

Abstract
Lignin is an attractive renewable feedstock for aromatic bulk and fine chemicals production, provided that suitable depolymerization procedures are developed. Here, we describe a tandem catalysis strategy for ether linkage cleavage within lignin, involving ether hydrolysis by water‐tolerant Lewis acids followed by aldehyde decarbonylation by a Rh complex. In situ decarbonylation of the reactive aldehydes limits loss of monomers by recondensation, a major issue in acid‐catalyzed lignin depolymerization. Rate of hydrolysis and decarbonylation were matched using lignin model compounds, allowing the method to be successfully applied to softwood, hardwood, and herbaceous dioxasolv lignins, as well as poplar sawdust, to give the anticipated decarbonylation products and, rather surprisingly, 4‐(1‐propenyl)phenols. Promisingly, product selectivity can be tuned by variation of the Lewis‐acid strength and lignin source.
Keywords: biomass, decarbonylation, lewis acid, lignin, tandem catalysis
Lignin is the only aromatic biomass component of substantial abundance and an ideal candidate for renewable aromatic bulk and fine chemicals production.1 Native lignins consist primarily of syringyl (S), guaiacyl (G), and p‐hydroxyphenyl (H)‐containing units,2 with structure varying with plant species and isolation method: grasses and softwoods typically are rich in H and/or G and hardwoods are rich in S. Different linkages are found in the macromolecule, of which up to 60 % are β‐O‐4 ethers.3 Isolation invariably changes the structure, with new carbon–carbon bonds forming by recondensation at the expense of the ether bonds (β‐O‐4 in particular), resulting in lignins more recalcitrant to further upgrading.2a Exactly this propensity to structural change, compounded by the structural complexity of lignin, hampers targeted (catalytic) lignin depolymerization, making its efficient conversion to a small number of valuable products extremely challenging. Some selective depolymerization strategies have nonetheless been reported,4 methods which typically rely on (excess) stoichiometric reagents to form stable end products before significant recondensation can occur. In addition, such strategies are often developed on lignin model compounds and translating the chemistry to actual lignins can prove particularly difficult.
Illustrative of the challenges listed above is lignin ether cleavage by acidolysis, one of the oldest catalytic methods for lignin depolymerization,5 typically giving low yields of monomeric species. Indeed, acid‐induced deformylation and hydrolysis of a β‐O‐4 ether result in styryl ether and aldehyde formation, both of which are highly susceptible to aldol condensation, affording higher molecular weight products (Figure 1).6 Only the fraction of the ethers that does not undergo deformylation gives rise to a mixture of the relatively stable Hibbert's ketones.7 Deuss et al. recently demonstrated that loss of monomers to recondensation could be limited by stoichiometrically trapping intermediate aldehydes formed in triflic acid‐catalyzed lignin depolymerization,8 for instance by acetalization with 1,2‐ethanediol. Preferably, a trapping strategy does not use sacrificial trapping reagents, is catalytic, and yields valuable aromatic monomers. Catalytic aldehyde decarbonylation is particularly promising in this respect, but also highly challenging with only two known systems showing reasonable efficiency.9 Decarbonylation was previously also attempted in the triflic acid‐catalyzed depolymerization, but only with limited success: whereas the decarbonylation worked on model compounds, the decarbonylation rate could not compete effectively with the condensation side‐reactions during actual lignin depolymerization.8 Another recent example of a trapping strategy made use of a combination of a Lewis acid metal triflate and Pd/C, with the latter catalyst serving to hydrogenate reactive intermediates formed by Lewis acid‐catalyzed elimination reactions.10 However, this method was not applied to actual lignin.
Figure 1.
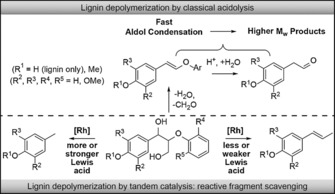
Lewis acid‐catalyzed cleavage of lignin, here represented as β‐O‐4 fragments, combined with Rh‐catalyzed decarbonylation prevents loss of monomers to recondensation.
