

Summary
Insulin secretion by pancreatic islet β-cells is critical for glucose homeostasis, and a blunted β-cell secretory response is an early deficit in type 2 diabetes. Here, we uncover a regulatory mechanism by which glucose recruits vascular-derived neurotrophins to control insulin secretion. Nerve Growth Factor (NGF), a classical trophic factor for nerve cells, is expressed in pancreatic vasculature while its TrkA receptor is localized to islet β-cells. High glucose rapidly enhances NGF secretion, and increases TrkA phosphorylation in mouse and human islets. Tissue-specific deletion of NGF, TrkA, or acute disruption of TrkA signaling impairs glucose tolerance and insulin secretion in mice. We show that internalized TrkA receptors promote insulin granule exocytosis via F-actin reorganization. Furthermore, NGF treatment augments glucose-induced insulin secretion in human islets. These findings reveal a non-neuronal role for neurotrophins, and identify a new regulatory pathway in insulin secretion that can be targeted to ameliorate β-cell dysfunction.
eTOC Blurb
Blunted insulin secretion by pancreatic β-cell contributes to type 2 diabetes. Houtz et al. reveal that glucose stimulates paracrine neurotrophin signaling between pancreatic vasculature and islet beta-cells to control insulin secretion. These findings identify a non-canonical role for neurotrophins, classical neuronal growth and survival factors, in insulin secretion and glucose homeostasis.
Introduction
Insulin-producing β-cells are a small population of endocrine cells that comprise less than 2% of the pancreas, yet their function is imperative to maintenance of blood glucose homeostasis. In response to elevated blood glucose, β-cells secrete the hormone insulin which triggers glucose uptake by peripheral tissues. Blunted insulin secretion by β-cells is one of the earliest features of type 2 diabetes, a disease affecting almost 300 million people worldwide (Guariguata et al., 2014), observed even in pre-diabetic individuals and thought to precede the onset of overt hyperglycemia (Weir and Bonner-Weir, 2004). Strikingly, it has been postulated that by the time a diagnosis of diabetes is made, patients have lost almost 80% of β-cell function (Defronzo, 2009). Although loss of β-cell secretory responses precede a reduction in β-cell mass in type 2 diabetes (Defronzo, 2009; Weir and Bonner-Weir, 2004), research has predominantly focused on mechanisms governing β-cell proliferation (Vetere et al., 2014; Wang et al., 2015).
While glucose is the primary stimulus for insulin secretion, β-cell secretion is potentiated by extrinsic signals that include fatty acids, amino acids, peptide hormones, and neurotransmitters that play essential roles (Henquin et al., 2003; Prentki et al., 2013; Rorsman and Braun, 2013). Glucose-stimulated insulin secretion (GSIS) involves the key steps of glucose uptake in β-cells, mitochondrial metabolism to alter the ATP/ADP ratio, closure of ATP-sensitive K+-channels and subsequent β-cell plasma-membrane depolarization, opening of voltage-gated Ca2+-channels, and Ca2+-dependent exocytosis of insulin granules (MacDonald et al., 2005). Extrinsic signals influence GSIS either by producing metabolic intermediates within β-cells, or by generating signaling second messengers that impinge on β-cell electrical activity and/or insulin exocytosis (Prentki et al., 2013; Rorsman and Braun, 2013). Because nutrient, hormonal, and neural inputs are precisely regulated by glucose itself, together they ensure insulin secretion remains glucose-dependent at much lower plasma glucose concentrations than are effective in vitro (Henquin et al., 2003; Prentki et al., 2013). Thus, a small meal-related increase in plasma glucose elicits a larger insulin response than predicted from in vitro dose-response curves, due to effects of glucose on non-glucose stimuli. Conversely, the exquisite glucose-dependence of these non-glucose insulin secretagogues also protects against hypoglycemia (Henquin et al., 2003).
Neurotrophins are soluble peptide factors known predominantly for their functions in neuronal survival, axon growth, and synaptic communication (Huang and Reichardt, 2001). Although neurotrophins and their Trk receptor tyrosine kinases are expressed in non-neuronal tissues including the pancreas (Tessarollo, 1998), little is known about their in vivo functions outside of the nervous system. Autocrine signaling by the classical neurotrophin, Nerve Growth Factor (NGF), has been implicated in β-cell survival and secretion in cell cultures (Navarro-Tableros et al., 2004; Rosenbaum et al., 2001). However, whether neurotrophin signaling is essential for β-cell function in vivo where intercellular communications between β-cells and neighboring cell types are preserved, has not been addressed. The need to understand the physiological relevance of neurotrophins in islet function is underscored by evidence that altered neurotrophin secretion and/or signaling could contribute to the etiology of diabetes (Bullo et al., 2007; Kim et al., 2009; Schreiber et al., 2005). In humans, mutations in the TrkA gene encoding for the NGF receptor, cause a form of hereditary peripheral neuropathy called congenital insensitivity to pain and anhidrosis (CIPA) (Indo et al., 1996). Children with CIPA show decreased insulin secretion in response to a glucose challenge, suggesting that NGF signaling may play a role in insulin responses in humans (Schreiber et al., 2005). Furthermore, altered circulating NGF levels have been noted in type 2 diabetes (Bullo et al., 2007; Kim et al., 2009), although whether this reflects a cause or effect in disease pathogenesis remains undefined.
Here, we uncover a fundamental role for neurotrophin signaling in controlling glucose-stimulated insulin secretion, and elucidate the cellular underpinnings. We found that NGF is robustly expressed in pancreatic vascular contractile cells, whereas its TrkA receptor is localized to islet β-cells. Elevated glucose rapidly increases NGF secretion, and stimulates TrkA phosphorylation in islets. Vascular-specific NGF deletion, pancreas-specific TrkA deletion, or acute inactivation of TrkA impairs glucose tolerance and attenuates GSIS in mice. TrkA activity promotes insulin granule localization to the β-cell plasma membrane via disassembly of a rigid F-actin barrier. Furthermore, Trk-mediated endosomal signaling, a critical determinant of neurotrophin actions in neurons, is conserved in β-cells and functionally important for insulin secretion. Finally, NGF potentiates GSIS in human islets. These findings elucidate a new pathway by which glucose promotes NGF/TrkA-mediated actin reorganization to trigger insulin secretion in β-cells.
Results
Pancreatic TrkA receptors are essential for glucose homeostasis and insulin secretion
We recently reported that NGF signaling indirectly influences islet architecture and functional maturation by recruiting sympathetic nerves to developing islets (Borden et al., 2013). However, both NGF and its TrkA receptors are reportedly expressed in rat β-cells (Kanaka-Gantenbein et al., 1995; Rosenbaum et al., 1998), suggesting a cell-autonomous requirement for NGF signaling. To identify an intrinsic role for NGF signaling in pancreatic cells, we first defined TrkA localization in the mouse pancreas. Since available TrkA antibodies were inadequate for immunohistochemical analyses in the pancreas, we employed a ligand binding assay where mouse pancreatic tissue sections were incubated with biotinylated NGF followed by detection with Alexa-546-conjugated streptavidin. Highest levels of biotinylated NGF binding were observed in islets and co-localized with insulin immunostaining (Figure 1A). TrkA expression in β-cells was also confirmed by immunoblotting of FAC-sorted β-cells from MIP-GFP transgenic mice (Figure S1A), which express a GFP reporter under the insulin promoter (Hara et al., 2003). In addition to TrkA, NGF also binds the p75 receptor. Immunohistochemistry with a p75 antibody revealed p75 expression in nerve fibers surrounding islets, but not in endocrine cells (Figure 1B). Ligand binding was not detected in nerve fibers innervating the pancreas (Figure 1A), likely due to low levels of TrkA in mature axons (Miller et al., 1994), and the lower affinity of NGF for p75 (Rodriguez-Tebar et al., 1990). Together, these results suggest that NGF signaling in islets is primarily transduced by β-cell-localized TrkA receptors.
Figure 1. Pdx1-Cre;TrkAf/f mice are glucose intolerant and have reduced insulin secretion.

(A) Biotinylated NGF (b-NGF) binding in insulin-positive β-cells. (B) p75 expression in nerve fibers at the islet perimeter. (C,D) b-NGF binding is reduced in Pdx1-Cre;TrkAf/f islets. Scale bar, 100 μm for (A–D). (E) Loss of TrkA in Pdx-Cre;TrkAf/f islets. Representative images in (A–E) are from at least 3 animals per genotype. (F,G) Pdx1-Cre;TrkAf/f mice have normal fasted blood glucose, but have high fed glucose. Means ± SEM for n=13 control, 14 mutant mice for (F), n=19 control, 15 mutant mice for (G). (H,I) Pdx1-Cre;TrkAf/f mice are glucose intolerant and have reduced insulin secretion. Means ± SEM for n=13 control, 14 mutant mice for (H), n=4 mice per genotype for (I). *p<0.05, **p<0.01, ***p<0.001, t-test for (F,G, H, and I).
