

Abstract
Background
In Japan, a variety of traditional dietary habits and daily routines have developed in many regions. The effects of these behaviors, and the regional differences in the composition of the gut microbiota, are yet to be sufficiently studied. To characterize the Japanese gut microbiota and identify the factors shaping its composition, we conducted 16S metagenomics analysis of fecal samples collected from healthy Japanese adults residing in various regions of Japan. Each participant also completed a 94-question lifestyle questionnaire.
Results
We collected fecal samples from 516 healthy Japanese adults (325 females, 191 males; age, 21–88). Heatmap and biplot analyses based on the bacterial family composition of the fecal microbiota showed that subjects’ region of residence or gender were not strongly correlated with the general composition of the fecal microbiota. Although clustering analysis for the whole cohort did not reveal any distinct clusters, two enterotype-like clusters were observed in the male, but not the female, subjects.
In the whole subject population, the scores for bowel movement frequency were significantly correlated with the abundances of Christensenellaceae, Mogibacteriaceae, and Rikenellaceae in the fecal microbiota (P < 0.001). These three bacterial families were also significantly more abundant (P < 0.05 or 0.01) in lean subjects (body mass index (BMI) < 25) than in obese subjects (BMI > 30), which is consistent with previously published results. However, a previously reported correlation between BMI and bowel movement frequency was not observed. In addition, the abundances of these three families were positively correlated with each other and comprised a correlative network with 14 other bacterial families.
Conclusions
The present study showed that the composition of the fecal microbiota of healthy Japanese adults at the national level was not strongly correlated with subjects’ area of residence or gender. In addition, enterotype partitioning was ambiguous in this cohort of healthy Japanese adults. Finally, the results implied that the abundances of Christensenellaceae, Mogibacteriaceae, and Rikenellaceae, along with several other bacterial components that together comprised a correlative network, contributed to a phenotype characterized by a high frequency of bowel movements and a lean body type.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-016-0898-x) contains supplementary material, which is available to authorized users.
Keywords: Human gut microbiota, Healthy Japanese adult, Bowel movement frequency, Body mass index (BMI), 16S metagenomics, Dietary habits and daily routine
Background
The human gut microbiota is a highly complex ecosystem composed of a large number of bacteria (1010–1011 cells/g feces) from several hundreds of bacterial species [1]. Recently, the composition of the human gut microbiota has been linked to host health and to the development of diseases such as obesity and inflammatory bowel disease [2, 3]. Therefore, modulation of the gut microbiota may be a useful means of developing personalized healthcare strategies [4].
Although variations exist in the composition of the gut microbiota from one person to another, common patterns in the bacterial ecosystem of the gut microbiota have been identified, which are called enterotypes. Arumugam et al. [5] examined the fecal microbiota of subjects from several countries and found three robust enterotypes enriched in Bacteroides, Prevotella, or Ruminococcus that were independent of nationality or region of residence. Subsequent studies have confirmed the existence of at least two enterotypes in which either Prevotella or Bacteroides species are predominant [6]. Host parameters such as age, gender, and body mass index (BMI) are reported to be linked to the composition of the gut microbiota [7–11], and differences in dietary habits have been shown to affect the bacterial diversity of the human gut microbiota. Together, these results partially explain why differences in the composition of the gut microbiota are observed in persons residing in different geographical regions [6, 12–14].
In Japan, a variety of regionally distinct traditional dietary habits and daily routines have developed [15]. Although the effects of dietary habits and daily routines on the composition of the fecal microbiota have been studied in small cohorts of Japanese individuals [13], these effects have not been studied at the national level. Here, we determined the abundances and prevalences of bacterial families in the fecal microbiota of a group of healthy Japanese adults residing in various regions of Japan by using a 16S metagenomic approach. We then examined whether there were any correlations between the subjects’ dietary habits and daily routines and the bacterial composition of their gut microbiota by using the results of a lifestyle questionnaire completed by the subjects.
Methods
Subjects, fecal sampling, and DNA extraction
Healthy adult Japanese volunteers residing in the seven regions of Japan (Hokkaido, Tohoku, Kanto, Chubu, Kansai, Chugoku-Shikoku, and Kyushu) were recruited into the study. Fecal sampling and DNA extraction were conducted by using the methods of Jin et al. [16].
Lifestyle questionnaire
Each subject was asked to complete a questionnaire containing 94 questions about physical characteristics, dietary habits, daily routines, level of stress, and physical and mental health. To assess the subjects’ level of stress, we used 20 questions from the Center for Epidemiologic Studies Depression Scale test [17]. Additional file 1: Table S1 shows an English translation of the original Japanese questionnaire, together with the scoring system used for each question.
Illumina sequencing and microbiological analyses
The detailed procedures for the Illumina sequencing and microbiological analyses are described in the Additional file 2. Briefly, the V1–V2 hypervariable region of the bacterial 16S rRNA gene sequence was determined by using an Illumina Miseq gene and small genome sequencer (Illumina, CA, USA). By using the Quantitative Insights Into Microbial Ecology (QIIME) bioinformatics pipeline [18], 5,290,023 high-quality reads (10,252 ± 2406 reads/sample) were generated (Additional file 1: Table S2) and assigned to operational taxonomic units (OTUs). Representative sequences for each OTU were then aligned by using the MUSCLE multiple sequence alignment algorithm [19], taxonomically classified by using RDP Classifier v2.2 [20], and then used to construct an OTU table.
Analyses of microbial diversity
Based on the abundances of the bacterial families identified in the fecal microbiota, a heatmap and biplots were constructed, and a cluster analysis was performed by using R program version 3.1.3 [21]. The heatmap was constructed by summing the OTU table for each bacterial family. A dendrogram based on the heatmap was then drawn by using the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) algorithm based on Jensen-Shannon divergence. Biplots were constructed by plotting PC1–PC2 scores obtained through a principal component analysis (PCA). Clusters were identified by conducting a principal coordinate analysis (PCoA) with the partitioning around medoids (PAM) algorithm by following the methods of Arumugam et al. [5]. Clusters with the highest Calinski-Harabasz (CH) index [22] were validated by using the methods of Koren et al. [23] to calculate prediction strength (PS) [24] and silhouette index (SI) [25]. The following thresholds were used for PS: no strong clustering (PS < 0.9), strong clustering (PS ≥ 0.9). The following thresholds were used for SI: no substantial structure (SI ≤ 0.25), weak structure (0.25 < SI ≤ 0.5), reasonable structure (0.5 < SI ≤ 0.75), and strong structure (SI > 0.75).
