the bacterium. This suggests that not all immune-regulatory effects induced by B. longum 35624 are mediated solely by exopolysaccharide. The bifidobacterial cell wall is a complex arrangement of macromolecules, consisting of a thick peptidoglycan layer that surrounds the cytoplasmic membrane, which is decorated with other glycopolymers, such as (lipo)teichoic acids, polysaccharides, and proteins, all of which may influence the immune response (45, 46) A few examples include the cell wall-associated proteins p40 and p75 from L. casei subsp. rhamnosus GG, the S-layer protein from Lactobacillus acidophilus, and the STp peptide from Lactobacillus plantarum (47–49).
TH17 cells are a subset of CD4+ T helper cells that mediate protective immunity to extracellular bacterial and fungal pathogens, predominantly at epithelial surfaces (50). Polarization of naive T cells into TH17 cells occurs following T-cell antigen receptor recognition of a major histocompatibility complex (MHC) class II-bound peptide in the presence of cytokines, including transforming growth factor β1 (TGF-β1), IL-6, or IL-1β (51, 52). While TH17 cells are required for protective immunity, these cells massively infiltrate the inflamed intestine of inflammatory bowel disease patients, where they produce IL-17 and other cytokines, triggering and amplifying the inflammatory process (53). Our data suggest that the B. longum 35624 strain-associated exopolysaccharide prevents the induction of a TH17 response to this bacterium. Multiple mechanisms may be involved in this process. For example, the exaggerated induction of cytokines, including IL-6, from dendritic cells may support TH17 lymphocyte polarization and development. Support for this hypothesis can be seen when we restimulate OVA-specific T cells with OVA and observe increased secretion of IL-17 when the lungs were previously exposed to sEPSneg. These OVA-specific T cells are not reacting to bifidobacterium-associated antigens, but more IL-17 is secreted upon OVA challenge, suggesting that it is the cytokine microenvironment provided by innate cells such as dendritic cells that is supporting excessive TH17 development. The observation that the addition of purified exopolysaccharide to sEPSneg-stimulated PBMCs suppresses the exaggerated IL-12p70 and IFN-γ secretion but not IL-17 secretion also suggests that multiple mechanisms may be involved. Future studies will determine whether it is the exopolysaccharide itself that can directly inhibit TH17 responses by binding to host receptors or whether the exopolysaccharide is simply masking TH17-promoting molecules on the surface of this bacterium.
In conclusion, we have identified a novel immunoregulatory activity associated with the presence of a exopolysaccharide in the human commensal B. longum strain 35624. Our findings suggest that this exopolysaccharide is required to prevent a potent tissue-damaging TH17 response to a commensal bacterium. Accordingly, our data on the B. longum 35624-associated exopolysaccharide corroborate and expand the published concept that exopolysaccharides produced by certain lactic acid bacteria and bifidobacteria may elicit immune-modulatory activities (54) that are important for appropriate host-microbe communication.
Supplementary Material
ACKNOWLEDGMENTS
35624 is a trademark of Alimentary Health Ltd., Cork, Ireland. We thank Patrycja Konieczna for her technical assistance.
These studies were directly supported by a European Union Marie Curie training network entitled TEAM-EPIC. In addition, the authors are supported by Swiss National Foundation grants (project numbers CRSII3_154488 and 310030_144219), Christine Kühne Center for Allergy Research and Education, and Science Foundation Ireland (SFI) (grant no. SFI/12/RC/2273). M.O.M. is a recipient of an HRB postdoctoral fellowship (grant no. PDTM/20011/9). E.S. was supported by an EAACI research fellowship award 2012.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02238-16.