To address the intrinsic role of TrkA in endocrine cells, we mated floxed TrkA (TrkAf/f) mice (Chen et al., 2005) with Pdx1-Cre transgenic mice (Hingorani and Tuveson, 2003), where Pdx1 is a transcription factor expressed in pancreatic progenitor cells. Pdx1-Cre;TrkAf/f mice would be expected to have an early and pancreas-wide deletion of TrkA. Pdx1-Cre;TrkAf/f mice survived to adulthood, had no gross morphological abnormalities, and had normal body weight (Figure S1B). Ligand binding and TrkA immunoblotting demonstrated reduced TrkA protein levels in mutant islets (Figures 1C–E), indicative of efficient TrkA deletion. Immunostaining with islet hormone markers, insulin and glucagon, revealed normal islet formation and cyto-architecture in neonatal Pdx1-Cre;TrkAf/f mice (Figure S1C). However, mutant islets showed a modest decrease in size by one month of age (Figures S1D,E), and this may be due, in part, to the trophic effects of NGF signaling in β-cells (Navarro-Tableros et al., 2004). When we assessed metabolic parameters at the whole animal level, 1.5–2 month-old Pdx1-Cre;TrkAf/f mice were slightly hyperglycemic when fed ad libitum, although fasted blood glucose levels were normal (Figures 1F,G). However, Pdx1-Cre;TrkAf/f mice showed significant glucose intolerance and decreased circulating insulin levels after glucose administration, compared to control TrkAf/f mice (Figures 1H,I). Although Cre recombinase activity has been reported in extra-pancreatic tissues, including the hypothalamus, in Pdx1-Cre transgenic mice (Song et al., 2010), there is little overlap with TrkA expression (Fagan et al., 1997). Additionally, the (Tg(Pdx1-CreTuv) transgenic mice that we employed have not been reported to carry a human growth hormone mini-gene, commonly found in several Cre lines, that elicits metabolic defects (Brouwers et al., 2014). Together, these results suggest that loss of TrkA in pancreatic tissues causes the defects in glucose homeostasis and insulin secretion in Pdx1-Cre;TrkAf/f mice.
TrkA kinase activity is acutely required for GSIS
In Pdx1-Cre;TrkAf/f mice, deletion of TrkA is initiated at early embryonic stages. Thus, the observations of impaired glucose tolerance and reduced insulin secretion in these mice could stem from developmental anomalies and/or acute deficits in insulin secretion. To distinguish between developmental versus acute effects of TrkA signaling, we employed a chemical-genetic approach to inducibly silence TrkA kinase activity in mature mice at 2 months of age. TrkAF592A knock-in mice express receptors with a mutated ATP binding pocket that can be selectively, rapidly and reversibly inhibited by a small molecule membrane-permeable inhibitor, 1NMPP1 (Chen et al., 2005) (Figure 2A). Although TrkAF592A mice have been reported to be hypomorphic (https://www.jax.org/strain/022362), these mice are viable and fertile, and TrkA signaling is effectively blocked by application of 1NMPP1 (Chen et al., 2005). In TrkAF592A mice, we always compared effects of 1NMPP1 administration to vehicle (DMSO) treatment in litter-mates. 1NMPP1 treatment attenuated TrkA phosphorylation in isolated TrkAF592A islets, but had no effect in wild-type islets (Figures 2B,C). Importantly, acutely disrupting TrkA activity in vivo by injecting 1NMPP1 (20 ng/g body weight, intra-peritoneally), 20 minutes prior to a glucose challenge, elicited glucose intolerance in adult TrkAF592A mice (Figure 2D), similar to Pdx1-Cre;TrkAf/f mice. The area under the curve (AUC) values of plasma glucose was significantly higher with 1NMPP1 treatment compared to vehicle (DMSO) injection (Figure 2E). Furthermore, TrkAF592A mice treated with 1NMPP1 showed reduced insulin secretion in the first phase of a glucose challenge, as well as dampened insulin levels in the sustained second phase (Figure 2F). Consistently, 1NMPP1 treatment also significantly decreased GSIS in TrkAF592A islets in static insulin secretion assays (Figure 2G).
Figure 2. Acute TrkA inactivation impairs glucose tolerance and insulin secretion.

(A) Chemical-genetic approach to silence TrkA kinase activity in TrkAF592A mice. (B,C) 1NMPP1 decreases TrkA phosphorylation in TrkAF592A islets, but has no effect in wild-type islets. Quantification of phospho-TrkA levels normalized to total TrkA. Values are relative to vehicle-treated islets. Means ± SEM from 3 experiments. (D) 1NMPP1 injection, prior to a glucose challenge, impairs glucose tolerance in TrkAF592A mice. n=6 vehicle- and n=8 1NMPP1-injected TrkAF592A mice. (E) Area under the curve (AUC) for glucose tolerance. (F) 1NMPP1 injection impairs glucose-induced insulin secretion in TrkAF592A mice. n=5 mice each for DMSO- and 1NMPP1-injections. (G) 1NMPP1-mediated attenuation of GSIS in isolated TrkAF592A islets. Means ± SEM from n=6 experiments. (H) Total islet insulin content is normal with 1NMPP1 treatment. Means ± SEM from n=5 experiments. (I) Normal insulin sensitivity in TrkAF592A mice treated with 1NMPP1. Means ± SEM from n=5 mice each for 1NMPP1 and vehicle injections. *p<0.05, **p<0.01,***p<0.001, n.s. not significant, t-test for (C, D, E, F, H, and I) and two- way ANOVA with Bonferroni post-hoc test for (G)
There were no differences in total islet insulin content and insulin sensitivity between 1NMPP1- and vehicle-injections in TrkAF592A mice (Figures 2H,I). Thus, glucose intolerance in 1NMPP1-treated mice likely does not arise from defects in insulin biosynthesis and insulin responsiveness. Notably, 1NMPP1 treatment did not alter glucose tolerance or insulin secretion in wild-type animals (Figures S2A–C), highlighting specific effects of 1NMPP1 in the context of the TrkAF592A mutation.
Together, these results indicate that TrkA signaling acutely regulates glucose homeostasis and insulin secretion, in a manner independent of developmental effects.
TrkA signaling promotes insulin granule exocytosis via F-actin remodeling
Regulatory control of insulin secretion can occur at the level of glucose uptake and metabolism in β-cells, β-cell membrane depolarization, and insulin granule mobilization and exocytosis (MacDonald et al., 2005). A key question is, which step in this stimulus-secretion coupling pathway is affected by TrkA signaling? Exposure to high extracellular K+ is a well- established strategy to depolarize the β-cell plasma membrane, thus by-passing the need for glucose uptake and metabolism to trigger insulin secretion (Hatlapatka et al., 2009). Elevated potassium chloride (KCl) results in the opening of voltage-dependent Ca2+ channels to promote the exocytosis of secretion-ready insulin granules (Hatlapatka et al., 2009). To determine if the requirement for TrkA signaling in insulin secretion was downstream of glucose sensing and metabolism, we assessed the effects of 1NMPP1 on KCl-induced insulin secretion. 1NMPP1-mediated inhibition of TrkA activity abolished insulin secretion induced by high KCl (30mM) in TrkAF592A islets (Figure 3A). Thus, TrkA signaling is required in the step(s) of the insulin secretory pathway that is distal to membrane depolarization.
Figure 3. TrkA signaling regulates insulin exocytosis via actin remodeling.

(A) TrkA activity is necessary for KCl-induced insulin secretion. 1NMPP1 suppresses insulin secretion in response to high KCl (30 min) in TrkAF592A islets. Means ± SEM from n=5 experiments. (B–E) TrkA signaling is necessary for glucose-induced surface localization of insulin granules. Docked insulin granules in β-cells are indicated by red arrowheads. Scale bar, 1 μm. (F) Quantification of docked insulin granules per micron of plasma membrane. Means ± SEM from n=3 mice each for vehicle and 1NMPP1 injections. (G–J) 16.7 mM glucose (30 min) decreases F-actin in TrkAF592A β-cells. Glucose-dependent F-actin remodeling was prevented by 1NMPP1. F-actin is marked by Alexa-546-phalloidin and β-cells were identified by insulin immunostaining. Scale bar, 5μm. (K) Average fluorescence intensity for F-actin. Means ± SEM from n=7 experiments. (L) 1NMPP1 attenuated glucose-induced increase in Rac1 activity. Means ± SEM from n=5 experiments. (M) Cytochalasin D corrected the GSIS defect in 1NMPP1-treated TrkAF592A islets. Means ± SEM from n=7 experiments. *p<0.05, **p<0.01, ***p<0.001, n.s. not significant, two-way ANOVA with Bonferroni post-test for (A, F, K, L and M).
To further probe the requirement for TrkA activity in insulin secretion, we employed ultra-structural analyses. Electron microscopy revealed an increase in insulin granule localization at the β-cell plasma membrane in response to an in vivo glucose challenge compared to the fasted state in TrkAF592A mice (Figures 3B,C, and F). However, 1NMPP1 injection, 20 minutes prior to glucose administration, suppressed glucose-induced recruitment of insulin granules to the cell periphery, but had no effect on insulin docking under basal conditions (Figures 3D,E, and F). 1NMPP1-treated β-cells had fewer docked insulin granules (quantified as granules within 50 nm of the plasma membrane) in the presence of high glucose. Since islet insulin content was unaffected by 1NMPP1 treatment (see Figure 2H), the reduction in surface-localized insulin granules is not due to defects in insulin biogenesis. Together, these results suggest that TrkA activity is necessary for glucose-dependent insulin granule positioning at the β-cell surface.