We here report an innovative and highly efficient tandem catalytic approach involving lignin hydrolysis/decarbonylation (Figure 1), using water‐tolerant Lewis acids [M(OTf)3, M=Sc, Y, In, Ga, and Yb].11 Rh‐catalyzed decarbonylation then effectively scavenges the aldehyde hydrolysis products to afford the desired decarbonylated 4‐methylphenols in reactions with model compounds, various dioxasolv lignins, as well as poplar sawdust. Remarkably, a second alternative trapping pathway is observed with the actual lignins, leading instead to the formation of 4‐(1‐propenyl)phenols (Figure 1), which are attractive high‐value/low‐volume lignin products. Crucially, we show that the trapping pathway can be controlled by variation of the Lewis‐acid strength. The tandem catalytic reaction was first developed on β‐O‐4 model compounds with Sc(OTf)3 as the Lewis acid12 in combination with an in situ prepared [Rh(cod)Cl]2/dppp decarbonylation catalyst (cod=1,5‐cyclooctadiene, dppp=bis‐1,3‐(diphenylphosphino) propane).9a A 9:1 mixture of 1,4‐dioxane and water as the solvent ensured good hydrolysis activity, without the Rh catalyst being negatively impacted by the excess water. Cleavage of the simple model compound 1 a (Table 1, entry 1) gave significant amounts of the expected products phenol and toluene at 200 °C. Phenol is liberated by hydrolysis of the intermediate enol ether and toluene is subsequently obtained by decarbonylation of 2‐phenylacetaldehyde (6 a). Although the latter intermediate was not observed in detectable amounts, 2‐phenylethanol was found (12 %), as well as dehydrogenated (α‐ketone) starting material, suggesting that catalytic transfer hydrogenation is also possible if an appropriate hydrogen donor is available.13 With only the Lewis acid, full conversion was observed with a reasonable phenol yield, but neither toluene nor 6 a could be detected. Instead, higher molecular weight (Mw) stilbenes and 2‐phenylnaphthalene (7 a) were found instead (Table 1, entry 2). With Sc(OTf)3 also being an excellent aldol condensation catalyst,14 7 a is likely formed by self‐condensation of 6 a, followed by intramolecular cyclization. These results clearly demonstrate the value of the tandem approach, with rapid removal of the reactive aldehydes by decarbonylation preventing undesired side reactions.
Table 1.
Results of the tandem catalytic cleavage of β‐O‐4 model compounds 1 a–1 d and major products observed.[a]
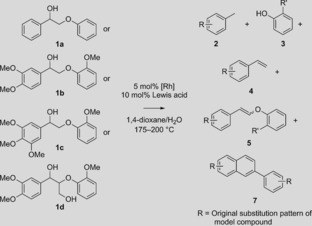
| Entry | β‐O‐4 | LA[b] | T | Conv.[c] | Product yield [%] | ||||
|---|---|---|---|---|---|---|---|---|---|
| model | [mol %] | [°C] | [%] | 2 | 3 | 4 | 5 | 7 | |
| 1 | 1a | 10 | 200 | 57 | 24 | 47 | – | 2 | – |
| 2 | 1a | 10[d] | 200 | 100 | ‐ | 63 | – | – | 9 |
| 3 | 1b | 10 | 200 | 96 | 65 | 77 | – | – | 1 |
| 4 | 1b | 10 | 175 | 99 | 37 | 81 | – | – | 17 |
| 5 | 1b | 5 | 175 | 99 | 66 | 85 | – | – | 4 |
| 6 | 1b | 2.5 | 175 | 92 | 36 | 41 | 3 | 44 | – |
| 7 | 1b | 5[e] | 175 | 99 | 5 | 84 | – | – | 31 |
| 8 | 1b | 2.5[e] | 175 | 98 | 45 | 61 | – | 25 | – |
| 9 | 1c | 5 | 175 | 97 | 67 | 71 | 3 | 4 | – |
| 10 | 1c | 2.5 | 175 | 39 | 7 | 12 | 8 | 16 | – |
| 11 | 1d | 5 | 175 | 100 | 51 | 88 | – | – | 3 |
[a] Conditions: 2.4 mmol substrate, 2.5 mol % [Rh(cod)Cl]2, 2:1 dppp/Rh, Sc(OTf)3 [or In(OTf)3] as listed; 175–200 °C, 2 h, 22 mL 9:1 1,4‐dioxane/water; yields determined by GC. [b] Lewis acid. [c] Conversion (%). [d] Only Sc(OTf)3, no [Rh(cod)Cl]2. [e] In(OTf)3.