Statistical analysis
All statistical analyses were conducted by using R program version 3.1.3. [21]. To compare average microbial abundances and questionnaire scores between two groups, Welch’s t test and the Mann-Whitney U test were used for numerical and ordinal data, respectively. To compare the prevalence between two groups, Fisher’s exact test was performed. To compare the prevalence among multiple groups, Holm’s correction was applied to the tests described above. Correlations among and between bacterial families and questionnaire scores were evaluated by using Kendall’s Tau-b test.
Data deposition
The 16S metagenomic sequences determined in this study (accession number DRA005068) and the representative sequences assigned to the OTUs (accession numbers LC180363-LC183482) have been deposited in the DNA Data Bank of Japan.
Results
Subjects
A total of 516 healthy adult volunteers (325 females, 191 males; age, 21–88 years) was recruited into the study. The number of volunteers recruited from each region in Japan was as follows: Hokkaido, n = 38; Tohoku, n = 40; Kanto, n = 193; Chubu, n = 49; Kansai, n = 49; Chugoku-Shikoku, n = 48, and Kyushu, n = 99. The raw physical characteristics data for the cohort are shown in Additional file 1: Table S2. The physical characteristics of the cohort stratified by region, age, and gender are shown in Table 1. The gender ratios for each subjects’ area of residence were also shown in Additional file 3: Figure S1. Although there were no significant differences in age among the regions, there were differences in the gender ratio between some of the regions.
Table 1.
Subject characteristics stratified by area of residence
| Subject characteristics | Whole | Hokkaido | Tohoku | Kanto | Chubu | Kansai | Chugoku Shikoku | Kyushu |
|---|---|---|---|---|---|---|---|---|
| Subject No. | 516 | 38 | 40 | 193 | 49 | 49 | 48 | 99 |
| Age | 52.4 ± 13.4 | 50.0 ± 13.8 | 49.7 ± 17.1 | 53.5 ± 12.3 | 53.7 ± 14.2 | 54.8 ± 16.1 | 53.3 ± 10.0 | 50.0 ± 13.1 |
| Gender ratio (Female : Male) | 325 : 191 | 14 : 24a | 26 : 14ab | 122 : 71ab | 31 : 18ab | 19 : 30a | 36 : 12b | 77 : 22b |
Significance was calculated among area of residences
In each row, scores with the same letter in their superscripts or without superscripts were not significantly different (P ≥ 0.05) from each other
Taxonomic analysis of Illumina sequencing data
A total of 5,290,023 high-quality reads was generated by the Illumina sequencing (Additional file 1: Table S2). These high-quality reads were assigned to 3119 OTUs that were taxonomically classified into 12 phyla, 24 classes, 39 orders, 66 families, 141 genera, 347 species, and a group of unclassified OTUs (Additional file 1: Tables S3 and S4). Good’s coverage estimate, which is an index of sampling completeness, was greater than 95% for all subjects (average, 97.9% ± 0.7%), indicating that almost all of the microbial content of the fecal samples was covered (Additional file 1: Table S2).
Microbial diversity
The most predominant bacterial families in the fecal samples were Bacteroidaceae (mean abundance ± SD: 33.1% ± 19.0%; prevalence: 100%), Lachnospiraceae (17.6% ± 10.1%; 100%), Ruminococcaceae (15.8% ± 9.3%; 100%), and Prevotellaceae (9.1% ± 18.0%; 73.3%) (Additional file 1: Table S5). The heatmap constructed did not reveal any distinct clusters based on area of residence or gender (Fig. 1).
Fig. 1.
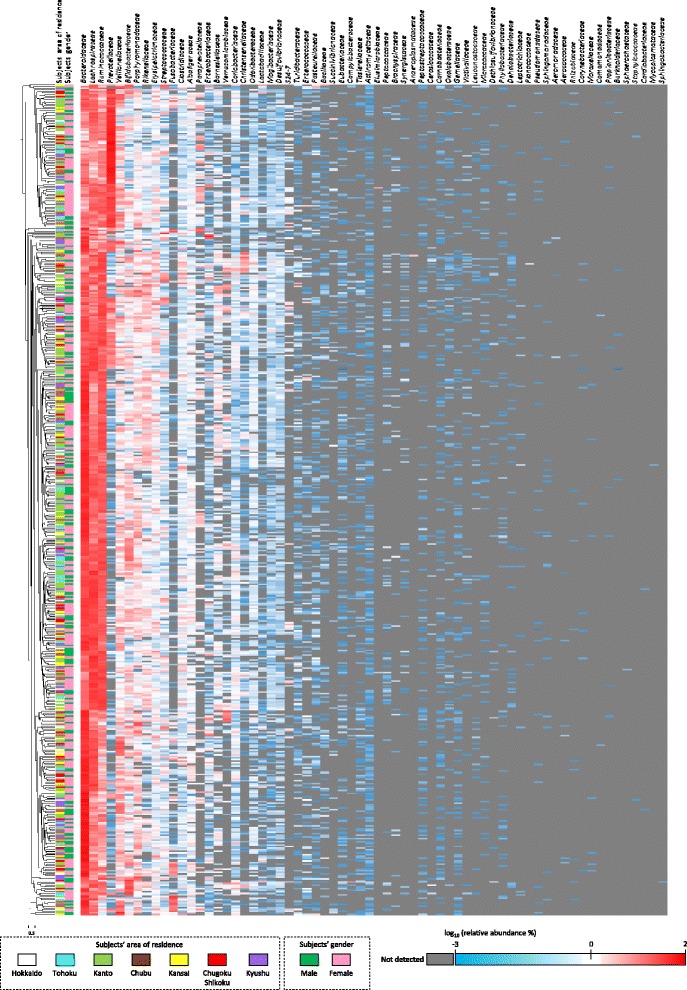
Heatmap of the abundances of the 66 bacterial families identified in the fecal microbiota. The data for each subject were aligned to a dendrogram constructed by using the UPGMA algorithm based on the Jensen-Shannon divergence. The color scale at the bottom of the heatmap shows the abundance of each bacterial family (log10 %) and the horizontal scale bar showed Jensen-Shannon divergence (0.5). Subjects’ area of residence and gender are indicated by using color codes on the bottom of this figure
In the biplots constructed, the location of the plots in the PC1–PC2 dimension was affected mainly by the abundances of the bacterial families Bacteroidaceae, Lachnospiraceae, Ruminococcaceae, and Prevotellaceae (hereafter referred to as “the major PCA determinants”) (Fig. 2a). Samples enriched with Prevotellaceae were located in the PC1-positive/PC2-positive direction. Samples enriched with Bacteroidaceae were located in the PC1-negative/PC2-positive direction. Samples enriched with Lachnospiraceae and Ruminococcaceae were located in the PC2-negative direction. The effect of the fifth most abundant bacterial family, Bifidobacteriaceae, on plot location was much smaller than those of the major PCA determinants. No distinct clusters were observed when the biplot was colored for area of residence or gender (Fig. 2b and c); however, Lachnospiraceae was found to be significantly more abundant (P < 0.05), but not more prevalent, in the subjects residing in Hokkaido compared with those residing in Kyushu (Tables 2 and 3). Significant differences (P < 0.05) between areas of residence were also observed in the abundance or prevalence of six other bacterial families (i.e., Bacillaceae, Bifidobacteriaceae, Lachnospiraceae, Lactobacillaceae, Pasteurellaceae, and S24-7), but these differences were only observed between two of the seven areas of residence for each family. No significant differences in alpha diversity score were observed among the areas of residence (Additional file 1: Table S5).
