

ABSTRACT
Lipoxins are host anti-inflammatory molecules that play a vital role in restoring tissue homeostasis. The efficacy of lipoxins and their analog epilipoxins in treating inflammation and its associated diseases has been well documented. Kaposi's sarcoma (KS) and primary effusion lymphoma (PEL) are two well-known inflammation related diseases caused by Kaposi's sarcoma-associated herpesvirus (KSHV). Controlling inflammation is one of the strategies adopted to treat KS and PEL, a primary motivation for exploring and evaluating the therapeutic potential of using lipoxins. This study documents how KSHV manipulates and downregulates the secretion of the anti-inflammatory lipoxin A4 in host cells and the viral factors involved in this process using in vitro KS and PEL cells as models. The presence of the lipoxin A4 receptor/formyl peptidyl receptor (ALX/FPR) in KS patient tissue sections and in vitro KS and PEL cell models offers a novel possibility for treating KS and PEL with lipoxins. Treating de novo KSHV-infected endothelial cells with lipoxin and epilipoxin creates an anti-inflammatory environment by decreasing the levels of NF-κB, AKT, ERK1/2, COX-2, and 5-lipoxygenase. Lipoxin treatment on CRISPR/CAS9 technology-mediated ALX/FPR gene deletion revealed the importance of the lipoxin receptor ALX for effective lipoxin signaling. A viral microRNA (miRNA) cluster was identified as the primary factor contributing to the downregulation of lipoxin A4 secretion in host cells. The KSHV miRNA cluster probably targets enzyme 15-lipoxygenase, which is involved in lipoxin A4 synthesis. This study provides a new insight into the potential treatment of KS and PEL using nature's own anti-inflammatory molecule, lipoxin.
IMPORTANCE KSHV infection has been shown to upregulate several host proinflammatory factors, which aid in its survival and pathogenesis. The influence of KSHV infection on anti-inflammatory molecules is not well studied. Since current treatment methods for KS and PEL are fraught with unwanted side effects and low efficiency, the search for new therapeutics is therefore imperative. The use of nature's own molecule lipoxin as a drug is promising. This study opens up new domains in KSHV research focusing on how the virus modulates lipoxin secretion and warrants further investigation of the therapeutic potential of lipoxin using in vitro cell models for KS and PEL.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV), also termed human herpesvirus 8 (HHV-8), is etiologically associated with Kaposi's sarcoma (KS) and B-cell lymphoproliferative primary effusion lymphoma (PEL). KS is a proliferative angiogenic tumor of endothelial cells characterized by vascular red/purplish lesions in the skin (1–3). PEL, also known as body cavity lymphoma, is a non-Hodgkin's lymphoma primarily present in the body cavity (4). KS and PEL are a significant cause of death in HIV patients. The presence of a suppressed host immune system along with KSHV-coded immunomodulatory proteins contributes to KSHV infection, and the lifelong KSHV latency establishment is the primary factor for pathogenesis (5, 6). KSHV utilizes its latency cluster containing ORF73 (latency-associated nuclear antigen 1 [LANA-1]), ORF72 (viral cyclin [vCyclin]), ORF71 (K13/vFLIP), and ORFK12 (kaposins A, B, and C), as well as 12 distinct pre-microRNAs, to modulate the host immune system and maintain lifelong latency (7–9). KSHV also encodes several homologs of cytokines and chemokines to alter the immune response (6).
KSHV induces several proinflammatory host molecules such as COX-2/PGE2, 5-lipoxygenase, and LTB4 to establish latency and aid in its pathogenesis (10–14). Beside upregulating proinflammatory pathways, KSHV also modulates the immune system by downregulating anti-inflammatory pathways (15). Since altering the host immune system is the hallmark of KSHV infection and pathogenesis, it is important to understand the relationship between the various components of the host immune system and KSHV to design better therapeutics.
To date, there is no effective treatment for KS and PEL. Current treatment involves the use of chemotherapeutics that work by targeting DNA replication of all dividing cells. This approach has the following disadvantages: low efficacy, cytotoxic side effects, depletion of CD4, and risk of secondary malignancies. Above all, these anticancer drugs do not control viral replication and pathogenesis. Surgery is an expensive alternative effective for small size lesions the chance of disease relapse is high. Since KSHV in KS and PEL remains primarily in the latent form, antiviral drugs are not very effective in reducing viral load since they target only the lytic replicating virus (16–19). Hence, there is an emerging need to develop alternative treatment methods for KS and PEL.
Lipoxins are anti-inflammatory metabolites of the arachidonic acid pathway, which have been well studied by Serhan et al. (20). Lipoxins are synthesized from arachidonic acid by the action of a series of lipoxygenases such as 5-, 15-, and 12-lipoxygenase. Epilipoxins or epimers of lipoxin are other potent forms of lipoxins, which are synthesized under the action of aspirin on cyclooxygenase, a metabolite of the arachidonic acid pathway. Lipoxins bind to a G-protein-coupled receptor on the host cell surface known as the lipoxin A4 receptor/formyl peptidyl receptor (ALX/FPR) to exert their anti-inflammatory action (21).
Lipoxins have shown promising results in treating inflammation-related diseases, such as asthma, chronic obstructive pulmonary disease, renal fibrosis, and cancer (21). Lipoxins have been shown to alter levels of various transcription factors such as NF-κB, AP-1, PPARγ, and Nrf-2, as well as various cytoplasmic signaling molecules such as phosphatidylinositol 3-kinase, AKT, mTOR, Ras, JAK, and STAT to create an anti-inflammatory environment (21). Lipoxin targets are also shown to play an important role in viral pathogenesis and malignancies (21). A previous study from our laboratory showed that lipoxin and epilipoxin treatment induces anti-inflammatory and antiangiogenic effects in KS cells (22). Treatment of KS cells with lipoxin or epilipoxin resulted in a significant decrease in levels of proinflammatory COX-2/PGE-2, 5-lipoxygenase, and LTB4, along with a decrease in the secretion of the angiogenic factor VEGF-C (22).
KS and PEL are inflammation-associated diseases. Since lipoxins are described to act as anti-inflammatory and proresolving molecules, we hypothesized that KSHV manipulates lipoxin pathways for its own advantage. This study shows that KSHV infection downregulates lipoxin A4 secretion. The viral factors involved in the downregulation of lipoxin A4 secretion are also investigated. The presence of lipoxin receptor ALX/FPR in clinical specimens of KS and in vitro cell models advocated the use of lipoxins as a promising approach to target KS. Supplementing lipoxins to treat KSHV-associated malignancies would provide a novel and effective treatment option and would be tested in future studies.
MATERIALS AND METHODS
Cells.
Human microvascular dermal endothelial cells (HMVEC-d; CC-2543; Lonza, Walkersville, MD) were cultured in endothelial basal medium 2 (EBM-2) with growth factors (Lonza). Cells were typically used between five and seven passages. Lentiviral transduced EA-hy926 cells expressing K10/12 microRNA (miRNA) and enhanced green fluorescent protein (EGFP) were kindly provided by Paivi Ojala, Institute of Biotechnology, University of Helsinki, Helsinki, Finland. EA-hy926 cell lines were propagated in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and blasticidin (3 μg/ml). miR cluster mutant KSHV and wild-type (WT) KSHV producer iSLK cells from Rolf Renne, University of Florida, were propagated in DMEM (Gibco BRL, Grand Island, NY), 10% heat-inactivated FBS (HyClone, Logan, UT), and 1% penicillin-streptomycin (Gibco BRL). PEL cells (KSHV positive [KSHV+]/Epstein-Barr virus negative [EBV−]; BCBL-1 and BC-3 cells) and B cells were cultured in RPMI 1640 medium (Gibco BRL) with 10% heat-inactivated FBS (HyClone, Logan, UT), 2 mM l-glutamine (Gibco BRL), and penicillin-streptomycin (Gibco BRL). BC-3 was purchased from the American Type Culture Collection (ATCC), Manassas, VA. The BCBL-1 cell line was a gift from M. McGrath (University of California at San Francisco). KSHV-BJAB cells were a gift from Blossom Damania, University of North Carolina. KSHV-BJAB (KSHV+/EBV−) cells were cultured in PEL cell growth medium supplemented with hygromycin B (0.2 mg/ml; Sigma, St. Louis, MO) (23). Human osteosarcoma cells (U2OS) were obtained from the ATCC and grown in DMEM supplemented with GlutaMAX (Gibco), 10% fetal bovine serum, and 1% penicillin-streptomycin (Gibco). After informed consent was obtained in accordance with the Declaration of Helsinki, peripheral blood was collected in sodium heparin vacutainers (Becton Dickinson, Franklin Lakes, NJ). The peripheral blood mononuclear cell fraction was collected after differential density centrifugation over Ficoll-Paque Plus (GE Healthcare, Piscataway, NJ) by methods described previously (24). B cells were purified by using a Human Cell Isolation Kit II (Miltenyi Biotech) according to the manufacturer's protocol. All cell lines were routinely tested for mycoplasma by using a Mycoalert kit (Lonza, Allendale, NJ) according to the manufacturer's instructions and found to be negative.