Model compounds 1 b–1 d allowed the influence of additional functional groups to be assessed. Cleavage of G–G model 1 b thus afforded both 4‐methylveratrole (2 b) and guaiacol (3 b) in good yield (Table 1, entry 3; Figure 2). Interestingly, 1 b could also be readily cleaved at 175 °C; the high guaiacol yield (81 %) at this temperature indicated efficient hydrolysis, but the much lower yield of 4‐methylveratrole (2 b, 38 %) showed that the decarbonylation catalyst cannot keep up with aldehyde formation. Homoveratryl aldehyde (6 b) was observed in about 1 % yield. With decarbonylation being too slow, higher molecular weight species were formed, such as homoveratryl aldehyde‐derived 7 b (Scheme 1).15 Indeed, it is imperative that the kinetics of hydrolysis and decarbonylation are carefully matched. Lowering the amount of Sc(OTf)3 (Table 1, entries 4–6), as expected, resulted in an increase in 4‐methylveratrole (2 b) yield to 66 %, with a concomitant drop in condensation products. Such an effect was also observed in the tandem catalytic system employed by Marks and coworkers, where using a weaker Lewis acid enabled the hydrogenation catalyst to remove reactive intermediates more effectively, leading to higher yields.10 Further lowering the Sc(OTf)3 loading (entry 6) proved very revealing mechanistically, with cis and trans enol ether intermediates (5 b) being detected, the latter being predominant.16 The time profile of 1 b with 2.5 mol % Sc(OTf)3 indeed shows that 5 b is an intermediate en route to decarbonylation (Figure 2). That homoveratryl aldehyde (6 b) and condensation product 7 b are hardly detected, demonstrates the efficient coupling of the tandem reaction. The subtle interplay between the Lewis acid and Rh catalyst is evidenced by the stronger acid In(OTf)3 (Table 1, entries 7 and 8), which shows faster hydrolysis, but also accelerated aldehyde condensation at the expense of decarbonylation. Notably, small amounts (ca. 3 %) of 3,4‐dimethoxystyrene (4 b) were also observed on treatment of 1 b with 2.5 mol % Sc(OTf)3 (entry 6, see below for further discussion). The S–G model 1 c also afforded excellent conversion and good guaiacol (3 b) and 3,4,5‐trimethoxytoluene (2 c) yields with 5 mol % Sc(OTf)3 at 175 °C (entry 9). At only 2.5 mol % Sc(OTf)3 (entry 10), conversion was only 39 %, with only 7 % yield of 3,4,5‐trimethoxytoluene (2 c) and a 16 % yield of the enol ethers 5 c. Again, the formation of 8 % of a styrene derivative (3,4,5‐trimethoxystyrene, 4 c) was observed.
Figure 2.
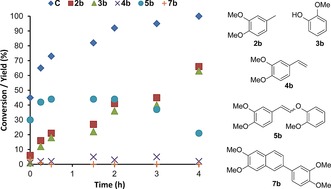
The reaction profile of 1 b as function of time [determined through individual reactions; 2.5 mol % Sc(OTf)3] shows the accumulation and subsequent decrease of 5 b as an intermediate. C=conversion.
Scheme 1.
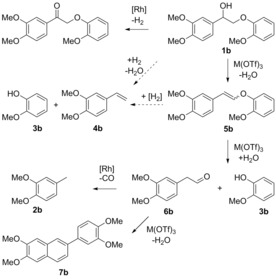
Proposed mechanism for the cleavage of β‐O‐4 models and possible pathways to styrene formation.
The formation of the styrene derivatives 4 is remarkable and has, to the best of our knowledge, not yet been reported. Whether the styrenes 4 b and 4 c are formed directly from the β‐O‐4 models or from the enol ethers is unclear at this point; nonetheless there must be a formal hydrogenolysis step involved in the mechanism. The alternative of styrene formation by transfer hydrogenation of the aldehyde (6 b/6 c), followed by acid‐catalyzed alcohol dehydration would not be in line with lower acid concentrations favoring styrene formation. Model compound 1 d (erythro/threo mixture) mimics the full glyceryl β‐O‐4 backbone and was also cleaved effectively (entry 11), albeit with slightly lower yields than with 1 b. Deformylation, as observed before during lignin model compound acidolysis,6 is thought to precede hydrolysis and decarbonylation. The mechanistic insights provided by the model compounds are summarized in Scheme 1. Initial Lewis acidcatalyzed dehydration of the α‐hydroxyl functionality in 1 b forms an isomeric mixture of enol ethers 5 b, followed by a slower Lewis acid‐catalyzed hydrolysis step to the corresponding acetaldehyde 6 b. A sufficiently active decarbonylation catalyst then gives the corresponding methyl‐substituted product 2 b, whilst in the absence thereof aldol condensation takes place instead to give 7 b. For the first time, an alternative pathway is also observed, tentatively originating from the enol ether 5 b, leading to the formation of a styrene derivative 4 b by insertion of a formal hydrogen equivalent.