Fig. 2.
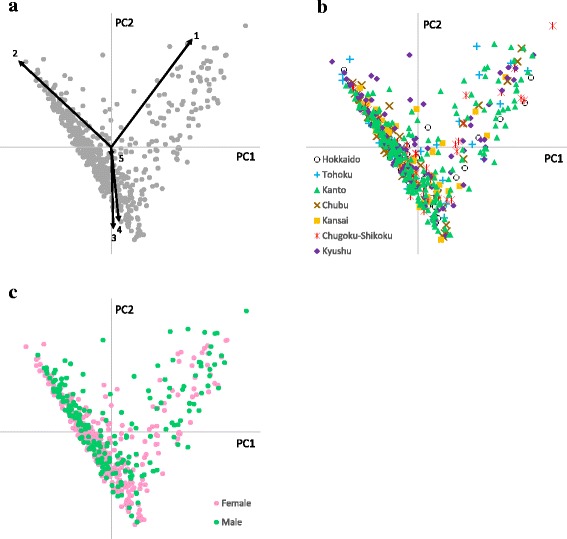
Biplots constructed based on the bacterial family composition of each subjects’ fecal microbiota. A principal component analysis of the bacterial family composition of each subjects’ fecal microbiota was performed, and a biplot in the PC1–PC2 dimension were constructed. The relative contributions of PC1 and PC2 were 54.1 and 25.7%, respectively. a The constructed biplot with an overlay showing the effects of the five most abundant bacterial families on plot location in the PC1–PC2 dimension (1. Prevotellaceae, 2. Bacteroidaceae, 3. Lachnospiraceae, 4. Ruminococcaceae, 5. Bifidobacteriaceae). b The biplot shown in (a) with the sample plots colored based on area of residence. c The biplot shown in (a) with the sample plots colored based on gender
Table 2.
Abundance of the major PCA determinants stratified by area of residence
| Bacterial family | Abundance (Mean% ± SD) | |||||||
|---|---|---|---|---|---|---|---|---|
| Whole (n = 516) |
Hokkaido (n = 38) |
Tohoku (n = 40) |
Kanto (n = 193) |
Chubu (n = 49) |
Kansai (n = 49) |
Chugoku Shikoku (n = 48) |
Kyushu (n = 99) |
|
| Bacteroidaceae | 33.1 ± 19.0 | 32.4 ± 19.8 | 32.4 ± 19.7 | 30.8 ± 18.0 | 39.2 ± 20.1 | 34.7 ± 17.4 | 30.8 ± 17.7 | 35.4 ± 20.5 |
| Lachnospiraceae | 17.6 ± 10.1 | 20.3 ± 7.7a | 18.4 ± 9.7ab | 18.7 ± 10.5ab | 15.3 ± 10.4ab | 17.8 ± 9.7ab | 18.1 ± 10.0ab | 15.1 ± 10.0b |
| Prevotellaceae | 9.1 ± 18.0 | 7.1 ± 16.8 | 10.0 ± 19.1 | 10.8 ± 19.3 | 5.9 ± 16.6 | 4.9 ± 11.1 | 10.4 ± 20.4 | 8.9 ± 17.1 |
| Ruminococcaceae | 15.8 ± 9.3 | 15.0 ± 10.3 | 16.5 ± 9.9 | 16.4 ± 8.9 | 15.3 ± 10.1 | 15.1 ± 9.9 | 15.0 ± 9.0 | 15.9 ± 8.8 |
Significance was calculated among area of residences
In each row, scores with the same letter in their superscripts or without superscripts were not significantly different (P ≥ 0.05) from each other
Table 3.
Prevalence of the major PCA determinants stratified by area of residence
| Bacterial family | Prevalence (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Whole (n = 516) |
Hokkaido (n = 38) |
Tohoku (n = 40) |
Kanto (n = 193) |
Chubu (n = 49) |
Kansai (n = 49) |
Chugoku Shikoku (n = 48) |
Kyushu (n = 99) |
|
| Bacteroidaceae | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| Lachnospiraceae | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| Prevotellaceae | 73.3 | 76.3 | 67.5 | 74.1 | 69.4 | 65.3 | 77.1 | 76.8 |
| Ruminococcaceae | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
Significance was calculated among area of residences
In each row, scores without superscripts were not significantly different (P ≥ 0.05) from each other
Among the major PCA determinants, the abundance of Prevotellaceae was significantly higher (P < 0.01) in male subjects, and the abundance of Ruminococcaceae was significantly higher (P < 0.01) in female subjects (Table 4). Overall, significant differences (P < 0.05) in the abundance or prevalence of 24 of the 66 identified bacterial families were observed between female and male subjects (Additional file 1: Table S6). Compared with the male subjects, the female subjects also had significantly higher alpha diversity scores (Chao1 index, P < 0.05; Shannon index, P < 0.01; phylogenetic diversity whole tree, P < 0.01) (Additional file 1: Table S6).
Table 4.
