

Abstract
Lysosomal disruption is increasingly regarded as a major pathogenic event in Parkinson disease (PD). A reduced number of intraneuronal lysosomes, decreased levels of lysosomal-associated proteins and accumulation of undegraded autophagosomes (AP) are observed in PD-derived samples, including fibroblasts, induced pluripotent stem cell-derived dopaminergic neurons, and post-mortem brain tissue. Mechanistic studies in toxic and genetic rodent PD models attribute PD-related lysosomal breakdown to abnormal lysosomal membrane permeabilization (LMP). However, the molecular mechanisms underlying PD-linked LMP and subsequent lysosomal defects remain virtually unknown, thereby precluding their potential therapeutic targeting. Here we show that the pro-apoptotic protein BAX (BCL2-associated X protein), which permeabilizes mitochondrial membranes in PD models and is activated in PD patients, translocates and internalizes into lysosomal membranes early following treatment with the parkinsonian neurotoxin MPTP, both in vitro and in vivo, within a time-frame correlating with LMP, lysosomal disruption, and autophagosome accumulation and preceding mitochondrial permeabilization and dopaminergic neurodegeneration. Supporting a direct permeabilizing effect of BAX on lysosomal membranes, recombinant BAX is able to induce LMP in purified mouse brain lysosomes and the latter can be prevented by pharmacological blockade of BAX channel activity. Furthermore, pharmacological BAX channel inhibition is able to prevent LMP, restore lysosomal levels, reverse AP accumulation, and attenuate mitochondrial permeabilization and overall nigrostriatal degeneration caused by MPTP, both in vitro and in vivo. Overall, our results reveal that PD-linked lysosomal impairment relies on BAX-induced LMP, and point to small molecules able to block BAX channel activity as potentially beneficial to attenuate both lysosomal defects and neurodegeneration occurring in PD.
Keywords: Parkinson disease, neurodegeneration, BAX channel inhibitor, lysosome, mitochondria, MPTP
Introduction
Mounting evidence indicates that impaired lysosomal function contributes to the pathogenesis of Parkinson disease.1 A reduced number of intraneuronal lysosomes, decreased levels of lysosomal-associated proteins, and accumulation of undegraded autophagosomes are observed in PD patients (i.e., post-mortem brain samples,2,3 fibroblasts,4,5 and induced pluripotent stem cell-derived dopaminergic neurons6) as well as in toxic and genetic rodent models of PD (i.e., mice treated with parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [MPTP]3 and rats overexpressing mutant SNCA/α-synuclein2). Mechanistic studies in the MPTP mouse model revealed that PD-linked lysosomal deficiency is secondary to abnormal lysosomal membrane permeabilization.3,7 In particular, MPTP-induced LMP results in a decreased number of lysosomes and impaired AP-lysosome fusion, leading to a defective clearance and subsequent accumulation of undegraded AP within affected neurons.3,7 In addition, LMP can directly participate in MPTP-induced dopaminergic cell death by the leakage of lysosomal proteases into the cytosol, some of which, such as CTSB/cathepsin B and CTSD, can remain active at neutral cytosolic pH and cause the digestion of vital proteins or the activation of additional hydrolases, including caspases.3,7 Supporting a pathogenic role for PD-related LMP, genetic, or pharmacological enhancement of lysosomal biogenesis and function with TFEB (transcription factor EB) or rapamycin, respectively, is able to restore lysosomal-mediated degradation, reverse AP accumulation, and attenuate dopaminergic neurodegeneration in toxic and genetic in vivo PD models.3,8,9 However, despite the accumulating evidence for a pathogenic role of LMP in the context of PD, the molecular mechanisms underlying PD-related LMP remain unknown.
In addition to LMP, mitochondrial outer membrane permeabilization (MOMP) constitutes another early pathogenic event contributing to dopaminergic cell death in toxic and genetic in vitro and in vivo PD models.10,11 PD-linked MOMP leads to CYCS (cytochrome c, somatic) release and subsequent caspase activation and relies on the formation of BAX-derived channels directly into mitochondrial membranes.10-13 Relevant to PD, activation of BAX, CASP9/caspase 9 and CASP3 have been observed in PD patients.2,3,6,14-16 Supporting a pathogenic role for BAX in PD-related neurodegeneration, genetic ablation of BAX in mutant mice completely abrogates MPTP-induced dopaminergic cell death.10,13 However, the feasibility and therapeutic potential of pharmacologically targeting BAX activity in the context of PD remain to be determined.
Remarkably, in some experimental in vitro settings unrelated to PD, LMP has been shown to require BAX activation.17-19 However, whether BAX can directly permeabilize lysosomal membranes, in analogy to its role in mitochondria, remains controversial.20 In addition, it is unknown whether such a potential effect of BAX on lysosomes could also occur in the brain and in an in vivo pathological situation.
Results
BAX translocates to lysosomal membranes early following MPTP treatment
The parkinsonian neurotoxin MPTP causes a PD-like syndrome in human and monkeys and reproduces in mice several PD-linked cellular alterations, such as inhibition of mitochondrial complex I, oxidative damage to lipids, DNA, and proteins (including SNCA), lysosomal impairment, and substantia nigra pars compacta (SNpc) dopaminergic cell death.21-23 It was previously shown that BAX permeabilizes mitochondrial membranes in this experimental model and plays an instrumental role in MPTP-induced dopaminergic cell death.10,11,13,21,24 Here, we assessed whether BAX may also be responsible for LMP and subsequent lysosomal deficiency occurring in this model of PD. To this purpose, we determined the presence of BAX by immunoblot in pure (mitochondria-free) lysosomal fractions isolated from the ventral midbrain region (which contains the SNpc) of saline- and MPTP-treated mice. Mice received 1 intraperitoneal injection of 30 mg/kg/day MPTP for 5 consecutive d and were euthanized early following MPTP treatment (i.e., 2 h after the last MPTP injection) in order to precede or coincide with the initiation of MPTP-induced LMP in these animals, which occurs within the first 24 h post-MPTP.3 In purified ventral midbrain lysosomes from saline-treated mice, BAX was barely detectable by immunoblot (Fig. 1A). In contrast, high levels of BAX were detected in ventral midbrain lysosomal fractions purified from MPTP-treated animals (Fig. 1A), thereby indicating a lysosomal translocation of BAX in this pathological situation. The detection of BAX in lysosomal fractions of MPTP-treated mice could not be attributed to a cross-contamination of these fractions by mitochondria (Fig. S1). Similar to the results in MPTP-treated mice, BAX lysosomal translocation was also observed in human dopaminergic BE(2)-M17 neuroblastoma cells treated with MPP+, MPTP’s active metabolite (Fig. 1B). Further fractionation of purified lysosomes into lysosomal membrane and matrix by hypotonic shock indicated that BAX was localized into lysosomal membranes, and not the lysosomal lumen, following MPTP treatment (Fig. 1C, left panel). This observation rules out the possibility of lysosomal BAX corresponding to BAX-decorated mitochondria being degraded by mitophagy within the lysosomal lumen. To confirm that lysosomal BAX was actually anchored and not merely loosely attached to lysosomal membranes, alkaline extraction experiments were performed in lysosomal membrane fractions from MPP+-treated cells. Following alkaline treatment, a significant amount of BAX remained in alkali-resistant lysosomal membrane pellet fractions (Fig. 1C, right panel), thereby indicating its internalization into lysosomal membranes. In both MPTP-treated mice and MPP+-treated cells, BAX lysosomal translocation correlated with the occurrence of LMP, as indicated by increased ectopic activities of the lysosomal enzymes ACP2 (acid phosphatase 2, lysosomal) and β-hexosaminidase (HEXA-HEXB) in cytosolic lysosomal-free fractions (indicative of their leakage from the lysosomal lumen) from MPTP-treated ventral midbrain mouse homogenates (Fig. 1D) and MPP+-treated cells (Fig. 1E).