The tandem strategy was then translated to actual lignin. With maximum monomer yield being approximately quadratic with the fraction of cleavable bonds,4c,4d a poplar sawdust dioxasolv lignin was isolated to ensure a relatively high abundance of the targeted β‐O‐4 linkage.4d Dioxasolv pulping furthermore ensures that 1) the α‐OH remains available for initial dehydration and 2) the isolated lignin will actually be soluble in (wet) 1,4‐dioxane, the solvent for the tandem reaction. The Mw was determined to be 3.0×103 Da by gel permeation chromatography (GPC). HSQC NMR analysis (Figure S1) showed 39 β‐O‐4, 3 β‐5, and 5 β‐β linkages per 100 aromatic units, with a S/G/H ratio of 2.1:1.0:0.0. In addition, 16 per 100 aromatic units of p‐hydroxybenzoate units were observed. This value is typical for poplar,17 but the actual content is likely lower as such units are systematically overestimated by 2 D NMR methods. Interestingly, Hibbert's ketone structures could be detected in the starting lignin, in line with previous organosolv lignin analyses.2b, 18
Generally, no char formation was observed and a very limited number of volatile products detected (Figure 3). Two of the major products were indeed the anticipated decarbonylation products, 4‐methylguaiacol (8 a) and 4‐methylsyringol (9 a), derived from S and G units in the lignin, showing that the model compound chemistry translates well to actual lignin. Surprisingly, significant quantities of iso‐eugenol (8 b) and 4‐(1‐propenyl)syringol (9 b) were also found, proposed to form analogously to the styrene derivatives 4 seen above for the β‐O‐4 model compounds. Formation of such compounds has been observed previously in reactive organosolv pulping, although they could not be obtained from isolated lignin.19 Compounds identified in smaller amounts included 4‐ethylguaiacol (8 d), 4‐ethylsyringol (9 d), (4‐guaiacyl)acetone (8 c), and (4‐syringyl)acetone (9 c). A potential mechanism of formation for the latter two may involve (transfer) hydrogenolysis of the terminal γ‐hydroxyl of the original Hibbert's ketone. Combined, all identified monomers comprised 9.5 wt % or approximately 12 mol % of the original lignin intake. Given a β‐O‐4 content of 39 %, which puts the actual maximum monomer yield at 20 mol %, the monomer yield thus corresponds to 50 % of the theoretical yield. GPC and HSQC NMR analysis of the reaction mixture (Figure S1–2, Table S1) showed the Mw drop to 7.7×102 Da and all β‐O‐4 bonds to be successfully cleaved. Signals for the β‐5 fragment also disappeared and the amount of β‐β linkages was also significantly reduced, suggesting that at least the ether linkages in these fragments are susceptible to cleavage as well. No β‐β linkage epimerization was observed.20 As a control experiment, lignin depolymerization without the Rh catalyst yielded no monomers, but a large amount of dark precipitate. With only the Rh complex, some monomers were observed, albeit in very small amounts (Table 2, entry 1). Possibly the Rh complex is also able to act as (weak) Lewis acid providing some hydrolysis activity.21
Figure 3.
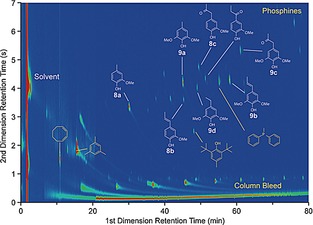
GC × GC–MS chromatogram of the volatile components after reaction {300 mg poplar dioxasolv lignin, 0.060 mmol [Rh(cod)Cl]2, 0.240 mmol dppp, 0.120 mmol (ScOTf)3; 175 °C, 2 h, 22 mL 1,4‐dioxane/water}. Lignin‐derived products in white; non‐lignin derived compounds in yellow.
Table 2.
Lignin depolymerization with different (amounts of) Lewis acids.