Abundance and prevalence of the major PCA determinants stratified by gender
| Bacterial family | Abundance (Mean% ± SD) | Prevalence (%) | ||||
|---|---|---|---|---|---|---|
| Female (n = 325) |
Male (n = 191) |
Significance | Female (n = 325) |
Male (n = 191) |
Significance | |
| Bacteroidaceae | 33.2 ± 18.5 | 32.9 ± 19.7 | NS | 100 | 100 | NS |
| Lachnospiraceae | 18.0 ± 10.4 | 17.0 ± 9.5 | NS | 100 | 100 | NS |
| Prevotellaceae | 9.7 ± 17.7 | 16.6 ± 22.6 | ** | 71.1 | 77.0 | NS |
| Ruminococcaceae | 17.9 ± 8.7 | 12.4 ± 9.2 | ** | 100 | 100 | NS |
NS: P ≥ 0.05, **: P < 0.01
In the cluster analysis, no statistically reliable clusters (i.e., clusters with PS > 0.9 and SI > 0.25) were found when the whole subjects’ data were used. To examine whether there were any trends in the cluster partitioning, cluster analyses were also conducted for each subjects’ area of residence and gender. When only the data for the male subjects were analyzed, two statistically reliable, weakly partitioned clusters (PS = 0.96, SI = 0.34) were observed (Table 5 and Fig. 3). Between these two clusters, significant differences in abundance or prevalence were observed for 21 of the 66 identified bacterial families (Additional file 1: Table S7). Among the major PCA determinants, the abundance and prevalence of Prevotellaceae was significantly higher (P < 0.01) in cluster 1 compared with the other families in the cluster, whereas the abundances of Bacteroidaceae (P < 0.01), Lachnospiraceae (P < 0.01), and Ruminococcaceae (P < 0.05) were significantly higher in cluster 2 compared with the other families in the cluster (Additional file 1: Table S7).
Table 5.
Statistical output for the cluster analysis
| Statistics | Whole | Female | Male | Hokkaido | Tohoku | Kanto | Chubu | Kansai | Chugoku Shikoku | Kyushu |
|---|---|---|---|---|---|---|---|---|---|---|
| Cluster no. showing max CH score | 3 | 3 | 2 | 2 | 2 | 3 | 4 | 2 | 2 | 3 |
| Max CH score | 328 | 183 | 208 | 21 | 30 | 186 | 32 | 33 | 44 | 60 |
| Prediction strength | 0.62 | 0.59 | 0.96 | 0.62 | 0.67 | 0.56 | 0.51 | 0.61 | 0.68 | 0.63 |
| Silhouette index | 0.20 | 0.20 | 0.34 | 0.32 | 0.36 | 0.21 | 0.20 | 0.16 | 0.36 | 0.20 |
Fig. 3.
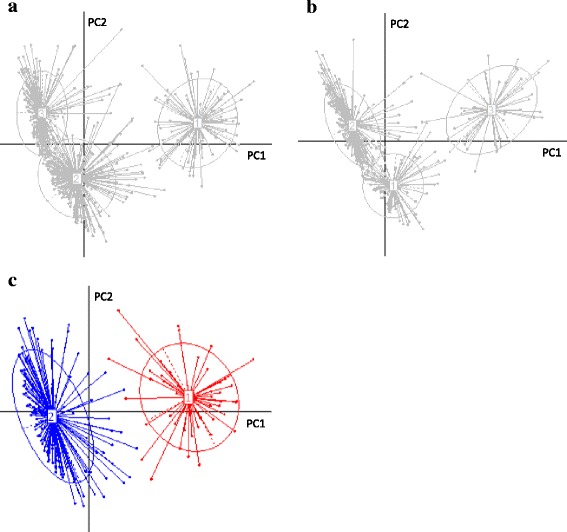
Cluster analysis based on the bacterial family compositions of the microbiota. Clusters were identified by conducting a PCoA with the PAM algorithm for the whole cohort (a) and for the female (b) and the male (c) subjects based on the bacterial family composition of the microbiota. The relative contributions of PC1 versus PC2 for (a), (b), and (c) were 35.4% versus 18.4%, 32.6% versus 18.8%, and 40.1% versus 17.2%, respectively. In (c), the different colors indicate that the clusters were statistically reliable
Questionnaire scores
The raw questionnaire data are presented in Additional file 1: Table S8. There were significant differences (P < 0.05) in scores for some of the questions among the areas of residence and between the genders (Additional file 1: Tables S9 and S10).
Correlations between microbial families abundance and questionnaire score
Significant differences (P < 0.01 or P < 0.05) were observed between the two clusters in the male subjects for the scores for one question about dietary habits and for four questions about physical and mental health (Table 6 and Additional file 1: Table S11). The scores for “Mushroom intake frequency” (Q 29) and “I feel like I am becoming quite healthy” (Q 94) were significantly higher in cluster 1 than in cluster 2, whereas the scores for “I was bothered by things that usually do not bother me within the past week” (Q 52), “A close relationship was negatively affected by a physical or mental health problem within the past month” (Q 81), and “I felt nervous within the past month” (Q 83) were significantly higher in cluster 2 than in cluster 1. However, there were no significant correlations between the scores to these questions and the abundance of any bacterial family.
Table 6.
Question scores that were significantly different between the two clusters identified in the male subjects
| Question | Cluster 1 (Mean ± SD) |
Cluster 2 (Mean ± SD) |
Significance | |
|---|---|---|---|---|
| Q 29 | Mushroom intake frequency | 2.6 ± 0.8 | 2.1 ± 0.6 | ** |
| Q 52 | I was bothered by things that usually do not bother me within the past week | 1.2 ± 0.5 | 1.5 ± 0.7 | * |
| Q 81 | A close relationship was negatively affected by a physical or mental health problem within the past month | 1.3 ± 0.6 | 1.5 ± 0.9 | * |
| Q 83 | I felt nervous within the past month | 1.8 ± 0.9 | 2.1 ± 1.0 | * |
| Q 94 | I feel like I am becoming quite healthy | 3.8 ± 0.9 | 3.5 ± 0.9 | * |
NS: P ≥ 0.05, *: P < 0.05, **: P < 0.01
There were significant weak negative correlations between bowel movement frequency (Q 7) and the abundance of Christensenellaceae, Mogibacteriaceae, and Rikenellaceae (P < 0.001, τ = −0.24, −0.28, and −0.25, respectively) (Table 7). There were significant positive correlations (P < 0.001 and τ = 0.39–0.49) among the abundances of these families (Fig. 4a). The abundance of at least one of these families was significantly correlated (P < 0.001 and |τ| > 0.2) with that of at least one of 14 other bacterial families (Fig. 4b), among which 11 families had a positive correlation (Barnesiellaceae, Cerasicoccaceae, Dehalobacteriaceae, Desulfovibrionaceae, Odoribacteraceae, Oxalobacteraceae, Peptococcaceae, Ruminococcaceae, Synergistaceae, Verrucomicrobiaceae, and Victivallaceae), and three families had a negative correlation (Bacteroidaceae, Fusobacteriaceae, and Veillonellaceae). In addition, there were significant weak positive correlations between the score for frequency of dairy product intake frequency (Q 13) and abundance of Lactobacillaceae (P < 0.001, τ = 0.25), and between the score for frequency of natto (traditional Japanese fermented soy beans) intake frequency (Q 21) and the abundance of Bacillaceae (P < 0.001, τ = 0.34) (Table 7).