Figure 1. BAX lysosomal translocation early following MPTP treatment in vitro and in vivo. (A and B) BAX immunoblot levels in purified lysosomal (Lys), cytosolic (Cyto) and total protein homogenate (Homg) fractions from ventral midbrain of saline- and MPTP-treated mice (A) and control and MPP+-treated BE(2)-M17 cells (B). Mice received daily injections of 30 mg/kg/day of MPTP for 5 consecutive d and were euthanized 2 h after the last MPTP injection. Cells were incubated with 250 μM MPP+ for 3 h. Quantifications correspond to BAX levels in lysosomal fractions. Lysosomal transmembrane protein LAMP1 is shown as a lysosomal marker. (C, left panel) BAX immunoblot levels in lysosomal membrane and lumen fractions obtained by hypotonic shock from ventral midbrain of saline- and MPTP-treated mice. (C, right panel) BAX immunoblot levels in lysosomal pellet fractions from MPP+-treated (250 μM for 3 h) BE(2)-M17 cells following alkaline extraction with Na2CO3 (0.1 M, pH 11.5, for 30 min). Alkali-resistant transmembrane protein LAMP1 and alkali-sensitive CTSB are shown as experimental controls. (D and E) Enzymatic activities of lysosomal enzymes ACP2 and β-hexosaminidase (β-hexo) in cytosolic, lysosomal-free fractions from the ventral midbrain of MPTP-treated mice, 2 h post-MPTP (D) and BE(2)-M17 cells treated with 250 μM MPP+ for 3 h (E). In all panels, data represent mean ± SEM. In (B and E), at least 3 independent experiments were performed. In (A,C, and D) each experiment corresponds to pooled ventral midbrains from 9 animals (either saline- or MPTP-treated). *P < 0.05, compared with respective control groups.
To further corroborate the lysosomal translocation of BAX, additional experiments were performed to colocalize BAX with either lysosomal or mitochondrial markers (Fig. 2). In MPP+-treated BE(2)-M17 cells, BAX colocalization with the lysosomal marker LAMP1 (lysosomal-associated membrane protein 1) was increased by 3 h post-MPP+ (Fig. 2A and B; Fig. S2), where it was maintained up to 24 h despite a decreased number of lysosomes at that time as a consequence of LMP (Fig. 2A and B). In contrast, colocalization of BAX with the mitochondrial marker TOMM20 (translocase of outer mitochondrial membrane 20 homolog [yeast]) was not observed at 3 h post-MPP+ and was only evident by 24 h (Fig. 2C and D), which coincides with CYCS release and cell death in this cellular model.3,10,11. Similar to the in vitro results, BAX was not yet translocated to mitochondrial fractions in MPTP-treated mice by the time of BAX lysosomal translocation (i.e., 2 h post-MPTP) (Fig. 1A; Fig. S3A). In these animals, BAX mitochondrial translocation and CYCS release are not conspicuously detected until later timepoints, by 2–4 d post-MPTP in vivo.10,11,13 Overall, our results indicate that BAX lysosomal translocation and LMP precede BAX mitochondrial translocation and MOMP in this experimental PD setting, both in vitro and in vivo.

Figure 2. BAX translocation to lysosomes and mitochondria following MPP+ treatment. (A) Colocalization of BAX (green) and lysosomes (immunostained with LAMP1 in red) in BE(2)-M17 cells treated with 250 μM MPP+ for either 3 h or 24 h (scale bars: 20 μm). (B) Quantification of BAX and LAMP1 colocalization calculated as the percentage of cytosolic area in which both signals colocalize. *P < 0.05, compared with untreated cells (0 h). (C) Colocalization of BAX (green) and mitochondria (immunostained with TOMM20 in red) in BE(2)-M17 cells treated with 250 μM MPP+ for either 3 h or 24 h (scale bars: 20 μm). (D) Quantification of BAX and TOMM20 colocalization calculated as the percentage of cytosolic area in which both signals colocalize. *P < 0.05, compared with untreated cells (0 h).
Remarkably, in some cellular settings unrelated to PD, LMP has been reported to precede and contribute to MOMP by a cathepsin-mediated cleavage of the BAX-activating protein BID (BH3 interacting domain death agonist) and subsequent enhancement of BAX mitochondrial translocation.25 It is thus possible that LMP might similarly contribute to BAX-dependent MOMP in our experimental PD settings. Ruling out this possibility, however, pharmacological inhibition of cathepsins did not interfere with BAX mitochondrial translocation and CYCS release in MPP+-treated cells (Fig. S3B). In addition, we have previously reported that BID is dispensable for MPTP-induced BAX activation and dopaminergic neurodegeneration.24 Therefore, while LMP is detected earlier than MOMP following MPTP/MPP+ treatment, MOMP seems to occur independently of LMP in this pathological situation. However, we cannot completely exclude the possibility of LMP and MOMP affecting each other in this model. In any event, both LMP and MOMP precede or coincide with dopaminergic cell death in these experimental PD settings.
Overall, our results indicate that, in addition to mitochondria, BAX translocates to, and internalizes into, lysosomal membranes early following MPTP treatment, both in vitro and in vivo. BAX lysosomal translocation parallels LMP and precedes MOMP and dopaminergic cell death in this model, thereby being compatible with lysosomal BAX playing a pathogenic role in this pathological experimental situation.
BAX induces LMP in purified brain lysosomes through its channel-forming activity
To determine whether BAX is required for MPTP-induced LMP, mutant mice deficient for BAX and their wild-type littermates were treated with MPTP as indicated above and euthanized 2 h after the last MPTP injection to assess the occurrence of LMP. In contrast to wild-type animals, ventral midbrain samples from BAX-deficient mice did not exhibit any leakage of lysosomal enzymes into the cytosol following MPTP treatment, indicating that BAX is indeed necessary for the permeabilization of lysosomal membranes induced by MPTP (Fig. 3A).