| Entry | Lewis acid | Monomers 8 and 9 yield [wt %] | Sum[b] | ||||
|---|---|---|---|---|---|---|---|
| type | amt [mmol] | a | b | c | d | [wt %] | |
| 1 | – | – | 0.2 | 1.4 | 0.0 | 0.1 | 2.6 |
| 2 | HOTf | 0.360 | 2.5 | 5.8 | 1.1 | 0.6 | 10.6 |
| 3 | Yb(OTf)3 | 0.120 | 3.2 | 5.6 | 1.3 | 0.7 | 11.4 |
| 4 | Sc(OTf)3 | 0.120 | 4.2 | 2.7 | 1.8 | 0.5 | 9.5 |
| 5 | In(OTf)3 | 0.120 | 5.0 | 0.6 | 2.7 | 1.0 | 9.7 |
| 6 | Ga(OTf)3 | 0.120 | 5.1 | 0.3 | 3.3 | 0.3 | 9.4 |
| 7 | Sc(OTf)3 | 0.240 | 4.2 | 0.8 | 2.2 | 0.4 | 7.9 |
| 8 | Sc(OTf)3 | 0.060 | 2.2 | 7.3 | 0.8 | 0.7 | 11.8 |
| 9 | Sc(OTf)3 | 0.030 | 1.2 | 9.0 | 0.4 | 0.5 | 12.1 |
| 10 | Yb(OTf)3 | 0.030 | 0.9 | 8.6 | 0.4 | 0.6 | 11.2 |
| 11 | Yb(OTf)3 [c] | 0.030 | 1.2 | 9.2 | 0.4 | 0.9 | 12.4 |
[a] Conditions: 300 mg poplar dioxasolv lignin, 0.060 mmol [Rh(cod)Cl]2, 0.240 mmol dppp, acid; 175 °C, 2 h, 22 mL 1,4‐dioxane/water (9:1); yields determined by GC. [b] Sum of all identified monomers (Table S5–13). [c] 4 h reaction.
The Lewis acids Yb(OTf)3, In(OTf)3 and Ga(OTf)3 were also investigated, as was triflic acid (3 equiv. compared to the metal triflates). Whereas the GPC traces of these reactions proved rather similar, a comparison of the monomer yield revealed some interesting trends (Table 2, entries 2–6). Yb(OTf)3 gave a relatively low amount of decarbonylation products (8 a, 9 a), but a relatively large amount of 4‐(1‐propenyl)phenols (8 b, 9 b), with a high total amount of monomers. Conversely, with In(OTf)3 or Ga(OTf)3, the selectivity was found to be the opposite. Stronger Lewis acids, as expressed by their hydrolysis constants (pKh),21 thus give more decarbonylation products, whereas the weaker acids favor 4‐(1‐propenyl)phenol formation (Figure 4 a). The differences seen with the triflic acid run, showing an even greater preference for the formation of 4‐(1‐propenyl)phenols, demonstrate that the metal triflates do not simply act as precursors to Brønsted acidity by metal triflate hydrolysis.
Figure 4.
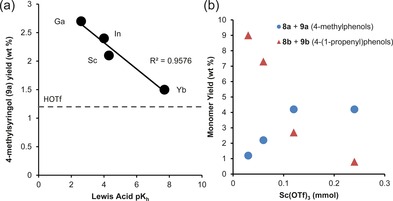
(a) Lewis acid strength21 correlates with 4‐methylsyringol (9 a) yields; (b) yields of 4‐methylphenols versus 4‐(1‐propenyl)phenols as a function of Sc(OTf)3 loading, demonstrating the ability to tune product distribution.
Likewise, variation of the amount of Lewis acid also allowed control over selectivity (Table 2, entries 4 and 7–9). With larger amounts of Sc(OTf)3, the relative yield of decarbonylation products was highest, while with lower Sc(OTf)3 concentrations the 4‐(1‐propenyl)phenols were again formed as the major products (Figure 4 b). This is in line with the model compound studies (Table 1) and suggests that for both lignin and the model compounds the formation of the decarbonylation products proceeds through an identical mechanism. Increased decarbonylation activity with more Lewis acid or a stronger one is then the result of faster enol ether hydrolysis. In turn, a weak acid or low loading favors a competitive (reductive) mechanism that leads to formation of the 4‐(1‐propenyl)phenols. The selectivity differences between the guaiacyl and syringylderived products are also in line with the model compound studies: the former tend to form decarbonylation products more rapidly, whilst the latter form more 4‐(1‐propenyl)phenols. Maximum yields of decarbonylation products (5.1 wt %) are obtained with the strongest Lewis acid gallium triflate. On the other hand, using only 0.030 mmol of scandium triflate the 4‐(1‐propenyl)phenol yield is the highest, with iso‐eugenol (8 b) and 4‐(1‐propenyl)syringol (9 b) being obtained instead in an overall yield of 9.0 wt %. Using a lower amount of the weakest Lewis acid [Yb(OTf)3; Table 2, entry 11], led both the highest yield of 4‐(1‐propenyl)phenols (8 b and 9 b, total yield of 9.2 wt %) as well as the highest overall monomer yield (12.4 wt %) after 4 h of reaction.