Since a brief (20 minute) exposure to 1NMPP1 was sufficient to elicit glucose intolerance and attenuate GSIS in TrkAF592A mice, we reasoned that TrkA signaling likely influences insulin secretion by mechanisms independent of gene transcription. Actin rearrangement is a critical acute determinant of GSIS (Kalwat and Thurmond, 2013). Actin microfilaments are organized as a dense meshwork beneath the β-cell plasma membrane that restricts insulin granule access to the docking and fusion machinery (Orci et al., 1972). Glucose stimulation rapidly promotes filamentous actin (F-actin) remodeling to mobilize insulin granules to the cell periphery (Kalwat and Thurmond, 2013; Thurmond et al., 2003). However, how glucose stimulation regulates actin reorganization has remained unclear.
Remodeling of the actin cytoskeleton is also a key cellular process by which neurotrophins control vesicular trafficking in neurons (Harrington and Ginty, 2013). Thus, we asked whether TrkA signaling promotes actin rearrangements in β-cells, a pre-requisite step in insulin granule positioning at the plasma membrane. Isolated TrkAF592A β-cells were pre-treated with 1NMPP1 for 20 minutes prior to stimulation with either low (2.8 mM) or high glucose (16.7 mM), and F- actin was visualized using Alexa-546-labeled phalloidin. Under basal conditions, both vehicle and 1NMPP1-treated β-cells showed a thick F-actin cortical ring (Figures 3G,I). High glucose treatment elicited a striking reduction in F-actin (Figures 3H,K), consistent with previous reports (Cai et al., 2012; Nevins and Thurmond, 2003). However, the glucose-induced dissolution of F- actin was prevented by 1NMPP1-mediated silencing of TrkA signaling (Figures 3J,K). Thus, TrkA activity is required for glucose-triggered actin reorganization in β-cells.
Glucose-mediated actin remodeling in β-cells depends on the activities of several actin-modulatory proteins including the Rac1 GTPase (Kalwat and Thurmond, 2013). In clonal β-cell lines and in islets, elevated glucose stimulates Rac1 activity (Kowluru, 2011). β-cell-specific deletion of Rac1 inhibits F-actin disassembly and impairs glucose tolerance and GSIS in mice (Asahara et al., 2013). Rac1-deficient β-cells also show impaired glucose-dependent recruitment of insulin granules to the β-cell membrane (Asahara et al., 2013), similar to that seen with TrkA inhibition. However, how elevated glucose activates Rac1 remains unclear. Using an ELISA-based immunoassay, we observed that high glucose treatment for 15 minutes significantly increased GTP-bound Rac1 levels (1.8 ± 0.15-fold increase) in TrkAF592A islets, which was suppressed by TrkA inhibition with 1NMPP1 (Figure 3L). These results suggest that TrkA kinase activity is required for glucose-mediated activation of Rac1.
We next reasoned that if indeed a perduring F-actin barrier was a key contributor to decreased GSIS upon TrkA inhibition, then forcing F-actin disassembly should alleviate impaired insulin secretion in 1NMPP1-treated islets. To test this prediction, we assessed insulin secretion in islets that were co-treated with 1NMPP1 and cytochalasin D, a cell-permeable fungal toxin that disrupts actin polymerization. As expected, 1NMPP1 treatment alone attenuated GSIS compared to vehicle (DMSO) in TrkAF592A islets (Figure 3M). Remarkably, normal insulin secretion in response to elevated glucose was observed in TrkAF592A islets treated with 1NMPP1 plus cytochalasin D (Figure 3M). Together, these results indicate that TrkA-mediated actin reorganization is a key mechanism contributing to glucose-induced insulin secretion.
We also noted that cytochalasin D treatment alone had a striking potentiating effect on GSIS, in agreement with previous studies (Lacy et al., 1973; van Obberghen et al., 1973), which was significantly higher than that observed in cytochalasin D plus 1NMPP1-treated islets (Figure 3M). That 1NMPP1 treatment dampened GSIS in the presence of cytochalasin D suggests that TrkA inactivation might diminish the efficacy of cytochalasin D, and/or that TrkA signaling utilizes additional mechanisms that are independent of the actin cytoskeleton to influence insulin granule exocytosis.
Endocytosed TrkA receptors mediate actin reorganization and insulin secretion
In neurons, endosomal signaling from intracellular TrkA receptors is a key determinant of actin remodeling (Harrington et al., 2011). NGF promotes endocytosis of its TrkA receptors in nerve endings into signaling endosomes that are retrogradely transported to neuronal cell bodies to activate trophic signaling (Ascano et al., 2012). Endosomal TrkA signaling overcomes a dense peripheral actin network in axons to “carve a path” for retrogradely moving vesicles (Harrington et al., 2011). These findings raise the possibility of an analogous mechanism in β-cells where neurotrophin signaling endosomes might mediate changes in F-actin necessary for insulin granule mobilization.
To address this possibility, we first asked whether TrkA receptors undergo ligand-dependent endocytosis in β-cells. We probed TrkA endocytosis using a cell surface biotinylation assay in MIN6 cells, a mouse insulinoma cell-line that exhibits many characteristics of primary β-cells, including GSIS, and have been widely used for biochemical analyses (Ishihara et al., 1993). NGF stimulation for 30 minutes markedly increased TrkA endocytosis compared to un-stimulated cells (Figures 4A,B). Furthermore, we visualized trafficking of surface TrkA receptors in primary β-cells using a well-established antibody-feeding assay (Ascano et al., 2009). Primary β-cells were infected with an adenoviral vector expressing FLAG-tagged chimeric receptors that have the extracellular domain of TrkB and the transmembrane and intracellular domains of TrkA (FLAG-TrkB:A). Chimeric Trk receptors respond to the TrkB ligand, Brain-Derived Neurotrophic Factor (BDNF), but retain the signaling properties of TrkA. Live-cell FLAG antibody feeding revealed predominantly surface localization of the receptors in the absence of ligand (Figure 4C). However, in BDNF-stimulated β-cells, Trk receptors accumulated in intracellular punctae that co-localized with Early Endosome Antigen 1 (EEA1), an early endosome marker (Figures 4D, E). Thus, ligand-mediated endocytosis of Trk receptors is a conserved mechanism that occurs in both β-cells and neurons.
Figure 4. TrkA endocytosis is necessary for glucose-induced actin changes and insulin secretion.

(A) NGF (100 ng/ml, 30 min) promotes TrkA internalization in MIN6 cells, assessed by a cell-surface biotinylation assay. Supernatants after neutravidin precipitation were probed for p85 for protein normalization. (B) Quantification of internalized TrkA. Means ± SEM from 4 experiments. (C,D) FLAG-TrkB:A chimeric receptors undergo BDNF-dependent internalization in β-cells, assessed by FLAG antibody feeding. Scale bar, 5 μm. (E) Trk internalization into early endosomes was determined by assessing EEA1 and FLAG co-localization. Means ± SEM from 3 experiments. (F, G) The PLCγ inhibitor, U73122, blocks NGF-dependent TrkA internalization in MIN6 cells. Total surface TrkA is shown in the “no stripping” lane. Means ± SEM from 7 experiments. (H–K) TrkA phosphorylation at Y794, the PLCγ docking site, is necessary for TrkA endocytosis. Scale bar, 5 μm. (L) Internal accumulation of FLAG-TrkA receptors was determined by assessing co-localization of FLAG with cytoplasmic GFP. Means ± SEM from 5 experiments. (M–Q) FLAG-TrkAWT, but not FLAG-TrkAY794F receptor, expression corrected 1NMPP1-mediated impairment of glucose-induced F-actin disassembly in isolated TrkAF592A β-cells. Scale bar, 5 μm. Means ± SEM from n=6 experiments. (R) Expression of wild-type FLAG-TrkA, but not FLAG-TrkAY794F receptors, rescued 1NMPP1-mediated impairment of GSIS in TrkAF592A islets. Means ± SEM from n=5 experiments. *p<0.05, **p<0.01, ***p<0.001, n.s. not significant, t-test for (B, E), one-way ANOVA and Tukey's post-hoc test for (G, L), and two-way ANOVA and Bonferroni post-hoc test for (Q, R).
In neurons, TrkA signaling triggers its own internalization by recruiting and activating the downstream effector, Phospholipase C gamma (PLCγ) (Bodmer et al., 2011). Employing cell surface biotinylation to monitor internalization, we found that a selective PLCγ inhibitor, U73122, markedly decreased endocytosis of endogenous TrkA receptors in MIN6 cells (Figures 4F,G). Phosphorylation of TrkA receptors at a specific tyrosine residue, Y794, is necessary for PLCγ recruitment (Stephens et al., 1994). To assess the requirement of Y794 phosphorylation for endocytosis, we performed antibody-feeding assays in MIN6 cells transfected with mutant FLAG-TrkAY794F or control FLAG-TrkA receptors. Upon NGF treatment, FLAG-TrkA receptors were found in intracellular punctae, whereas mutant FLAG-TrkAY794F receptors remained largely at the cell surface (Figures 4H, 4I, 4J, and 4L). Furthermore, FLAG-TrkAY499F receptors, mutated at a site necessary for recruitment of the adaptor protein, Shc, and coupling to downstream MAP kinase and phosphatidylinositol 3-kinase (PI-3K) effector pathways, were internalized normally in response to ligand (Figures 4K and 4L). Thus, TrkA phosphorylation at Y794 and activation of PLCγ are required for endocytosis in β-cells, as in neurons.