Drugs and reagents.
Lipoxin (catalog no. 90415 [5(S),6(R),15(R)-trihydroxy-7E,9E,11Z,13E-eicosatetraenoic acid]) and lipoxin A4 methyl ester (catalog no. 10033 [5(S),6(R),15(R)-TriHETE methyl ester]) were purchased from Cayman Chemical (Ann Arbor, MI), and ethanol was used as a solvent control for all experiments involving treatments with inhibitors.
Virus.
Induction of the KSHV lytic cycle in BCBL-1 cells, supernatant collection, and virus purification procedures were described previously (25). The virus preparations used in our studies represent mostly enveloped KSHV virion particles, and only enveloped virus particles were seen by electron microscopy, indicating the purity of virus preparations (24–27). Since every viral genome contains a single copy of the ORF73 gene, the number of viral DNA molecules could be calculated from the corresponding copy numbers of the ORF73 gene. Hence, we extracted the KSHV DNA from the purified virus. The copy numbers were quantitated by real-time DNA PCR using primers amplifying the KSHV ORF73 gene (24–27). We used 30 DNA copies/cell for infection in all of the experiments.
To produce stocks of miR cluster mutant KSHV and WT KSHV, iSLK cells harboring BAC16 were treated with doxycycline (1 μM; Sigma-Aldrich, St. Louis, MO) and sodium butyrate (3 mM; Sigma-Aldrich) for 72 h. Supernatants from the treated cells were collected, and cell debris was removed via low-speed centrifugation. Supernatants were then transferred to ultracentrifuge tubes, underlaid with 25% sucrose in TNE (150 mM NaCl, 10 mM Tris [pH 8.0], 2 mM EDTA [pH 8.0]), and centrifuged at 78,000 × g for 2 h at 4°C. The resulting virion pellets were resuspended in TNE (28).
Immunofluorescence assay.
Confluent HMVEC-d cells in eight-well chamber slides (Nalge Nunc International, Naperville, IL) were infected with KSHV (30 DNA copies/cell) for 72 h and treated with lipoxin for 20 min. For ALX immunostaining, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.4% Triton X-100, and stained with ALX rabbit polyclonal antibody (Cayman Chemical) overnight at 4°C. To observe the cell surface expression of ALX/FPR receptors, the cells were fixed with 4% paraformaldehyde and stained with ALX/FPRL-1 rabbit polyclonal antibody overnight. The cells were washed and incubated with 1:200 dilution of Alexa 594-coupled anti-rabbit antibody (Molecular Probes, Eugene, OR) for 1 h at room temperature. Nuclei were visualized by using DAPI (4′,6′-diamidino-2-phenylindole; excitation wavelength at 358 nm/emission wavelength at 416 nm; Molecular Probes) as a counter stain. Stained cells were washed and viewed with appropriate filters under a fluorescence microscope using the Nikon Metamorph digital imaging system.
Confluent U20S cells in eight-well chamber slides were infected with KSHV (30 DNA copies/cell) for 72 h. For LANA-1 staining, cells were fixed with 4% paraformaldehyde, permeabilized with 0.4% Triton X-100 and stained with LANA-1 rabbit polyclonal antibody (from Bala Chandran, Rosalind Franklin University of Medicine and Science) overnight at 4°C. Cells were washed and incubated with 1:200 dilution of Alexa 594-coupled anti-rabbit antibody (Molecular Probes) for 1 h at room temperature. Nuclei were visualized by using DAPI as a counterstain. Stained cells were washed and viewed with appropriate filters under a fluorescence microscope with the Nikon Metamorph digital imaging system.
Immunohistochemistry and light microcopy.
Various tissue sections from diverse populations, including both healthy subjects and subjects with Kaposi's sarcoma, were obtained from the AIDS and Cancer Specimen Resource (ACSR; San Francisco, CA). Tissue sections were deparaffinized with Histochoice clearing reagent and hydrated with water before microwave treatment in 1 mM EDTA (pH 8.0; Sigma-Aldrich) for 15 min for antigen retrieval. Sections were blocked with blocking solution (2% donkey serum and 0.3% Triton X-100 in phosphate-buffered saline), followed by incubation with the primary antibody against ALX/FPRL-1 (Abcam, catalog no. 63022) overnight at 4°C. These sections were incubated with rat polymer HRP (Biocare Medical) for 15 min and developed using diaminobenzidine (DAB) reagent. This step was followed by light microscopy imaging at 4× and 60×. Based on visual comparison of the intensity of ALX staining, a staining score was assigned to each section, which was used to make statistical conclusions from the tissue sections.
Western blot analysis.
Cells were lysed in radioimmunoprecipitation assay lysis buffer (Thermo Scientific, catalog no. 89901), along with protease inhibitor cocktail (Thermo Scientific, catalog no. 78410). Lysates were then sonicated and centrifuged at 10,000 × g for 10 min. The cell extracts were quantitated by a bicinchoninic acid (BCA) protein assay (Thermo Scientific, catalog no. 23227), and equal amounts of protein (20 μg/lane) were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and electrotransferred to 0.45-μm-pore-size nitrocellulose membranes. The membranes were blocked with 5% bovine serum albumin, probed with the antibodies ALXR/FPR (38 kDa, catalog no. 63022; Abcam), p-FAK (125 kDa, catalog no. 3283; Cell Signaling), T-FAK (125 kDa, catalog no. 3285; Cell Signaling), p-Src (60 kDa, catalog no. 2101; Cell Signaling), T-Src (60 kDa, catalog no. 2123; Cell Signaling), p-ERK1/2 (42 and 44 kDa, catalog no. 9101; Cell Signaling), ERK2 (42 kDa, catalog no. 9108; Cell Signaling), p-P65 (65 kDa, catalog no. 3285; Cell Signaling), T-P65 (65 kDa, catalog no. 3031; Cell Signaling), p-AKT (60 kDa, catalog no. 9271; Cell Signaling), T-AKT (60 kDa, catalog no. 9272; Cell Signaling), anti-α tubulin clone DM (50 kDa, catalog no. T9026; Sigma), and anti β-actin clone AC-15 (42 kDa, catalog no. A5441; Sigma), and then visualized by using an enhanced chemiluminescence detection system (25).
ELISA for lipoxin A4 detection.
Lipoxin A4 levels were measured by lipoxin A4 enzyme-linked immunosorbent assay (ELISA; Neogen Corp., catalog no. 407010) according to the manufacturer's instructions. Data are expressed as the amount of lipoxin A4 secreted (pg/ml).
Lentiviral transduction.
Lentiviral infection was done as described by Vart et al. (29). Vesicular stomatitis virus-G envelope-pseudotyped lentiviruses expressing viral genes (v-FLIP/K13/ORF71, v-Cyclin/ORF72, and LANA-1/ORF73) were produced using a four-plasmid transfection system and tested as described previously (30). Lentiviruses expressing pSIN (empty vector) or GFP were used as controls. HMVEC-d cells were split at 24 h after lentivirus infection and allowed to grow until 80 to 90% confluence. The cells were serum starved, and supernatants were collected after 48 h, spun at 1,000 rpm for 10 min at 4°C to remove the particulates, and used for measuring lipoxin A4 levels by ELISA.
TaqMan gene expression analysis for 15- and 12-lipoxygenase.
30 KSHV DNA per cell were used for the infection of HMVEC-d cells for different times. The gene expression of 15- and 12-lipoxygenase was analyzed using TaqMan gene expression assays (24) by the methods described in the manufacturer's protocol. PCR amplifications without cDNA were performed as negative controls.
