

Abstract
Background and Purpose
Valproic acid (VPA), a widely used epilepsy and bipolar disorder treatment, provides acute protection against haemorrhagic shock‐induced mortality in a range of in vivo models through an unknown mechanism. In the liver, this effect occurs with a concomitant protection against a decrease in GSK3β‐Ser9 phosphorylation. Here, we developed an in vitro model to investigate this protective effect of VPA and define a molecular mechanism.
Experimental Approach
The human hepatocarcinoma cell line (Huh7) was exposed to conditions occurring during haemorrhagic shock (hypoxia, hypercapnia and hypothermia) to investigate the changes in GSK3β‐Ser9 phosphorylation for a 4 h period following treatment with VPA, related congeners, PPAR agonists, antagonists and siRNA.
Key Results
Huh7 cells undergoing combined hypoxia, hypercapnia, and hypothermia reproduced the reduced GSK3β‐Ser9 phosphorylation shown in vivo during haemorrhagic shock, and this change was blocked by VPA. The protective effect occurred through upstream PTEN and Akt signalling, and prevented downstream β‐catenin degradation while increasing histone 2/3 acetylation. This effect was reproduced by several VPA‐related compounds with known PPARγ agonist activity, independent of histone deacetylase (HDAC) inhibitory activity. Specific pharmacological inhibition (by T0070907) or knockdown of PPARγ blocked the protective effect of VPA against these signalling changes and apoptosis. In addition, specific activation of PPARγ using ciglitazone reproduced the changes induced by VPA in haemorrhagic shock‐like conditions.
Conclusion and Implications
Changes in GSK3β‐Ser9 phosphorylation in in vivo haemorrhagic shock models can be modelled in vitro, and this has identified a role for PPARγ activation in the protective role of VPA.
Abbreviations
- 2eVPA
2‐ene‐VPA
- 2POA
2‐propyloctanoic acid
- H2/H3/H4
histone 2/3/4
- SA
sebacic acid
- siRNA
small interfering RNA
- VPA
valproic acid
- VPD
valpromide
Tables of Links
| TARGETS | |
|---|---|
| Nuclear hormone receptors a | Enzymes b |
| PPARα | Akt (PKB) |
| PPARβ/δ | Caspase 3 |
| PPARγ | Caspase 7 |
| GSK3β | |
| HDAC | |
| PTEN |
| LIGANDS | |
|---|---|
| β‐catenin | GW6471 |
| β‐tubulin | Octanoic acid |
| ATP | T0070907 |
| Ciglitazone | Valproic acid |
| Decanoic acid |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,2013b).
Introduction
Haemorrhagic shock is the significant loss of intravascular blood volume leading to reduced tissue perfusion and resulting in reduced oxygen (hypoxia), build‐up of carbon dioxide (hypercapnia) and overall reduction in body temperature (hypothermia) (Angele et al., 2008). Decreased tissue perfusion due to blood loss leads to a reduction in oxygen available for cellular uptake, the rate of which remains constant (Kheirbek et al., 2009). This oxygen deprivation induces a switch from aerobic to anaerobic cellular metabolism (Shoemaker, 1996), during which carbon dioxide accumulates in cells, causing acidosis. As ATP consumption continues to exceed production, it is eventually depleted resulting in cell death (Keller et al., 2003; Kheirbek et al., 2009). The slowing of ATP metabolism causes spontaneous hypothermia, the occurrence of which is independently associated with an increased likelihood of the patient dying (Rossaint et al., 2006). Approximately 40% of early human deaths due to trauma are caused by haemorrhage and haemorrhagic shock (Kauvar et al., 2006), and 62% of these deaths occur in the first four hours (Frey et al., 2006). Thus, timely management of this pathological state is central to saving lives (Lecky et al., 2002).
Treatment of haemorrhage currently relies on fluid transfusion and blood component reconstitution, including red blood cells, platelets and spray‐dried plasma (Gutierrez et al., 2004; Alam et al., 2009). However, these components need to be stored and transported under specific conditions, often require matching to the patient and carry the risk of disease transmission (Kauvar and Wade, 2005). Thus, there is a need for new approaches to stabilizing patients during the critical 4 h period post‐injury. Recent investigations into pharmacological resuscitation have demonstrated that valproic acid (VPA; 2‐propylpentanoic acid), a branched short‐chain fatty acid that is a well‐established treatment for a multitude of conditions including epilepsy and bipolar disorder (Isoherranen et al., 2003; Bialer and Yagen, 2007), prevents death in animal models following haemorrhagic shock (Shults et al., 2008; Alam et al., 2009).
Most haemorrhagic shock research is performed using whole animal models (e.g. Alam et al., 2009; Hwabejire et al., 2014), a necessary approach for establishing the efficacy of any given intervention in attenuating the whole‐organism reaction to blood loss. However, the exclusive use of animal models severely limits the possibilities for detailed investigation into cellular events during haemorrhagic shock and pharmacological resuscitation. Isolating and reproducing the regulation of signalling pathways becomes a time‐consuming and difficult process due to the complexity of the multi‐organ response involved. These issues have limited the investigation of the molecular mechanisms behind the pathology and any pharmacological (therapeutic) intervention, indicating a necessity for a simple model system for the study of signalling changes involved. One of these changes has been observed in the activity of the key enzyme, glycogen synthase kinase 3β (GSK3β), which shows reduced phosphorylation at serine 9 (pGSK3β‐Ser9) giving elevated activity in vivo in the liver during haemorrhagic shock (Alam et al., 2009). Two upstream regulators of GSK3β signalling, PTEN and Akt, also show concurrent deactivation (Hwabejire et al., 2014), while β‐catenin degradation, a downstream effect of GSKβ activity, is increased. These studies have also shown that VPA prevents this decrease in pGSK3β‐Ser9 (Alam et al., 2009; Hwabejire et al., 2014). An in vitro model of haemorrhagic shock signalling may provide a useful model for investigating the mechanism of action of VPA in haemorrhagic shock.
Although VPA has a wide variety of therapeutic roles (Terbach and Williams 2009), its molecular mechanisms remain mostly unclear. One well‐documented direct effect of VPA is as a histone deacetylase (HDAC) inhibitor (Göttlicher et al., 2001; Terbach and Williams, 2009), which is likely to be the cause of its teratogenicity (Jentink et al., 2010), but may also underpin its anticancer activity (Gurvich et al., 2004; Duenas‐Gonzalez et al., 2008). We have recently shown that VPA also acts through the prevention of a reduction in phosphoinositide signalling during seizures (Chang et al., 2014; Xu et al., 2007) and in the regulation of inositol phosphates in bipolar disorder (Williams et al., 2002). In addition, we have shown that VPA regulates fatty acid levels (Elphick et al., 2012), and others have shown that it acts as a ligand of PPAR (Lampen et al., 1999), of which PPARγ has been implicated in the direct regulation of PTEN (Patel et al., 2001). A therapeutic role for this latter mechanism is unclear.
In this work, we established an in vitro model for molecular signalling in haemorrhagic shock, based on the regulation of pGSK3β‐Ser9 as a molecular marker for the signalling changes observed in the liver during haemorrhagic shock. Using a combination of hypoxia, hypercapnia and hyperthermia, we showed a reduction in pGSK3β‐Ser9 and that VPA prevents this reduction. We characterized the molecular pathway leading to this effect and further demonstrated that congeners of VPA and unrelated structures that are well‐characterized PPARγ agonists were also effective at reducing pGSK3β‐Ser9. These data suggest that pharmacological protection against haemorrhagic shock signalling may be through PPARγ activation.