Figure 3. BAX mediates LMP in vivo and in purified brain lysosomes. (A) Enzymatic activities of lysosomal enzymes ACP2 (left panel) and CTSD (right panel) in cytosolic, lysosomal-free fractions from the ventral midbrain of MPTP-treated wild-type (WT) or BAX-deficient (KO) mice, 2 h post-MPTP, expressed as percentage of total enzymatic activity (lysosomal+cytosolic). *P < 0.05, compared with WT saline-injected mice; #P < 0.05, compared with WT MPTP-treated mice. (B) Enzymatic activities of ACP2 (left panel) and β-hexosaminidase (right panel) in supernatant fractions from fresh purified mouse brain lysosomes after incubation with recombinant BAX (100 nM for 30 min), in the presence or the absence of Bci (2 μM). (C) Enzymatic activities of ACP2 (left panel) and β-hexosaminidase (right panel) in supernatant fractions from fresh purified mouse brain lysosomes after incubation with either H2O2 (1 mM) or NH4Cl (40 mM) for 30 min, in the presence or the absence of Bci (2 μM). In all panels, data represent mean ± SEM. In (B and C), at least 3 independent experiments were performed. In (A), n = 4 animals per group.
Because the effect of BAX on MPTP-induced LMP could be indirect, we next determined whether BAX is able to directly permeabilize brain lysosomal membranes using purified mouse brain lysosomes. Incubation of freshly isolated intact mouse brain lysosomes with recombinant oligomeric BAX (100 nM for 30 min) resulted in the release of lysosomal enzymes ACP2 and β-hexosaminidase from lysosomal pellets to supernatant fractions (Fig. 3B), thus indicating the ability of BAX to directly permeabilize brain lysosomal membranes. To further demonstrate BAX capacity to directly induce LMP, additional experiments were performed using a compound capable of blocking the channel-forming activity of BAX in lipid membranes. This compound, named BAX channel inhibitor (Bci), is a member of a family of molecules possessing specific BAX channel inhibitory activity that were previously identified by medium-throughput screening from a large collection of low molecular weight compounds to inhibit BAX-induced permeabilization in liposomes, planar lipid membranes, and purified mitochondria.26,27 From a mechanistic point of view, this family of compounds has been shown by electrophysiology to function as genuine channel blockers without affecting BAX conformational changes required for BAX oligomerization and activation.26 In addition, the activity of these compounds is specific for BAX, as indicated by: i) their absence of activity on a subset of receptors, ion transporters, and channels, such as VDAC1 (voltage-dependent anion channel 1), SCNs (sodium channel, voltage gated), Na+-K+ ATPase or SLC9/Na+-H+ antiporter26 and ii) their lack of protective effects in BAX-deficient cells.26 In purified mouse brain lysosomes, treatment with Bci markedly attenuated BAX-induced LMP (Fig. 3B), thereby indicating that the latter occurred through BAX channel-forming activity on lysosomal membranes. In contrast, Bci was unable to attenuate LMP caused by other known BAX-independent LMP inducers such as H2O2 or NH4Cl (Fig. 3C). Taken together, our results indicate that BAX is able to directly permeabilize brain lysosomes through its channel-forming properties.
BAX channel inhibition prevents the pathogenic effects of PD-linked LMP and MOMP in vitro
After having demonstrated in purified brain lysosomes the ability of BAX to directly permeabilize lysosomal membranes, we next explored whether BAX may be responsible for LMP in a PD-related in vitro setting. Similar to BE(2)-M17 cells, MPP+ induces LMP in human dopaminergic SH-SY5Y neuroblastoma cells, as indicated by increased ectopic activities of lysosomal enzymes ACP2 and β-hexosaminidase in lysosome-free cytosolic fractions extracted from these cells (Fig. 4A and B). We have previously shown by a combination of different experimental approaches that MPP+-induced LMP disrupts lysosomal integrity and leads to a decreased number of lysosomes, impaired AP-lysosome fusion and defective clearance and subsequent accumulation of undegraded AP.3,7 In agreement with our previous observation, MPP+-induced LMP in SH-SY5Y cells is associated with: i) a reduced amount of lysosomes, as illustrated by decreased immunoblot levels of lysosomal structural marker LAMP1 (Fig. 4C) and ii) accumulation of undegraded AP, as shown by increased immunoblot levels of the AP marker microtubule-associated protein 1A/1B-light chain 3-II (LC3-II) (Fig. 4D). MPP+-induced LMP, lysosomal deficiency, and AP accumulation were all prevented by Bci, thereby indicating their dependence on BAX channel-forming activity (Fig. 4A–D).

Figure 4. Pharmacological inhibition of BAX channel activity in MPP+-treated cells. (A andB) Enzymatic activities of (A) ACP2 and (B) β-hexosaminidase in lysosomal-free cytosolic fractions from MPP+-treated SH-SY5Y cells, in the presence or the absence of Bci. (C) LAMP1 immunoblot levels in total protein homogenates from MPP+-treated cells, in the presence or the absence of Bci. (D) LC3-II immunoblot levels in total protein homogenates from MPP+-treated SH-SY5Y cells, in the presence or the absence of Bci. (E) ACP2 activity in lysosomal-free cytosolic fractions from MPP+-treated SH-SY5Y cells, in the presence or the absence of Bci and/or Tempol. (F) Immunoblot levels of CYCS in cytosolic and mitochondrial fractions from MPP+-treated SH-SY5Y cells, in the presence or the absence of Bci. (G) Quantification of cell death by flow cytometry following propidium iodide staining in MPP+-treated SH-SY5Y cells, in the presence or the absence of Bci. (H) Cell death quantified by flow cytometry after propidium iodide staining in MPP+-treated SH-SY5Y cells in the presence or the absence of leupeptin/E64d and/or pan-caspase inhibitor z-VAD-fmk. In all panels, data represent mean ± SEM from at least 3 independent experiments. Treatments were performed for 24 h (MPP+, 250 μM; Bci, 2 μM; Tempol, 500 μM; leupeptin, 100 μM; E64d, 10 μg/μl; z-VAD-fmk, 50 μM). *P < 0.05, compared with control cells, #P < 0.05 compared with MPP+-treated cells, $P < 0.05 compared with MPP+-intoxicated cells treated with either leupeptin/E64d or z-VAD-fmk.
Among the different stimuli reported to induce LMP in other experimental situations, one of the most frequently implicated has been increased production of reactive oxygen species (ROS).25 Enhanced ROS production, in turn, is a major pathogenic event following complex I inhibition with MPP+/MPTP.23,28 It is thus possible that ROS may be also directly contributing to LMP in our experimental system. Along this line, we have previously reported that MPP+-induced LMP in cultured cells occurred downstream of increased mitochondria-derived ROS production.3 The latter observation, however, is also compatible with a direct effect of BAX on PD-linked LMP, since: i) MPTP-induced BAX activation also occurs downstream of increased mitochondria-derived ROS production, as it relies on the coordinated activation of TRP53 (tumor protein p53) and MAPK/c-Jun N-terminal kinase molecular pathways secondary to mitochondria-dependent oxidative DNA damage;10,11 ii) a combined treatment with Bci and the free-radical scavenger superoxide dismutase mimetic compound tempol did not further attenuate MPP+-induced LMP compared with the administration of either compound separately, thereby indicating that ROS and BAX are acting within the same molecular pathway to induce LMP (Fig. 4E); iii) Bci is able to prevent BAX-induced, but not ROS-induced, LMP in purified brain lysosomes (Fig. 3). Taken together, these observations support a pathogenic scenario in which PD-linked LMP is being directly mediated by BAX, with ROS acting upstream of BAX activation. However, the possibility that part of the MPP+-derived ROS might also be directly contributing to LMP in this model cannot be completely ruled out.