Generation of ALX/FPR-negative U2OS cells.
A pool of three plasmids was obtained from Santa Cruz Biotechnology, Inc., Santa Cruz, CA. Each plasmid has ALX/FPR-guided RNA designed for maximum knockout efficiency, Cas9 RNase, and GFP. Using Lipofectamine (Life Technologies, catalog no. 11668), U2OS cells were transfected with 1 μg of plasmid. At 48 h posttransfection, GFP-positive cells were sorted individually into 96-well plates containing complete growth media. The lack of ALX/FPR expression in each clone was screened by Western blotting.
RESULTS
KSHV downregulates lipoxin secretion.
To study the influence of KSHV on lipoxin secretion during primary infection, serum-starved HMVEC-d cells were infected with KSHV (30 DNA copies/cell) for different durations (8, 24, and 72 h). Lipoxin A4 levels were measured from the supernatants of infected and uninfected HMVEC-d cells (Fig. 1a). KSHV infection was found to decrease lipoxin A4 secretion over time. After being infected with KSHV for 8 h, a 57% decrease in the secretion of lipoxin was observed compared to the control uninfected cells. Within 72 h after infection, the lipoxin A4 secretion levels further decreased by 74% compared to the uninfected controls (Fig. 1a).
FIG 1.

Lipoxin A4 secretion in KS and PEL cell models. (a) HMVEC-d cells grown to 80 to 90% confluence were serum starved for the entire duration of the study and infected with 30 DNA copies of KSHV/cell for different time points. At different time points postinfection (8, 24, and 72 h), supernatants were collected, and ELISA was performed according to the manufacturer's instructions. Uninfected HMVEC-d cells were used as a control. (b) Healthy B cells were used as a control, and BCBL-1 cells and BC-3 cells were used as PEL model cell lines. All cell lines were serum starved for 24 h, and lipoxin A4 secretion in the conditioned media was measured by ELISA according to the manufacturer's protocol. (c) KSHV BJAB and BJAB cells were serum starved for 24 h. Lipoxin A4 secretion in KSHV BJAB and BJAB cells were measured by ELISA. The percent inhibition was calculated by considering uninfected cells as 100%.
To study lipoxin secretion in latently infected B cells, PEL cells were used. Lipoxin A4 levels were measured from the supernatants of BCBL-1 and BC-3 cells while healthy B cells were used as control. KSHV-infected B cell lines, including BCBL-1 and BC-3, showed a decreased amount of lipoxin A4 secretion compared to control healthy B cells (Fig. 1b). Similarly, compared to BJAB cells, KSHV-BJAB cells had a 66% reduced amount of lipoxin secretion (Fig. 1c). These results suggest the downregulation of lipoxin and support its role in KSHV pathogenesis and the resulting malignancy.
To exclude the influence of serum starvation on lipoxin secretion, supernatants from HMVEC-d cells grown in complete media and serum-free media were collected. The reduction in lipoxin A4 secretion observed during serum starvation was only 5%, which suggests that serum starvation has negligible influence on the downregulation of lipoxin A4 secretion (data not shown).
Expression of lipoxin receptor ALX/FPR on human KS tissue sections.
The presence of lipoxin receptor ALX/FPR in KS tissue sections was analyzed to study the possibility of treating KS with lipoxin. An array of 224 tissue sections from KS patients and healthy controls were obtained from AIDS and Cancer Specimen Resource (ACSR). The ACSR KS TMA tissue section had 224 sections from skin, mouth, anus, lymph node, small bowel, tonsils/adenoids, and the head and neck region from normal and KS patients. The tissue array represented sections from several infected organs from an ethnically diverse population. The tissue section array was stained for ALX/FPR and observed under a light microscope at ×60 magnification (Fig. 2a). Regions in brown indicate the presence of ALX/FPR (Fig. 2a). Based on the brown color staining, a staining score was assigned for ALX/FPR staining. Statistical analyses were performed to evaluate apparent differences in the occurrence of ALX/FPR receptor in KS patients and healthy controls. Statistically insignificant difference in expression of ALX/FPR receptor between KS-positive tissue sections and normal healthy tissue sections was observed (Student t test: t [dF = 1.28] = −0.34, P = 0.74). Statistically insignificant difference in ALX/FPR expression was observed among the different patient populations [one-way analysis of variance: F(3,24.78) = 1.763, P = 0.18]. These results demonstrate that ALX/FPR is abundant in both KS and healthy tissue sections and also present in a diverse patient population. This suggests that lipoxins can potentially be used in treating KS.
FIG 2.

Expression of lipoxin receptor ALX/FPR on human KS tissue sections and in vitro cell models of KS and PEL. (a) A KS tissue section array from the ACSR was immunohistochemically stained for ALX/FPR, and microscopy was performed. The brown color indicates ALX/FPR staining. (b) Lysates from HMVEC-d KSHV-infected cells (30 DNA copies/cell) for the indicated time points (8, 24, and 48 h) and uninfected control HMVEC-d cells were Western blotted for ALX/FPR, stripped, and immunoblotted for tubulin. (c) In vitro PEL models BCBL-1 and BC-3 cells were fixed and stained for ALX/FPR using an Alexa 594-labeled antibody and, as a control, stained for IgG isotype.
ALX/FPR status in in vitro KS and PEL cell lines.
After ALX/FPR was observed abundantly on KS patient tissue sections, it was quantified in in vitro KS and PEL cell models (Fig. 2b). For the KS cell model, HMVEC-d cells were de novo infected with 30 DNA copies/cell of KSHV for various durations, whereas uninfected HMVEC-d cells served as controls. Western blots were performed using cell lysates of infected and uninfected HMVEC-d cells to observe changes in protein levels of ALX/FPR. The results show that there is no difference in the protein levels of ALX/FPR upon infection. Flow cytometry is another technique that was used to study the expression levels of ALX/FPR (Fig. 2c). In vitro PEL cell models BCBL-1 and BC-3 cell were fixed and stained for ALX/FPR using an Alexa 594 label. They were costained for IgG isotype, which served as a control. Flow cytometry results also confirm the presence of ALX/FPR on infected B cells. These studies reconfirm the presence of receptors on infected cells and strongly suggest the possibility of treating KS and PEL with lipoxin.
Immunofluorescence analysis of ALX/FPR in long term latently infected HMVEC-d cells treated with solvent or lipoxin.
To exert its anti-inflammatory activity, lipoxin binds to its receptor ALX/FPR on the cell surface. Upon lipoxin activation, the receptor undergoes internalization to the perinuclear space. To evaluate the impact of KSHV infection on functionality of the ALX/FPR receptor, lipoxin and solvent (ethanol) stimulation of long-term latently infected HMVEC-d cells was performed. Rapid perinuclear translocation of ALX/FPR in cells stimulated with lipoxin was observed (Fig. 3). On the other hand, cells that were solvent treated showed a dispersed distribution of ALX/FPR (Fig. 3). This observation shows that the functionality of the ALX/FPR receptor remains unaltered upon KSHV infection.
FIG 3.

Immunofluorescence analysis of ALX/FPR in long-term latently infected HMVEC-d cells treated with solvent (ethanol) or lipoxin. Latently infected HMVEC-d cells were treated with either lipoxin (50 nM) or solvent (ethanol) for 20 min. Cells were fixed, permeabilized, and incubated overnight with primary ALX rabbit antibody. The cells were then washed and incubated in secondary anti-rabbit Alexa 594 for 2 h and mounted with mounting medium containing nuclear stain DAPI (blue). Immunofluorescence microscopy was performed at ×40 magnification.
Influence of lipoxin treatment on host signaling molecules.
The mechanism of action of lipoxins was explored as a result of two main observations: (i) the presence of ALX/FPR receptor on KS tissue sections and in vitro cell models of KS and PEL (Fig. 2) and (ii) the functionality of the receptor being unaltered upon KSHV infection (Fig. 3). Latently infected HMVEC-d cells were treated with lipoxin A4, epilipoxin A4 or solvent (ethanol). Changes in levels of various host cell-signaling proteins, which are known to play an important role in KS pathogenesis, were evaluated. These host cell signaling proteins are involved in entry, pathogenesis, and latency establishment of KSHV, with FAK, Src, and AKT phosphorylation being essential for KSHV entry (11, 31). ERK and AKT promote infection and increase survival of the infected cells (32, 33). P65 (NF-κB) helps in viral entry and latency establishment (34). Treatment with either lipoxin A4 or epilipoxin A4 downregulated activation of ERK, NF-κB, and AKT pathways compared to solvent treatment (Fig. 4a).