Methods
Huh7 cell culture
Huh7 (Japanese Collection of Research Bioresources Cell Bank, no. JCRB0403, Japan) cells were cultured in DMEM high glucose culture medium (Sigma‐Aldrich Co. LLC. no. D5796) supplemented with 10% FBS (Invitrogen), 1× penicillin/streptomycin (Sigma) and non‐essential amino acids (Sigma) in Normoxic conditions (37°C, 5% CO2). Cells were passaged at 70–80% confluency using 0.05% Trypsin in PBS (Severn Biotech). Cells were used experimentally up to passage 10. For treatment, cells were seeded into 6‐well plates at 2 × 105 cells per well and allowed to recover for 48 h. Treatment compounds were added directly into culture medium. Cells were treated for 4 h either under standard conditions or in stress conditions (2% O2, 10% CO2, 32°C; combined hypoxia, hypercapnia and hypothermia) with a vehicle control (DMSO unless otherwise indicated) or compound of interest: 2‐ene‐VPA (2VPA; MolPort), 2‐propyloctanoic acid (2POA; Sigma), ciglitazone (Tocris), decanoic acid (Sigma), GSK3787 (Tocris), GW6471 (Tocris), octanoic acid (Sigma), sebacic acid (SA; Sigma), T0070907 (Tocris), VPA (Sigma, vehicle dH2O), valpromide (VPD; Katwijk Chemie, The Netherlands).
Protein analysis
Protein extract in RIPA buffer (Sigma) was boiled (95°C, 10 min) in SDS loading buffer (0.8 ml 2M Tris pH 6.8, 3 ml 80% glycerol, 5 ml 10% SDS, 1.25 ml β‐mercaptoethanol; all reagents from Sigma), loaded into a 12.5% acrylamide/bisacrylamide (Sigma) gel, separated by SDS‐PAGE and transferred to a PVDF membrane (Merck Millipore) via Western blot. Membranes were blocked in 5% BSA V (Sigma) in TBST (Severn Biotech) for 1 h. Antibody was added directly to blocking buffer (1:1000), and membrane was incubated at 4°C overnight. All primary antibodies were provided by Cell Signaling Technology: GSK3β (no. 12456), pGSK3β‐Ser9 (no. 5558), Akt (no. 9272), pAkt‐Ser473 (no. 4060), PPARγ (no. 2443), PTEN (no. 9188), Ser380/Thr382/383 pPTEN (no. 9549), β‐catenin (no. 8480), acetylated lysine (no. 9441), β‐actin (no. 4970), β‐tubulin (no. 2128). Membranes were washed in TBST and incubated with secondary antibody (Li‐Cor no. 926‐32211 Goat anti‐Rabbit) in Odyssey Blocking Buffer (Li‐Cor no. 927‐50000) for 1 h at room temperature. Membranes were visualized and quantified using the Odyssey Sa system (Li‐Cor), which directly quantifies fluorescence and, therefore, protein abundance in a linear manner. Both phosphorylated and total protein levels were corrected for loading using β‐tubulin/β‐actin levels, and relative phosphorylation was calculated as the ratio of corrected phosphorylated‐to‐total protein.
Apoptosis assay
Huh7 cells were analysed for apoptosis using ApoTox Glo (Promega) according to the manufacturer's instructions. Briefly, the assay provides a luminogenic substrate, which when cleaved by caspase‐3/7 yields quantifiable luminescence to indicate the presence of apoptotic signalling.
HDAC inhibition assay
HDAC inhibition assays were performed using a fluorimetric in vitro histone deacetylase assay (Merck Millipore), according to the manufacturer's instructions, using human‐derived HeLa cell enzyme extract (Enzo) in a 1:10 dilution as described previously (Chang et al., 2015). Briefly, HeLa deacetylases act upon a substrate to sensitize it to a developer, the binding of which produces quantifiable luminescence. The presence of an HDAC inhibitor decreases HDAC activity therefore yielding decreased fluorescence.
PPARγ siRNA knockdown
Four mixed specific PPARγ siRNAs and negative control siRNA (Qiagen nos. GS5468 and SI03650325 respectively) were used in conjunction with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocols. Briefly, cells were seeded into 6‐well plates and cultured to 70% confluence (48 h). Cells were transfected in 250 µl unsupplemented culture medium (DMEM high glucose, as before) with all four PPARγ siRNAs or the negative control siRNA for 6 h, after which 750 µl DMEM containing 10% FBS was added to each well. Cells were rested overnight (16 h), at which point medium was replaced with fresh DMEM (containing 10% FBS) and allowed to grow for a further 24 h before the experiments.
Lactate dehydrogenase (LDH) release assay
Huh7 cells were underwent stress conditions (2% O2, 10% CO2, 32°C) with or without VPA (0.75 mM), and LDH release was measured using an LDH Cytotoxicity Assay (Pierce) and according to the manufacturer's instructions.
Statistical analyses
Results are expressed as means ± SEM. Data were analysed using one‐way ANOVA or Student's t‐test as appropriate. Error bars depict SEM. P values >0.05 were considered non‐significant, 0.01–0.05 significant (*), 0.001–0.01 very significant (**) and <0.001 highly significant (***).
Results
Developing an in vitro model of haemorrhagic shock
To establish an in vitro model for the analysis of haemorrhagic shock signalling, we employed a human liver cell line (Huh7). Cells were exposed to the haemorrhagic shock‐like conditions of hypoxia (2% O2), hypercapnia (10% CO2) and hypothermia (32°C) over a 4 h period. Quantitative analysis of GSK3β‐Ser9 phosphorylation status, shown to be regulated in in vivo models (Alam et al., 2009; Hwabejire et al., 2014), was employed as a read‐out for haemorrhagic shock‐like conditions (Figure 1A). Simultaneous exposure of the cells to all three stress conditions triggered a significant 50 ± 5% reduction of pGSK3β‐Ser9 levels compared with control conditions, an effect which was not evident in individual or paired conditions. As phosphorylation at this site inhibits GSK3β activity, these results suggest an increase in enzymatic activity under haemorrhagic shock‐like conditions. VPA treatment caused a dose‐dependent protection against the reduction in pGSK3β‐Ser9 levels (58 ± 5% at 0.1 mM; 91 ± 10% at 0.5 mM; 128 ± 16% at 0.75 mM VPA compared with control conditions; Figure 1B) at concentrations found in patients treated with VPA (0.4–0.7 mM (DSM IV, 2000)) consistent with in vivo data. VPA did not alter pGSK3β‐Ser9 levels in cells in the absence of haemorrhagic shock‐like conditions, suggesting this VPA‐induced effect was dependent upon these stress conditions.
Figure 1.