In addition to LMP, MPP+ also induces MOMP in SH-SY5Y cells, as indicated by the release of mitochondrial CYCS to the cytosol in these cells (Fig. 4F). Consistent with its known dependence on BAX activity, MPP+-induced CYCS release was also markedly attenuated by Bci (Fig. 4F). Taken together, our results indicate that BAX underlies not only mitochondrial permeabilization, as was previously known, but also lysosomal permeabilization caused by MPP+. Because both LMP and MOMP can individually lead to cell death (i.e., by the release of cathepsins and CYCS, respectively), the pharmacological inhibition of LMP and MOMP by Bci in MPP+-treated cells is associated with an overall attenuation of MPP+-induced cell death, as measured by flow cytometry after propidium iodide staining (Fig. 4G). While the relative contribution of either LMP or MOMP to overall cell death may be difficult to decipher in this experimental setting, in which both pathogenic events occur, a combined treatment with cathepsin inhibitors (leupeptin/E64d) and a pan-caspase inhibitor (zVAD-fmk) resulted in a greater attenuation of MPP+-induced cell death than the administration of either compound separately (Fig. 4H), thereby indicating that both LMP and MOMP are contributing to overall MPP+-induced cell death.
Our results indicate that BAX channel activity, in addition to MOMP, also underlies PD-linked LMP in an in vitro experimental setting. Importantly, pharmacological inhibition of BAX channel-forming activity is able to prevent both LMP and MOMP, restore lysosomal deficiency, reverse AP accumulation, and attenuate overall cell death in this experimental system.
BAX channel inhibition restores lysosomal deficiency and nigrostriatal dopaminergic neurodegeneration in MPTP-treated mice
To determine the relevance of our in vitro results to an in vivo situation, we next assessed the effects of Bci in MPTP-treated mice. To this purpose, Bci was administered stereotaxically into the brain of MPTP-treated mice by means of a permanent cannula placed in the right lateral ventricle, which allowed repeated administrations of the compound. Bci was administered twice, by means of an infusion pump, at 2 and 48 h after the last MPTP injection, in order to cover the extent of MPTP-induced BAX activation, LMP and MOMP in this model, all of which occur within the first 4 d post-MPTP.3,10,11,13 Animals were euthanized either at early time-points (i.e., within the first 4 d post-MPTP) to assess LMP and MOMP or at later time-points (i.e., 21 d post-MPTP, once the dopaminergic lesion is stabilized in this model29) to assess dopaminergic nigrostriatal denervation. Similar to the in vitro results reported above, Bci was able to prevent both LMP and MOMP in MPTP-treated mice as indicated, respectively, by decreased releases of lysosomal CTSB and mitochondrial CYCS into cytosolic ventral midbrain fractions of these animals (Fig. 5A and B). Supporting a pathogenic role for BAX channel-mediated LMP on MPTP-induced lysosomal deficiency, Bci treatment restored lysosomal levels and reversed AP accumulation in these animals, as assessed by LAMP1 and LC3-II immunoblotting, respectively (Fig. 5C and D). In addition, blockade of LMP and MOMP by Bci in MPTP-treated mice was associated with a significant attenuation of overall dopaminergic neurodegeneration, both at the level of SNpc dopaminergic neuron cell bodies as well as striatal dopaminergic terminals, as determined by stereological neuron cell counts and optical densitometry, respectively (Fig. 5E).

Figure 5. Pharmacological inhibition of BAX channel activity in MPTP-treated mice. (A and B) Immunoblot levels of (A) lysosomal-processed CTSB and (B) CYCS in cytosolic ventral midbrain fractions from saline- and MPTP-injected mice, treated or not with Bci, at day 4 (4 d) after the last MPTP injection. (C and D) Immunoblot levels of (C) LAMP1 and (D) LC3-II in total protein fractions from the ventral midbrain of saline- and MPTP-injected mice, treated or not with Bci, at day 1 (1 d) after the last MPTP injection. (E) Quantification of substantia nigra pars compacta (SNpc) TH-positive neurons by stereology (left panel) and assessment of dopaminergic innervation in the striatum (Str) by optical densitometry (right panel) in saline- and MPTP-injected mice, treated or not with Bci, at day 21 (21 d) after the last MPTP injection. On average, n = 7 for saline-injected (control) mice, n = 12 for MPTP-injected mice, n = 7 for MPTP + Bci-treated mice. Data represent mean ± SEM *P < 0.05, compared with saline-injected animals, #P < 0.05 compared with MPTP-injected animals. Scale bars: 500 μm.
These results indicate that BAX channel activity underlies PD-linked LMP in vivo and demonstrate the feasibility and therapeutic potential of pharmacologically targeting BAX-induced membrane permeabilization, both at the level of lysosomes and mitochondria, in an in vivo situation related to PD.
Discussion
Abnormal permeabilization of lysosomal and mitochondrial membranes has been previously identified as early pathogenic features of PD, contributing to both autophagy impairment and SNpc dopaminergic neurodegeneration occurring in this disease.3,7,8,10,11 However, the feasibility and therapeutic potential of pharmacologically targeting PD-linked LMP and MOMP had remained so far unknown due in part to the lack of specific molecular pharmacological tools and the incomplete knowledge on the molecular mechanisms underlying LMP.
Here we show that the pro-cell death agonist BAX, in addition to its known role in permeabilizing mitochondrial membranes, is also able to permeabilize brain lysosomes in experimental PD-related models, both in vitro and in vivo. Furthermore, we reveal that the ability of BAX to permeabilize brain lysosomal membranes relies on its channel-forming capacity. More importantly, from a potential therapeutic point of view, we demonstrated that pharmacological blockade of BAX channel-mediated lysosomal and mitochondrial permeabilization results in an overall attenuation of PD-linked lysosomal alterations and dopaminergic neurodegeneration, both in vitro and in vivo (Fig. 6). Remarkably, in contrast to many published neuroprotective studies in MPTP-treated mice in which therapeutic manipulations were performed prior to, or concomitant with, MPTP treatment, here we show that the therapeutic in vivo effects of Bci are obtained when this compound is administered post-MPTP treatment, once the pathogenic neuronal changes are already in motion, thereby mimicking a situation more comparable to PD.