FIG 4.

Levels of various host cell proteins and arachidonic acid metabolites during treatment of de novo KSHV-infected HMVEC-d cells with lipoxin and epilipoxin. (a) Lysates from untreated latently KSHV-infected HMVEC-d cells (30 DNA copies/cell) or from lipoxin A4 (100 nM)- or epilipoxin A4 (100 nM)-treated latently KSHV-infected HMVEC-d cells (30 DNA copies/cell) were Western blotted with the indicated antibodies, stripped, and immunoblotted for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as a loading control. (b) Lysates from latently KSHV-infected HMVEC-d cells (30 DNA copies/cell) or from lipoxin (100 nM)- or epilipoxin (100 nM)-treated latently KSHV-infected HMVEC-d cells (30 DNA copies/cell) were Western blotted for COX-2 and 5-lipoxygenase, stripped, and immunoblotted for β-actin.
Lipoxin treatment affects the levels of various enzymes of the arachidonic acid pathway.
Previous studies have identified COX-2 and 5-lipoxygenase, both enzymes of the arachidonic acid pathway, as being upregulated during KSHV infection and linked with its pathogenesis and associated tumor development (25, 35). Treating de novo KSHV-infected HMVEC-d cells with lipoxin A4 and epilipoxin reduced protein levels of COX-2 and 5-lipoxygenase compared to untreated (Fig. 4b).
ALX/FPR is vital for lipoxin signaling.
The observation of ALX/FPR in KS patient tissue sections and in vitro KS and PEL cell models led to the investigation of lipoxin's therapeutic potential. Lipoxin treatment decreased the levels of several host cell-signaling molecules, which play an important role in pathogenesis and disease progression. We have shown earlier that lipoxin or epilipoxin treatment on HMVEC-d cells helps to lower the levels of AKT, ERK1/2, and P65 (NF-κB). The importance of ALX/FPR in lipoxin signaling was then studied. Osteosarcoma U2OS cells were genetically modified to mute the expression of ALX/FPR using CRISPR/CAS9 technology. Western blotting experiments confirmed the knockdown of ALX/FPR in mutated U20S cells, while WT U20S and a few positive clones of CRISPR/CAS9 gene editing still show higher levels of ALX/FPR (Fig. 5a).
FIG 5.
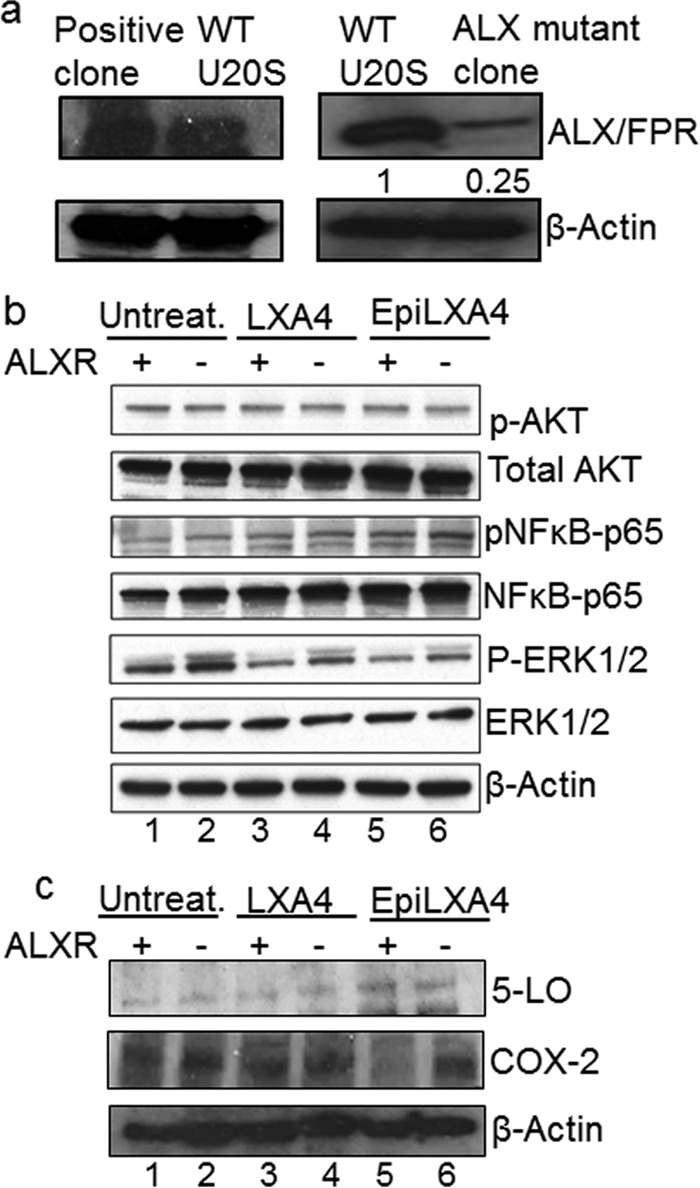
ALX/FPR is vital for lipoxin signaling. (a) Immunofluorescence analysis of KSHV-infected U20S cells. U20S cells were infected with KSHV (30 DNA copies/cell) for 24 h. The cells were fixed, permeabilized, and incubated overnight with primary LANA-1 rabbit antibody. The cells were then washed and incubated in secondary anti-rabbit Alexa 594 for 2 h and mounted with mounting medium containing the nuclear stain DAPI (blue). Immunofluorescence microscopy was performed at ×40 magnification. (b) CRISPR/CAS9 plasmids were transfected in U2OS cells. At 24 h after transfection, the cells were sorted for the presence of GFP. Individual cells were grown in 96-well plates and then transferred to 24-well plates. The cells were then screened for the presence of ALX/FPR by Western blotting. Actin was used as a loading control. (b) Lysates from latently infected (48 h) WT U2OS cells and ALX mutant U2OS cells, which were also treated (100 nM lipoxin or 100 nM epilipoxin), were Western blotted with antibodies as indicated, stripped, and immunoblotted for β-actin.
The mutated cells and WT U20S cells were latently infected with KSHV and then treated with lipoxin or epilipoxin or, as a control, cells were treated with solvent (ethanol). Cell lysates were Western blotted for various host cell-signaling proteins such as NF-κB, AKT, and ERK (Fig. 5b). Changes in levels of arachidonic acid pathway enzymes such as COX-2 and 5-lipoxygenase were also evaluated by Western blotting (Fig. 5c). In the absence of ALX receptor, U20S treated with lipoxin show a 1.7-fold increase in pERK level compared to its control WT U20S cells treated with lipoxin. NF-κB levels in U20S ALX mutant cells increased by 1.79-fold on epilipoxin treatment compared to its control WT U20S cells treated with epilipoxin. No change in the levels of 5-lipoxygenase was observed, while there was a 1.75-fold increase in the level of COX-2 in U20S ALX mutant cells treated with epilipoxin. This observation of increased levels of proinflammatory proteins despite lipoxin/epilipoxin treatment could be attributed to the absence of ALX receptor.
Viral factors that affect lipoxin secretion.
Identifying the viral factor responsible for the downregulation of lipoxin is critical to understanding the relationship between lipoxin and KSHV. Since KS and PEL are associated with the latency phase of virus, an investigation on the latency factor contributing to lipoxin secretion was conducted. HMVEC-d cells were transduced with lentivirus expressing v-Cyclin, v-FLIP, and LANA-1. Lentivirus with background plasmid pSIN was transduced and used as an empty vector control. Lentivirus expressing GFP was also used as a control. No significant change in lipoxin secretion was observed when latency genes were expressed in HMVEC-d cells (Fig. 6).
FIG 6.