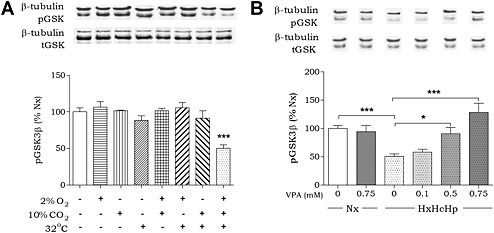
Developing an in vitro model of in vivo haemorrhagic shock. (A) Huh7 cells were exposed to hypoxia, hypercapnia and/or hypothermia for 4 h as indicated and analysed for pGSK3β‐Ser9 levels and normalized to Nx. (B) Huh7 cells were exposed to normoxic conditions (37°C, 5% CO2; Nx), or combined hypoxia, hypercapnia and hypothermia (HxHcHp), treated with VPA (0.75 mM) as indicated, analysed for pGSK3β‐Ser9 levels and normalized to Nx. Data were quantified from at least triplicate experiments with technical triplicates (n > 9) ± SEM. Data were analysed using one‐way ANOVA and post hoc Tukey test.
Defining the haemorrhagic shock signalling pathway
Because the in vitro haemorrhagic shock model reproduced the in vivo reduction in pGSK3β‐Ser9 levels and VPA‐dependent protection (Alam et al., 2009), we next examined the signalling pathway involved in this effect (Figure 2A). Phosphorylation of GSK3β at Ser9 is catalysed by Akt (Delcommenne et al., 1998). Monitoring Akt activity, using pAkt‐Ser473 levels as an indication of enhanced activity (Hanada et al., 2004), suggests that haemorrhagic shock‐like conditions result in a significant reduction in pAkt‐Ser473 and thus activity (60 ± 6% compared with control conditions) (Figure 2C). This reduction was partially blocked by VPA (87 ± 5% at 0.75 mM compared with control). This VPA‐induced effect was only seen under haemorrhagic shock‐like conditions. Phosphorylation of Akt‐Ser473 is dependent upon the production of phosphoinositide 3,4,5‐trisphosphate (Delcommenne et al., 1998), a key signalling molecule that is degraded by the phospholipid phosphatase, PTEN. Inhibitory regulation of PTEN activity is coordinated by phosphorylation at Ser380/Thr382/383, resulting in enhanced Akt activity (Sun et al., 1999). Monitoring pPTEN levels by quantitative analysis under haemorrhagic shock‐like conditions indicated a significant reduction in phosphorylation (69 ± 7% of control conditions) (Figure 2B), which was reversed by VPA (89 ± 4% at 0.75 mM compared with control).
Figure 2.

Defining the haemorrhagic shock signalling pathway. Huh7 cells were incubated for 4 h in the presence of hypoxia (2% O2), hypercapnia (10% CO2) and hypothermia (32°C) (HxHcHp), and treated with VPA as indicated. All data are shown as mean ± SEM normalized to normoxic conditions (Nx). Phosphorylation levels are presented as percentage of untreated control and corrected for loading with loading control indicated. (A) An overview of PI3K signalling pathway regulation by haemorrhagic shock signalling (stress response) and VPA treatment (VPA response). (B) Protein extract was analysed for PTEN phosphorylation levels at Ser380/Thr382/383. (C) Protein extract was analysed for Akt phosphorylation levels at Ser473. (D) Protein extract was analysed for total β‐catenin levels. (E) Protein extract was analysed for histone (H)2/3 and H4 acetylation using acetylated lysine antibody. Data were quantified from at least triplicate experiments with technical triplicates (n ≥ 9) ± SEM. Data were analysed using one‐way ANOVA and post hoc Tukey test (B, C and D) or using two‐way ANOVA and post hoc Bonferroni tests (E).
GSK3β plays a key role in regulating cellular function through a range of targets including β‐catenin, which is primed for degradation through phosphorylation (Rubinfeld et al., 1996). We, therefore, assessed the effect of haemorrhagic shock‐like conditions on β‐catenin levels. These conditions reduced β‐catenin levels (63 ± 7% of control), a result consistent with enhanced GSK3β activity. VPA prevented this decrease in β‐catenin abundance (91 ± 5% of control conditions) in agreement with a protective effect of VPA against the reduction in pGSK3β‐sSer9 levels under haemorrhagic shock‐like conditions.
The mechanism of action of VPA in the prevention of haemorrhagic shock‐induced lethality has been proposed to depend upon HDAC inhibition (Alam et al., 2009). To evaluate whether the molecular mechanism of VPA in this model system relies on HDAC regulation, we monitored histone acetylation under control and haemorrhagic shock‐like conditions in the presence and absence of VPA (0.75 mM). In control conditions, VPA did not alter histone 2/3 (H2/H3) acetylation (100 ± 6% of untreated control) but caused a significant increase in histone 4 (H4) acetylation (406 ± 38% of untreated control). Haemorrhagic shock‐like conditions alone did not alter H2/H3 or H4 acetylation levels and also did not affect the VPA‐dependent increase in H4 acetylation. However, VPA gave rise to a significant increase in H2/H3 acetylation levels (681 ± 121% of control; Figure 2E) during haemorrhagic shock‐like conditions. As VPA‐induced H2/H3 acetylation only occurred in these conditions, the mechanism of this effect is likely to be dependent on the presence of hypoxia, hypercapnia and hypothermia and, therefore, is likely to be specific to the pathological environment under investigation.
PPARγ agonists attenuate haemorrhagic shock‐like signalling independently of HDAC inhibitory activity
We extended our analysis of haemorrhagic shock‐like conditions in regulating pGSK3β‐Ser9 levels by investigating the efficacy of a range of VPA congeners (Figure 3A). We employed two straight chain fatty acids, octanoic acid and decanoic acid, the latter of which shows enhanced seizure control compared with VPA (Chang et al., 2012; Chang et al., 2014); two acids showing the same branching structure as VPA, 2POA and 2eVPA, which both also show seizure control (Palaty and Abbott 1995; Chang et al., 2013); VPD, the amide derivative of VPA (Bialer, 1991); and a key metabolite of decanoic acid and SA (Gregersen et al., 1983). All compounds were initially tested at 0.25 and 0.75 mM for efficacy in preventing the reduction in pGSK3β‐Ser9 levels caused by haemorrhagic shock‐like conditions. Octanoic acid, 2POA and VPD had no effect (Figure 3B), but decanoic acid, 2eVPA and SA prevented the decrease in pGSK3β‐Ser9. These active compounds were reassessed at 0.1 mM (Figure 3C). Serum levels in patients taking a decanoic acid‐related diet are around 0.157 mM (Gregersen et al., 1983) suggesting that this concentration is therapeutically relevant (Hughes et al., 2014). Decanoic acid and 2eVPA showed enhanced potency over VPA, replicating its effect on pGSK3β‐Ser9 at a 7.5‐fold reduced dose and showing a typical biphasic response with optimal efficacy at 0.1 mM. Interestingly, both compounds have been reported to provide a strong activation of PPAR activity, above that of VPA (Lampen et al., 2001).
Figure 3.