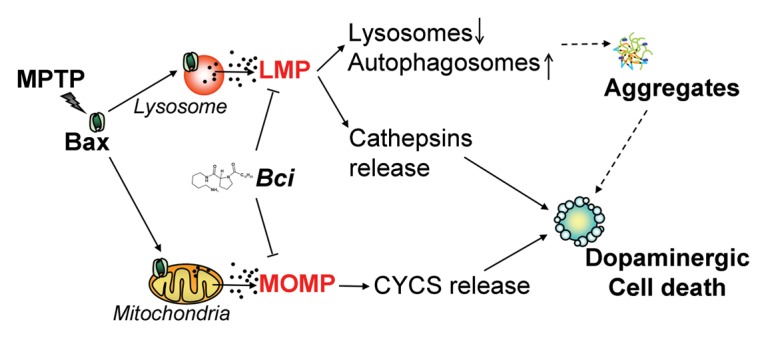
Figure 6. Proposed lysosomal and mitochondrial pathogenic role of BAX and therapeutic action of Bci in experimental PD. Early following MPTP treatment, activated BAX translocates to both lysosomal and mitochondrial membranes causing LMP and MOMP, respectively. BAX-induced LMP leads to a decreased number of lysosomes and subsequent accumulation of undegraded AP, eventually leading to the formation of protein aggregates. Besides impairing lysosomal-mediated degradation, BAX-induced LMP directly contributes to dopaminergic cell death by the leakage of lysosomal proteases into the cytosol, some of which, such as CTSB and CTSD, can remain active at neutral cytosolic pH and cause the digestion of vital proteins or the activation of additional hydrolases, including caspases. In turn, MOMP results in CYCS release and activation of caspase-dependent dopaminergic cell death. Pharmacological blockade of BAX channel activity with Bci is able to prevent both LMP and MOMP, restore lysosomal levels, reverse AP accumulation, and attenuate overall nigrostriatal dopaminergic neurodegeneration caused by MPTP. See main text for details. Dashed arrows correspond to currently unknown mechanisms.
While LMP dependence on BAX activation was previously observed in some non-neuronal cell systems and purified liver lysosomes,17-19 a potential direct effect of BAX on lysosomal membranes has remained so far controversial.20 In addition, whether such an effect of BAX could also occur in brain lysosomes and in an in vivo situation had not been previously addressed. In human fibroblasts, liver cells, and KMCH cholangiocarcinoma cell lines exposed to various pro-apoptotic stimuli, BAX was shown to immunocolocalize with lysosomes, which was attributed to a translocation and possible direct effect of BAX on lysosomal membranes.17-19 However, other studies failed to convincingly demonstrate a lysosomal localization of BAX during apoptosis and speculated instead about the possibility of lysosomal BAX actually corresponding to an autophagic sequestration of BAX-containing damaged mitochondria within the lysosomal lumen.20 Here, using purified lysosomal membrane and lumenal fractions, we demonstrate that BAX is translocated and internalized into lysosomal membranes (and not just passively located in the lysosomal lumen) following MPP+/MPTP treatment, which is compatible with BAX playing a direct permeabilizing role on lysosomal membranes. Further supporting the latter, we also show here that i) recombinant BAX is able to permeabilize purified brain lysosomes in the absence of any other factor, ii) pharmacological inhibition of BAX channel activity with Bci restores LMP induced by either recombinant BAX (but not other stimuli) in purified brain lysosomes or by MPP+/MPTP in cell cultures and mice, and iii) MPTP-induced LMP is prevented by genetic ablation of BAX in mutant mice. Taken together, these results strongly support the concept that BAX is directly permeabilizing brain lysosomes in this experimental PD situation.
Remarkably, BAX-dependent LMP was detected earlier than BAX-mediated MOMP in MPTP-treated mice, which is compatible with LMP occurring upstream of MOMP in this experimental model. This observation concurs with previous reports in other experimental in vitro paradigms, such as TP53 (tumor protein p53)- and TNFSF10/TRAIL (tumor necrosis factor [ligand] superfamily, member 10)-induced apoptosis in non-neuronal cell lines,30,31 and may reflect an apparent higher lability of lysosomal membranes compared with mitochondria.25 Alternatively, LMP and MOMP may be occurring concomitantly downstream of BAX activation, but the sensitivity of the techniques used to detect LMP (i.e., enzymatic activities of ACP2 and β-hexosaminidase) and MOMP (i.e., immunoblot of CYCS) may be different. In any event, the relationship between LMP and MOMP has proven complex and dependent on the pathological situation.32,33 In some cellular settings, LMP precedes and contributes to the activation of mitochondria-dependent apoptosis25 whereas in other situations LMP occurs downstream of mitochondrial permeabilization and it amplified rather than initiated the apoptotic process.20 In our study, PD-linked LMP and MOMP occurred independently of each other downstream of BAX activation and both preceded and contributed to overall dopaminergic cell death.
The results presented here also shed light on the molecular mechanisms underlying BAX-induced membrane permeabilization in the context of PD, which may have important therapeutic implications for the development of new therapeutic tools. In relation to BAX-mediated MOMP, 2 potential distinct mechanisms have been generally proposed, one involving the opening of the so-called mitochondrial permeability transition pore complex (PTPC) and another dependent on the formation of BAX-derived channels directly into mitochondrial membranes.12 We had previously observed that BAX-induced CYCS release in MPTP-treated mice was independent of the PTPC, as it was unresponsive to the PTPC blocker cyclosporin A and to the genetic ablation of one of the critical components of the PTPC, PPID/cyclophilin D.11 Here, our results with Bci indicate that BAX-induced membrane permeabilization, in both mitochondria and lysosomes, relies instead on the formation of BAX-derived channels directly into mitochondrial and lysosomal membranes. Along this line, it has been reported that BAX channel activity can be enhanced at acidic pH,26 which further strengthens a potential role of BAX-derived channels on lysosomes, as the latter are acidic organelles. In this context, it would be interesting to determine whether pro-survival BCL2 (B-cell CLL/lymphoma 2) proteins, the overexpression of which is able to protect against MPTP-induced dopaminergic neurodegeneration,34-36 could reverse BAX lysosomal localization by retrotranslocation to the cytosol, as has been shown for mitochondrial BAX.37
In summary, our study indicates that the pathogenic effects of BAX in the context of PD are much more widespread than initially thought, as they are not limited to MOMP and activation of mitochondria-dependent apoptotic pathways, as has been previously reported,10,11,13,21 but also involve the pathogenic permeabilization of lysosomal membranes, the latter underlying the lysosomal defects occurring in this pathological situation and also contributing to overall dopaminergic cell death. Supporting the potential relevance of this scenario to PD, BAX activation, lysosomal disruption, AP accumulation, and mitochondrial permeabilization have all been observed in PD patients.2-6,14-16,38 In addition, other experimental animal models of PD, such as the one produced by viral vector-mediated overexpression of SNCA in the substantia nigra of adult rats,39 exhibit decreased lysosomal levels2 that can be restored by overexpression of TFEB,9 BECN19 or lysosomal-associated membrane protein 2A (LAMP2A),40 all of which results in an overall attenuation of dopaminergic cell death.9,40 From a potential therapeutic point of view, we demonstrate here the feasibility and beneficial effects of pharmacologically targeting BAX-induced membrane permeabilization, both at the level of lysosomes and mitochondria, in the context of PD. In particular, our results point to small molecules able to block the channel-forming activity of BAX, or their derivatives, as new molecular tools with potential therapeutic interest to attenuate both lysosomal impairment and neurodegeneration linked to PD.