REFERENCES
- 1.Donaldson GP, Lee SM, Mazmanian SK. 2016. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 14:20–32. doi: 10.1038/nrmicro3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marchesi JR, Adams DH, Fava F, Hermes GDA, Hirschfield GM, Hold G, Quraishi MN, Kinross J, Smidt H, Tuohy KM, Thomas LV, Zoetendal EG, Hart A. 2016. The gut microbiota and host health: a new clinical frontier. Gut 65:330–339. doi: 10.1136/gutjnl-2015-309990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frei R, Lauener RP, Crameri R, O'Mahony L. 2012. Microbiota and dietary interactions—an update to the hygiene hypothesis? Allergy 67:451–461. doi: 10.1111/j.1398-9995.2011.02783.x. [DOI] [PubMed] [Google Scholar]
- 4.Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, Marsland BJ. 2014. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 20:159–166. doi: 10.1038/nm.3444. [DOI] [PubMed] [Google Scholar]
- 5.Frei R, Akdis M, O'Mahony L. 2015. Prebiotics, probiotics, synbiotics, and the immune system: experimental data and clinical evidence. Curr Opin Gastroenterol 31:153–158. doi: 10.1097/MOG.0000000000000151. [DOI] [PubMed] [Google Scholar]
- 6.Tomkovich S, Jobin C. 2016. Microbiota and host immune responses: a love-hate relationship. Immunology 147:1–10. doi: 10.1111/imm.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Konieczna P, Akdis CA, Quigley EMM, Shanahan F, O'Mahony L. 2012. Portrait of an immunoregulatory Bifidobacterium. Gut Microbes 3:261–266. doi: 10.4161/gmic.20358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Mahony C, Scully P, O'Mahony D, Murphy S, O'Brien F, Lyons A, Sherlock G, MacSharry J, Kiely B, Shanahan F, O'Mahony L. 2008. Commensal-induced regulatory T cells mediate protection against pathogen-stimulated NF-κB activation. PLoS Pathog 4:e1000112. doi: 10.1371/journal.ppat.1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyons A, O'Mahony D, O'Brien F, MacSharry J, Sheil B, Ceddia M, Russell WM, Forsythe P, Bienenstock J, Kiely B, Shanahan F, O'Mahony L. 2010. Bacterial strain-specific induction of Foxp3+ T regulatory cells is protective in murine allergy models. Clin Exp Allergy 40:811–819. doi: 10.1111/j.1365-2222.2009.03437.x. [DOI] [PubMed] [Google Scholar]
- 10.McCarthy J, O'Mahony L, O'Callaghan L, Sheil B, Vaughan EE, Fitzsimons N, Fitzgibbon J, O'Sullivan GC, Kiely B, Collins JK, Shanahan F. 2003. Double blind, placebo controlled trial of two probiotic strains in interleukin 10 knockout mice and mechanistic link with cytokine balance. Gut 52:975–980. doi: 10.1136/gut.52.7.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scully P, MacSharry J, O'Mahony D, Lyons A, O'Brien F, Murphy S, Shanahan F, O'Mahony L. 2013. Bifidobacterium infantis suppression of Peyer's patch MIP-1α and MIP-1β secretion during Salmonella infection correlates with increased local CD4+ CD25+ T cell numbers. Cell Immunol 281:134–140. doi: 10.1016/j.cellimm.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Symonds EL, O'Mahony C, Lapthorne S, O'Mahony D, Sharry JM, O'Mahony L, Shanahan F. 2012. Bifidobacterium infantis 35624 protects against salmonella-induced reductions in digestive enzyme activity in mice by attenuation of the host inflammatory response. Clin Transl Gastroenterol 3:e15. doi: 10.1038/ctg.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konieczna P, Groeger D, Ziegler M, Frei R, Ferstl R, Shanahan F, Quigley EMM, Kiely B, Akdis CA, O'Mahony L. 2012. Bifidobacterium infantis 35624 administration induces Foxp3 T regulatory cells in human peripheral blood: potential role for myeloid and plasmacytoid dendritic cells. Gut 61:354–366. doi: 10.1136/gutjnl-2011-300936. [DOI] [PubMed] [Google Scholar]