The tandem approach was also successfully applied to dioxasolv lignins isolated from brewer's spent grain (BSP) and pine sawdust, representative for grasses and softwoods, respectively. HSQC NMR analysis of the BSP lignin revealed a β‐O‐4 content of 42 per 100 aromatic units, an S/G/H ratio of 0.5:1.0:0.1 as well as p‐coumarate, ferulate, and tricin units, common in grasses (Figure S4, Table S2).17 Incomplete solubility in THF precluded GPC analysis for this lignin. The amount of monomers formed with BSP lignin was lower than with poplar lignin and some dark precipitate was detected. In line with its higher G content, a larger amount of guaiacyl‐substituted products were observed (Table 3, entry 1). HSQC analysis of the pine lignin revealed it to consist exclusively of G units, with 29 β‐O‐4, 12 β‐5, and 4 β‐β linkages per 100 aromatic units (Figure S6, Table S3). Correspondingly, after reaction only guaiacyl‐derived products are observed, further decreasing the complexity of the product mixture. The lower total monomer yield is in line with the lower β‐O‐4 content. On the other hand, at 5.1 wt % of 4‐methylguaiacol (8 a), the highest single product yield for a decarbonylation product is observed with this lignin. Consequently, lignin depolymerization selectivity can be tuned in two dimensions (Figure 5): by selection of a biomass source with an appropriate S/G ratio and by selection of the appropriate (amount of) Lewis acid catalyst.
Table 3.
Results of the depolymerization of other lignins and sawdust.[a]
| Entry | Substrate | Lewis acid | Monomer yield [wt %] | |||||
|---|---|---|---|---|---|---|---|---|
| type | amt [mmol] | 8 a | 9 a | 8 b | 9 b | Sum[b] | ||
| 1 | BSP | Sc(OTf)3 | 0.120 | 1.7 | 0.7 | 0.4 | 0.8 | 5.2 |
| 2 | pine | Sc(OTf)3 | 0.120 | 5.1 | 0.0 | 0.6 | 0.0 | 6.8 |
| 3 | sawdust | Sc(OTf)3 | 0.120 | 2.1 | 1.4 | 0.7 | 2.9 | 9.8 |
| 4 | sawdust | Ga(OTf)3 | 0.120 | 2.4 | 1.6 | 0.3 | 1.4 | 7.1 |
| 5 | sawdust[c] | Yb(OTf)3 | 0.030 | 0.0 | 0.1 | 1.6 | 3.0 | 7.6 |
[a] Conditions: 300 mg lignin or 2.0 g poplar sawdust, 0.060 mmol [Rh(cod)Cl]2, 0.240 mmol dppp, acid; 175 °C, 2 h, 22 mL 1,4‐dioxane/water. [b] Sum of all identified monomers (see Tables S14–18 for complete listing). [c] 4 h reaction.
Figure 5.
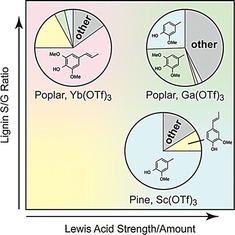
Three protypical reactions demonstrate the ability to tune the product distribution of lignin depolymerization. Lignins with an appropriate S/G ratio affords either S or G (9 or 8) products, whereas the choice of Lewis acid adjusts the selectivity between methylphenols (a) or 1‐propenylphenols (b).
Reactive lignin upgrading directly on the biomass has recently gained interest,18, 22 as this avoids the generation of more recalcitrant lignins by recondensation.22a Indeed, high yields of small aromatics have been obtained with this ‘lignin‐first’ approach.22 Our reaction was therefore also performed directly on ball‐milled poplar sawdust (Klason lignin 22.6 wt %). Gratifyingly, under standard conditions using 2.0 g sawdust, lignin depolymerization products identical to those from the isolated lignins were obtained, as well as sugar‐derived furfural and hydroxymethylfurfural. The monomer yield obtained with the whole biomass (accounting for the lignin content) is comparable to that observed for the isolated lignin (Table 3, entries 3–5). This is somewhat surprising as, based on the higher amount of β‐O‐4 linkages in the unprocessed biomass, one would expect larger amounts of liberated monomers. The similar monomer yield might be a result of condensation reactions of the aromatics with (liberated) sugar compounds. Likewise, reducing sugars may be competitive substrates, inhibiting the decarbonylation reaction. Nonetheless, in line with the results on isolated lignin, changing the Lewis acid co‐catalyst again allowed the product distribution to be tuned (Table 3, entries 3–5): when using Ga(OTf)3 predominantly the decarbonylation products 4‐methylguaiacol (8 a) and 4‐methylsyringol (9 a) were observed; whereas Yb(OTf)3 gave a larger amount of the 4‐(1‐propenyl)phenols.