Materials and Methods
Cell cultures
Human neuroblastoma cell line BE(2)-M17 was provided by B Wolozin (Boston University School of Medicine) and was grown in Opti-MEM (Life Technologies, 31985-047) plus 10% fetal bovine serum (Sigma-Aldrich, D0899) supplemented with 1% penicillin/streptomycin (Sigma-Aldrich, P0781). SH-SY5Y human dopaminergic neuroblastoma cells were obtained from ATCC (CRL-2266) and maintained at 37 °C in 5% CO2 in MEM/Ham’s F12 medium (Sigma-Aldrich, N4888), supplemented with 10% fetal calf serum (Life Technologies, 26140-079) and 1% penicillin/streptomycin. For drug treatments, cells were grown at 70–80% confluence and treated for either 3 or 24 h, as indicated in the text. SH-SY5Y cells were treated with 250 μM MPP+ (Sigma-Aldrich, D048) in the presence or the absence of a 2 μM BAX channel inhibitor compound (Bci, provided by B.A., Merck-Serono) and/or 500 μM Tempol (Sigma-Aldrich, 176141), and in the presence or the absence of 100 μM leupeptin (Sigma-Aldrich, L2884) plus 10 μg/μl E64d (Sigma-Aldrich, E8640) and/or pan-caspase inhibitor 50 μM z-VAD-fmk (Promega, G7232). Cell death was quantified by flow cytometry after propidium iodide staining, as previously described.3
Animals
Eight- to 10-wk-old male C57Bl/6 mice received 1 intraperitoneal injection of MPTP-HCl per day (30 mg/kg/day of free base; Sigma-Aldrich, M0896) for 5 consecutive d. Control mice received saline injections only. For stereotaxic delivery of Bci in selected experimental groups, a permanent cannula was placed in the right lateral ventricle (AP: 0.26; L: -0.75; DV: -2.5). At 2 and 48 h after the last MPTP injection, 2.5 µl of a 39.2 mM solution (in saline) of Bci was administered at a flow rate of 1 µl/min by means of an infusion pump. Mice were euthanized at different time-points after MPTP injections, as indicated in the text. An average of 7 to 12 mice was used in each group.
Subcellular fractionation
i) Purification of lysosomes from mouse ventral midbrain
Fresh ventral midbrain samples from 9 saline- or MPTP-treated mice were dissected and pooled avoiding freezing. The tissue was homogenized in buffered sucrose (Sigma-Aldrich S9378; 4 mM HEPES, 150 mM sucrose, pH 7.4) and following differential centrifugation the cytosolic fraction (supernatant fraction after 100,000 × g centrifugation) and the light mitochondrial-lysosomal enriched fraction were obtained. Pure lysosomal and mitochondrial fractions were isolated from light mitochondrial-lysosomal enriched fractions after discontinuous Nycodenz (Progen Biotechnik, 1002424) density gradient centrifugation following the procedure described elsewhere.41 Lysosomal lumenal contents and membranes were isolated after hypotonic shock and centrifugation at 100,000 × g for 30 min.41 For selected experiments with total mouse brain homogenates, lysosomal purification was performed as previously described.3
ii) Purification of lysosomes from cell lines
Lysosomes from cultured cells were isolated as described.42 Briefly, collected cells were washed in sucrose 0.25 M (pH 7.2) and disrupted in a nitrogen cavitation chamber (Kontes Glass Company, NJ, USA), followed by homogenization in a Teflon-glass homogenizer and centrifugation (2,500 × g for 15 min); the mitochondria-lysosomal enriched fraction was collected after 17,000 × g centrifugation and the cytosolic fraction was collected as the supernatant fraction after 100,000 × g centrifugation. The lysosomal-mitochondrial enriched fraction was then loaded in 2 subsequent discontinuous Nycodenz, sucrose or percoll (GE Healthcare, 17-0891-01) density gradient from which lysosomal, heavy mitochondrial and light mitochondrial fractions were isolated.42
iii) Purification of mitochondria from cell lines and mouse brains
In cultured cells, additional protein extractions of mitochondrial and cytosolic fractions were performed with the Mitochondria Isolation Kit for cultured cells (Pierce, 89874), following manufacturer recommendations, or as described in section ii above. In mice, mitochondrial and cytosolic fractions were obtained at the indicated time points from fresh ventral midbrain tissue from saline- and MPTP-injected mice, as previously described.10,11
In all protocols, the purity of the different fractions was confirmed by immunoblot with lysosomal (LAMP1, CTSB) or mitochondrial (HSPD1, COX4I1) markers and by electron microscopy.
Antibodies
The following antibodies were used: ACTB/β-actin (mouse monoclonal, Sigma-Aldrich, A5441), BAX (rabbit polyclonal, Cell Signaling, 2772), BAX 6A7 (mouse monoclonal, Abcam, ab5714), CTSB/cathepsin B (rabbit polyclonal, Calbiochem, 219408), CYCS/cytochrome c (mouse monoclonal, BD PharMingen, 556433), COX4I1/cytochrome c oxidase subunit IV (mouse monoclonal, Molecular Probes, A21348), HSPD1/HSP60 (goat polyclonal, Santa Cruz, sc-1052), LAMP1 (mouse monoclonal, DSHB, 1D4B or rabbit polyclonal, Genetex, GTX19294), LC3 (rabbit polyclonal, Novus Biologicals, NB100-2220), TH/tyrosine hydroxylase (rabbit polyclonal, Calbiochem, 657012).
β-hexosaminidase and acid phosphatase enzymatic assays
β-Hexosaminidase activity was measured as described.42 Briefly, pre-warmed (37 °C) substrate (8 mM 4-methyllumbelliferyl-N-acetyl-B-D-glucopyranoside) solution and sample (cytosolic or lysosomal fractions) were mixed in a 96-well plate and incubated at 37 °C for 30 min. The reaction was stopped by adding stop solution (2 M glycine, 2 M Na2CO3) and absorption was measured at 405 nm. ACP2/acid phosphatase and CTSD activities of the cytosolic and lysosomal fraction were determined with the Acid Phosphatase Assay Kit from Sigma (CS0740) and the Cathepsin D Activity Assay Kit from Abcam (ab65302), respectively, according to the manufacturer’s instructions.