To investigate the effects of endocytosis-deficient Trk receptors on actin remodeling and insulin secretion in primary β-cells and islets, we generated adenoviral vectors expressing mutant FLAG-TrkAY794F or control FLAG-TrkA receptors, that were also doxycycline-inducible to precisely control expression. FLAG-tagged receptors were expressed in a doxycycline-dependent manner and also appeared normally on the cell surface in infected MIN6 cells (Figure S3A). To assess the effects of ectopic TrkA receptors on glucose-induced actin remodeling, isolated β-cells from TrkAF592A mice were infected with FLAG-TrkA adenoviruses, treated with doxycycline (100 ng/ml) for 24–30 hr, and then treated with 1NMPP1 for 20 minutes to silence endogenous TrkA activity. The FLAG-TrkA receptors are impervious to 1NMPP1 since they do not harbor the F592A mutation. β-cells were then exposed to low (2.8 mM) or high (16.7 mM) glucose, and F-actin visualized with phalloidin labeling. We found that ectopic expression of control FLAG-TrkA receptors promoted glucose-dependent actin reorganization in 1NMPP1-treated cells (Figures 4M, 4N and 4Q). In contrast, mutant FLAG-TrkAY794F receptors were unable to rescue the 1NMPP1-mediated impairment in actin reorganization (Figures 4O–Q). In insulin secretion assays, control FLAG-TrkA receptors elicited robust insulin secretion in response to glucose, despite the presence of 1NMPP1 (Figure 4R). However, GSIS was suppressed in islets expressing mutant FLAG-TrkAY794F receptors (Figure 4R). Furthermore, PLCγ inhibition also decreased GSIS in islets, similar to the effects of the non-internalizable TrkA receptors (Figure S3B). These results suggest that TrkA endocytosis, via PLCγ activity, is required for glucose-stimulated actin remodeling and insulin secretion.
NGF expression in the vasculature, but not pancreatic anlage, is necessary for glucose homeostasis, GSIS, and actin remodeling
Our findings that TrkA receptors in β-cells are required for GSIS prompted us to address the cellular source of NGF in the pancreas. Previous studies have reported NGF expression in β-cells (Rosenbaum et al., 1998), and antibody-mediated neutralization of endogenous NGF attenuated GSIS in dissociated β-cell cultures (Rosenbaum et al., 2001). To address whether β-cell-derived NGF is essential for glucose homeostasis and GSIS in vivo, we crossed mice carrying a floxed NGF allele (NGFf/f mice) (Muller et al., 2012) with Pdx1-Cre mice. Surprisingly, conditional loss of NGF from all pancreatic cell types including β-cells did not impair glucose tolerance (Figure 5A), and isolated Pdx1-Cre;NGFf/f islets showed normal insulin secretion in response to a glucose challenge (Figure 5B). These results suggest that autocrine NGF signaling in β-cells is not a major contributing factor to glucose homeostasis at the whole animal level and for GSIS in intact islets.
Figure 5. Vascular-specific NGF deletion impairs glucose homeostasis, GSIS, and actin remodeling.

(A) Normal glucose tolerance in Pdx1-Cre;NGFf/f mice. Values are means ± SEM from n=10 mice per genotype. (B) GSIS is unaffected in Pdx1-Cre;NGFf/f islets. Means ± SEM from n=11 NGFf/f and n=7 Pdx1-Cre;NGFf/f animals. (C) X-gal staining reports NGF expression in large diameter blood vessels outside islets (red arrows), and also within islets (red arrowheads) in NGFLacZ/+ mice. An islet is outlined in dashed lines, and shown below in higher magnification. Scale bar, 100 μm (D,E) Co-localization of β-gal with smooth muscle actin (SMA), and PDGFRβ, a pericyte marker. Insulin staining is in blue. Inset shows intra-islet pericytes. Scale bars, 100 μm for (D,E) and 50 μm for inset in E. Representative images in (C–E) are from analyzing five NGFLacZ/+ mice. (F) Vascular-specific NGF loss impairs glucose tolerance in tamoxifen-injected Myh11-CreERT2;NGFf/f mice. Means ± SEM for n=7 mice each for vehicle and tamoxifen injections. (G) GSIS is attenuated in tamoxifen-injected Myh11-CreERT2;NGFf/f islets. Means ± SEM from n=8 mice each for vehicle and tamoxifen injection. (H–L) Cultured β-cells from tamoxifen-treated Myh11-CreERT2;NGFf/f mice show persistent cortical F-actin in high glucose. Scale bar, 5 μm. Means ± SEM from n=7 experiments. *p<0.05, **p<0.01, n.s. not significant, two-way ANOVA and Bonferroni post-hoc test for (B, G, and L) and t-test for (F).
To obtain a thorough profile of NGF expression in the pancreas, we next used NGFLacZ/+ knock-in mice where the LacZ transgene reports NGF expression (Liu et al., 2012). Using X-gal staining, we observed robust LacZ expression in large diameter blood vessels outside islets, and in intra-islet micro-vasculature (Figure 5C). To further define vascular cell types that express NGF, we performed double immunofluorescence for β-galactosidase (β-gal) and vascular markers. We observed β-gal immunofluorescence in vascular smooth muscle cells (VSMCs) labeled by smooth muscle actin (SMA) in large arteries outside islets (Figure 5D), and in smooth muscle cell-like pericytes labeled by PDGFRβ within the islet micro-vasculature (Figure 5E). Reporter expression was not detected in pancreatic endothelial cells, ducts, or exocrine tissue (Figures S4A,B). We did not detect LacZ expression in β-cells in tissue sections, in agreement with the lack of metabolic phenotypes in Pdx1-Cre;NGFf/f mice. However, low LacZ expression was seen in β-cells in dissociated islet cultures (Figures S4C,D), consistent with previous findings (Rosenbaum et al., 1998). In dissociated islet cultures, prominent NGF expression was observed in intra-islet pericytes that are also present (Figures S4E,E’). Together, these results suggest vascular contractile cells as a likely source of NGF in the pancreas.
To address the role of vascular-derived NGF in glucose homeostasis and insulin secretion, we generated mutant mice to inducibly delete NGF from vascular contractile cells. We crossed NGFf/f mice to Myh11-CreERT2 transgenic mice, in which tamoxifen-induced Cre expression is specific to vascular smooth muscle cells including pericytes (Heinze et al., 2014; Wirth et al., 2008). To visualize tamoxifen-induced Cre activity in NGF-expressing cell types, we generated Myh11-CreERT2;R26-EYFP mice which were then mated to NGFLacZ/+ mice. Cre reporter fluorescence was observed in NGF-expressing large-diameter blood vessels outside islets and in intra-islet pericytes but not β-cells, upon tamoxifen injection in NGFLacZ/+;Myh11-CreERT2;R26-YFP mice (Figure S4F). qPCR analysis showed significant NGF depletion in islets isolated from tamoxifen-injected-Myh11-CreERT2;NGFf/f mice compared to vehicle treated controls (Figure S4G). When we evaluated metabolic parameters, tamoxifen-injected Myh11-CreERT2;NGFf/f mice were found to be glucose intolerant two weeks after induction of Cre activity (Figure 5F). Furthermore, isolated islets from tamoxifen-injected mice had attenuated GSIS ex vivo, compared to vehicle treatment (Figure 5G). These results indicate that glucose homeostasis and GSIS relies on NGF derived from pancreatic vascular contractile cells.
We next asked whether vascular-specific inactivation of NGF influences glucose-induced cellular events in β-cells. Thus, we examined actin rearrangements in response to high glucose in dissociated islet cultures from vehicle- and tamoxifen-injected Myh11-CreERT2;NGFf/f mice. Tamoxifen injection would ablate NGF from intra-islet pericytes that are present in dissociated islet cultures, but not from β-cells. Visualization of F-actin with Alexa-546-labeled phalloidin revealed that while high glucose elicited cortical actin dissolution in β-cells from vehicle-injected mice (Figures 5H, 5I, and 5L), this effect of glucose was abrogated in β-cells from tamoxifen-injected mice (Figures 5J, 5K, and 5L), similar to our observations with TrkA inactivation. Since islet pericytes are the only cell types in the dissociated cultures that would be targeted by tamoxifen-induced Cre activity, these results indicate that NGF produced by intra-islet pericytes is necessary for glucose-induced actin remodeling in β-cells. Thus, although cultured β-cells express low levels of NGF, this expression is insufficient to mediate glucose-dependent cellular changes in the absence of vascular-derived NGF.