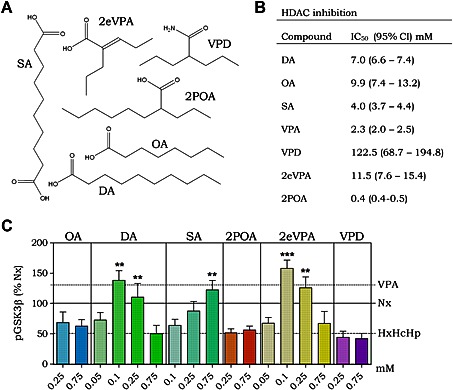
PPARγ agonists provide protection against haemorrhagic shock signalling. Huh7 cells were incubated for 4 h in the presence of hypoxia (2% O2), hypercapnia (10% CO2) and hypothermia (32°C) (HxHcHp) and treated with compounds as indicated. (A) Six congeners of valproic acid were investigated for their effect on the pathway of interest. (B) All compounds were assessed for HDAC inhibitory activity using a commercial assay (Merck) to establish IC50 values. Mean values were obtained using the Hill's equation. (C) Huh7 cells were treated with octanoic acid (OA), 2POA, VPD, SA, 2eVPA and decanoic acid (DA, between 0.05 and 0.75 mM as indicated), for 4 h while undergoing stress conditions (2% O2, 10% CO2, 32°C), and protein extract was analysed for pGSK3β‐Ser9 levels. Data were analysed using one‐way ANOVA and post hoc Tukey test. Mean values of previous data (Figure 1B) are shown as horizontal lines for ease of comparison. Data were quantified from at least triplicate experiments with technical triplicates (n ≥ 9) ± SEM and were normalized to results in Nx. ** P > 0.01 and *** P > 0.001 indicate significance compared with HxHcHp.
Having found novel compounds showing activity in attenuating signalling changes in haemorrhagic shock‐like conditions, we then evaluated these compounds for HDAC inhibitory activity. Compounds were assessed in an established HDAC inhibition assay (Chang et al., 2015), which uses HeLa cell enzyme extract as the source of HDAC activity, to define an IC50 for efficacy comparison (Figure 3D and Supporting Information Figure S1). The compounds investigated were observed to include some with both increased and reduced potency than VPA in inhibiting HDAC activity. However, there was no correlation between a compound's HDAC inhibitory activity and its efficacy at preventing the haemorrhagic shock‐induced decrease in pGSK3β‐Ser9.
Attenuation of haemorrhagic shock‐like signalling depends on PPARγ activity
Our data suggest that PPAR agonists may reproduce the therapeutic mechanism of the protective effect of VPA against signalling events caused by haemorrhagic shock‐like conditions. To verify a role for PPAR activation in this system, we treated cells undergoing haemorrhagic shock‐like conditions with VPA (0.75 mM) in the presence of selective PPARα, PPARβ/δ and PPARγ inhibitors. Selective inhibitors for PPARα (GW6471; 50 μM; Abu Aboud et al., 2013) and PPARβ/δ (GSK3787; 10 μM; Palkar et al., 2010) did not inhibit the effect of VPA on pGSK3β‐Ser9 (Figure 4A). However, the PPARγ inhibitor, T0070907 (50 μM; An et al., 2014), blocked the VPA‐induced increase in pGSK3β‐Ser9 (Figure 4A). This is consistent with VPA modulating signalling by altering transcriptional activity, where the protective effect was blocked by the application of a general transcription in inhibitor actinomycin D (1 µg∙ml−1; Supporting Information Figure S2). These data suggest that the VPA‐dependent regulation of pGSK3β‐Ser9 levels is mediated by PPARγ activity.
Figure 4.
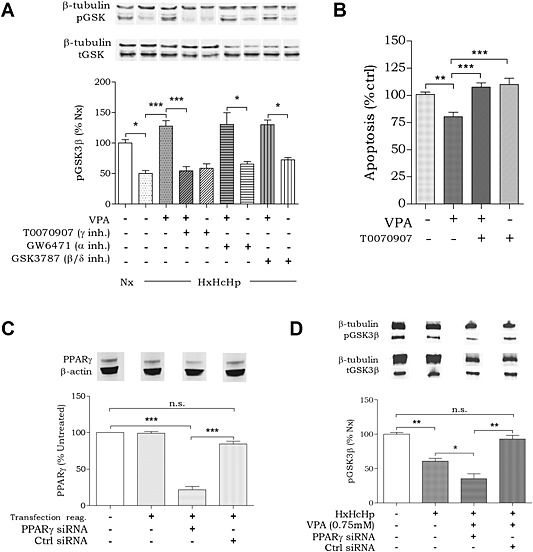
Protection against haemorrhagic shock‐like signalling depends on PPARγ activity. Huh7 cells were incubated for 4 h in the presence of hypoxia (2% O2), hypercapnia (10% CO2) and hypothermia (32°C) (HxHcHp). (A) Cells were treated with VPA (0.75 mM) and PPAR inhibitors (T0070907 and GW6471 50 μM; GSK3787 10 μM), and protein extract was analysed for pGSK3β‐Ser9 levels and β‐tubulin loading control. (B) Apoptotic signalling in Huh7 cells in response to VPA (0.75 mM) and/or PPARγ inhibitor T0070907 (50 μM) was analysed using a commercial assay (Promega). (C) PPARγ was knocked down in Huh7 cells using four commercially produced (Qiagen) variants of PPARγ siRNA. A scrambled siRNA (Ctrl) was used as negative control. Huh7 cells were transfected in standard cell culture conditions (5% CO2, 37°C). (D) Cells with and without PPARγ knockdown were tested for pGSK3 regulation under equivalent haemorrhagic shock‐like and treatment conditions. Data were quantified from at least triplicate experiments with technical triplicates (n ≥ 9) ± SEM. Data were normalized to untreated (A and B), untransfected (C) and Nx (D) and were analysed using one‐way ANOVA and post hoc Tukey test.
We next investigated whether VPA‐induced PPARγ activation is related to cell survival. Here, we monitored apoptotic signalling using an in‐cell reporter assay (ApoTox Glo), which provides a luminogenic substrate for caspase‐3/7 cleavage in cells. We tested cells in haemorrhagic shock‐like conditions in the absence or presence of VPA (0.75 mM) and following treatment with the PPARγ‐specific inhibitor T0070907 (50 μM; Figure 4B). VPA treatment reduced apoptotic signalling to 79 ± 4% (compared with untreated control) and reduced LDH release (Supporting Information Figure S2) suggesting a protective effect on cell survival. Apoptotic signalling protection was prevented by the addition of T0070907 (108 ± 4% compared with untreated). These data suggest that pharmacological inhibition of PPARγ acts to block the effect of VPA in attenuating haemorrhagic shock‐like signalling relating to cell survival.
Because pharmacological inhibitors may produce off‐target effects, we employed a genetic approach to deplete PPARγ levels, and then investigated the effect of VPA. Treating cells with four individual PPARγ siRNAs in combination significantly reduced PPARγ protein abundance to 22 ± 5% of untreated cells (Figure 4C), whereas scrambled (Ctrl) siRNA did not. We then assessed changes in pGSK3β‐Ser9 levels in these cells under haemorrhagic shock‐like conditions (61 ± 4% compared with control conditions; Figure 4D), in the presence and absence of VPA. Cells treated with scrambled siRNA still showed the VPA‐dependent protection against the pGSK3β‐Ser9 reduction (93 ± 5% compared with control; Figure 4D) seen earlier (Figure 1B). However, treatment with the PPARγ‐specific siRNAs inhibited the VPA‐dependent effect on pGSK3β‐Ser9 levels, resulting in a further reduction in pGSK3β‐Ser9 levels (35 ± 7% compared with control; Figure 4D). These data further confirm the essential role for PPARγ activation in protection against haemorrhagic shock‐dependent signalling changes.