Alkaline extraction
Lysosomes isolated from cultured cells were broken by hypotonic shock as described above and lysosomal membranes were incubated in MOPS buffer (10 mM MOPS, pH 7.3, 0.3 M sucrose) or 0.1 M Na2CO3 (pH 11.5) for 30 min on ice and centrifuged at 100,000 × g for 30 min and pellet and supernatant fractions were separately recovered.
Immunocytochemistry
BE(2)-M17 cells grown on coverslips were fixed with a 3% formaldehyde solution, blocked, and then incubated with the indicated primary and corresponding fluorescence-conjugated secondary antibodies. Images were acquired with an Espectral FV1000 Olympus confocal microscope. Quantification of the colocalization was performed using ImageJ (NIH).
Electron microscopy
Isolated pure lysosomal and mitochondrial fractions were pelleted and fixed in sodium cacodylate buffer (0.1 M; pH 7.4), 2.5% glutaraldehyde, and 0.25 M sucrose at 4 °C, then post-fixed in 1% osmium tetroxide for 1 h at 4 °C. The samples were dehydrated, embedded in plastic and cut into in 70-nm sections for microscopy. Sections were subsequently post-stained with 5% uranyl acetate and viewed with a JEOL JEM 1010 electron microscope.
Quantitative morphology
The total number of TH-positive SNpc neurons was counted in the different groups of mice at the indicated timepoints after the last MPTP or saline injection by stereology using the optical fractionator method (StereoInvestigator; MBF Bioscience, Williston, VT), as previously described.11 The extent of striatal dopaminergic denervation was assessed by optical densitometry in TH-immunostained striatal sections.11
Statistical analysis
All values are expressed as the mean ± SEM. Differences among means were analyzed with one-way ANOVA. When ANOVA showed significant differences, pairwise comparisons between means were tested by Student-Newman-Keuls post hoc testing. In all analyses, the null hypothesis was rejected at the 0.05 level.
Supplementary Material
Glossary
Abbreviations:
- AP
autophagosome
- BAX
BCL2-associated X protein
- Bci
BAX channel inhibitor
- LAMP1
lysosomal-associated membrane protein 1
- LC3-II
microtubule-associated protein 1A/1B-light chain 3-II
- LMP
lysosomal membrane permeabilization
- MOMP
mitochondrial outer membrane permeabilization
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PD
Parkinson disease
- PTPC
permeability transition pore complex
- ROS
reactive oxygen species
- SNpc
substantia nigra pars compacta
- TH
tyrosine hydroxylase
- TOMM20
translocase of outer mitochondrial membrane 20 homolog (yeast)
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank A Parent, E Pérez, B Rodríguez and M Humà for their technical assistance and P Boya (CIB-CSIC, Spain) for her insightful comments. This work was supported by European Commission Marie Curie Excellence Grant and Marie Curie International Reintegration Grant (MV), Fundació la Caixa, Spain (MV), FIS-ISCIII, Spain (MV, JB, MM-V, CP), MICINN, Spain (MV, MM-V), and Ramón y Cajal Program from MICINN (CP). BD received a fellowship award from the Foundation Bettencourt-Schueller (France).
References
- 1.Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M, Bezard E. . Lysosomal impairment in Parkinson’s disease. Mov Disord 2013; 28:725 - 32; http://dx.doi.org/ 10.1002/mds.25462; PMID: 23580333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. . Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 2009; 35:385 - 98; http://dx.doi.org/ 10.1016/j.nbd.2009.05.023; PMID: 19505575 [DOI] [PubMed] [Google Scholar]
- 3.Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, Vila M. . Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci 2010; 30:12535 - 44; http://dx.doi.org/ 10.1523/JNEUROSCI.1920-10.2010; PMID: 20844148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. . Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 2012; 109:9611 - 6; http://dx.doi.org/ 10.1073/pnas.1112368109; PMID: 22647602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. . Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci 2012; 32:4240 - 6; http://dx.doi.org/ 10.1523/JNEUROSCI.5575-11.2012; PMID: 22442086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sánchez-Danés A, Richaud-Patin Y, Carballo-Carbajal I, Jiménez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, et al. . Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 2012; 4:380 - 95; http://dx.doi.org/ 10.1002/emmm.201200215; PMID: 22407749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vila M, Bové J, Dehay B, Rodríguez-Muela N, Boya P. . Lysosomal membrane permeabilization in Parkinson disease. Autophagy 2011; 7:98 - 100; http://dx.doi.org/ 10.4161/auto.7.1.13933; PMID: 21045565 [DOI] [PubMed] [Google Scholar]
- 8.Bové J, Martínez-Vicente M, Vila M. . Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 2011; 12:437 - 52; http://dx.doi.org/ 10.1038/nrn3068; PMID: 21772323 [DOI] [PubMed] [Google Scholar]
- 9.Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Björklund A. . TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci U S A 2013; 110:E1817 - 26; http://dx.doi.org/ 10.1073/pnas.1305623110; PMID: 23610405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perier C, Tieu K, Guégan C, Caspersen C, Jackson-Lewis V, Carelli V, Martinuzzi A, Hirano M, Przedborski S, Vila M. . Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci U S A 2005; 102:19126 - 31; http://dx.doi.org/ 10.1073/pnas.0508215102; PMID: 16365298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perier C, Bové J, Wu DC, Dehay B, Choi DK, Jackson-Lewis V, Rathke-Hartlieb S, Bouillet P, Strasser A, Schulz JB, et al. . Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson’s disease. Proc Natl Acad Sci U S A 2007; 104:8161 - 6; http://dx.doi.org/ 10.1073/pnas.0609874104; PMID: 17483459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galluzzi L, Blomgren K, Kroemer G. . Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci 2009; 10:481 - 94; http://dx.doi.org/ 10.1038/nrn2665; PMID: 19543220 [DOI] [PubMed] [Google Scholar]
- 13.Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S. . Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A 2001; 98:2837 - 42; http://dx.doi.org/ 10.1073/pnas.051633998; PMID: 11226327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, et al. . Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc Natl Acad Sci U S A 2000; 97:2875 - 80; http://dx.doi.org/ 10.1073/pnas.040556597; PMID: 10688892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartmann A, Michel PP, Troadec JD, Mouatt-Prigent A, Faucheux BA, Ruberg M, Agid Y, Hirsch EC. . Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson’s disease?. J Neurochem 2001; 76:1785 - 93; http://dx.doi.org/ 10.1046/j.1471-4159.2001.00160.x; PMID: 11259496 [DOI] [PubMed] [Google Scholar]