Table 7.
Question scores that were significantly correlated with bacterial family abundance
| Question | Bacterial family | P | τ | Abundance (Mean% ± SD) (n = 516) |
Prevalence (%) (n = 516) |
|
|---|---|---|---|---|---|---|
| Q 7 | Bowel movement frequency | Christensenellaceae | <0.001 | −0.24 | 0.39 ± 1.54 | 56.8 |
| Q 7 | Bowel movement frequency | Mogibacteriaceae | <0.001 | −0.28 | 0.18 ± 0.27 | 82.4 |
| Q 7 | Bowel movement frequency | Rikenellaceae | <0.001 | −0.25 | 1.51 ± 1.82 | 90.9 |
| Q 13 | Dairy product intake frequency | Lactobacillaceae | <0.001 | 0.25 | 0.19 ± 1.35 | 52.5 |
| Q 21 | Natto intake frequency | Bacillaceae | <0.001 | 0.34 | 0.03 ± 0.36 | 26.6 |
Combinations showing significant correlation (P < 0.001 and |τ| > 0.2) were listed
Fig. 4.
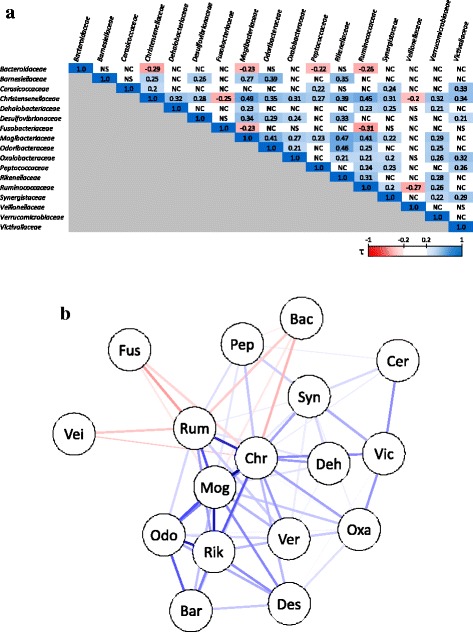
Correlations between Christensenellaceae, Mogibacteriaceae, Rikenellaceae, and 14 other bacterial families. Correlations between the abundance of Christensenellaceae, Mogibacteriaceae, Rikenellaceae, and 14 other bacterial families are shown as a heatmap (a) and network (b). a Kendall’s Tau-b ratio is shown for combinations with P < 0.001 and |τ| > 0.2. The strength of the shading within each cell indicates the strength of the positive (blue) or negative (red) correlation for a given bacterial family combination. NS, P ≥ 0.001; NC, |τ| ≤ 0.2. b Nodes for two correlated families (P < 0.001 and |τ| > 0.2) are shown connected by a line. The color, strength, and thickness of the lines indicate the strength of the positive (blue) or negative (red) correlation between the families. Bac, Bacteroidaceae; Bar, Barnesiellaceae; Cer, Cerasicoccaceae; Chr, Christensenellaceae; Deh, Dehalobacteriaceae; Des, Desulfovibrionaceae; Fus, Fusobacteriaceae; Mog, Mogibacteriaceae; Odo, Odoribacteraceae; Oxa, Oxalobacteraceae; Pep, Peptococcaceae; Rik, Rikenellaceae; Rum, Ruminococcaceae; Syn, Synergistaceae; Vei, Veillonellaceae; Ver, Verrucomicrobiaceae; Vic, Victivallaceae
The abundance of two OTUs belonging to Christensenellaceae (OTUs 844 and 2858) and of one OTU belonging to Mogibacteriaceae (OTU 417) had a weak negative correlation with the score for bowel movement frequency (Q 7, τ = −0.21, −0.22, and −0.23, respectively) (Table 8). The representative sequences for these OTUs had low identities with the sequences of known species (80.7–81.9%, 80.6–81.4%, and 82.0–87.5%, respectively) and therefore could not be identified at the species level (Additional file 1: Table S12). However, the abundance of one species belonging to family Lactobacillaceae (OTU 318) had a weak positive correlation with the score for frequency of dairy product intake frequency (Q 13, τ = 0.36), and the abundance of one species belonging to family Bacillaceae (OTU 547) had a weak positive correlation with the score for intake frequency of natto (Q 21, τ = 0.36). The representative sequences for these OTUs had a high identity with known sequences of species belonging to the Lactobacillus casei [26] and Bacillus subtilis subgroups [27], respectively.
Table 8.
Correlations between question score and operational taxonomic unit abundance
| Question | OTU ID | Bacterial family | P | τ | Abundance (Mean% ± SD) (n = 516) |
Prevalence (%) (n = 516) |
|
|---|---|---|---|---|---|---|---|
| Q 7 | Bowel movement frequency | 844 | Christensenellaceae | <0.001 | −0.21 | 0.05 ± 0.03 | 14.3 |
| Q 7 | Bowel movement frequency | 2858 | Christensenellaceae | <0.001 | −0.22 | 0.03 ± 0.01 | 11.6 |
| Q 7 | Bowel movement frequency | 417 | Mogibacteriaceae | <0.001 | −0.23 | 0.04 ± 0.03 | 18.0 |
| Q 13 | Dairy product intake frequency | 318 | Lactobacillaceae | <0.001 | 0.36 | 0.09 ± 0.08 | 24.8 |
| Q 21 | Natto intake frequency | 547 | Bacillaceae | <0.001 | 0.36 | 0.05 ± 0.03 | 25.2 |
Combinations showing significant correlation (P < 0.001 and |τ| > 0.2) were listed
Correlations between BMI and bowel movement frequency or abundance of the three bacterial families correlated with bowel movement frequency
Although bowel movement frequency in lean (BMI < 25, n = 425) and in obese (BMI > 30, n = 13) subjects was comparable, the abundances of the three bacterial families correlated with bowel movement frequency were significantly higher in lean subjects than in obese subjects (Christensenellaceae and Mogibacteriaceae, P < 0.01; Rikenellaceae, P < 0.05). The prevalence of Christensenellaceae was also significantly higher in lean subjects than in obese patients (P < 0.05) (Table 9).
Table 9.