- 14.Groeger D, O'Mahony L, Murphy EF, Bourke JF, Dinan TG, Kiely B, Shanahan F, Quigley EMM. 2013. Bifidobacterium infantis 35624 modulates host inflammatory processes beyond the gut. Gut Microbes 4:325–339. doi: 10.4161/gmic.25487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konieczna P, Ferstl R, Ziegler M, Frei R, Nehrbass D, Lauener RP, Akdis CA, O'Mahony L. 2013. Immunomodulation by Bifidobacterium infantis 35624 in the murine lamina propria requires retinoic acid-dependent and independent mechanisms. PLoS One 8:e62617. doi: 10.1371/journal.pone.0062617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sibartie S, O'Hara AM, Ryan J, Fanning A, O'Mahony J, O'Neill S, Sheil B, O'Mahony L, Shanahan F. 2009. Modulation of pathogen-induced CCL20 secretion from HT-29 human intestinal epithelial cells by commensal bacteria. BMC Immunol 10:54. doi: 10.1186/1471-2172-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Mahony L, O'Callaghan L, McCarthy J, Shilling D, Scully P, Sibartie S, Kavanagh E, Kirwan WO, Redmond HP, Collins JK, Shanahan F. 2006. Differential cytokine response from dendritic cells to commensal and pathogenic bacteria in different lymphoid compartments in humans. Am J Physiol Gastrointest Liver Physiol 290:G839–G845. doi: 10.1152/ajpgi.00112.2005. [DOI] [PubMed] [Google Scholar]
- 18.Round JL, Mazmanian SK. 2010. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A 107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dasgupta S, Erturk-Hasdemir D, Ochoa-Reparaz J, Reinecker HC, Kasper DL. 2014. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe 15:413–423. doi: 10.1016/j.chom.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones SE, Paynich ML, Kearns DB, Knight KL. 2014. Protection from intestinal inflammation by bacterial exopolysaccharides. J Immunol 192:4813–4820. doi: 10.4049/jimmunol.1303369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fanning S, Hall LJ, Cronin M, Zomer A, MacSharry J, Goulding D, Motherway MO, Shanahan F, Nally K, Dougan G, van Sinderen D. 2012. Bifidobacterial surface-exopolysaccharide facilitates commensal-host interaction through immune modulation and pathogen protection. Proc Natl Acad Sci U S A 109:2108–2113. doi: 10.1073/pnas.1115621109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rossi O, Khan MT, Schwarzer M, Hudcovic T, Srutkova D, Duncan SH, Stolte EH, Kozakova H, Flint HJ, Samsom JN, Harmsen HJ, Wells JM. 2015. Faecalibacterium prausnitzii strain HTF-F and its extracellular polymeric matrix attenuate clinical parameters in DSS-induced colitis. PLoS One 10:e0123013. doi: 10.1371/journal.pone.0123013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altmann FAF, Kosma P, O'Callaghan A, Leahy S, Bottacini F, Molloy E, Plattner S, Schiavi E, Gleinser M, Groeger D, Grant R, Rodriguez Perez N, Healy S, Svehla E, Windwarder M, Hofinger A, O'Connell Motherway M, Akdis CA, Xu J, Roper J, van Sinderen D, O'Mahony L. 2016. Genome analysis and characterisation of the exopolysaccharide produced by Bifidobacterium longum subsp. longum 35624. PLoS One 11:e0162983. doi: 10.1371/journal.pone.0162983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Riordan K, Fitzgerald GF. 1998. Evaluation of bifidobacteria for the production of antimicrobial compounds and assessment of performance in cottage cheese at refrigeration temperature. J Appl Microbiol 85:103–114. doi: 10.1046/j.1365-2672.1998.00474.x. [DOI] [PubMed] [Google Scholar]
- 25.Maze A, O'Connell-Motherway M, Fitzgerald GF, Deutscher J, van Sinderen D. 2007. Identification and characterization of a fructose phosphotransferase system in Bifidobacterium breve UCC2003. Appl Environ Microbiol 73:545–553. doi: 10.1128/AEM.01496-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Law J, Buist G, Haandrikman A, Kok J, Venema G, Leenhouts K. 1995. A system to generate chromosomal mutations in Lactococcus lactis which allows fast analysis of targeted genes. J Bacteriol 177:7011–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alvarez-Martín P, O'Connell-Motherway M, van Sinderen D, Mayo B. 2007. Functional analysis of the pBC1 replicon from Bifidobacterium catenulatum L48. Appl Microbiol Biotechnol 76:1395–1402. doi: 10.1007/s00253-007-1115-5. [DOI] [PubMed] [Google Scholar]