A PPARγ‐specific agonist shows potent therapeutic efficacy in haemorrhagic shock‐like conditions
As specific PPARγ agonists have been used as medical treatments, we investigated a role for one of these in our model of haemorrhagic shock. Here, the specific PPARγ activator ciglitazone, like VPA, caused a dose‐dependent protection against the reduction in pGSK3β‐Ser9 levels under haemorrhagic shock‐like conditions (122 ± 16% at 20 μM; 127 ± 20% at 40 μM; 164 ± 22% at 80 μM compared with untreated; Figure 5A). Furthermore, we showd that apoptotic signalling triggered by haemorrhagic shock‐like conditions was reduced by ciglitazone (77 ± 6% at 60 μM compared with untreated), as well as 2eVPA (73 ± 7% at 0.1 mM) and decanoic acid (76 ± 4% at 0.1 mM) (Figure 5B) in a similar manner to that of VPA (Figure 4B). These data strongly support a role for activation of PPARγ as a therapeutic treatment for haemorrhagic shock and propose that currently licensed medical treatments such as ciglitazone may provide enhanced protection compared with VPA in the treatment of haemorrhagic shock.
Figure 5.

PPARγ ligands show enhanced potency compared with VPA at protecting against haemorrhagic shock‐induced signalling. Huh7 cells were incubated for 4 h in the presence of hypoxia (2% O2), hypercapnia (10% CO2) and hypothermia (32°C) (HxHcHp). (A) Huh7 cells were treated with the PPARγ ligand ciglitazone, and protein extract was analysed for pGSK3β‐Ser9 levels and β‐tubulin loading control. (B) Apoptotic signalling in Huh7 cells in response to PPARγ ligands 2eVPA, decanoic acid (DA) and ciglitazone was analysed using a commercial assay (Promega). Data were quantified from at least triplicate experiments with technical triplicates (n ≥ 9) ± SEM and normalized to Nx 0 (A) and untreated control (B). Data were analysed using one‐way ANOVA and post hoc Tukey test.
Discussion
Identifying the molecular mechanisms of pharmacological treatments to prevent haemorrhagic shock‐related mortality may significantly reduce the incidence of patient death. VPA has been demonstrated to be effective in this role in multiple animal studies (Gutierrez et al., 2004; Shults et al., 2008; Alam et al., 2009), yet its mechanism has remained unclear. Here, we established an in vitro model for haemorrhagic shock signalling to reproduce the decrease in pGSK3β‐Ser9 levels shown in animal experiments (Figure 6). We showed that VPA protects against this signalling change in this model as it does in vivo, and that this effect is consistent with those observed in the regulation of upstream effectors and downstream targets of GSK3β. Furthermore, we identified a range of compounds that provide enhanced potency compared with VPA in this model. In addition, we have shown that the mechanism of VPA in this model depends on the activation of PPARγ. This discovery may provide a rare example of a defined therapeutic mechanism for VPA in one of its many roles (Terbach and Williams 2009). Further investigations based on this discovery may lead to the development of efficacious therapeutic compounds to save lives in the treatment of massive blood loss.
Figure 6.
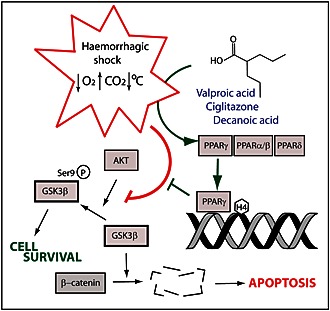
Schematic representing changes occurring during haemorrhagic shock‐like conditions, leading to reduced pGSK3β‐Ser9 levels. Valproic acid and other PPARγ activators block this change.
In developing an in vitro model for haemorrhagic shock research, we have taken into account three stressors, which occur at a cellular level during blood loss: hypoxia, a reduction in oxygen levels; hypercapnia, an increase in carbon dioxide levels; and hypothermia, a decrease in temperature. This multiparameter approach to inducing haemorrhagic shock‐like stress conditions is a novel one and has not been employed in previous studies. Haemorrhagic or ischaemic studies have often relied exclusively on low oxygen as a model system for research (Tramontano et al., 2003), although it has been suggested that hypercapnia is essential to accurately model these conditions (Hotter et al., 2004). The third component of the ‘lethal triad’ (Angele et al., 2008) of haemorrhagic shock included in the present study, hypothermia (Kheirbek et al., 2009), is rarely included in in vitro studies. Spontaneous hypothermia during blood loss is independently associated with significantly reduced survival rate, and mild hypothermia is commonly seen during massive blood loss (Kheirbek et al., 2009).
Studies into haemorrhagic shock have increasingly focused on cytosolic changes in protein activity in an effort to discover a target for pharmacological resuscitation (Li et al., 2008; Hwabejire et al., 2014). The PI3K pathway, in particular, has been repeatedly implicated in survival‐relevant signalling changes, both in haemorrhagic shock and in neuroprotection (Kitagishi and Matsuda, 2013). In a recent in vivo haemorrhagic shock study, VPA treatment was shown to dose‐dependently activate the PI3K pathway (Hwabejire et al., 2014) and reverse the decrease in phosphorylation of both Akt and PTEN caused by haemorrhagic shock that regulate GSK3β activity (Delcommenne et al., 1998). However, our data have replicated these signalling changes in our Huh7 model, where cells exposed to hypoxia, hypercapnia and hypothermia showed decreased phosphorylation of PTEN, Akt and GSK3β in the manner observed in vivo in porcine liver (Alam et al., 2009; Hwabejire et al., 2014), rodent brain (Li et al., 2008) and rodent kidney (Zacharias et al., 2011) during haemorrhagic shock. As it does in vivo, VPA acted dose‐dependently in our model in blocking the reduction of GSK3β phosphorylation (Alam et al., 2009; Hwabejire et al., 2014). Thus, although further studies will be needed to translate our data from Huh7 cells to primary cells, including both hepatocytes and other cells types, as cellular responses in primary cells may not be conserved, our study provides for the first time, the recreation of this in vivo effect in vitro.
The serine/threonine kinase, GSK3β, plays a central role in a range of normal cells functions and has been associated with both the pathology and treatment of a long list of diseases (Jope et al., 2007). GSK3β activity has been implicated as a target for bipolar disorder treatments (Valvezan and Klein 2012), in diabetes (Eldar‐Finkelman and Krebs 1997), Huntington's disease (Carmichael et al., 2002) and Alzheimer's disease (Hooper et al., 2008). On a cellular level, GSK3β phosphorylates a range of substrates including β‐catenin, which it primes for ubiquitylation and subsequent degradation (Sakanaka, 2002). The accumulation of β‐catenin is generally associated with a pro‐survival phenotype in haemorrhagic shock‐like conditions (Alam et al., 2009; Shults et al., 2008), consistent with an important role for GSK3β in this pathology. VPA has been extensively debated as a regulator of GSK3β signalling for over a decade, with some studies suggesting both direct and indirect inhibitory effects (Chen et al., 1999; Hall et al., 2002) yet other studies suggesting no direct effect (Phiel et al., 2001; Ryves et al., 2005). No studies, to our knowledge, have described a mechanism for an effect of VPA on GSK3β activity. Our data suggest that VPA acts to regulate GSK3β through an indirect mechanism, and, most importantly, only in defined (stress) conditions, which may explain the divergent effects discussed in the literature. Further studies will be necessary to investigate this mechanism in other disease models, but it is likely that the discovery of this context‐dependent regulation of GSK3β by VPA will have implications for a long list of conditions.