Bowel movement frequency and correlated bacterial families in lean and obese subjects
| Questionnaire score (Mean ± SD) or abundance (Mean% ± SD) | Prevalence (%) | |||||
|---|---|---|---|---|---|---|
| Lean group (BMI < 25) (n = 425) |
Obese group (BMI > 30) (n = 13) |
Significance | Lean group (BMI < 25) (n = 425) |
Obese group (BMI > 30) (n = 13) |
Significance | |
| Bowel movement frequency | 3.53 ± 0.77 | 3.54 ± 0.78 | NS | - | - | - |
| Christensenellaceae | 0.77 ± 2.14 | 0.01 ± 0.004 | ** | 59.3 | 23.1 | * |
| Mogibacteriaceae | 0.23 ± 0.29 | 0.06 ± 0.05 | ** | 84.7 | 69.2 | NS |
| Rikenellaceae | 1.77 ± 1.86 | 0.90 ± 0.96 | * | 91.5 | 84.6 | NS |
NS: P ≥ 0.05, *: P < 0.05, **: P < 0.01
Discussion
Microbial diversity in the fecal microbiota
In the biplot analysis based on the bacterial family composition of the fecal microbiota, the location of the plots in the PC1–PC2 dimension was affected mainly by the abundances of Prevotellaceae, Bacteroidaceae, Lachnospiraceae, and Ruminococcaceae, which are the same families that were found to be driving the enterotypes identified in a previous study [5] (Fig. 2a). Furthermore, no distinct clusters were found in the data from the whole subjects (Table 5 and Fig. 3a), which is consistent with previous studies [23, 28]. This suggests that the more sample data is used, the less distinct the enterotype partitioning becomes.
Association between area of residence and the bacterial composition of the fecal microbiota
No distinct clusters were found for area of residence in the heatmap of bacterial family composition (Fig. 1) or in the biplots of the PC1–PC2 dimension (Fig. 2b). Furthermore, differences were found for the abundances of only six bacterial families between only two of the seven regions (Table 2 and Additional file 1: Table S5), suggesting that area of residence does not strongly affect the composition of the fecal microbiota in the healthy adults living in Japan. Therefore, the dietary habits and daily routines that are common in the various regions of Japan today (Additional file 1: Table S9) are not sufficiently distinct as to lead to clear microbial diversity in the gut, which is in contrast to the current situation in Thailand [14] and Mongolia [10].
Cluster analysis based on bacterial family composition and gender
Although no significant differences in the composition of the fecal microbiota were found between the female and male subjects in the heatmap and biplots (Figs 1 and 2c), statistically reliable clusters were observed in the male subjects but not in the female subjects (Table 5 and Fig. 3b, c). Furthermore, significant differences were found between female and male subjects in the abundance or prevalence of the 66 identified microbial families (Additional file 1: Table S6). Given that the distributions of the plots constructed by the PCA and PCoA were comparable, the position of the plots in the PCoA should reflect the abundances of the major PCA determinants (Figs 2a and 3). In male subjects, the abundance and prevalence of Prevotellaceae, which was the sole strong determinant of a sample plot being located in the PC1-positive/PC2-positive direction in the PCA analysis, were both significantly higher (P < 0.01) than in female subjects (Table 4), which may account for the distinct partitioning between cluster 1 and cluster 2 in the male subjects. Recent studies have demonstrated that the composition of the gut microbiota is gender-specific (microgenderome) and that there is a gender-specific, bidirectional relationship between the host and the gut microbiota that is mediated via sex hormones [7, 29]. This suggests that the differences in the composition of the fecal microbiota observed between the female and male subjects in the present study are a result of a gender-specific relationship between the host and their gut microbiota. Since there were significant differences in some of the question scores between the female and male subjects (Additional file 1: Table S10), it is also possible that differences in lifestyle between the female and male subjects contributed to the differences observed in the bacterial composition of the fecal microbiota.
Meaning of the clusters observed in the male subjects
Although the scores for one question about food intake frequency and for four questions about physical and mental health were significantly different between the two clusters in the male subjects (Table 6), these scores were not correlated with the abundance of any bacterial family. This suggests that food intake frequency does not strongly affect the composition of the fecal microbiota and that the composition of the fecal microbiota does not strongly affect the host’s physical and mental health. Indeed, the differences observed in the scores for the questions related to physical and mental health may simply be a result of environmental or genetic factors.
Effects of dietary habits on the bacterial composition of the fecal microbiota
Among the questions examining dietary habits, the frequencies of dairy product and natto intake were significantly correlated (P < 0.001) with the abundances of one Lactobacillaceae species and of one Bacillaceae species (Table 7). Given that some species of Lactobacillus, which is the main genus in family Lactobacillaceae, are used in the manufacture of dairy products [30], and that natto is a product made by fermenting soybeans with B. subtilis [31], it is not surprising that the frequencies of intake of these products were positively correlated with the abundances of these bacterial families. Indeed, the OTUs assigned to these two species were classified into the L. casei subgroup to which some probiotic strains belong [32] and the B. subtilis subgroup. In the present study, the frequency of intake of only two kinds of foodstuffs were correlated with the abundance of a bacterial family, implying that the human gut microbiota is affected more by the amount of particular foods consumed rather than by the frequency of intake.
Relationship between BMI and bacterial family abundance or bowel movement frequency
The abundances of Christensenellaceae, Mogibacteriaceae, and Rikenellaceae were negatively correlated with bowel movement frequency (Table 7). Furthermore, the abundances of these bacterial families were significantly higher in lean subjects (BMI < 25) than in obese subjects (BMI > 30) (Table 9), which is consistent with previous results [9]. Furthermore, the abundances of Christensenellaceae, Mogibacteriaceae, and Rikenellaceae were correlated with each other and formed part of a positively correlated network together with 11 other bacterial families. Three negatively correlated families (Bacteroidaceae, Fusobacteriaceae, and Veillonellaceae) were significantly correlated with four of the components of this network (Christensenellaceae, Mogibacteriaceae, Peptococcaceae, and Ruminococcaceae) (Fig. 4), indicating that BMI and bowel movement frequency could be controlled by modulating the abundances of Christensenellaceae, Mogibacteriaceae, and Rikenellaceae. As previous study reported, the abundances of these families associated with lower level of triglyceride and higher level of HDL cholesterol both of which correlate to lower BMI [33]. The abundances of Christensenellaceae and Rikenellaceae were highly associated with lower triglyceride, while the abundance Mogibacteriaceae of was highly associated with the higher level of HDL, implying their underlining mechanisms to reduce BMI were different. In addition, a cultured member of Christensenellaceae reduced weight and adiposity gains with amending obese associated microflora when inoculated to germ-free mice [9]. This result suggested that Christensenellaceae was one of the functional key factors contributing to lean body type in the correlation network. However, since there is little published work and this study was based on the 16S rRNA gene profiles, therefore, the further investigation should be needed to explain the functional role of these bacterial families to modify the host’s bowel movement frequency. Although these three families did not have a direct correlation in a study by Goodrich et al. [9], they were positively correlated with each other, as in our study, in the Yatsunenko dataset described in the same report, which suggests that these correlations are cohort-dependent.