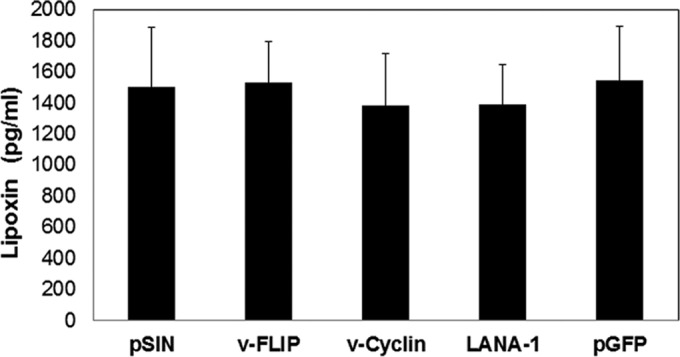
Measuring lipoxin secretion in lentivirus-transduced HMVEC-d cells. Lentivirus expressing viral latency genes, including v-FLIP, v-Cyclin, and LANA-1, and control lentivirus expressing pSIN (empty vector) or GFP were transduced into HMVEC-d cells. Lentivirus-transduced HMVEC-d cells were split after 24 h and grown to 80% confluence. The lentivirus-transduced cells were serum starved for 48 h, and the supernatants were subjected to lipoxin ELISA in accordance with the manufacturer's protocol.
Lipoxin secretion is downregulated by the KSHV miRNA cluster.
The latency cluster also contains a group of KSHV 12 pre-miRNA. To evaluate the effect of KSHV miRNA on lipoxin secretion, EA.hy endothelial cells were modified lentivirally to express KSHV miRNA. This cell lines expresses all the intronic miRNA; miRNA K12-1-9 and K11 and has been referred to as K10/12. EA.hy cells expressing GFP were used as controls. These cells have been previously used in a study to show the effect of KSHV miRNAs on apoptosis (36). Briefly, miRNA (K10/12) was cloned into pDONR 207 and transferred to pLENTI6/V5-DEST. Control plasmid was pLENTI6/V5-EGFP (36). The Vira Power Lentiviral Gateway Expression system was used to transduce EA.hy cells (36). Blasticidin was added for selection. These cells were verified for KSHV miRNA expression by RT-PCR and deep sequencing (36). Lipoxin secretion was measured from the supernatants of these cell lines. A 43% reduction in the amount of lipoxin secretion in the cell line expressing KSHV miRNA was observed. This suggests the possible involvement of KSHV miRNA in the downregulation of the lipoxin synthesis pathway (Fig. 7).
FIG 7.
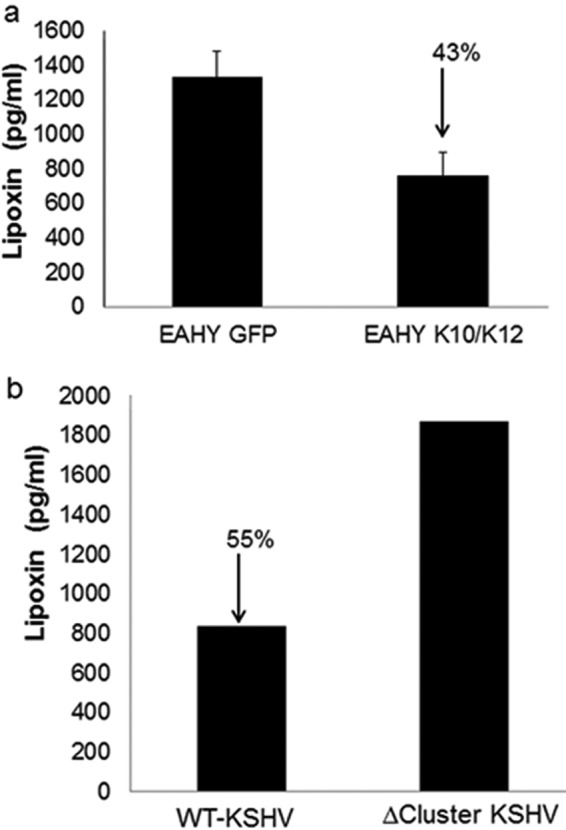
KSHV miRNA cluster downregulates lipoxin secretion. (a) Lipoxin A4 secretion in cells expressing KSHV miRNA cluster versus EGFP. EAhy-K10/K12 and EAhy-GFP cells at 80 to 90% confluence were serum starved for 8 h. Supernatants were collected from cells growing in serum-free medium, and ELISA was performed according to the manufacturer's instructions. (b) Lipoxin A4 secretion in HMVEC-d cells with de novo-infected miRNA cluster-deleted KSHV and WT KSHV was measured by ELISA.
KSHV miRNA could potentially degrade host mRNAs of enzymes involved in lipoxin synthesis. Evaluating the levels of protein is a method used to study the expression of mRNA. Lipoxin synthesis occurs with the help of enzymes 15-lipoxygenase and 12-lipoxygenase. To identify the target mRNA, the protein levels of the enzymes (15-lipoxygenase and 12-lipoxygenase) involved in the lipoxin synthesis pathway were measured in EAhy K10/K12 and EAhy GFP cells. A significant reduction in the protein levels of 15-lipoxygenase was observed in cell lysate obtained from cells expressing KSHV miRNA compared to cells expressing GFP. No change was observed in the protein levels of 12-lipoxygenase (Fig. 8a). In addition, gene expression analysis of HMVEC-d cells de novo infected with KSHV for 8, 24, or 48 h also show a reduction in the levels of 15-lipoxygenase and 12-lipoxygenase. 15-Lipoxygenase gene expression was reduced by 50% 8 h after KSHV infection while 12-lipoxygenase gene expression was reduced by 30%, 8 h after infection (Fig. 8b). To verify the contribution of the KSHV miRNA cluster on lipoxin secretion, a loss of function assay was performed. miRNA cluster mutant recombinant KSHV and WT KSHV was prepared from producer iSLK cells, followed by their de novo infection in HMVEC-d cells for 24 h. A higher level of lipoxin secretion was observed in the miRNA cluster mutant KSHV than in WT KSHV.
FIG 8.

Identifying targets of lipoxin downregulation. (a) Cellular target of KSHV miRNA. Western blot analysis of cellular proteins from EA-hy926 cells expressing miRNA or GFP and probed for enzymes was performed. LO, lipoxygenase involved in the lipoxin synthesis pathway. (b) HMVEC-d cells were propagated to 80 to 90% confluence and infected with KSHV (30 DNA copies/cell) for 24 h. Gene expression analysis of 15-lipoxygenase and 12-lipoxygenase from de novo KSHV-infected HMVEC-d cells was performed using a TaqMan gene expression assay.
DISCUSSION
Studies have shown that KSHV infection alters the inflammatory microenvironment to its own advantage. Several proinflammatory metabolites of the arachidonic acid pathway, such as COX-2/PGE2 and 5-lipoxygenase/LTB4, were upregulated on KSHV infection (14, 35). To study the KSHV infection mediated alteration of the anti-inflammatory lipoxin secretion pathway, an ELISA was performed on supernatants from KSHV-infected and uninfected HMVEC-d cells. The results show that KSHV infection downregulates the secretion of lipoxin A4. All cells used in this study were serum starved for the entire duration of the study, and serum does not influence lipoxin secretion. Similar downregulation of lipoxin secretion was observed in the PEL cell models BC-3, BCBL-1, and KSHV-BJAB. These observations suggest that KSHV infection contributes to the proinflammatory microenvironment by downregulating anti-inflammatory lipoxin A4 secretion for its own advantage.
Lipoxins bind to several receptors in various cell types. One of the most common and well-studied receptors is ALX/FPR (37). Other receptors include G-protein-coupled receptor 32, aryl hydrocarbon receptor (38, 39), and estrogen receptor (40). Since lipoxins exert their anti-inflammatory activity by binding to the G-protein-coupled receptor AXL/FPR, this was investigated in patient tissue sections and in vitro KSHV-infected cells. Immunohistochemistry results from KS patient tissue samples and healthy control population show the ubiquitous presence of the ALX/FPR receptor. Statistical analysis shows the presence of the ALX/FPR receptor in KS patients belonging to various ethnicities, including Hispanic, African-American, Caucasian, and Asian patients. In vitro cell models for KS and PEL also show the expression of ALX/FPR. These results pave the way for future studies to identify the therapeutic potential of lipoxin in treating KS and PEL.