In conclusion, an efficient tandem catalysis approach for cleavage of β‐O‐4 fragments in lignin models and (isolated) lignins has been developed. This strategy addresses the key challenge of loss of yield of mono‐aromatics owing to lignin recondensation. Building on the mechanistic insight provided by the model compound studies, the anticipated decarbonylation products could be obtained in good yields when the rate of both reactions was appropriately matched. Importantly, the selectivity towards either 4‐methylphenols or 4‐(1‐propenyl)phenols can be controlled by varying the amount and strength of the Lewis acid catalyst. By using a lignin with an appropriate S/G ratio and selection of the right Lewis acid, each of the four major products can then be specifically targeted. Trapping of reactive intermediates in catalytic lignin depolymerization is thus shown to be a very versatile approach. Further detailed mechanistic studies on the remarkable cleavage of the β‐O‐4 aryl alkyl ethers to 4‐(1‐propenyl)phenols are currently under way.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
P.C.A.B. thanks the Netherlands Organization for Scientific Research (NWO) for a Vernieuwingsimpuls Veni Grant. The CatchBio program is acknowledged for financial support. N.J.W. thanks EPSRC for funding by the EP/J018139/1, EP/K00445X/1 grants and an EPSRC Doctoral Prize Fellowship to C.S.L. Saumya Dabral and Prof. Carsten Bolm (RWTH Aachen) are gratefully acknowledged for providing an authentic sample of 5 b. Dr. Wouter Huijgen (ECN) is acknowledged for Klason lignin determination. SuBiCat Initial Training Network, Call FP7‐PEOPLE‐2013‐ITN, #607044) is gratefully acknowledged.
R. Jastrzebski, S. Constant, C. S. Lancefield, N. J. Westwood, B. M. Weckhuysen, P. C. A. Bruijnincx, ChemSusChem 2016, 9, 2074.
References
- 1.
- 1a. Zakzeski J., Bruijnincx P. C. A., Jongerius A. L., Weckhuysen B. M., Chem. Rev. 2010, 110, 3552–3599; [DOI] [PubMed] [Google Scholar]
- 1b. Ragauskas A. J., Beckham G. T., Biddy M. J., Chandra R., Chen F., Davis M. F., Davison B. H., Dixon R. A., Gilna P., Keller M., Langan P., Naskar A. K., Saddler J. N., Tschaplinski T. J., Tuskan G. A., Wyman C. E., Science 2014, 344, 1246843; [DOI] [PubMed] [Google Scholar]
- 1c.R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx, B. M. Weckhuysen, Angew. Chem. Int. Ed 2016, DOI: 10.1002/anie.201510351. [DOI] [PMC free article] [PubMed]
- 2.
- 2a. Chakar F. S., Ragauskas A. J., Ind. Crops Prod. 2004, 20, 131–141; [Google Scholar]
- 2b. Constant S., Wienk H. L. J., Frissen A. E., de Peinder P., Boelens R., van Es D. S., Grisel R. J. H., Weckhuysen B. M., Huijgen W. J. J., Gosselink R. J. A., Bruijnincx P. C. A., Green. Chem. 2016, 18, 2651–2665. [Google Scholar]
- 3. Ralph J., Lundquist K., Brunow G., Lu F., Kim H., Schatz P. F., Marita J. M., Hatfield R. D., Ralph S. A., Christensen J. H., Boerjan W., Phytochem. Rev. 2004, 3, 29–60. [Google Scholar]
- 4.
- 4a. Son S., Toste F. D., Angew. Chem. Int. Ed. 2010, 49, 3791–3794; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3879–3882; [Google Scholar]
- 4b. Rahimi A., Ulbrich A., Coon J. J., Stahl S. S., Nature 2014, 515, 249–252; [DOI] [PubMed] [Google Scholar]
- 4c. Feghali E., Carrot G., Thuéry P., Genre C., Cantat T., Energy Environ. Sci. 2015, 8, 2734–2743; [Google Scholar]
- 4d. Lancefield C. S., Ojo O. S., Tran F., Westwood N. J., Angew. Chem. Int. Ed. 2015, 54, 258–262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 260–264; [Google Scholar]
- 4e. Deuss P. J., Barta K., Coord. Chem. Rev. 2016, 306, 510–532. [Google Scholar]
- 5. Adler E., Wood Sci. Technol. 1977, 11, 169–218. [Google Scholar]
- 6.