A positive correlation between BMI and bowel movement frequency has been previously reported in a large cohort of European subjects, and it was also reported that obese subjects had a significantly higher bowel movement frequency compared with lean subjects [34]. However, in the present study, no significant differences in bowel movement frequency were found between lean and obese subjects (P = 0.74, τ = 0.01) (Table 9), suggesting that the correlation between bowel movement frequency and the abundance of Christensenellaceae, Mogibacteriaceae, and Rikenellaceae observed in the present study was not solely a result of BMI. The abundance of OTUs belonging to the families Christensenellaceae (OTUs 844 and 2858) and Mogibacteriaceae (OTU 417) were also correlated with bowel movement frequency. However, given that the abundances of these OTUs were relatively low compared with those of the other OTUs in the same bacterial family, the correlation between the abundance of these bacterial families and bowel movement frequency was likely not a result of the abundance of these specific OTUs but of the combined abundance of all of the OTUs in the families. A similar relationship with bowel movement frequency was found for Rikenellaceae: however, no OTUs belonging to this family were correlated with bowel movement frequency.
Conclusions
In the present study, we used a 16S metagenomics method to characterize the fecal microbiota of 516 healthy Japanese adults living in various regions of Japan. Based on the bacterial family composition of the fecal microbiota, subjects’ region of residence and gender were not strongly correlated with the general composition of the fecal microbiota. Clustering analysis showed that the enterotype partitioning was ambiguous in the whole cohort. However, two enterotype-like clusters were observed in the male, but not the female, subjects, suggesting that the composition of the fecal microbiota has a gender-specific component. The bacterial compositions of the microbiota were then compared with scores obtained from a lifestyle questionnaire completed by each subject. Significant correlations were found between bowel movement frequency and the abundances of Christensenellaceae, Mogibacteriaceae, and Rikenellaceae. Furthermore, the abundance of these bacterial families was significantly higher in lean subjects than in obese subjects. The abundances of these families were positively correlated with each other and comprised a correlative network with 14 other bacterial families. Together, our results present an overview of the microbial composition of the fecal microbiota of healthy Japanese adults residing in Japan and implied that the abundances of Christensenellaceae, Mogibacteriaceae, and Rikenellaceae contributed subject’s high bowel movement frequency and lean phenotype together with those of some other bacterial components comprising correlative network with them.
Acknowledgments
The authors would like to thank Mutsumi Nakamura, Yoshiko Benno, and Jong-Sik Jin of the Benno Laboratory, RIKEN Innovation Center, for collecting the fecal samples and questionnaire data. We would also like to thank Junji Fujimoto, Kazunori Matsuda, Takuya Akiyama, and Agata Gawad of the Yakult Honsha European Research Center ESV as well as Takahiro Matsuki of the Yakult Central Institute for their invaluable suggestions regarding this study, and Yasuhisa Shimakawa and Toru Odani of the Yakult Central Institute for their support and encouragement during the course of these research activities.
Funding
None.
Availability of data and materials
The 16S metagenomic sequences determined in this study (accession number DRA005068) and the representative sequences assigned to the OTUs (accession numbers LC180363-LC183482) have been deposited in the DNA Data Bank of Japan. All data supporting the results of our study are included within the article and Additional file 1.
Authors’ contributions
KO contributed to designing the study, conducted most of the experiments, analyzed and interpreted the data, wrote the manuscript, and is the corresponding author. MT collected the fecal samples and extracted the fecal DNA, and collected the questionnaire data. TB conducted part of the statistical analysis and interpretation of the questionnaire data. OC and YB oversaw the study, supervised the project, and reviewed the manuscript. KW oversaw the study, contributed to the designing of the study, interpreted the data, and edited the manuscript. All authors read and approved the final manuscript.
Authors’ information
Present address of KO: Yakult Honsha European Research Center for Microbiology ESV, Technologiepark 4, 9052 Zwijnaarde, Belgium.
Present address of KW: Department of Animal Science and Technology, National Taiwan University, No. 50, Lane 155, Sec 3, Keelung Rd., Taipei 10673, Taiwan, R.O.C.
Competing interests
Kaihei Oki and Osamu Chonan are employed by Yakult Honsha European Research Center for Microbiology (East Flanders, Belgium) and Yakult Central Institute (Tokyo, Japan), respectively, which are institutions affiliated with Yakult Honsha Co., Ltd (Tokyo, Japan). Koichi Watanabe was also employed by Yakult Central Institute at the time this study was conducted, but he has subsequently left the company. There are no patents, products in development, or marketed products to declare. This does not alter our adherence to all of BioMed Central’s policies on sharing data and materials.
Consent for publication
Written informed consents for publication were also obtained from all of the volunteers who provided fecal samples for this study.
Ethics approval and consent to participate
This study was approved by the ethics committee of RIKEN Research, Wako, Saitama (Reference number: Wako 3 27-22). Written informed consents for participation were obtained from all of the volunteers who provided fecal samples for this study. We entered in the database and analyzed all the fecal sample and questionnaire data anonymously and are publishing the data anonymously by using personal numbers. The methods were carried out in accordance with all relevant guidelines.
Additional files
English translation of the lifestyle questionnaire and the scoring system used for each question. Table S2. Subject characteristics and the results of the Illumina sequencing. Table S3. Operational taxonomic unit table for the whole subjects (% in whole reads). Table S4. Basic Local Alignment Search Tool (BLAST) results and representative sequences for the operational taxonomic units. Table S5. Abundance and prevalence of the 66 identified bacterial families and alpha-diversity scores stratified by area of residence. Table S6. Abundance and prevalence of the 66 identified bacterial families and alpha-diversity scores stratified by gender. Table S7. Abundance and prevalence of the 66 identified bacterial families and alpha-diversity scores in the two clusters identified in the male subjects. Table S8. Raw questionnaire data. Table S9. Questionnaire scores stratified by area of residence. Table S10. Questionnaire scores stratified by gender. Table S11. Questionnaire scores in the two clusters identified in the male subjects. Table S12. Detailed Basic Local Alignment Search Tool (BLAST) search results for the operational taxonomic units shown in Table 8. Table S13. Quantitative Insights Into Microbial Ecology (QIIME) bioinformatics pipeline scripts used in the present study. (XLSX 8232 kb)
Detailed methods for the Illumina sequencing and microbiological analyses. (DOCX 36 kb)
Distribution of subjects’ genders stratified by area of residence. Gender ratios of female to male represent in brackets. Among the areas sharing the same letter above their bar graphs, significant difference was not observed (P ≥ 0.05) in the gender ratio. (PPTX 144 kb)
Contributor Information
Kaihei Oki, Phone: +32 9 241 1134, Email: kaihei.oki@yher.be.