Elevated glucose acutely enhances NGF secretion and TrkA phosphorylation
Alterations in NGF levels have been noted in serum, nerves, and peripheral tissues in diabetic humans and animal models (Bullo et al., 2007; Kim et al., 2009; Meloni et al., 2012). Additionally, elevated glucose enhances NGF biosynthesis and secretion in cultured β-cells and islets (Pingitore et al., 2016; Rosenbaum et al., 1998). These results, together with the observed expression patterns of NGF and TrkA in the pancreas, prompted us to ask if the NGF signaling pathway might be regulated by glucose in vivo. To address this question, we measured serum NGF levels in mice that were fasted overnight and subjected to a glucose challenge administered intra-peritoneally. We observed a significant elevation (1.5-fold increase) in circulating NGF levels within 15 minutes of the glucose challenge (Figure 6A), suggesting that NGF secretion is acutely regulated by glucose in vivo. We were unable to directly profile local release of NGF from islets in response to glucose, because the levels were below the detection limit of our immunoassay, perhaps in part, due to the entrapment of secreted NGF in the extracellular matrix and/or because NGF-expressing pericytes comprise a small fraction of the islet cell population. However, in dissociated aortic tissue cultures, an abundant source of vascular smooth muscle cells, we found that NGF secretion was significantly enhanced (1.48-fold increase) by 15 minutes of exposure to elevated glucose (Figure 6B), similar to the increase in serum NGF observed with an in vivo glucose challenge. Together, these results highlight a direct and acute effect of glucose on vascular smooth muscle cells, the primary NGF-expressing cell types in the pancreas, to elicit NGF release.
Figure 6. NGF signaling is acutely regulated by glucose, and exogenous NGF enhances insulin secretion in human islets.

(A) Circulating NGF levels are rapidly increased 15 min after a glucose injection. Values are the mean ± SEM from n=14 mice. (B) 16.7 mM glucose (15 min) enhances NGF secretion from cultured vascular smooth muscle cells. Means ± SEM from n=3 experiments. (C, D) TrkA phosphorylation is increased by high glucose (15 min) in mouse islets. Means ± SEM from n=3 experiments. (E,F) 16.7 mM glucose (15 min) enhances TrkA phosphorylation in human islets. *p<0.05, t-test. (G) NGF treatment potentiates GSIS in human islets. Islets were treated with either 2.8 or 16.7 mM glucose in the presence or absence of NGF (100 ng/ml) for 30 min. *p<0.05, **p<0.01, t-test for (A, B, D, F, G).
Given that elevated glucose acutely enhanced NGF secretion, we next asked if glucose influences TrkA activity in islets. Thus, we assessed TrkA phosphorylation in isolated islets treated with high glucose (16.7mM). 15 minutes of exposure to elevated glucose stimulated a robust increase (11.69 ± 1.9-fold) in islet TrkA phosphorylation levels, compared to basal conditions (2.8 mM glucose) (Figures 6C, D). NGF binds with high affinity to TrkA receptors with a reported Kd of 10−11M (i.e. 130 pg/ml) (Bibel and Barde, 2000; Sutter et al., 1979). Given that our measurements of circulating NGF was ~200 pg/ml, it is reasonable that modest increases in NGF levels with glucose stimulation would elicit large alterations in TrkA activity. Together, these findings indicate that NGF secretion and signaling in pancreatic islets are regulated by glucose, and suggest an instructive role for the NGF pathway in facilitating GSIS.
We next addressed whether glucose-mediated regulation of the NGF pathway occurs in human islets. Similar to our findings in mouse islets, we found that elevated glucose significantly increased TrkA phosphorylation in human islets (Figure 6E, F), although the fold change (1.74 ± 0.27-fold) was modest compared to that in mouse islets. Although human and mouse NGF have similar binding affinities for TrkA (Altar et al., 1991), that human islets had higher basal TrkA phosphorylation compared to mouse islets could reflect differences in basal NGF levels, tyrosine phosphatase activity, age of islets, or manner of islet isolation and culture. Nevertheless, these findings support the notion that the ability of elevated glucose to activate NGF signaling in islets is conserved.
NGF enhances glucose-dependent insulin secretion in cultured rodent β-cells and islets (Pingitore et al., 2016; Rosenbaum et al., 2001). Whether NGF has a similar effect on human islets has not been addressed. We found that NGF treatment augmented GSIS in human islets, but basal insulin secretion was unaffected (Figure 6G). These results support a physiological role for NGF in potentiating GSIS, although NGF is insufficient by itself to trigger insulin secretion.
Discussion
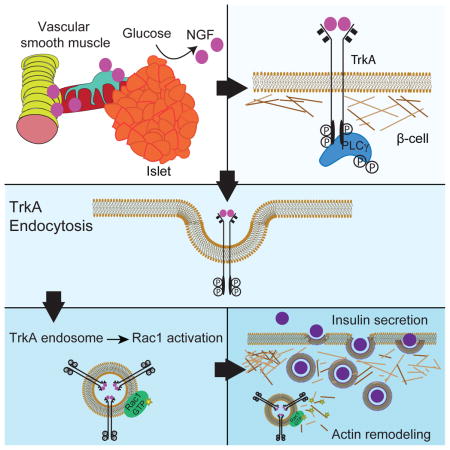
Here, we reveal a mechanism by which neurotrophin signaling influences endocrine functions. Our findings suggest a feed-forward model whereby NGF, secreted by the pancreatic vasculature in response to glucose, activates β-cell TrkA receptors to acutely promote glucose-stimulated insulin secretion in β-cells (Figure 7). TrkA signaling, specifically by internalized receptors, overcomes a peripheral F-actin barrier to boost insulin granule exocytosis in β-cells. Together, these findings identify a new regulatory pathway essential for insulin secretion and blood glucose homeostasis.
Figure 7. Neurotrophin signaling acutely promotes glucose-induced insulin secretion via actin reorganization in β-cells.

(1) NGF is secreted by pancreatic vascular smooth muscle cells and intra-islet pericytes in response to elevated glucose. (2) Vascular-derived NGF activates TrkA receptors on islet β-cells. (3) TrkA phosphorylation on Y794 leads to association and activation of the downstream effector, PLCγ, which triggers receptor internalization. (4) Endosomal signaling from internalized TrkA receptors recruits and activates the actin modulatory protein Rac1, to (5) remodel a peripheral F-actin barrier, and (6) promote insulin granule exocytosis.
The spatial pattern of NGF and TrkA expression in the pancreas, and effects of vascular- specific NGF deletion on GSIS support a paracrine effect of vascular-derived NGF on neighboring β-cells. The NGF expression pattern also implies that signals derived from large diameter blood vessels juxtaposed to islets and/or intra-islet pericytes should be sufficient to support adult islet function. Consistent with this notion, regression of intra-islet endothelial cells due to loss of VEGF signaling did not perturb adult insulin secretion, and elicited only modest elevations in blood glucose (D'Hoker et al., 2013; Reinert et al., 2013). In particular, impaired GSIS in isolated islets ex vivo from tamoxifen-injected Myh11-CreERT2;NGFf/f mice highlights an essential role for NGF produced by intra-islet pericytes in β-cell function. Recently, diphtheria toxin-mediated killing of islet pericytes was found to impair GSIS in mice (Sasson et al., 2016), supporting the significance of these cell types within the islet micro-environment. Although β-cells express NGF, we show that loss of β-cell-derived NGF does not compromise glucose homeostasis in vivo. Notably, NGF expressed in β-cells cannot compensate for loss of vascular-derived NGF in GSIS and actin remodeling. The reasons for differences between our findings and previous reports of autocrine NGF signaling in insulin secretion (Pingitore et al., 2016; Rosenbaum et al., 2001) remain unclear, although the possibility remains that dissociated islet cultures employed in previous studies may have included NGF-expressing pericytes, as we noted in this study. Together, our results support the notion that, while autocrine NGF signaling may be relevant for β-cell development or survival (Navarro-Tableros et al., 2004), paracrine NGF signaling is predominantly required for GSIS and glucose homeostasis.
We identify TrkA signaling as a key mechanism by which glucose stimulation overcomes a dense F-actin meshwork to mobilize insulin granules to the β-cell membrane for efficient secretion. The disparate roles of actin in regulated exocytosis have been well-documented with evidence to support both negative and positive effects. On the one hand, F-actin tracks are needed for mobilizing vesicles from deeper reserve pools to the plasma membrane (Schuh, 2011). On the other hand, cortical F-actin acts as a physical barrier to limit exocytosis in diverse cell types (Porat-Shliom et al., 2013). In β-cells, the barrier function of the cytoskeleton has been proposed to be critical for maintaining low levels of insulin release under basal conditions (Kalwat and Thurmond, 2013). Importantly, a dense cytoskeletal meshwork at the cell periphery presents a layer of regulation for controlled insulin release under elevated glucose (Zhu et al., 2015). Each individual β-cell contains approximately 10,000 secretory granules of insulin but only a fraction (several hundreds) are released at a time in response to high glucose, emphasizing the precise regulation of release probability of insulin granules (Rorsman and Renstrom, 2003). The relevance of actin dynamics in glucose homeostasis is exemplified by genetic studies in mice where deletion of Rac1, or its effector, p21-activated kinase (PAK), elicits glucose intolerance and diminished insulin secretion (Asahara et al., 2013; Wang et al., 2011). Notably, a 10-fold increase in total cellular actin has been observed in islets from type 2 diabetes patients (Ostenson et al., 2006). Although a number of actin regulators of glucose-stimulated insulin secretion including Rac1, Cdc42, PAK, Focal Adhesion Kinase (FAK), cofilin, and gelsolin, have been identified in β-cells (Kalwat and Thurmond, 2013; Thurmond et al., 2003), the events upstream of these actin-modulatory proteins have remained unclear. Our results, together with a previous study in the MIN6 cell line implicating EphA receptors in actin dynamics and insulin secretion (Konstantinova et al., 2007), highlight receptor tyrosine kinase signaling as a critical link in relaying a glucose signal to the β-cell cytoskeleton.