The pro‐survival effect of VPA in treating haemorrhagic shock is widely considered to be due to an HDAC inhibitory effect (Shults et al., 2008; Alam et al., 2009; Zacharias et al., 2011). This activity has been associated with a variety of biological processes, both adverse and therapeutic. For instance, VPA‐dependent HDAC inhibition has been demonstrated to be the cause of teratogenic changes in mammals (Gotfryd et al., 2010; Jentink et al., 2010), which lead to major congenital malformations (e.g. neural tube defects, hypospadias and skeletal abnormalities) in humans (Tomson and Battino 2008). However, HDAC inhibition has been shown to contribute to beneficial therapeutic effects such as in the treatment of cancer (Duenas‐Gonzalez et al., 2008; Gotfryd et al., 2010). Our study has confirmed an effect of VPA on elevating histone 4 acetylation in haemorrhagic shock‐like conditions, but this change is equally observed in response to VPA during normal cell culture conditions. In contrast, we have also shown that VPA treatment gives rise to a fourfold increase in histone 2/3 acetylation levels that is only seen under haemorrhagic shock‐like conditions. This is consistent with in vivo studies where the acetylation of lysine residue H3K9 is used as a marker for histone acetylation (Alam et al., 2009), but indicates that any histone‐mediated attenuating effects are specific to a certain subset of this class. Our results suggest that the action of VPA in haemorrhagic shock‐like signalling does not have a generalized effect on HDAC activity, but instead regulates histone 2/3 deactetylation specifically.
As the molecular mechanisms of VPA in the treatment of epilepsy and other conditions have remained unclear until recently (Chang et al., 2014), many congeners of VPA have been developed in search of improved therapeutic profiles. These compounds, often with known potency against molecular targets such as HDAC inhibition (Lampen et al., 2001; Eikel et al., 2006) or PPAR activation (Lampen et al., 1999), have then been used in a wide range of disease models potentially affected by VPA (Isoherranen et al., 2003; Bialer and Yagen, 2007; Chang et al., 2012). In the experiments described here, six different VPA congeners with a range of HDAC inhibitory activities (Lampen et al., 2001) were employed. We found that compound efficacy in attenuating the decrease in pGSK3β‐Ser9 manifested independently of HDAC inhibitory activity, with 2eVPA, decanoic acid and SA improving (2eVPA and decanoic acid) or mimicking (SA) the protective effect of VPA. These three compounds are all activators of PPARs (Lampen et al., 1999), suggesting PPAR activity may be a key component of the mechanism of VPA in modulating haemorrhagic shock‐like signalling. These findings imply that a direct HDAC inhibitory effect of VPA is unlikely to cause the signalling changes observed in this haemorrhagic shock model, but PPAR activation may.
The PPAR family, part of the ligand‐activated nuclear receptor superfamily, comprises a range of cytoplasmic receptors for fatty acids that function through nuclear transcription (Kota et al., 2005). All three PPAR isoforms (α, β/δ and γ) possess a number of conserved domains, including a DNA‐binding domain, which interact with PPAR response elements (PPREs) in target gene promoters (Berger, 2002) alongside a domain which confers target specificity (Kliewer et al., 1995). PPARs have been implicated in a wide variety of cellular and molecular processes, while PPARγ has been studied in insulin sensitization, cancer and inflammation (Kota et al., 2005). Our data, for the first time, strongly suggests a mechanism for VPA in protection against haemorrhagic shock‐like signalling through PPARγ activation. We show this mechanism by blocking the effect of VPA using specific PPARγ inhibitors as well as targeted siRNA knockdown. We also show that treating cells with a specific PPARγ ligand (Ciglitazone) reproduces the response caused by VPA. We further show that the VPA‐dependent activation of PPARγ protects against apoptotic signalling under haemorrhagic shock‐like conditions, increasing cell survival. This mechanism is supported by evidence provided in an earlier study, where a PPARγ ligand structurally unrelated to VPA was shown to reduce organ injury in a rodent model of haemorrhagic shock, an effect attenuated by a PPARγ inhibitor (Abdelrahman et al., 2004). Our study is therefore the first to describe the mechanism of VPA in protection against haemorrhagic shock‐like signalling through PPARγ activation.
In this study, we have developed an in vitro model of haemorrhagic shock to investigate the mechanism of VPA in attenuating in vivo haemorrhagic shock‐related signalling and lethality (Gutierrez et al., 2004; Shults et al., 2008; Alam et al., 2009). By combining hypoxia, hypercapnia and hypothermia, we have reproduced a haemorrhagic shock‐like environment sufficient to cause a reduction in pGSK3β‐Ser9, which is prevented by VPA treatment in a manner analogous to that observed in vivo (Alam et al., 2009; Hwabejire et al., 2014). We have also used this model to identify PPARγ activity as an essential component in the VPA mechanism of action, although other regulated pathways may also contribute to this effect (Elphick et al., 2012; Chang et al., 2014). The discovery of this mechanism and the efficacy of PPARγ‐specific ligands (e.g. ciglitazone) as VPA‐replacing therapeutic intervention provide an immediate investigative target to translate to in vivo models, and then to more clinical settings. The further investigation of potent PPARγ ligands as a means of pharmacological resuscitation in the treatment of haemorrhagic shock may ultimately provide life‐saving therapeutics.
Author contributions
A. M. E. Z., R. L. R. and R. S. B. W. designed the research. A. M. E. Z. performed the research. D. B. contributed new reagents/analytics tools. A. M. E. Z. and R. S. B. W. analysed data. A. M. E. Z. and R. S. B. W. wrote the paper.
Conflict of interest
Authors declare that they have not any conflict of interest.
Supporting information
Figure S1 Compounds structurally similar to VPA show variable HDAC inhibitory activity. Enzyme extract from HeLa cells was treated with decanoic acid (A), sebacic acid (B), valpromide (C), 2eVPA (D), and 2‐propyloctanoic acid (E) at concentrations between 0.5 and 10 mM, proportion of deacetylated assay substrate measured (fluorescence) and compared with an uninhibited control. Data are quantified from at least triplicate experiments with technical triplicates (n ≥ 9) ⨦ SEM.
Figure S2 VPA acts through a transcriptional mechanism and reduces LDH release. Huh7 cells were incubated for 4 h in the presence of hypoxia (2% O2), hypercapnia (10% CO2), and hypothermia (32°C) (HxHcHp). (A) Cells were treated with VPA (0.75 mM) in the presence or absence of transcription inhibitor actinomycin D, and protein extract was analysed for pGSK3β‐Ser9 levels and β‐tubulin loading control. (B) Huh7 cells, again under stress conditions, with or without VPA (0.75mM) were assayed for LDH release using LDH Cytotoxicity Assay. Data are quantified from at least triplicate experiments with technical triplicates (n ≥ 9) ⨦ SEM. *P > 0.05, ***P > 0.001.
Supporting info item
Supporting info item
Acknowledgements
This project was supported by a School of Biological Sciences studentship to A. M. E. Z.
Zuckermann, A. M. E. , La Ragione, R. M. , Baines, D. L. , and Williams, R. S. B. (2015) Valproic acid protects against haemorrhagic shock‐induced signalling changes via PPARγ activation in an in vitro model. British Journal of Pharmacology, 172: 5306–5317. doi: 10.1111/bph.13320.