Mutsumi Toyama, Email: touyama0101@riken.jp.
Taihei Banno, Email: taihei.banno@riken.jp.
Osamu Chonan, Email: osamu-chonan@yakult.co.jp.
Yoshimi Benno, Email: benno828@riken.jp.
Koichi Watanabe, Email: koichi_wtnb@yahoo.co.jp.
References
- 1.Guarner F, Malagelada J-R. Gut flora in health and disease. Lancet. 2003;360:512–519. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- 2.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 3.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinross JM, Darzi AW, Nicholson JK. Gut microbiome-host interactions in health and disease. Genome Med. 2011;3:14. doi: 10.1186/gm228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende D, et al. Enterotypes of the human gut microbiome. Nature. 2011;273:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flak MB, Neves JF, Blumberg RS. Immunology. Welcome to the microgenderome. Science. 2013;339:1044–1045. doi: 10.1126/science.1236226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Guo Z, Lim AAQ, Zheng Y, Koh EY, Ho D, et al. Mongolians core gut microbiota and its correlation with seasonal dietary changes. Sci Rep. 2014;4:5001. doi: 10.1038/srep05001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Odamaki T, Kato K, Sugahara H, Hashikura N, Takahashi S, Xiao J-Z, et al. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 2016;16:90. doi: 10.1186/s12866-016-0708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakayama J, Watanabe K, Jiang J, Matsuda K, Chao S-H, Haryono P, et al. Diversity in gut bacterial community of school-age children in Asia. Sci Rep. 2015;5:8397. doi: 10.1038/srep08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.La-Ongkham O, Nakphaichit M, Leelavatcharamas V, Keawsompong S, Nitisinprasert S. Distinct gut microbiota of healthy children from two different geographic regions of Thailand. Arch Microbiol. 2015;197:561–573. doi: 10.1007/s00203-015-1089-0. [DOI] [PubMed] [Google Scholar]
- 15.Japan ministry of Health, Labour and Welfare. National health and nutrition survey in 2014. http://www.e-stat.go.jp/SG1/estat/GL08020103.do?_toGL08020103_&listID=000001151595&requestSender=estat. Accessed 22 Nov 2016.
- 16.Jin JS, Touyama M, Kibe R, Tanaka Y, Benno Y, Kobayashi T, et al. Analysis of the human intestinal microbiota from 92 volunteers after ingestion of identical meals. Benef Microbes. 2013;4:187–193. doi: 10.3920/BM2012.0045. [DOI] [PubMed] [Google Scholar]
- 17.Radloff LS. The CES-D Scale: A self-report depression scale for research in the general population. Appl Psychol Meas. 1977;1:385–401. doi: 10.1177/014662167700100306. [DOI] [Google Scholar]
- 18.Caporaso J, Kuczynski J, Stombaugh J, Bittinger K, Bushman F, Costello E, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, et al. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007;35:D169–D172. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The R foundation. The R Project for Statistical Computing. https://www.r-project.org/. Accessed 22 Nov 2016.
- 22.Caliński T, Harabasz J. A dendrite method for cluster analysis. Commun Stat Methods. 2007;3:1–27. doi: 10.1080/03610927408827101. [DOI] [Google Scholar]
- 23.Koren O, Knights D, Gonzalez A, Waldron L, Segata N, Knight R, et al. A guide to enterotypes across the human body: Meta-analysis of microbial community structures in human microbiome datasets. Eisen JA, editor. PLoS Comput Biol. 2013;9:e1002863. doi: 10.1371/journal.pcbi.1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tibshirani R, Walther G. Cluster validation by prediction strength. J Comput Graph Stat. 2005;14:511–528. doi: 10.1198/106186005X59243. [DOI] [Google Scholar]
- 25.Rousseeuw PJ. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J Comput Appl Math. 1987;20:53–65. doi: 10.1016/0377-0427(87)90125-7. [DOI] [Google Scholar]
- 26.Felis GE, Dellaglio F. Taxonomy of lactobacilli and bifidobacteria. Curr Issues Intest Microbiol. 2007;8:44–61. [PubMed] [Google Scholar]
- 27.Ash C, Farrow JAE, Wallbanks S, Collins MD. Phylogenetic heterogeneity of the genus Bacillus revealed by comparative analysis of small-subunit-ribosomal RNA sequences. Lett Appl Microbiol. 2008;13:202–206. doi: 10.1111/j.1472-765X.1991.tb00608.x. [DOI] [Google Scholar]
- 28.Knights D, Ward TL, McKinlay CE, Miller H, Gonzalez A, McDonald D, et al. Rethinking “enterotypes”. Cell Host Microbe. 2014;16:433–437. doi: 10.1016/j.chom.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallis A, Butt H, Ball M, Lewis DP, Bruck D. Support for the microgenderome: Associations in a human clinical population. Sci Rep. 2016;6:19171. doi: 10.1038/srep19171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Özer BH, Kirmaci HA. Functional milks and dairy beverages. Int J Dairy Technol. 2010;63:1–15. doi: 10.1111/j.1471-0307.2009.00547.x. [DOI] [Google Scholar]
- 31.Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 32.Coudeyras S, Marchandin H, Fajon C, Forestier C. Taxonomic and strain-specific identification of the probiotic strain Lactobacillus rhamnosus 35 within the Lactobacillus casei group. Appl Environ Microbiol. 2008;74:2679–2689. doi: 10.1128/AEM.02286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu J, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, Dekens JAM, et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ Res. 2015;117:817–824. doi: 10.1161/CIRCRESAHA.115.306807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanjoaquin MA, Appleby PN, Spencer EA, Key TJ. Nutrition and lifestyle in relation to bowel movement frequency: a cross-sectional study of 20 630 men and women in EPIC–Oxford. Public Health Nutr. 2007;7:77–83. doi: 10.1079/phn2003522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The 16S metagenomic sequences determined in this study (accession number DRA005068) and the representative sequences assigned to the OTUs (accession numbers LC180363-LC183482) have been deposited in the DNA Data Bank of Japan. All data supporting the results of our study are included within the article and Additional file 1.