Binding of lipoxin to ALX/FPR leads to its internalization in a time-dependent manner (41, 42). Studies in neutrophils and HUVEC cells have shown that ALX/FPR translocates to the perinuclear space upon lipoxin stimulation. This translocation has been shown as a vital requirement for lipoxin's anti-inflammatory activity (43–45). To determine whether this relocation of ALX/FPR is still maintained in cells infected by KSHV, a lipoxin stimulation experiment using de novo-infected HMVEC-d cells was performed. Similar to previously reported studies, a time-dependent translocation of the ALX/FPR receptor in de novo KSHV-infected HMVEC-d cells was observed. This result suggests that KSHV infection does not affect the functionality of ALX/FPR.
The presence of the ALX/FPR receptor on infected cells reinforced the concept of treating KSHV-infected cells with lipoxin and epilipoxin. Western blotting was performed to understand the impact of lipoxin treatment on various cellular signaling molecules known to influence KSHV pathogenesis. For example, ERK and AKT pathways have previously been shown to promote KSHV infection and increase the survival of infected cells and the development of malignancy (32, 33). NF-κB has been reported to promote an inflammatory microenvironment and help in viral entry and the establishment of latency (34, 46). Our studies show a decrease in the levels of these host proteins upon lipoxin and epilipoxin treatment. Previous studies from our research group have shown that several proinflammatory factors of the arachidonic acid pathway, such as COX-2, PGE2, 5-lipoxygenase, and LTB4, are upregulated during KSHV infection (10, 14, 35). These proinflammatory arachidonic acid metabolites have been shown to enhance KSHV latency, pathogenesis, and immune evasion and to promote an inflammatory microenvironment (10, 14, 35). Treatment with lipoxin and epilipoxin decreased the levels of these proinflammatory molecules, resulting in a more anti-inflammatory microenvironment.
To determine whether the ALX/FPR receptor is vital for lipoxin signaling, a cell line with a deleted ALX gene was developed. CRISPR/CAS9 technology was used to create ALX-negative cells. This system is naturally found in bacteria as a protective defense mechanism against foreign genetic material (47, 48). U20S cells were selected as a model cell line to perform this gene mutation study for several reasons. (i) U20S cells are easily infected with KSHV and express the latent gene LANA-1 (data not shown), and several other studies have used U20S to study KSHV infection (49, 50). (ii) WT U20S cells express ALX/FPR (Fig. 5a). (iii) U20S cells have been widely used to perform CRISPR/Cas9 gene modification studies (51, 52). Plasmids containing the GFP gene, guide RNA and the Cas9 gene were transfected into U2OS cells. GFP serves as a transfection control and also helps in sorting cells. Guide RNA has a sequence commentary to the ALX/FPR gene. Once transfected, guide RNA binds to its cDNA sequence. When the guide RNA forms a duplex with host DNA, CAS9 enzymatic activity is activated. The Cas9 enzyme creates a double-stranded break in the DNA, which is repaired by an error-prone host repair mechanism. This creates a defective gene. To screen for cells with a mutated ALX gene, transfected cells are flow sorted to isolate GFP-positive cells. Individual cells were grown and screened for mutation. Western blots of cell lysates isolated from clones show the absence of ALX/FPR.
ALX mutant U20S cells and wild-type U20S cells were treated with 100 nM lipoxin, 100 nM epilipoxin, or ethanol for 72 h. Lysates from the treated cells were Western blotted for NF-κB, AKT, and ERK1/2 to elusidate the role of ALX in lipoxin signaling. Phosphorylated forms of the above proteins give an indication of the level of activity. The pervious study shows that the levels of NF-κB, AKT, and ERK1/2 in latently infected HMVEC-d cells (cells expressing ALX receptor) decrease upon treatment with lipoxin or epilipoxin (Fig. 4a). In latently infected U20S ALX mutant cells, treatment with lipoxin/epilipoxin yielded an increase in the levels of NF-κB and ERK1/2 compared to treated wild-type U20S cells (latently infected). We speculate that this observation is related to the absence of the ALX receptor. Lipoxin via interaction with the ALX receptor downregulates NF-κB and ERK1/2, which are also involved in KSHV pathogenesis. Similar studies with HMVEC-d cells are required to further understand ALX-mediated lipoxin signaling. This result highlights the importance of the ALX receptor for lipoxin signaling. The AKT levels seem to be unaltered in U20S ALX mutant cells and WT U20S cells. This observation could either be attributed to the nature of the cell type used or to lipoxin signaling via other receptors.
Latency plays an important role in KSHV infection. The majority of the KSHV genome is in latent form in KS and PEL. The latency cluster of the KSHV genome consists of three main genes, including LANA-1, v-Cyclin, and v-FLIP, in addition to a group of 12 pre-miRNA (53–56). In order to study the role of latency factors on lipoxin secretion, latency genes were lentivirus transduced in HMVEC-d cells, and the lipoxin secretion was measured. The results show that there is no effect of latency genes on lipoxin secretion.
Latency is also controlled by a group of 12 pre-miRNA located in the K12 region of the KSHV genome (57). KSHV miR12-1 to KSHV miR12-9 and KSHV miR12-11 have been located in the intronic region of K12 gene. Although KSHV miR12 is found in the 3′ untranslated region (UTR) of the coding region of the K12 gene and KSHV miR 12-10 is found in the coding region of the K12 gene. For this study, we have focused on the miRNAs located in the intronic region of the K12 gene. Cell lines expressing KSHV miRNA or GFP were compared to determine the effect of miRNA on lipoxin secretion. An ELISA performed on the supernatants of these cell lines showed that there was a 43% decrease in the amount of lipoxin secretion when these miRNAs were expressed (Fig. 7a). To validate the role of miRNA in lipoxin secretion, KSHV virions from iSLK producer cells with WT and miRNA cluster mutant were isolated. The recombinant virus was then used to de novo infect HMVEC-d cells. Lipoxin A4 ELISA was performed on the supernatant of the HMVEC-d cells infected with recombinant WT KSHV and miRNA cluster-deleted KSHV. No change in lipoxin secretion was observed in miRNA cluster-deleted KSHV infection, while WT KSHV infection did reduce lipoxin secretion (Fig. 7b). These results indicate that KSHV miRNA could play an important role in regulating genes that control lipoxin secretion.
miRNAs are small noncoding RNAs that can posttranslationally alter gene expression by targeting selective mRNAs for degradation. miRNA targets and binds to the 3′ UTR region of mRNA and degrades it (58). Lipoxin synthesis involves three main enzymes: the 5-, 12-, and 15-lipoxygenases (59). If a decrease in lipoxin secretion occurs, it is most likely due to the degradation of enzymes that are involved in synthesis. A Western blot analysis was performed on cell lysates from EAhy-926 cells expressing KSHV miRNA or GFP. Lysates were probed with antibodies for 12- and 15-lipoxygenases. KSHV miRNAs could impact lipoxin secretion since a significant reduction in the protein level of 15-lipoxygenase was observed in cell lysate obtained from cells expressing KSHV miRNA compared to cells expressing GFP. This study gives an overview of the potential mechanism of viral miRNA involved in downregulating lipoxin synthesis. Further studies are in progress to identify individual miRNAs involved in the process and other possible host pathways targeted. Extensive studies have been performed to identify targets for KSHV miRNAs (60). None of these studies show the influence of KSHV miRNA on 15-lipoxygenase. However, the indirect action of KSHV miRNA on 15-lipoxygenase cannot be disregarded. KSHV miRNA has been shown to upregulate cellular NF-κB levels (61). In this regard, a study on Alzheimer's disease has shown that upregulated NF-κB activates cellular miRNA to degrade 15-lipoxygenase (62). Although KSHV miRNAs do not directly influence 15-lipoxygenase levels, the upregulated NF-κB could contribute to a downregulation of 15-lipoxygenase. In addition, gene expression analysis of de novo KSHV-infected HMVEC-d cells shows a downregulation of the levels of 15-lipoxygenase and 12-lipoxygenase (Fig. 8b), suggesting additional unknown mechanism(s) of miRNA-mediated lipoxin downregulation, such as targeting transcription factors of 15-lipoxygenase and 12-lipoxygenase genes or direct interaction with 15- and 12-lipoxygenase genes could be possible.