Although neurons and β-cells have distinct developmental origins (Pictet et al., 1976), they share remarkable similarities in terms of electrical properties, ion channel composition, and exocytosis machinery involved in regulated secretion (Arntfield and van der Kooy, 2011). In this study, we describe a non-canonical role for neurotrophins outside of the nervous system, and also define a conserved mechanism for neurotrophin actions via signaling endosomes in non-polarized β-cells. TrkA receptors, that were incapable of internalizing, failed to stimulate actin reorganization and insulin secretion in response to glucose. In neurons, TrkA internalization in axon terminals and subsequent endosomal signaling is obligatory for long-distance communication between distal axons, the site of action of target-derived NGF, and neuronal cell bodies, as recently reviewed in (Cosker and Segal, 2014). In β-cells, TrkA endocytosis may allow access to intracellular signaling effectors including actin modulatory proteins, prolong receptor activation, or allow localized responses at specific sub-cellular domains (Sorkin and von Zastrow, 2009). Interestingly, β-cell specific deletion of dynamin 2 resulted in a striking increase in F-actin density and impaired insulin secretion and glucose tolerance (Fan et al., 2015), similar to our observations with TrkA inactivation, raising the possibility that TrkA receptors may be a cargo for dynamin 2-mediated endocytosis in β-cells. Together, our findings suggest TrkA-harboring endosomes as a unique locus for regulatory control of insulin granule mobilization in β-cells.
Intriguingly, we observed that although cytochalasin D normalized GSIS in 1NMPP1-treated islets, the secretory response in these islets was still blunted compared to the effects of cytochalasin D alone. These findings suggest that TrkA activity might be necessary to render the actin network more susceptible to cytochalasin D-mediated actin disassembly, perhaps by affecting actin-modulatory proteins that influence the ability of cytochalasin D to cap barbed ends or bind actin monomers (Cooper, 1987). Additionally, these observations could suggest contributions of actin-independent TrkA signaling mechanisms to GSIS. Two candidate TrkA pathways that might influence GSIS are the Ca2+ and cAMP second messenger systems. NGF- dependent insulin secretion in cultured rat β-cells was shown to be dependent on Ca2+ influx through voltage-gated Ca2+ channels (Rosenbaum et al., 2001). Furthermore, cAMP signaling via Epac2, a guanine nucleotide exchange factor and its target small GTPase, Rap1, is a key contributing mechanism to insulin exocytosis (Shibasaki et al., 2007). In neurons, Rap1 is localized to TrkA-containing endosomes (Wu et al., 2001), making it an attractive candidate endosomal effector of TrkA that might elicit insulin secretion. Monitoring TrkA-mediated Ca2+ and cAMP activity in β-cells and assessing the consequences of manipulating pathway effectors on insulin secretion and glucose homeostasis will be of interest in future studies.
Our study revealed an unexpected physiological cross-talk between nutrient and neurotrophin signaling. Elevated glucose rapidly increased NGF secretion in vivo, and enhanced TrkA phosphorylation in both mouse and human islets. These results suggest an instructive mechanism by which glucose recruits the NGF signaling axis to augment insulin secretion. This is similar to the effects of glucose on intestinal hormones and neurotransmitters that subsequently act in concert to tightly regulate a post-prandial insulin secretory response. Future studies will be of interest in elucidating the glucose sensing and signaling mechanisms that underlie glucose-mediated neurotrophin secretion in pancreatic vascular cells.
Previous studies on neurotrophin regulation of metabolism have primarily focused on central hypothalamic circuits that control appetite and energy balance (Xu and Xie, 2016). Despite expression of neurotrophins and their receptors in peripheral metabolic tissues, little is known about peripheral mechanisms by which neurotrophins influence metabolism. Recently, p75 receptors in adipocytes were found to regulate energy expenditure and obesity, although this effect was independent of the p75 extracellular domain and neurotrophin binding (Baeza-Raja et al., 2016). Here, we report a direct role for NGF/TrkA signaling within pancreatic endocrine cells in controlling insulin secretion and blood glucose homeostasis. These findings are of clinical relevance given that TrkA mutations have been linked to impaired glucose-stimulated insulin secretion in humans (Schreiber et al., 2005). Our results that NGF treatment potentiated GSIS in human islets suggest the potential utility of neurotrophins and small molecule receptor agonists in the treatment of type 2 diabetes.
Experimental Procedures
Details of immunohistochemistry, EM, receptor trafficking analyses, plasmids, adenoviral vectors, drug treatments, antibodies, insulin and NGF ELISAs, metabolic analyses and insulin secretion can be found in Supplemental Experimental Procedures.
Mice and cell lines
Procedures relating to animal care and treatment conformed to Johns Hopkins University Animal Care and Use Committee (ACUC) and NIH guidelines. Mice were housed in a standard 12:12 light-dark cycle. Mice were maintained on a C57BL/6 background, or mixed C57BL/6 and 129P, or C57BL/6 and FVB backgrounds. Both sexes were used for analyses at 1–2 months of age. TrkAF592A (TrkAf/f), Myh11-cre/ERT2, R26-EYFP and MIP-GFP mice were from Jackson Laboratory, and Pdx1-Cre mice (Tg(Pdx1-CreTuv) were obtained from NCI Frederick. NGFLacZ/+ mice were gifted by Dr. David Ginty (Harvard) and NGFf/f mice were previously generated in the Minichiello laboratory (Muller et al., 2012). MIN6 cells were obtained from Dr. Jun-ichi Miyazaki (Osaka University) and Dr. Donald Steiner (University of Chicago).
In vivo metabolic analyses
For glucose tolerance tests, 1–2-month-old mice were fasted overnight, with a blood glucose reading the evening before the assay serving as fed blood glucose measurement. Mice were injected with glucose (2g/kg, i.p). Blood glucose measurements were made from tail blood using OneTouch Ultra glucometer (Gu et al., 2010). For acute 1NMPP1 treatments, mice received i.p. injections with 20ng/g 1NMPP1 or DMSO, 20 min prior to glucose administration. For in vivo insulin and NGF secretion, mice were fasted overnight before being injected with glucose (3 g/kg, i.p.). Blood was collected from the tail, spun down, and the plasma fractions subjected to either insulin (Crystal Chem, 90080) or NGF ELISA (Millipore, CYT304). Reactions were assessed using a Tecan infinite 200 plate reader. For insulin sensitivity, mice were separated into individual cages with food the evening prior to the assay. The next morning, mice were treated with 0.75 U/kg of insulin (Novolin-R; Novo Nordisk), and blood glucose measurements were made from tail blood (Bruning et al., 1997).
Islet insulin secretion
Islets pooled from 4–5 mice were allowed to recover overnight in RPMI 1640 media containing 5% FBS and 5 U/l penicillin/streptomycin. Groups of 5–10 islets of similar size were handpicked into 24-well dishes, washed in KRHB containing 2.8 mM glucose and allowed to stabilize for 1 hr. Islets were pre-incubated with vehicle (DMSO) alone, 1NMPP1 (20μM), cytochalasin D (25μM) alone or 1NMPP1 (20 μM) plus cytochalasin D (25μM) for 20 minutes. Islets were then incubated in 2.8 mM or 16.7 mM glucose, or KCl (30 mM) in KRHB buffer for another 30 min. Supernatant fractions were removed, islets lysed in acid ethanol, and both cellular and supernatant fractions were subjected to insulin ELISA (Crystal Chem). For total insulin, islets were lysed with acid ethanol, and insulin content determined by ELISA and normalized to DNA, measured from the same lysates using a PicoGreen kit (Invitrogen). To assess effects of endocytosis-defective TrkA receptors on GSIS, TrkAF592A islets were incubated with high-titer adenoviruses expressing either FLAG-TrkAY794F or control FLAG-TrkA receptors, and treated with doxycycline (100 ng/ml) in RPMI 1640 media containing 5% FBS and 5 U/l penicillin/streptomycin for 30 hr. Islets were then incubated in 1NMPP1 (20μM) for 20 min to silence endogenous TrkA receptors, and insulin secretion was measured in low or high glucose conditions.
Human islets were obtained from 10 cadaver donors through the Integrated Islet Distribution Program (IIDP) funded by NIDDK. Islets were allowed to recover overnight in CMRL 1066 medium containing 10% human serum albumin, 5 U/L penicillin/5μg/L streptomycin, 2mM GlutaMAX, and 1mM sodium pyruvate, washed in KRHB containing 2.8 mM glucose and allowed to stabilize for 1 hr. Insulin secretion was measured using ELISA (ALPCO) as described above.
Statistical analyses
All graphs and statistical analyses were done using GraphPad Prism software. Except where noted, Student’s t tests were performed assuming Gaussian distribution, two-tailed, unpaired, and a confidence interval of 95%. One-way or two-way ANOVA analyses were performed when more than two groups were compared.