References
- Abdelrahman M, Collin M, Thiemermann C (2004). The peroxisome proliferator‐activated receptor‐γ ligand 15‐deoxyd12,14 prostaglandin J2 reduces the organ injury in hemorrhagic shock. Shock 22: 555–561. [DOI] [PubMed] [Google Scholar]
- Abu Aboud O, Wettersten HI, Weiss RH (2013). Inhibition of PPARα induces cell cycle arrest and apoptosis, and synergizes with glycolysis inhibition in kidney cancer cells. PLoS One 8: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam HB, Shuja F, Butt MU, Duggan M, Li Y, Zacharias N et al. (2009). Surviving blood loss without blood transfusion in a swine poly‐trauma model. Surgery 146: 325–333. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013a). The concise guide to PHARMACOLOGY 2013/14: nuclear hormone receptors. Br J Pharmacol 170: 1652–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013b). The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Z, Muthusami S, Yu JR, Park WY (2014). T0070907, a PPAR γ inhibitor induced G2/M arrest enhances the effect of radiation in human cervical cancer cells through mitotic catastrophe. Reprod Sci 21: 1352–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angele MK, Schneider CP, Chaudry IH (2008). Bench‐to‐bedside review: latest results in hemorrhagic shock. Crit Care 12: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialer M (1991). Clinical pharmacology of valpromide. Clin Pharmacokinet 20: 114–122. [DOI] [PubMed] [Google Scholar]
- Bialer M, Yagen B (2007). Valproic acid: second generation. Neurotherapeutics 4: 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL (2002). Histone modifications in transcriptional regulation. Curr Opin Genet Dev 12: 142–148. [DOI] [PubMed] [Google Scholar]
- Carmichael J, Sugars KL, Bao YP, Rubinsztein DC (2002). Glycogen synthase kinase‐3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington's disease mutation. J Biol Chem 277: 33791–33798. [DOI] [PubMed] [Google Scholar]
- Chang P, Orabi B, Deranieh R, Dham M, Hoeller O, Shimshoni J et al. (2012). The antiepileptic drug valproic acid and other medium‐chain fatty acids acutely reduce phosphoinositide levels independently of inositol in Dictyostelium. Dis Model Mech 5: 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P, Terbach N, Plant N, Chen PE, Walker MC, Williams RSB (2013). Seizure control by ketogenic diet‐associated medium chain fatty acids. Neuropharmacology 69: 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P, Walker MC, Williams RSB (2014). Seizure‐induced reduction in PIP3 levels contributes to seizure‐activity and is rescued by valproic acid. Neurobiol Dis 62: 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P, Zuckermann AME, Williams S, Close AJ, Cano‐Jaimez M, Mcevoy JP et al. (2015). Seizure control by derivatives of medium chain fatty acids associated with the ketogenic diet show novel branching‐point structure for enhanced potency. J Pharmacol Exp Ther 352: 43–52. [DOI] [PubMed] [Google Scholar]
- Chen G, Huang LD, Jiang YM, Manji HK (1999). The mood‐stabilizing agent valproate inhibits the activity of glycogen synthase kinase‐3. J Neurochem 72: 1327–1330. [DOI] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S (1998). Phosphoinositide‐3‐OH kinase‐dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin‐linked kinase. Proc Natl Acad Sci U S A 95: 11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duenas‐Gonzalez A, Candelaria M, Perez‐Plascencia C, Perez‐Cardenas E, de la Cruz‐Hernandez E, Herrera LA (2008). Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat Rev 34: 206–222. [DOI] [PubMed] [Google Scholar]
- DSM IV (2000). American Psychiatric Association: Diagnostic and statistical manual of mental disorders. American Psychiatric Association: Washington DC. [Google Scholar]
- Eikel D, Lampen A, Nau H (2006). Teratogenic effects mediated by inhibition of histone deacetylases: evidence from quantitative structure activity relationships of 20 valproic acid derivatives. Chem Res Toxicol 19: 272–278. [DOI] [PubMed] [Google Scholar]
- Eldar‐Finkelman H, Krebs EG (1997). Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci U S A 94: 9660–9664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elphick LM, Pawolleck N, Guschina IA, Chaieb L, Eikel D, Nau H et al. (2012). Conserved valproic‐acid‐induced lipid droplet formation in Dictyostelium and human hepatocytes identifies structurally active compounds. Dis Model Mech 5: 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey KP, Egleston BL, Salkever DS, Scharfstein DO (2006). A national evaluation of the effect of trauma‐center care on mortality. N Engl J Med 354: 366–378. [DOI] [PubMed] [Google Scholar]
- Gotfryd K1, Skladchikova G, Lepekhin EA, Berezin V, Bock E, Walmod PS (2010). Cell type‐specific anti‐cancer properties of valproic acid: independent effects on HDAC activity and Erk1/2 phosphorylation. BMC Cancer 10: 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen N, Mortensen PB, Kølvraa S (1983). On the biologic origin of C6‐C10‐dicarboxylic and C6‐C10‐omega‐1‐hydroxy monocarboxylic acids in human and rat with acyl‐CoA dehydrogenation deficiencies: in vitro studies on the omega‐ and omega‐1‐oxidation of medium‐chain (C6‐C12) fatty acids in human and rat liver. Pediatr Res 17: 828–834. [DOI] [PubMed] [Google Scholar]
- Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S et al. (2001). Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20: 6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS (2004). Histone deacetylase is a target of valproic acid‐mediated cellular differentiation. Cancer Res 64: 1079–1086. [DOI] [PubMed] [Google Scholar]
- Gutierrez G, Reines HD, Wulf‐Gutierrez ME (2004). Clinical review: hemorrhagic shock. Crit Care 8: 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AC, Brennan A, Goold RG, Cleverly K, Lucas FR, Gordon‐Weeks PR et al. (2002). Valproate regulates GSK‐3‐mediated axonal remodeling and synapsin I clustering in developing neurons. Mol Cell Neurosci 20: 257–270. [DOI] [PubMed] [Google Scholar]
- Hanada M, Feng J, Hemmings BA (2004). Structure, regulation and function of PKB/AKT – a major therapeutic target. Biochim Biophys Acta 1697: 3–16. [DOI] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S (2008). The GSK3 hypothesis of Alzheimer's disease. J Neurochem 104: 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotter G, Palacios L, Sola A (2004). Low O2 and high CO2 in LLC‐PK1 cells culture mimics renal ischemia‐induced apoptosis. Lab Investig 84: 213–220. [DOI] [PubMed] [Google Scholar]
- Hughes SD, Kanabus M, Anderson G, Hargreaves IP, Rutherford T, O'Donnell M et al. (2014). The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J Neurochem 129: 426–433. [DOI] [PubMed] [Google Scholar]
- Hwabejire JO, Lu J, Liu B, Li Y, Halaweish I, Alam HB (2014). Valproic acid for the treatment of hemorrhagic shock: a dose‐optimization study. J Surg Res 186: 363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoherranen N, Yagen B, Bialer M (2003). New CNS‐active drugs which are second‐generation valproic acid: can they lead to the development of a magic bullet? Curr Opin Neurol 16: 203–211. [DOI] [PubMed] [Google Scholar]
- Jentink J, Loane MA, Dolk H, Barisic I, Garne E, Morris JK et al. (2010). Valproic acid use in pregnancy and congenital malformations. N Engl J Med 363: 2185–2193. [DOI] [PubMed] [Google Scholar]