- 28.Masuko T, Minami A, Iwasaki N, Majima T, Nishimura SI, Lee YC. 2005. Carbohydrate analysis by a phenol-sulphuric acid method in microplate format. Anal Biochem 339:69–72. doi: 10.1016/j.ab.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Arnold IC, Hutchings C, Kondova I, Hey A, Powrie F, Beverley P, Tchilian E. 2015. Helicobacter hepaticus infection in BALB/c mice abolishes subunit-vaccine-induced protection against M. tuberculosis. Vaccine 33:1808–1814. doi: 10.1016/j.vaccine.2015.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Connell Motherway M, O'Driscoll J, Fitzgerald GF, Van Sinderen D. 2009. Overcoming the restriction barrier to plasmid transformation and targeted mutagenesis in Bifidobacterium breve UCC2003. Microb Biotechnol 2:321–332. doi: 10.1111/j.1751-7915.2008.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hidalgo-Cantabrana C, Sanchez B, Alvarez-Martin P, Lopez P, Martinez-Alvarez N, Delley M, Marti M, Varela E, Suarez A, Antolin M, Guarner F, Berger B, Ruas-Madiedo P, Margolles A. 2015. A single mutation in the gene responsible for the mucoid phenotype of Bifidobacterium animalis subsp. lactis confers surface and functional characteristics. Appl Environ Microbiol 81:7960–7968. doi: 10.1128/AEM.02095-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Egan M, O'Connell Motherway M, Ventura M, van Sinderen D. 2014. Metabolism of sialic acid by Bifidobacterium breve UCC2003. Appl Environ Microbiol 80:4414–4426. doi: 10.1128/AEM.01114-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Connell Motherway M, Kinsella M, Fitzgerald GF, van Sinderen D. 2013. Transcriptional and functional characterization of genetic elements involved in galacto-oligosaccharide utilization by Bifidobacterium breve UCC2003. Microb Biotechnol 6:67–79. doi: 10.1111/1751-7915.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Kleij H, O'Mahony C, Shanahan F, O'Mahony L, Bienenstock J. 2008. Protective effects of Lactobacillus reuteri and Bifidobacterium infantis in murine models for colitis do not involve the vagus nerve. Am J Physiol Regul Integr Comp Physiol 295:R1131–R1137. doi: 10.1152/ajpregu.90434.2008. [DOI] [PubMed] [Google Scholar]
- 35.Lu S, Li H, Gao R, Gao X, Xu F, Wang Q, Lu G, Xia D, Zhou J. 2016. IL-17A, but not IL-17F, is indispensable for airway vascular remodeling induced by exaggerated Th17 cell responses in prolonged ovalbumin-challenged mice. J Mol Med (Berl) 194:3557–3566. [DOI] [PubMed] [Google Scholar]
- 36.Salazar N, Ruas-Madiedo P, Kolida S, Collins M, Rastall R, Gibson G, de los Reyes-Gavilán CG. 2009. Exopolysaccharides produced by Bifidobacterium longum IPLA E44 and Bifidobacterium animalis subsp. lactis IPLA R1 modify the composition and metabolic activity of human faecal microbiota in pH-controlled batch cultures. Int J Food Microbiol 135:260–267. doi: 10.1016/j.ijfoodmicro.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 37.Alp G, Aslim B. 2010. Relationship between the resistance to bile salts and low pH with exopolysaccharide (EPS) production of Bifidobacterium spp. isolated from infants feces and breast milk. Anaerobe 16:101–105. doi: 10.1016/j.anaerobe.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Salazar N, Gueimonde M, de los Reyes-Gavilán CG, Ruas-Madiedo P. 2016. Exopolysaccharides produced by lactic acid bacteria and bifidobacteria as fermentable substrates by the intestinal microbiota. Crit Rev Food Sci Nutr 56:1440–1453. doi: 10.1080/10408398.2013.770728. [DOI] [PubMed] [Google Scholar]