Supplementary Material
Highlights.
Glucose acutely stimulates NGF secretion and signaling in the pancreas
Vascular NGF and beta-cell TrkA receptors are essential for glucose homeostasis
Internalized TrkA receptors promote insulin exocytosis via F-actin reorganization
NGF augments glucose-stimulated insulin secretion in human islets
Acknowledgments
We thank Mehboob Hussain, Seth Blackshaw, Steve Leach, Samer Hattar, Haiqing Zhao, and Robert Johnston for helpful discussions. We thank David Ginty for NGFLacZ/+ mice, and Jun-ichi Miyazaki and Donald Steiner for MIN6 cells. This work was supported by NIH R01 (DK108267) to R.K., and in part by P30 DK079637 to JHU-UMD-Diabetes Research Center. J.H., A.C., and P.B. were supported by an NIH training grant (T32GM007231).
Footnotes
Author Contributions
J.H. and R.K. designed the study, analyzed data, and wrote the manuscript. J.H., P.B., and. A.C performed experiments. L.M provided mice, contributed to discussions, and manuscript edits.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altar CA, Burton LE, Bennett GL, Dugich-Djordjevic M. Recombinant human nerve growth factor is biologically active and labels novel high-affinity binding sites in rat brain. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:281–285. doi: 10.1073/pnas.88.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arntfield ME, van der Kooy D. beta-Cell evolution: How the pancreas borrowed from the brain: The shared toolbox of genes expressed by neural and pancreatic endocrine cells may reflect their evolutionary relationship. BioEssays : news and reviews in molecular, cellular and developmental biology. 2011;33:582–587. doi: 10.1002/bies.201100015. [DOI] [PubMed] [Google Scholar]
- Asahara S, Shibutani Y, Teruyama K, Inoue HY, Kawada Y, Etoh H, Matsuda T, Kimura-Koyanagi M, Hashimoto N, Sakahara M, et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia. 2013;56:1088–1097. doi: 10.1007/s00125-013-2849-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Bodmer D, Kuruvilla R. Endocytic trafficking of neurotrophins in neural development. Trends in cell biology. 2012;22:266–273. doi: 10.1016/j.tcb.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Richmond A, Borden P, Kuruvilla R. Axonal targeting of Trk receptors via transcytosis regulates sensitivity to neurotrophin responses. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:11674–11685. doi: 10.1523/JNEUROSCI.1542-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza-Raja B, Sachs BD, Li P, Christian F, Vagena E, Davalos D, Le Moan N, Ryu JK, Sikorski SL, Chan JP, et al. p75 Neurotrophin Receptor Regulates Energy Balance in Obesity. Cell reports. 2016;14:255–268. doi: 10.1016/j.celrep.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel M, Barde YA. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes & development. 2000;14:2919–2937. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- Bodmer D, Ascano M, Kuruvilla R. Isoform-specific dephosphorylation of dynamin1 by calcineurin couples neurotrophin receptor endocytosis to axonal growth. Neuron. 2011;70:1085–1099. doi: 10.1016/j.neuron.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden P, Houtz J, Leach SD, Kuruvilla R. Sympathetic Innervation during Development Is Necessary for Pancreatic Islet Architecture and Functional Maturation. Cell reports. 2013;4:287–301. doi: 10.1016/j.celrep.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers B, de Faudeur G, Osipovich AB, Goyvaerts L, Lemaire K, Boesmans L, Cauwelier EJ, Granvik M, Pruniau VP, Van Lommel L, et al. Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell metabolism. 2014;20:979–990. doi: 10.1016/j.cmet.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JC, Winnay J, Cheatham B, Kahn CR. Differential signaling by insulin receptor substrate 1 (IRS-1) and IRS-2 in IRS-1-deficient cells. Molecular and cellular biology. 1997;17:1513–1521. doi: 10.1128/mcb.17.3.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullo M, Peeraully MR, Trayhurn P, Folch J, Salas-Salvado J. Circulating nerve growth factor levels in relation to obesity and the metabolic syndrome in women. European journal of endocrinology/European Federation of Endocrine Societies. 2007;157:303–310. doi: 10.1530/EJE-06-0716. [DOI] [PubMed] [Google Scholar]
- Cai EP, Casimir M, Schroer SA, Luk CT, Shi SY, Choi D, Dai XQ, Hajmrle C, Spigelman AF, Zhu D, et al. In vivo role of focal adhesion kinase in regulating pancreatic beta-cell mass and function through insulin signaling, actin dynamics, and granule trafficking. Diabetes. 2012;61:1708–1718. doi: 10.2337/db11-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. The Journal of cell biology. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosker KE, Segal RA. Neuronal signaling through endocytosis. Cold Spring Harbor perspectives in biology. 2014:6. doi: 10.1101/cshperspect.a020669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hoker J, De Leu N, Heremans Y, Baeyens L, Minami K, Ying C, Lavens A, Chintinne M, Stange G, Magenheim J, et al. Conditional hypovascularization and hypoxia in islets do not overtly influence adult beta-cell mass or function. Diabetes. 2013;62:4165–4173. doi: 10.2337/db12-1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Garber M, Barbacid M, Silos-Santiago I, Holtzman DM. A role for TrkA during maturation of striatal and basal forebrain cholinergic neurons in vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17:7644–7654. doi: 10.1523/JNEUROSCI.17-20-07644.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Ji C, Wu Y, Ferguson SM, Tamarina N, Philipson LH, Lou X. Dynamin 2 regulates biphasic insulin secretion and plasma glucose homeostasis. The Journal of clinical investigation. 2015;125:4026–4041. doi: 10.1172/JCI80652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Stein GH, Pan N, Goebbels S, Hornberg H, Nave KA, Herrera P, White P, Kaestner KH, Sussel L, et al. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell metabolism. 2010;11:298–310. doi: 10.1016/j.cmet.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes research and clinical practice. 2014;103:137–149. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Hara M, Wang X, Kawamura T, Bindokas VP, Dizon RF, Alcoser SY, Magnuson MA, Bell GI. Transgenic mice with green fluorescent protein-labeled pancreatic beta -cells. American journal of physiology Endocrinology and metabolism. 2003;284:E177–183. doi: 10.1152/ajpendo.00321.2002. [DOI] [PubMed] [Google Scholar]
- Harrington AW, Ginty DD. Long-distance retrograde neurotrophic factor signalling in neurons. Nature reviews Neuroscience. 2013;14:177–187. doi: 10.1038/nrn3253. [DOI] [PubMed] [Google Scholar]
- Harrington AW, St Hillaire C, Zweifel LS, Glebova NO, Philippidou P, Halegoua S, Ginty DD. Recruitment of actin modifiers to TrkA endosomes governs retrograde NGF signaling and survival. Cell. 2011;146:421–434. doi: 10.1016/j.cell.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatlapatka K, Willenborg M, Rustenbeck I. Plasma membrane depolarization as a determinant of the first phase of insulin secretion. American journal of physiology Endocrinology and metabolism. 2009;297:E315–322. doi: 10.1152/ajpendo.90981.2008. [DOI] [PubMed] [Google Scholar]
- Heinze C, Seniuk A, Sokolov MV, Huebner AK, Klementowicz AE, Szijarto IA, Schleifenbaum J, Vitzthum H, Gollasch M, Ehmke H, et al. Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. The Journal of clinical investigation. 2014;124:675–686. doi: 10.1172/JCI70025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC, Ravier MA, Nenquin M, Jonas JC, Gilon P. Hierarchy of the beta-cell signals controlling insulin secretion. European journal of clinical investigation. 2003;33:742–750. doi: 10.1046/j.1365-2362.2003.01207.x. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Tuveson DA. Targeting oncogene dependence and resistance. Cancer cell. 2003;3:414–417. doi: 10.1016/s1535-6108(03)00115-6. [DOI] [PubMed] [Google Scholar]
- Huang E, Reichardt L. Neurotrophins: Roles in neuronal development and function. Annu Rev Neurosci. 2001;24:667–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indo Y, Tsuruta M, Hayashida Y, Karim MA, Ohta K, Kawano T, Mitsubuchi H, Tonoki H, Awaya Y, Matsuda I. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nature genetics. 1996;13:485–488. doi: 10.1038/ng0896-485. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, Kikuchi M, Yazaki Y, Miyazaki JI, Oka Y. Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia. 1993;36:1139–1145. doi: 10.1007/BF00401058. [DOI] [PubMed] [Google Scholar]
- Kalwat MA, Thurmond DC. Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet beta cells. Experimental & molecular medicine. 2013;45:e37. doi: 10.1038/emm.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaka-Gantenbein C, Tazi A, Czernichow P, Scharfmann R. In vivo presence of the high affinity nerve growth factor receptor Trk-A in the rat pancreas: differential localization during pancreatic development. Endocrinology. 1995;136:761–769. doi: 10.1210/endo.136.2.7835308. [DOI] [PubMed] [Google Scholar]
- Kim HC, Cho YJ, Ahn CW, Park KS, Kim JC, Nam JS, Im YS, Lee JE, Lee SC, Lee HK. Nerve growth factor and expression of its receptors in patients with diabetic neuropathy. Diabetic medicine : a journal of the British Diabetic Association. 2009;26:1228–1234. doi: 10.1111/j.1464-5491.2009.02856.x. [DOI] [PubMed] [Google Scholar]