- 16.Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK. . Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease. J Neurosci 2001; 21:9519 - 28; PMID: 11739563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. . Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol 2006; 290:G1339 - 46; http://dx.doi.org/ 10.1152/ajpgi.00509.2005; PMID: 16484678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Werneburg NW, Guicciardi ME, Bronk SF, Kaufmann SH, Gores GJ. . Tumor necrosis factor-related apoptosis-inducing ligand activates a lysosomal pathway of apoptosis that is regulated by Bcl-2 proteins. J Biol Chem 2007; 282:28960 - 70; http://dx.doi.org/ 10.1074/jbc.M705671200; PMID: 17686764 [DOI] [PubMed] [Google Scholar]
- 19.Kågedal K, Johansson AC, Johansson U, Heimlich G, Roberg K, Wang NS, Jürgensmeier JM, Ollinger K. . Lysosomal membrane permeabilization during apoptosis--involvement of Bax?. Int J Exp Pathol 2005; 86:309 - 21; http://dx.doi.org/ 10.1111/j.0959-9673.2005.00442.x; PMID: 16191103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oberle C, Huai J, Reinheckel T, Tacke M, Rassner M, Ekert PG, Buellesbach J, Borner C. . Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ 2010; 17:1167 - 78; http://dx.doi.org/ 10.1038/cdd.2009.214; PMID: 20094062 [DOI] [PubMed] [Google Scholar]
- 21.Perier C, Bové J, Vila M. . Mitochondria and programmed cell death in Parkinson’s disease: apoptosis and beyond. Antioxid Redox Signal 2012; 16:883 - 95; http://dx.doi.org/ 10.1089/ars.2011.4074; PMID: 21619488 [DOI] [PubMed] [Google Scholar]
- 22.Perier C, Vila M. . Mitochondrial biology and Parkinson’s disease. Cold Spring Harb Perspect Med 2012; 2:a009332; http://dx.doi.org/ 10.1101/cshperspect.a009332; PMID: 22355801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dauer W, Przedborski S. . Parkinson’s disease: mechanisms and models. Neuron 2003; 39:889 - 909; http://dx.doi.org/ 10.1016/S0896-6273(03)00568-3; PMID: 12971891 [DOI] [PubMed] [Google Scholar]
- 24.Ramonet D, Perier C, Recasens A, Dehay B, Bové J, Costa V, Scorrano L, Vila M. . Optic atrophy 1 mediates mitochondria remodeling and dopaminergic neurodegeneration linked to complex I deficiency. Cell Death Differ 2013; 20:77 - 85; http://dx.doi.org/ 10.1038/cdd.2012.95; PMID: 22858546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boya P. . Lysosomal function and dysfunction: mechanism and disease. Antioxid Redox Signal 2012; 17:766 - 74; http://dx.doi.org/ 10.1089/ars.2011.4405; PMID: 22098160 [DOI] [PubMed] [Google Scholar]
- 26.Hetz C, Vitte PA, Bombrun A, Rostovtseva TK, Montessuit S, Hiver A, Schwarz MK, Church DJ, Korsmeyer SJ, Martinou JC, et al. . Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem 2005; 280:42960 - 70; http://dx.doi.org/ 10.1074/jbc.M505843200; PMID: 16219766 [DOI] [PubMed] [Google Scholar]
- 27.Peixoto PM, Ryu SY, Bombrun A, Antonsson B, Kinnally KW. . MAC inhibitors suppress mitochondrial apoptosis. Biochem J 2009; 423:381 - 7; http://dx.doi.org/ 10.1042/BJ20090664; PMID: 19691447 [DOI] [PubMed] [Google Scholar]
- 28.Perier C, Bové J, Dehay B, Jackson-Lewis V, Rabinovitch PS, Przedborski S, Vila M. . Apoptosis-inducing factor deficiency sensitizes dopaminergic neurons to parkinsonian neurotoxins. Ann Neurol 2010; 68:184 - 92; PMID: 20695011 [DOI] [PubMed] [Google Scholar]
- 29.Jackson-Lewis V, Przedborski S. . Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc 2007; 2:141 - 51; http://dx.doi.org/ 10.1038/nprot.2006.342; PMID: 17401348 [DOI] [PubMed] [Google Scholar]
- 30.González-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquère S, Eskelinen EL, Pierron G, Saftig P, Kroemer G. . The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci 2005; 118:3091 - 102; http://dx.doi.org/ 10.1242/jcs.02447; PMID: 15985464 [DOI] [PubMed] [Google Scholar]
- 31.Kroemer G, Levine B. . Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004 - 10; http://dx.doi.org/ 10.1038/nrm2529; PMID: 18971948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Offen D, Beart PM, Cheung NS, Pascoe CJ, Hochman A, Gorodin S, Melamed E, Bernard R, Bernard O. . Transgenic mice expressing human Bcl-2 in their neurons are resistant to 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine neurotoxicity. Proc Natl Acad Sci U S A 1998; 95:5789 - 94; http://dx.doi.org/ 10.1073/pnas.95.10.5789; PMID: 9576963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang L, Matthews RT, Schulz JB, Klockgether T, Liao AW, Martinou JC, Penney JB Jr., Hyman BT, Beal MF. . 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyride neurotoxicity is attenuated in mice overexpressing Bcl-2. J Neurosci 1998; 18:8145 - 52; PMID: 9763461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dietz GP, Stockhausen KV, Dietz B, Falkenburger BH, Valbuena P, Opazo F, Lingor P, Meuer K, Weishaupt JH, Schulz JB, et al. . Membrane-permeable Bcl-xL prevents MPTP-induced dopaminergic neuronal loss in the substantia nigra. J Neurochem 2008; 104:757 - 65; PMID: 17995935 [DOI] [PubMed] [Google Scholar]
- 35.Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N, Youle RJ. . Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011; 145:104 - 16; http://dx.doi.org/ 10.1016/j.cell.2011.02.034; PMID: 21458670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vila M, Przedborski S. . Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci 2003; 4:365 - 75; http://dx.doi.org/ 10.1038/nrn1100; PMID: 12728264 [DOI] [PubMed] [Google Scholar]
- 37.Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Björklund A. . Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci 2002; 22:2780 - 91; PMID: 11923443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, Kirik D, Stefanis L. . Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013; 136:2130 - 46; http://dx.doi.org/ 10.1093/brain/awt131; PMID: 23757764 [DOI] [PubMed] [Google Scholar]
- 39.Yuan XM, Li W, Dalen H, Lotem J, Kama R, Sachs L, Brunk UT. . Lysosomal destabilization in p53-induced apoptosis. Proc Natl Acad Sci U S A 2002; 99:6286 - 91; http://dx.doi.org/ 10.1073/pnas.092135599; PMID: 11959917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Werneburg NW, Guicciardi ME, Bronk SF, Gores GJ. . Tumor necrosis factor-alpha-associated lysosomal permeabilization is cathepsin B dependent. Am J Physiol Gastrointest Liver Physiol 2002; 283:G947 - 56; PMID: 12223355 [DOI] [PubMed] [Google Scholar]
- 41.Cuervo AM, Dice JF, Knecht E. . A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem 1997; 272:5606 - 15; http://dx.doi.org/ 10.1074/jbc.272.9.5606; PMID: 9038169 [DOI] [PubMed] [Google Scholar]
- 42.Storrie B, Madden EA. . Isolation of subcellular organelles. Methods Enzymol 1990; 182:203 - 25; http://dx.doi.org/ 10.1016/0076-6879(90)82018-W; PMID: 2156127 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.