- 39.Conover MS, Sloan GP, Love CF, Sukumar N, Deora R. 2010. The Bps polysaccharide of Bordetella pertussis promotes colonization and biofilm formation in the nose by functioning as an adhesin. Mol Microbiol 77:1439–1455. doi: 10.1111/j.1365-2958.2010.07297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu CL, Wang YZ, Jin ML, Yang XQ. 2009. Preparation, characterization and immunomodulatory activity of selenium-enriched exopolysaccharide produced by bacterium Enterobacter cloacae Z0206. Bioresour Technol 100:2095–2097. doi: 10.1016/j.biortech.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 41.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. 2011. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. 2005. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Mazmanian SK, Round JL, Kasper DL. 2008. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 44.Yasuda E, Serata M, Sako T. 2008. Suppressive effect on activation of macrophages by Lactobacillus casei strain Shirota genes determining the synthesis of cell wall-associated polysaccharides. Appl Environ Microbiol 74:4746–4755. doi: 10.1128/AEM.00412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chapot-Chartier M-P, Kulakauskas S. 2014. Cell wall structure and function in lactic acid bacteria. Microb Cell Fact 13:S9. doi: 10.1186/1475-2859-13-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruiz L, Hevia A, Bernardo D, Margolles A, Sánchez B. 2014. Extracellular molecular effectors mediating probiotic attributes. FEMS Microbiol Lett 359:1–11. doi: 10.1111/1574-6968.12576. [DOI] [PubMed] [Google Scholar]
- 47.Konstantinov SR, Smidt H, de Vos WM, Bruijns SCM, Singh SK, Valence F, Molle D, Lortal S, Altermann E, Klaenhammer TR, van Kooyk Y. 2008. S layer protein A of Lactobacillus acidophilus NCFM regulates immature dendritic cell and T cell functions. Proc Natl Acad Sci U S A 105:19474–19479. doi: 10.1073/pnas.0810305105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Al-Hassi HO, Mann ER, Sanchez B, English NR, Peake STC, Landy J, Man R, Urdaci M, Hart AL, Fernandez-Salazar L, Lee GH, Garrote JA, Arranz E, Margolles A, Stagg AJ, Knight SC, Bernardo D. 2014. Altered human gut dendritic cell properties in ulcerative colitis are reversed by Lactobacillus plantarum extracellular encrypted peptide STp. Mol Nutr Food Res 58:1132–1143. doi: 10.1002/mnfr.201300596. [DOI] [PubMed] [Google Scholar]
- 49.Yan F, Cao H, Cover TL, Washington MK, Shi Y, Liu L, Chaturvedi R, Peek RM, Wilson KT, Polk DB. 2011. Colon-specific delivery of a probiotic-derived soluble protein ameliorates intestinal inflammation in mice through an EGFR-dependent mechanism. J Clin Invest 121:2242–2253. doi: 10.1172/JCI44031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korn T, Bettelli E, Oukka M, Kuchroo VK. 2009. IL-17 and Th17 cells. Annu Rev Immunol 27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 51.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. 2006. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 52.Akdis M, Burgler S, Crameri R, Eiwegger T, Fujita H, Gomez E, Klunker S, Meyer N, O'Mahony L, Palomares O, Rhyner C, Ouaked N, Schaffartzik A, Van De Veen W, Zeller S, Zimmermann M, Akdis CA. 2011. Interleukins, from 1 to 37, and interferon-γ: receptors, functions, and roles in diseases. J Allergy Clin Immunol 127:701–721. doi: 10.1016/j.jaci.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 53.Gálvez J. 2014. Role of Th17 cells in the pathogenesis of human IBD. ISRN Inflamm 2014:928461. doi: 10.1155/2014/928461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hidalgo-Cantabrana C, Lopez P, Gueimonde M, de Los Reyes-Gavilan CG, Suarez A, Margolles A, Ruas-Madiedo P. 2012. Immune modulation capability of exopolysaccharides synthesised by lactic acid bacteria and bifidobacteria. Probiotics Antimicrob Proteins 4:227–237. doi: 10.1007/s12602-012-9110-2. [DOI] [PubMed] [Google Scholar]
- 55.Margolles A, Florez AB, Moreno JA, van Sinderen D, de los Reyes-Gavilan CG. 2006. Two membrane proteins from Bifidobacterium breve UCC2003 constitute an ABC-type multidrug transporter. Microbiology 152:3497–3505. doi: 10.1099/mic.0.29097-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.