- Jope RS, Yuskaitis CJ, Beurel E (2007). Glycogen synthase kinase‐3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem Res 32: 577–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauvar DS, Wade CE (2005). The epidemiology and modern management of traumatic hemorrhage: US and international perspectives. Crit Care 9: S1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauvar DS, Lefering R, Wade CE (2006). Impact of hemorrhage on trauma outcome: an overview of epidemiology, clinical presentations, and therapeutic considerations. J Trauma 60: S3–11. [DOI] [PubMed] [Google Scholar]
- Keller ME, Aihara R, LaMorte WW, Hirsch EF (2003). Organ‐specific changes in high‐energy phosphates after hemorrhagic shock and resuscitation in the rat. J Am Coll Surg 196: 685–690. [DOI] [PubMed] [Google Scholar]
- Kheirbek T, Kochanek AR, Alam HB (2009). Hypothermia in bleeding trauma: a friend or a foe? Scand J Trauma Resusc Emerg Med 17: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagishi Y, Matsuda S (2013). Diets involved in PPAR and PI3K/AKT/PTEN pathway may contribute to neuroprotection in a traumatic brain injury. Alzheimers Res Ther 5: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM (1995). A prostaglandin J2 metabolite binds peroxisome proliferator‐activated receptor gamma and promotes adipocyte differentiation. Cell 83: 813–819. [DOI] [PubMed] [Google Scholar]
- Kota BP, Huang THW, Roufogalis BD (2005). An overview on biological mechanisms of PPARs. Pharmacol Res 51: 85–94. [DOI] [PubMed] [Google Scholar]
- Lampen A, Siehler S, Ellerbeck U, Goettlicher M, Nau H (1999). New molecular bioassays for the estimation of the teratogenic potency of valproic acid derivatives in vitro: activation of the peroxisomal proliferator‐activated receptor δ (PPARδ). Toxicol Appl Pharmacol 160: 238–249. [DOI] [PubMed] [Google Scholar]
- Lampen A, Carlberg C, Nau H (2001). Peroxisome proliferator‐activated receptor delta is a specific sensor for teratogenic valproic acid derivatives. Eur J Pharmacol 431: 25–33. [DOI] [PubMed] [Google Scholar]
- Lecky FE, Woodford M, Bouamra O, Yates DW (2002). Trauma Audit and Research Network. Lack of change in trauma care in England and Wales since 1994. Emerg Med J 19: 520–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu B, Sailhamer EA, Yuan Z, Shults C, Velmahos GC et al. (2008). Cell protective mechanism of valproic acid in lethal hemorrhagic shock. Surgery 144: 217–224. [DOI] [PubMed] [Google Scholar]
- Palaty J, Abbott FS (1995). Structure‐activity relationships of unsaturated analogs of valproic acid. J Med Chem 38: 3398–3406. [DOI] [PubMed] [Google Scholar]
- Palkar PS, Borland MG, Naruhn S, Ferry CH, Lee C, Sk UH et al. (2010). Cellular and pharmacological selectivity of the peroxisome proliferator‐activated receptor‐β/δ antagonist GSK3787. Mol Pharmacol 78: 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH (2001). Tumor suppressor and anti‐inflammatory actions of PPARγ agonists are mediated via upregulation of PTEN. Curr Biol 11: 764–768. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ; NC‐IUPHAR. (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42(Database Issue): D1098‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS (2001). Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276: 36734–36741. [DOI] [PubMed] [Google Scholar]
- Rossaint R, Cerny V, Coats TJ, Duranteau J, Fernández‐Mondéjar E, Gordini G, et al. (2006). Key issues in advanced bleeding care in trauma. Shock 26: 322–331. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P (1996). Binding of GSK3beta to the APC‐beta‐catenin complex and regulation of complex assembly. Science 272: 1023–1026. [DOI] [PubMed] [Google Scholar]
- Ryves W, Dalton EC, Harwood AJ, Williams RSB (2005). GSK‐3 activity in neocortical cells is inhibited by lithium but not carbamazepine or valproic acid. Bipolar Disord 7: 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakanaka C (2002). Phosphorylation and regulation of β‐catenin by casein kinase Iε. J Biochem 132: 697–703. [DOI] [PubMed] [Google Scholar]
- Shults C, Sailhamer EA, Li Y, Liu B, Tabbara M, Butt MU et al. (2008). Surviving blood loss without fluid resuscitation. J Trauma 64: 629–638. [DOI] [PubMed] [Google Scholar]
- Shoemaker WC (1996). Oxygen Transport and Oxygen Metabolism in Shock and Critical Illness. Crit Care Clin 12: 939–969. [DOI] [PubMed] [Google Scholar]
- Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J et al. (1999). PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,‐trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A 96: 6199–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terbach N, Williams RSB (2009). Structure‐function studies for the panacea, valproic acid. Biochem Soc Trans 37: 1126–1132. [DOI] [PubMed] [Google Scholar]
- Tomson T, Battino D (2008). Teratogenic effects of antiepileptic drugs. Seizure 17: 166–171. [DOI] [PubMed] [Google Scholar]
- Tramontano AF, Muniyappa R, Black AD, Blendea MC, Cohen I, Deng L et al. (2003). Erythropoietin protects cardiac myocytes from hypoxia‐induced apoptosis through an Akt‐dependent pathway. Biochem Biophys Res Commun 308: 990–994. [DOI] [PubMed] [Google Scholar]
- Valvezan AJ, Klein PS (2012). GSK‐3 and Wnt signaling in neurogenesis and bipolar disorder. Front Mol Neurosci 5: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RSB, Cheng L, Mudge AW, Harwood AJ (2002). A common mechanism of action for three mood‐stabilizing drugs. Nature 417: 292–295. [DOI] [PubMed] [Google Scholar]
- Xu X, Müller‐Taubenberger A, Adley KE, Pawolleck N, Lee VWY, Wiedemann C et al. (2007). Attenuation of phospholipid signaling provides a novel mechanism for the action of valproic acid. Eukaryot Cell 6: 899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias N, Sailhamer EA, Li Y, Liu B, Butt MU, Shuja F et al. (2011). Histone deacetylase inhibitors prevent apoptosis following lethal hemorrhagic shock in rodent kidney cells. Resuscitation 82: 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Compounds structurally similar to VPA show variable HDAC inhibitory activity. Enzyme extract from HeLa cells was treated with decanoic acid (A), sebacic acid (B), valpromide (C), 2eVPA (D), and 2‐propyloctanoic acid (E) at concentrations between 0.5 and 10 mM, proportion of deacetylated assay substrate measured (fluorescence) and compared with an uninhibited control. Data are quantified from at least triplicate experiments with technical triplicates (n ≥ 9) ⨦ SEM.
Figure S2 VPA acts through a transcriptional mechanism and reduces LDH release. Huh7 cells were incubated for 4 h in the presence of hypoxia (2% O2), hypercapnia (10% CO2), and hypothermia (32°C) (HxHcHp). (A) Cells were treated with VPA (0.75 mM) in the presence or absence of transcription inhibitor actinomycin D, and protein extract was analysed for pGSK3β‐Ser9 levels and β‐tubulin loading control. (B) Huh7 cells, again under stress conditions, with or without VPA (0.75mM) were assayed for LDH release using LDH Cytotoxicity Assay. Data are quantified from at least triplicate experiments with technical triplicates (n ≥ 9) ⨦ SEM. *P > 0.05, ***P > 0.001.
Supporting info item
Supporting info item