- Konstantinova I, Nikolova G, Ohara-Imaizumi M, Meda P, Kucera T, Zarbalis K, Wurst W, Nagamatsu S, Lammert E. EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell. 2007;129:359–370. doi: 10.1016/j.cell.2007.02.044. [DOI] [PubMed] [Google Scholar]
- Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet beta-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochemical pharmacology. 2011;81:965–975. doi: 10.1016/j.bcp.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy PE, Klein NJ, Fink CJ. Effect of cytochalasin B on the biphasic release of insulin in perifused rat islets. Endocrinology. 1973;92:1458–1468. doi: 10.1210/endo-92-5-1458. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rutlin M, Huang S, Barrick CA, Wang F, Jones KR, Tessarollo L, Ginty DD. Sexually dimorphic BDNF signaling directs sensory innervation of the mammary gland. Science. 2012;338:1357–1360. doi: 10.1126/science.1228258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald PE, Joseph JW, Rorsman P. Glucose-sensing mechanisms in pancreatic beta-cells. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2005;360:2211–2225. doi: 10.1098/rstb.2005.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloni M, Descamps B, Caporali A, Zentilin L, Floris I, Giacca M, Emanueli C. Nerve growth factor gene therapy using adeno-associated viral vectors prevents cardiomyopathy in type 1 diabetic mice. Diabetes. 2012;61:229–240. doi: 10.2337/db11-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FD, Speelman A, Mathew TC, Fabian J, Chang E, Pozniak C, Toma JG. Nerve growth factor derived from terminals selectively increases the ratio of p75 to trkA NGF receptors on mature sympathetic neurons. Developmental biology. 1994;161:206–217. doi: 10.1006/dbio.1994.1021. [DOI] [PubMed] [Google Scholar]
- Muller M, Triaca V, Besusso D, Costanzi M, Horn JM, Koudelka J, Geibel M, Cestari V, Minichiello L. Loss of NGF-TrkA signaling from the CNS is not sufficient to induce cognitive impairments in young adult or intermediate-aged mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:14885–14898. doi: 10.1523/JNEUROSCI.2849-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Tableros V, Sanchez-Soto MC, Garcia S, Hiriart M. Autocrine regulation of single pancreatic beta-cell survival. Diabetes. 2004;53:2018–2023. doi: 10.2337/diabetes.53.8.2018. [DOI] [PubMed] [Google Scholar]
- Nevins AK, Thurmond DC. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. American journal of physiology Cell physiology. 2003;285:C698–710. doi: 10.1152/ajpcell.00093.2003. [DOI] [PubMed] [Google Scholar]
- Orci L, Gabbay KH, Malaisse WJ. Pancreatic beta-cell web: its possible role in insulin secretion. Science. 1972;175:1128–1130. doi: 10.1126/science.175.4026.1128. [DOI] [PubMed] [Google Scholar]
- Ostenson CG, Gaisano H, Sheu L, Tibell A, Bartfai T. Impaired gene and protein expression of exocytotic soluble N-ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients. Diabetes. 2006;55:435–440. doi: 10.2337/diabetes.55.02.06.db04-1575. [DOI] [PubMed] [Google Scholar]
- Pictet RL, Rall LB, Phelps P, Rutter WJ. The neural crest and the origin of the insulin-producing and other gastrointestinal hormone-producing cells. Science. 1976;191:191–192. doi: 10.1126/science.1108195. [DOI] [PubMed] [Google Scholar]
- Pingitore A, Caroleo MC, Cione E, Castanera Gonzalez R, Huang GC, Persaud SJ. Fine tuning of insulin secretion by release of nerve growth factor from mouse and human islet beta-cells. Molecular and cellular endocrinology. 2016;436:23–32. doi: 10.1016/j.mce.2016.07.014. [DOI] [PubMed] [Google Scholar]
- Porat-Shliom N, Milberg O, Masedunskas A, Weigert R. Multiple roles for the actin cytoskeleton during regulated exocytosis. Cellular and molecular life sciences : CMLS. 2013;70:2099–2121. doi: 10.1007/s00018-012-1156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell metabolism. 2013;18:162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Reinert RB, Brissova M, Shostak A, Pan FC, Poffenberger G, Cai Q, Hundemer GL, Kantz J, Thompson CS, Dai C, et al. Vascular endothelial growth factor-a and islet vascularization are necessary in developing, but not adult, pancreatic islets. Diabetes. 2013;62:4154–4164. doi: 10.2337/db13-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Tebar A, Dechant G, Barde YA. Binding of brain-derived neurotrophic factor to the nerve growth factor receptor. Neuron. 1990;4:487–492. doi: 10.1016/0896-6273(90)90107-q. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annual review of physiology. 2013;75:155–179. doi: 10.1146/annurev-physiol-030212-183754. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Renstrom E. Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46:1029–1045. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- Rosenbaum T, Sanchez-Soto MC, Hiriart M. Nerve growth factor increases insulin secretion and barium current in pancreatic beta-cells. Diabetes. 2001;50:1755–1762. doi: 10.2337/diabetes.50.8.1755. [DOI] [PubMed] [Google Scholar]
- Rosenbaum T, Vidaltamayo R, Sanchez-Soto MC, Zentella A, Hiriart M. Pancreatic beta cells synthesize and secrete nerve growth factor. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:7784–7788. doi: 10.1073/pnas.95.13.7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasson A, Rachi E, Sakhneny L, Baer D, Lisnyansky M, Epshtein A, Landsman L. Islet pericytes are required for beta-cell maturity. Diabetes. 2016 doi: 10.2337/db16-0365. [DOI] [PubMed] [Google Scholar]
- Schreiber R, Levy J, Loewenthal N, Pinsk V, Hershkovitz E. Decreased first phase insulin response in children with congenital insensitivity to pain with anhidrosis. Journal of pediatric endocrinology & metabolism : JPEM. 2005;18:873–877. doi: 10.1515/jpem.2005.18.9.873. [DOI] [PubMed] [Google Scholar]
- Schuh M. An actin-dependent mechanism for long-range vesicle transport. Nature cell biology. 2011;13:1431–1436. doi: 10.1038/ncb2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, et al. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19333–19338. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Xu Y, Hu X, Choi B, Tong Q. Brain expression of Cre recombinase driven by pancreas-specific promoters. Genesis. 2010;48:628–634. doi: 10.1002/dvg.20672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nature reviews Molecular cell biology. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens RM, Loeb DM, Copeland TD, Pawson T, Greene LA, Kaplan DR. Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma 1 to mediate NGF responses. Neuron. 1994;12:691–705. doi: 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- Sutter A, Riopelle RJ, Harris-Warrick RM, Shooter EM. Nerve growth factor receptors. Characterization of two distinct classes of binding sites on chick embryo sensory ganglia cells. The Journal of biological chemistry. 1979;254:5972–5982. [PubMed] [Google Scholar]
- Tessarollo L. Pleiotropic functions of neurotrophins in development. Cytokine & growth factor reviews. 1998;9:125–137. doi: 10.1016/s1359-6101(98)00003-3. [DOI] [PubMed] [Google Scholar]
- Thurmond DC, Gonelle-Gispert C, Furukawa M, Halban PA, Pessin JE. Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol Endocrinol. 2003;17:732–742. doi: 10.1210/me.2002-0333. [DOI] [PubMed] [Google Scholar]
- van Obberghen E, Somers G, Devis G, Vaughan GD, Malaisse-Lagae F, Orci L, Malaisse WJ. Dynamics of insulin release and microtubular-microfilamentous system. I. Effect of cytochalasin B. The Journal of clinical investigation. 1973;52:1041–1051. doi: 10.1172/JCI107269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetere A, Choudhary A, Burns SM, Wagner BK. Targeting the pancreatic beta-cell to treat diabetes. Nature reviews Drug discovery. 2014;13:278–289. doi: 10.1038/nrd4231. [DOI] [PubMed] [Google Scholar]
- Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, Garcia-Ocana A, Stewart AF. Diabetes mellitus--advances and challenges in human beta-cell proliferation. Nature reviews Endocrinology. 2015;11:201–212. doi: 10.1038/nrendo.2015.9. [DOI] [PubMed] [Google Scholar]
- Wang Z, Oh E, Clapp DW, Chernoff J, Thurmond DC. Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. The Journal of biological chemistry. 2011;286:41359–41367. doi: 10.1074/jbc.M111.291500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 2004;53(Suppl 3):S16–21. doi: 10.2337/diabetes.53.suppl_3.s16. [DOI] [PubMed] [Google Scholar]
- Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nature medicine. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- Wu C, Lai CF, Mobley WC. Nerve growth factor activates persistent Rap1 signaling in endosomes. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:5406–5416. doi: 10.1523/JNEUROSCI.21-15-05406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Xie X. Neurotrophic factor control of satiety and body weight. Nature reviews Neuroscience. 2016;17:282–292. doi: 10.1038/nrn.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Hu R, Brissova M, Stein RW, Powers AC, Gu G, Kaverina I. Microtubules Negatively Regulate Insulin Secretion in Pancreatic beta Cells. Developmental cell. 2015;34:656–668. doi: 10.1016/j.devcel.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.