Apart from the latency cluster, viral proteins could also be involved in downregulating lipoxin secretion. To study the role of KSHV proteins in lipoxin secretion, replication-deficient, UV-irradiated KSHV was used, and we found no change in the level of lipoxin secretion (results not shown).
The present study adds to the understanding of inflammatory microenvironment modulation by KSHV infection (Fig. 9). Apart from upregulating several proinflammatory molecules, KSHV infection downregulates anti-inflammatory lipoxin secretion. The presence of lipoxin receptor ALX/FPR in KS patient tissue sections and in vitro KS and PEl cell models has enabled treating KSHV infection with lipoxin. Supplementing lipoxin decreases the levels of proinflammatory host cell proteins, such as ERK and NF-κB, and metabolites, such as COX-2 and PGE2, which are believed to help in KSHV entry, pathogenesis, and latency establishment. The presence of ALX/FPR on infected cells is a vital component for lipoxin signaling. Viral microRNA has been identified to decrease lipoxin secretion by the enzyme 15-lipoxygenase degradation.
FIG 9.

Mechanistic pathway of KSHV infection negatively affecting lipoxin secretion. KSHV infection apart from upregulating several proinflammatory cell signaling pathways and enzymes, such as NF-κB, AKT, FAK, Src, ERK, COX-2, and 5-lipoxygenase (5-LO), also downregulate anti-inflammatory lipoxins. The KSHV miRNA cluster targets enzyme 15-LO to reduce lipoxin secretion. The ALX/FPR receptor has been shown to be vital for lipoxin signaling since blocking this receptor elevates the levels of NF-κB, ERK, and COX-2. Treating cells with lipoxins reduces the levels of KSHV infection-induced NF-κB, AKT, and ERK.
Considering the effectiveness of current treatment strategies for KSHV-associated malignancies, there is an urgent need to look for alternative therapeutics. These in vitro studies show the efficacy of lipoxin treatment. This study opens up further investigation on the role of KSHV miRNA on lipoxin synthesis. The therapeutic potential of lipoxin in in vivo using animal models is yet to be explored.
ACKNOWLEDGMENTS
We gratefully acknowledge Keith Philibert for suggestions and critically reading the manuscript. We thank Robert Dickinson from the Rosalind Franklin University of Medicine and Science (RFUMS) Cell Sorting Core Facility for help in fluorescence-activated cell-sorting analyses. We thank Steven Miller (RFUMS) for help in statistical analysis. We are grateful to the NIH AIDS Research and Reference Reagent Program for normal and KS specimens. We thank Paivi Ojala (Imperial College of London) for EAhy cell lines, Blossom Damania (University of North Carolina) for KSHV-BJAB cells, and Rolf Renne and his lab members (University of Florida) for providing iSLK producer cells.
We are grateful for NIH-funded grant R01CA192970 and to RFUMS for start-up funds to N.S.-W. We also acknowledge financial support from the H. M. Bligh Cancer Research Fund. The funders had no role in study, design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. 1999. Distribution of human herpesvirus 8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A 96:4546–4551. doi: 10.1073/pnas.96.8.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muralidhar S, Veytsmann G, Chandran B, Ablashi D, Doniger J, Rosenthal LJ. 2000. Characterization of the human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus) oncogene, kaposin (ORF K12). J Clin Virol 16:203–213. doi: 10.1016/S1386-6532(99)00081-5. [DOI] [PubMed] [Google Scholar]
- 3.Schulz TF. 1999. Epidemiology of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. Adv Cancer Res 76:121–160. doi: 10.1016/S0065-230X(08)60775-7. [DOI] [PubMed] [Google Scholar]
- 4.Chen YB, Rahemtullah A, Hochberg E. 2007. Primary effusion lymphoma. Oncologist 12:569–576. doi: 10.1634/theoncologist.12-5-569. [DOI] [PubMed] [Google Scholar]
- 5.Euvrard S, Kanitakis J, Claudy A. 2003. Skin cancers after organ transplantation. N Engl J Med 348:1681–1691. doi: 10.1056/NEJMra022137. [DOI] [PubMed] [Google Scholar]
- 6.Lee HR, Lee S, Chaudhary PM, Gill P, Jung JU. 2010. Immune evasion by Kaposi's sarcoma-associated herpesvirus. Future Microbiol 5:1349–1365. doi: 10.2217/fmb.10.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J Virol 72:8309–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McClure LV, Sullivan CS. 2008. Kaposi's sarcoma herpes virus taps into a host microRNA regulatory network. Cell Host Microbe 3:1–3. doi: 10.1016/j.chom.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol 71:715–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.George Paul A, Sharma-Walia N, Kerur N, White C, Chandran B. 2010. Piracy of prostaglandin E2/EP receptor-mediated signaling by Kaposi's sarcoma-associated herpes virus (HHV-8) for latency gene expression: strategy of a successful pathogen. Cancer Res 70:3697–3708. doi: 10.1158/1538-7445.AM10-3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naranatt PP, Akula SM, Zien CA, Krishnan HH, Chandran B. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J Virol 77:1524–1539. doi: 10.1128/JVI.77.2.1524-1539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naranatt PP, Krishnan HH, Svojanovsky SR, Bloomer C, Mathur S, Chandran B. 2004. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res 64:72–84. doi: 10.1158/0008-5472.CAN-03-2767. [DOI] [PubMed] [Google Scholar]
- 13.Paul AG, Chandran B, Sharma-Walia N. 2013. Concurrent targeting of eicosanoid receptor 1/eicosanoid receptor 4 receptors and COX-2 induces synergistic apoptosis in Kaposi's sarcoma-associated herpesvirus and Epstein-Barr virus associated non-Hodgkin lymphoma cell lines. Transl Res 161:447–468. doi: 10.1016/j.trsl.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paul AG, Chandran B, Sharma-Walia N. 2013. Cyclooxygenase-2-prostaglandin E2-eicosanoid receptor inflammatory axis: a key player in Kaposi's sarcoma-associated herpes virus associated malignancies. Transl Res 162:77–92. doi: 10.1016/j.trsl.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontana JM, Mygatt JG, Conant KL, Parsons CH, Kaleeba JA. 2014. Kaposi's sarcoma-associated herpesvirus subversion of the anti-inflammatory response in human skin cells reveals correlates of latency and disease pathogenesis. J Skin Cancer 2014:246076. doi: 10.1155/2014/246076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arav-Boger R. 2009. Treatment for Kaposi sarcoma herpesvirus: great challenges with promising accomplishments. Virus Genes 38:195–203. doi: 10.1007/s11262-008-0325-y. [DOI] [PubMed] [Google Scholar]
- 17.Coen N, Duraffour S, Snoeck R, Andrei G. 2014. KSHV targeted therapy: an update on inhibitors of viral lytic replication. Viruses 6:4731–4759. doi: 10.3390/v6114731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scolaro MJ, Gunnill LB, Pope LE, Khalil MH, Katz DH, Berg JE. 2001. The antiviral drug docosanol as a treatment for Kaposi's sarcoma lesions in HIV type 1-infected patients: a pilot clinical study. AIDS Res Hum Retroviruses 17:35–43. doi: 10.1089/088922201750056762. [DOI] [PubMed] [Google Scholar]
- 19.Uldrick TS, Whitby D. 2011. Update on KSHV epidemiology, Kaposi sarcoma pathogenesis, and treatment of Kaposi sarcoma. Cancer Lett 305:150–162. doi: 10.1016/j.canlet.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serhan CN, Hamberg M, Samuelsson B. 1984. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci U S A 81:5335–5339. doi: 10.1073/pnas.81.17.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandrasekharan JA, Sharma-Walia N. 2015. Lipoxins: nature's way to resolve inflammation. J Inflamm Res 8:181–192. doi: 10.2147/JIR.S90380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marginean A, Sharma-Walia N. 2015. Lipoxins exert anti-angiogenic and anti-inflammatory effects on Kaposi's sarcoma cells. Transl Res 166:111–133. doi: 10.1016/j.trsl.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 23.Nun TK, Kroll DJ, Oberlies NH, Soejarto DD, Case RJ, Piskaut P, Matainaho T, Hilscher C, Wang L, Dittmer DP, Gao SJ, Damania B. 2007. Development of a fluorescence-based assay to screen antiviral drugs against Kaposi's sarcoma associated herpesvirus. Molecular cancer therapeutics 6:2360–2370. doi: 10.1158/1535-7163.MCT-07-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L, Chandran B. 2004. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J Virol 78:4207–4223. doi: 10.1128/JVI.78.8.4207-4223.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma-Walia N, Raghu H, Sadagopan S, Sivakumar R, Veettil MV, Naranatt PP, Smith MM, Chandran B. 2006. Cyclooxygenase 2 induced by Kaposi's sarcoma-associated herpesvirus early during in vitro infection of target cells plays a role in the maintenance of latent viral gene expression. J Virol 80:6534–6552. doi: 10.1128/JVI.00231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J Virol 79:10308–10329. doi: 10.1128/JVI.79.16.10308-10329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma-Walia N, Paul AG, Bottero V, Sadagopan S, Veettil MV, Kerur N, Chandran B. 2010. Kaposi's sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog 6:e1000777. doi: 10.1371/journal.ppat.1000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain V, Plaisance-Bonstaff K, Sangani R, Lanier C, Dolce A, Hu J, Brulois K, Haecker I, Turner P, Renne R, Krueger B. 2016. A toolbox for herpesvirus miRNA research: construction of a complete set of KSHV miRNA deletion mutants. Viruses 8:E54. doi: 10.3390/v8020054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vart RJ, Nikitenko LL, Lagos D, Trotter MW, Cannon M, Bourboulia D, Gratrix F, Takeuchi Y, Boshoff C. 2007. Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoietin-2 expression in lymphatic endothelial cells. Cancer Res 67:4042–4051. doi: 10.1158/0008-5472.CAN-06-3321. [DOI] [PubMed] [Google Scholar]
- 30.Sharma-Walia N, Patel K, Chandran K, Marginean A, Bottero V, Kerur N, Paul AG. 2012. COX-2/PGE2: molecular ambassadors of Kaposi's sarcoma-associated herpes virus oncoprotein-v-FLIP. Oncogenesis 1:e5. doi: 10.1038/oncsis.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veettil MV, Bandyopadhyay C, Dutta D, Chandran B. 2014. Interaction of KSHV with host cell surface receptors and cell entry. Viruses 6:4024–4046. doi: 10.3390/v6104024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhatt AP, Damania B. 2013. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front Immunol 3:401. doi: 10.3389/fimmu.2012.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan H, Xie J, Ye F, Gao SJ. 2006. Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J Virol 80:5371–5382. doi: 10.1128/JVI.02299-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadagopan S, Sharma-Walia N, Veettil MV, Raghu H, Sivakumar R, Bottero V, Chandran B. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-κB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J Virol 81:3949–3968. doi: 10.1128/JVI.02333-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma-Walia N, Chandran K, Patel K, Veettil MV, Marginean A. 2014. The Kaposi's sarcoma-associated herpesvirus (KSHV)-induced 5-lipoxygenase-leukotriene B4 cascade plays key roles in KSHV latency, monocyte recruitment, and lipogenesis. J Virol 88:2131–2156. doi: 10.1128/JVI.02786-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suffert G, Malterer G, Hausser J, Viiliainen J, Fender A, Contrant M, Ivacevic T, Benes V, Gros F, Voinnet O, Zavolan M, Ojala PM, Haas JG, Pfeffer S. 2011. Kaposi's sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS Pathog 7:e1002405. doi: 10.1371/journal.ppat.1002405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serhan CN, Levy BD, Clish CB, Gronert K, Chiang N. 2000. Lipoxins, aspirin-triggered 15-epi-lipoxin stable analogs and their receptors in anti-inflammation: a window for therapeutic opportunity. Ernst Schering Res Found Workshop 2000:143–185. [DOI] [PubMed] [Google Scholar]
- 38.Gronert K, Martinsson-Niskanen T, Ravasi S, Chiang N, Serhan CN. 2001. Selectivity of recombinant human leukotriene D4, leukotriene B4, and lipoxin A4 receptors with aspirin-triggered 15-epi-LXA4 and regulation of vascular and inflammatory responses. Am J Pathol 158:3–9. doi: 10.1016/S0002-9440(10)63937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaldach CM, Riby J, Bjeldanes LF. 1999. Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry 38:7594–7600. doi: 10.1021/bi982861e. [DOI] [PubMed] [Google Scholar]
- 40.Russell R, Gori I, Pellegrini C, Kumar R, Achtari C, Canny GO. 2011. Lipoxin A4 is a novel estrogen receptor modulator. FASEB J 25:4326–4337. doi: 10.1096/fj.11-187658. [DOI] [PubMed] [Google Scholar]
- 41.Serhan CN. 2008. Systems approach with inflammatory exudates uncovers novel anti-inflammatory and pro-resolving mediators. Prostaglandins Leukot Essent Fatty Acids 79:157–163. doi: 10.1016/j.plefa.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Serhan CN, Chiang N. 2004. Novel endogenous small molecules as the checkpoint controllers in inflammation and resolution: entree for resoleomics. Rheum Dis Clin North Am 30:69–95. doi: 10.1016/S0889-857X(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 43.Maderna P, Cottell DC, Toivonen T, Dufton N, Dalli J, Perretti M, Godson C. 2010. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J 24:4240–4249. doi: 10.1096/fj.10-159913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitchell S, Thomas G, Harvey K, Cottell D, Reville K, Berlasconi G, Petasis NA, Erwig L, Rees AJ, Savill J, Brady HR, Godson C. 2002. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J Am Soc Nephrol 13:2497–2507. doi: 10.1097/01.ASN.0000032417.73640.72. [DOI] [PubMed] [Google Scholar]
- 45.Serhan CN, Chiang N. 2008. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol 153(Suppl 1):S200–S215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawrence T. 2009. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol 1:a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sander JD, Joung JK. 2014. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng F, Weidner-Glunde M, Varjosalo M, Rainio EM, Lehtonen A, Schulz TF, Koskinen PJ, Taipale J, Ojala PM. 2009. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog 5:e1000324. doi: 10.1371/journal.ppat.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarek G, Kurki S, Enback J, Iotzova G, Haas J, Laakkonen P, Laiho M, Ojala PM. 2007. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J Clin Invest 117:1019–1028. doi: 10.1172/JCI30945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He Z, Proudfoot C, Mileham AJ, McLaren DG, Whitelaw CB, Lillico SG. 2015. Highly efficient targeted chromosome deletions using CRISPR/Cas9. Biotechnol Bioeng 112:1060–1064. doi: 10.1002/bit.25490. [DOI] [PubMed] [Google Scholar]
- 52.Liao S, Tammaro M, Yan H. 2015. Enriching CRISPR-Cas9 targeted cells by cotargeting the HPRT gene. Nucleic Acids Res 43:e134. doi: 10.1093/nar/gkv675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. 2005. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci U S A 102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jenner RG, Alba MM, Boshoff C, Kellam P. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J Virol 75:891–902. doi: 10.1128/JVI.75.2.891-902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. 2005. Identification of microRNAs of the herpesvirus family. Nat Methods 2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 56.Samols MA, Hu J, Skalsky RL, Renne R. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J Virol 79:9301–9305. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bellare P, Ganem D. 2009. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6:570–575. doi: 10.1016/j.chom.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 59.Maderna P, Godson C. 2009. Lipoxins: resolutionary road. Br J Pharmacol 158:947–959. doi: 10.1111/j.1476-5381.2009.00386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gottwein E, Corcoran DL, Mukherjee N, Skalsky RL, Hafner M, Nusbaum JD, Shamulailatpam P, Love CL, Dave SS, Tuschl T, Ohler U, Cullen BR. 2011. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 10:515–526. doi: 10.1016/j.chom.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao Z, Dou Y, Chen Y, Zheng Y. 2014. MicroRNA roles in the NF-κB signaling pathway during viral infections. Biomed Res Int 2014:436097. doi: 10.1155/2014/436097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao Y, Bhattacharjee S, Jones BM, Hill J, Dua P, Lukiw WJ. 2014. Regulation of neurotropic signaling by the inducible, NF-κB-sensitive miRNA-125b in Alzheimer's disease (AD) and in primary human neuronal-glial (HNG) cells. Mol Neurobiol 50:97–106. doi: 10.1007/s12035-013-8595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]