- 6a. Hoo L. H., Sarkanen K. V., Anderson C. D., J. Wood Chem. Technol. 1983, 3, 223–243; [Google Scholar]
- 6b. Yokoyama T., J. Wood Chem. Technol. 2014, 35, 27–42. [Google Scholar]
- 7. Kulka M., Hibbert H., J. Am. Chem. Soc. 1943, 65, 1180–1185. [Google Scholar]
- 8. Deuss P. J., Scott M., Tran F., Westwood N. J., de Vries J. G., Barta K., J. Am. Chem. Soc. 2015, 137, 7456–7467. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Kreis M., Palmelund A., Bunch L., Madsen R., Adv. Synth. Catal. 2006, 348, 2148–2154; [Google Scholar]
- 9b. Olsen E. P. K., Madsen R., Chem. Eur. J. 2012, 18, 16023–16029. [DOI] [PubMed] [Google Scholar]
- 10. Lohr T. L., Li Z., Marks T. J., ACS Catal. 2015, 5, 7004–7007. [Google Scholar]
- 11.
- 11a. Kobayashi S., Synlett 1994, 689–701; [Google Scholar]
- 11b. Prakash G. K. S., Mathew T., Olah G. A., Acc. Chem. Res. 2012, 45, 565–577; [DOI] [PubMed] [Google Scholar]
- 11c. Atesin A. C., Ray N. A., Stair P. C., Marks T. J., J. Am. Chem. Soc. 2012, 134, 14682–14685; [DOI] [PubMed] [Google Scholar]
- 11d. Yang L., Li Y., Savage P. E., Ind. Eng. Chem. Res. 2014, 53, 2633–2639. [Google Scholar]
- 12. Constant S., Basset C., Dumas C., Di Renzo F., Robitzer M., Barakat A., Quignard F., Ind. Crops Prod. 2015, 65, 180–189. [Google Scholar]
- 13.
- 13a. Hutschka F., Dedieu A., Eichberger M., Fornika R., Leitner W., J. Am. Chem. Soc. 1997, 119, 4432–4443; [Google Scholar]
- 13b. Pàmies O., Bäckvall J.-E., Chem. Eur. J. 2001, 7, 5052–5058. [DOI] [PubMed] [Google Scholar]
- 14. Ishikawa S., Hamada T., Kobayashi S., J. Am. Chem. Soc. 2004, 126, 12236–12237. [DOI] [PubMed] [Google Scholar]
- 15. Brunow G., Lundquist K., Acta Chem. Scand. Ser. B 1984, 38, 323–325. [Google Scholar]
- 16. Dabral S., Mottweiler J., Rinesch T., Bolm C., Green Chem. 2015, 17, 4908–4912. [Google Scholar]
- 17. Kim H., Ralph J., Org. Biomol. Chem. 2010, 8, 576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lancefield C. S., Westwood N. J., Green Chem. 2015, 17, 4980–4990. [Google Scholar]
- 19.
- 19a. Galkin M. V., Sawadjoon S., Rohde V., Dawange M., Samec J. S. M., ChemCatChem 2014, 6, 179–184; [Google Scholar]
- 19b. Galkin M. V., Samec J. S. M., ChemSusChem 2014, 7, 2154–2158. [DOI] [PubMed] [Google Scholar]
- 20. Tran F., Lancefield C. S., Kamer P. C. J., Lebl T., Westwood N. J., Green Chem. 2015, 17, 244–249. [Google Scholar]
- 21. Kobayashi S., Manabe K., Acc. Chem. Res. 2002, 35, 209–217. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Ferrini P., Rinaldi R., Angew. Chem. Int. Ed. 2014, 53, 8634–8639; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8778–8783; [Google Scholar]
- 22b. Van den Bosch S., Schutyser W., Vanholme R., Driessen T., Koelewijn S. F., Renders T., De Meester B., Huijgen W. J. J., Dehaen W., Courtin C. M., Largrain B., Boerjan W., Sels B. F., Energy Environ. Sci. 2015, 8, 1748–1763; [Google Scholar]
- 22c. Parsell T., Yohe S., Jarrell T., Klein I., Gencer E., Hewetson B., Choudhari H., Saha B., Meilan R., Mosier N., Ribeiro F., Delgass W. N., Chapple C., Kenttämaa H. I., Agrawal R., Abu-Omar M. M., Green Chem. 2015, 17, 1492–1499. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary