

Abstract
ST8SIA2 is a polysialyltransferase that attaches polysialic acid to the glycoproteins NCAM1 and CADM1. Polysialylation is involved in brain development and plasticity. ST8SIA2 is a schizophrenia candidate gene, and St8sia2 −/− mice exhibit schizophrenia‐like behavior. We sought to identify new pathological consequences of ST8SIA2 deficiency. Our proteomic analysis suggested myelin impairment in St8sia2 −/− mice. Histological and immune staining together with Western blot revealed that the onset of myelination was not delayed in St8sia2 −/− mice, but the content of myelin was lower. Ultrastructure analysis of the corpus callosum showed thinner myelin sheaths, smaller and irregularly shaped axons, and white matter lesions in adult St8sia2 −/− mice. Then we evaluated oligodendrocyte differentiation in vivo and in vitro. Fewer OLIG2+ cells in the cortex and corpus callosum, together with the higher percentage of undifferentiated oligodenroglia in St8sia2 −/− mice suggested an impairment in oligodendrocyte generation. Experiment on primary cultures of oligodendrocyte precursor cells (OPCs) confirmed a cell‐autonomous effect of ST8SIA2 in oligodendroglia, and demonstrated that OPC to oligodendrocyte transition is inhibited in St8sia2 −/− mice. Concluding, ST8SIA2‐mediated polysialylation influences on oligodendrocyte differentiation, and oligodendrocyte deficits in St8sia2 mice are a possible cause of the demyelination and degeneration of axons, resembling nerve fiber alterations in schizophrenia. GLIA 2016;65:34–49
Keywords: Schizophrenia, St8sia2, polysialic acid, myelin, oligodendrocyte
Introduction
The glycosyltransferase ST8SIA2 is a plausible factor for neurodevelopmental predisposition to schizophrenia. ST8SIA2 is part of a susceptibility region 15q23 (Maziade et al., 2005), and numerous studies link polymorphisms in ST8SIA2 to the disease (Arai et al., 2006; Gilabert‐Juan et al., 2013; McAuley et al., 2012; Tao et al., 2007; Yang et al., 2015). A possible role for ST8SIA2 in schizophrenia is supported by post‐mortem studies that have revealed decreases in the levels of ST8SIA2 enzymatic product, polysialic acid (polySia), in the brain of schizophrenics (Barbeau et al., 1995; Gilabert‐Juan et al., 2012). A recent study reported correlations between increases in polySia serum levels, structural brain alterations, and negative and cognitive symptoms in schizophrenia patients (Piras et al., 2015). Animal studies also support the association between ST8SIA2 and schizophrenia. St8sia2 −/− mice exhibited schizophrenia‐like behavior (Calandreau et al., 2010; Kröcher et al., 2015), enlarged lateral ventricles, decreased thalamic volume, and thalamocortical disconnectivity (Kröcher et al., 2015), reminiscent of the anomalies in schizophrenia (Mitelman et al., 2007; Wobrock et al., 2008; Woodward et al., 2012).
ST8SIA2 coordinates with another polysialyltransferase, ST8SIA4, in modifying acceptor proteins by attaching polysialic acid (Schnaar et al., 2014). Different phenotypes of St8sia2 −/− and St8sia4 −/− mice prove that the function of ST8SIA2 and ST8SIA4 are not fully redundant (Angata et al., 2004; Calandreau et al., 2010; Eckhardt et al., 2000; Hildebrandt et al., 2009; Kröcher et al., 2015). The expression of St8sia2 and St8sia4 is spatially regulated and largely overlapping (Angata et al., 2004; Hildebrandt et al., 1998; Oltmann‐Norden et al., 2008). However, St8sia2 and St8sia4 show different time‐specific patterns of expression, indicating the independent regulation of both genes. ST8SIA2 appears to predominate during embryonic and early postnatal development, whereas ST8SIA4 is the major enzyme in the adult brain. Moreover, ST8SIA2 and ST8SIA4 show different substrate preferences. The major carrier of polySia is neural cell adhesion molecule 1 (NCAM1) while less prominent carriers are cell adhesion molecule 1 (CADM1) and neuropilin‐2 (Galuska et al., 2010; Rollenhagen et al., 2012; Werneburg et al., 2015a; Werneburg et al., 2015b). In contrast to NCAM1, which can be polysialylated by ST8SIA2 and ST8SIA4, CADM1 is exclusively polysialylated by ST8SIA2 (Rollenhagen et al., 2012; Werneburg et al., 2015b).
PolySia weakens cell‐cell contacts by decreasing the adhesive properties of NCAM1 (Schnaar et al., 2014). Furthermore, polySia impacts cell signaling by modulating NCAM signals (Eggers et al., 2011; Seidenfaden et al., 2003), ion channel activities (Ahrens et al., 2011; Hammond et al., 2006; Vaithianathan et al., 2004), and accessibility of secreted bioactive molecules (Kiermaier et al., 2016; Sato and Kitajima 2013). NCAM1 polysialylation is vital for the migration and clustering of neural and glial precursors (Angata et al., 2007; Kröcher et al., 2014; Wang et al., 1994), the growth and fasciculation of nerve fibers (Kolkova et al., 2000; Schiff et al., 2011), and synapse formation and plasticity (Kochlamazashvili et al., 2010; Senkov et al., 2012).
Polysialylation has also been implicated in myelin formation. Myelination in the mammalian brain begins postnatally (Sowell et al., 2003) and is modulated by: electrical activity (Gibson et al., 2014; Wake et al., 2011), axon‐glia interactions and the extracellular matrix (Ahrendsen and Macklin 2013; White and Krämer‐Albers 2014). The loss of polySia on the membrane of oligodendrocytes and axons was proposed to be a prerequisite for myelin formation (Bakhti et al., 2013; Charles et al., 2000; Fewou et al., 2007; Franceschini et al., 2004; Jakovcevski et al., 2007; Prolo et al., 2009). Consistent with the need to downregulate polySia during myelination, St8sia4 −/− mice exhibited improvements in remyelination in the brain after cuprizone‐induced demyelination, although they displayed no obvious deficits in developmental myelination (Koutsoudaki et al., 2010). The myelin‐related phenotype of St8sia2 −/− mice has not yet been investigated, and the role of ST8SIA2 in myelination, myelin maintenance, and myelin repair is unclear.
We found that ST8SIA2 deficiency led to myelin deficits, thinning of axons and the age‐related degeneration of white matter. St8sia2 −/− oligodendrocyte precursor cells (OPCs) exhibited lower differentiation potential both in vivo and in vitro, which might explain the deterioration of myelin and axons in St8sia2 −/− mice.
Materials and Methods
Mice
St8sia2 and St8sia4 knockout mice on a C57BL/6 background were housed and fed under standard conditions. The mice were bred as heterozygotes to obtain wildtype (St8sia2+/+, St8sia4+/+) and knockout littermates. The mice were genotyped by polymerase chain reaction (PCR) using the following primers: ST1 (forward GAGACAGCAACTAGAGGAATAACA), ST2 (reverse CCTAGATGGGTTGGTGTTGC), and ST3 (reverse ACAGTTAGAACACCACCTTC) to amplify St8sia2 and LW13 (forward CTCAGTTCTGGCTATTTCTTTTGT), LW4 (reverse GAGCTCACAACGACTCTCCGAGC), and LW18 (revers ACCGCGAGGCGGTTTTCTCCGGC) to amplify St8sia4. All of the protocols for animal use were approved by the Polish Local Ethical Committee No. 1 in Warsaw.
Brain Fixing and Sectioning
Mice on postnatal day 15 (P15) and older were anesthetized with pentobarbital sodium and transcardially perfused with 0.1 M phosphate‐buffered saline (PBS) solution, pH 7.4, followed by 4.5% paraformaldehyde in PBS for immunochemistry or 4.5% paraformaldehyde and 2.5% glutaraldehyde in PBS for transmission electron microscopy (TEM). The brains were dissected, post‐fixed, cryoprotected with 30% sucrose in PBS for 24 h, and sectioned with a microtome (40 µm slices for immunohistochemistry, immunofluorescence or histology, 100 µm slices for electron microscopy).
Immunohistochemistry and Black‐Gold II Myelin Staining
Free‐floating slices were washed with PBST (PBS + 0.2% Triton X‐100) and quenched with 0.3% H2O2. They were then blocked in the serum of the appropriate species in PBST and incubated overnight with primary antibodies (anti‐MBP, 1 : 500, ProteinTech, catalog number 10458‐1‐AP; anti‐Neurofilament, 1 : 1,000, ProteinTech, catalog number 18934) at 4°C. The next day, the slices were incubated with biotinylated secondary antibody (Vector Labs) for 1 h at room temperature. Staining was visualized by reacting with 3,3‐diaminobenzidine substrate using the ABC kit (Vector Labs). For histological myelin staining, the Black Gold II Myelin Staining Kit (Millipore) was used according to the manufacturer's instructions. Quantifications were performed using ImagePro Plus software (Media Cybernetics).
Protein Extraction and Western Blot
Proteins from brain structures or cells were extracted with ice‐cold RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM ethylenediaminetetraacetic acid, 1 mM NaF, Complete Protease Cocktail and Phosphatase Inhibitor Cocktail 2), centrifuged, and stored at −80ºC until use. Proteins were separated in sodium dodecyl sulfate‐acrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked with non‐fat dry milk and incubated overnight with primary antibodies (anti‐GAPDH, SantaCruz, SC‐25772; anti‐Pan‐Cadherin, Thermo Scientific, MA1‐91128; anti‐PLP1/DM20, Merc Millipore, MAB388; anti‐MBP, ProteinTech, 10458‐1‐AP; anti‐CNPase, Merc Millipore, MAB326; anti‐MOBP, ProteinTech, 12690‐1‐AP; anti‐MOG, ProteinTech, 12690; anti‐polySia(mouse IgG2a, clone 735), gift from prof. Gerardy‐Schahn; anti‐OLIG2, Merc Millipore, AB9610; anti‐NG2 ProteinTech, 55027‐1‐AP; anti‐Neurofilamen M and H, ProteinTech, 18934; anti‐Neurofilament L, MP Biomedical, 10510; anti‐pMAPT gift from prof. Jaworski and prof Davis; anti‐pERK 1/2, Cell Signaling, 9021; anti‐pAKT, Cell Signaling, 9271; anti‐AKT, Cell Signaling, 9272S) at 4°C. After washing, the membranes were incubated with secondary antibodies (anti‐Rabbit IgG – peroxidase antibody, Sigma Aldrich, A0545; anti‐Goat IgG – peroxidase antibody, Sigma Aldrich, A5420; anti‐Mouse IgG – peroxidase antibody, Sigma Aldrich, A9044) for 2 h at room temperature. Staining was then visualized by chemiluminescence. The images were captured using an ImageQuant LAS 4000. Densitometric analyses were performed using Quantity One 1‐D software (BioRad).
Oligodendrocyte Precursor Cell Culture
Mixed glial cultures were set up according to the protocol of O'Meara et al. (2011), with minor modifications. The brain cortex from P2 mouse pups were dissected and digested in digestion solution (1.5 mg/mL papain, 360 μg/mL L‐cysteine, and 60 μg/mL DNaseI in MEM ‐ Modified Eagle Medium). The cell suspension was triturated with a Pasteur pipette, centrifuged at 300g, and seeded in poly‐L‐lysine‐coated tissue culture flasks and cultured in MGC medium (DMEM supplemented with: 4.5 mg/L glucose, fetal bovine serum (10%), Pen/Strep (1%) and GlutaMAX® (1x)). After 12 days, the flasks were transferred to an orbital shaker to remove microglia, followed by shaking for an additional 16 h to detach OPCs, which were then seeded on poly‐D‐lysine‐coated plates and cultured in SATO medium (DMEM supplemented with: B27 Supplement (1x), 4.5 g/L glucose, GlutaMAX® (1x), 1 mM sodium pyruvate, 100 μg/mL apotransferrin, 100 μg/mL BSA, 20 nM progesterone, 15 μg/mL putrescine, 30 nM sodium selenite, 5 μg/mL insulin, 50 μg/ml N‐acetyl‐l‐cysteine, 10 ng/mL biotin, Trace B element (1x), 25 μM Forskolin, 1% Pen/Strep) supplemented with either 10 ng/mL platelet‐derived growth factor (PDGF‐AA) and 20 ng/mL basic fibroblast growth factor (bFGF; maintenance conditions) or 45 nM 3,3',5‐triiodo‐L‐thyronine (T3; differentiation conditions).
Real‐Time Quantitative Polymerase Chain Reaction
RNA from OPCs was isolated with the RNeasy Kit (Qiagen) and transcribed to cDNA using the Transcriptor High Fidelity cDNA Synthesis Kit (Qiagen). The levels of transcripts were measured using the SYBR Green I Master Kit (Roche) and a LightCycler 480 Instrument II (Roche). Expression was measured with the following primers: Gapdh (forward, CAGTGGCAAAGTGGAGATTG; reverse, AATTGCCGTGAGTGGAGTC), Mbp (forward, AAATCGGCTCACAAGGGATTC; reverse CTCCCAGCTTAAAGATTTTGGAAA), Plp1 (forward, CTGCCAGTCTATTGCCTTCC; reverse, AGCATTCCATGGGAGAACAC), Mag (forward, GGTGTTGAGGGAGGCAGTTG; reverse, CGTTCTCTGCTAGGCAAGCA), Olig2 (forward, TGGCTTCAAGTCATCTTCCTC; reverse, GGCTCAGTCATCTGCTTCTT). Primers were designed using PrimerQuestSM.
Immunofluorescence
OPCs were fixed, washed with PBS, blocked in 3% donkey serum in PBST, and then incubated overnight with primary antibodies (anti‐PDGFR alpha, Abcam, ab6121; anti‐OLIG1, Merck Millipore, MAB5540, anti‐MBP, ProteinTech, 10458‐1‐AP) at 4°C. Free‐floating brain slices were washed with PBST, blocked in 5% donkey serum in PBST, and incubated overnight with primary antibodies (mouse anti‐OLIG2, 1:250, Merck Millipore, catalog number MABN50; rabbit anti‐OLIG2, Merck Millipore, catalog number AB9610; anti‐TCF7l2, 1:250, Merck Millipore, catalog number 05‐511; anti‐PDGFR alpha, Abcam, ab6121; anti‐OLIG1, Merck Millipore, MAB5540) at 4°C. On the next day, OPCs and brain slices were washing and they were incubated with Alexa Fluor‐conjugated secondary antibodies (anti‐Rabbit IgG (H + L), Alexa Fluor® 488 conjugate, ThermoFisher Scientific, A‐21206; anti‐Mouse IgG (H + L) Secondary Antibody, Alexa Fluor® 594 conjugate, ThermoFisher Scientific, A‐21203). Cell nuclei were counterstained with DAPI (OPCs) or Hoechst (brain slices). The images were captured with Nikon Eclipse Ni‐U fluorescence microscope or Axio Imager Z2 LSM 700 Zeiss Confocal Microscope.
Transmission Electron Microscopy
Slices were postfixed in 1% osmium tetroxide for 1 h at room temperature, dehydrated in an increasing series of ethanol concentrations, incubated with propylene oxide, and embedded in Epon (Sigma‐Aldrich). Sixty‐nanometer‐thick sections were prepared and stained in 1% uranyl acetate. The images were captured with a JEM 1400 transmission electron microscope (JEOL, Japan). The morphometric analysis was performed using ImageJ software. The diameters of the axons and myelinated fibers were calculated according to the following equation: diameter = perimeter/π, which allowed us to calculate the g‐ratio (ratio of the inner axonal diameter to the total outer diameter of a myelinated axon) for irregularly shaped fibers.
Statistical Analysis
Two‐tailed Student's t‐test was used to test for significance when comparing data for St8sia2 −/− and wildtype mice. Correlations were examined using Pearson correlation coefficient of linear regression, and ANCOVA was used to test for significance when comparing regression coefficients for St8sia2 −/− and wildtype mice. Values of P < 0.05 were considered statistically significant.
Results
The Levels of Myelin Proteins Are Lower in the Brain in St8sia2−/− Mice but Not in St8sia4−/− Mice
Our proteomic analysis revealed lower levels of several myelin proteins in St8sia2 −/− mice but not in St8sia4 −/− mice (Supp. Info. Table S1A, B). This result was corroborated by the Western blot analysis of the levels of proteolipid protein 1 (PLP1) in total protein lysates from different parts of the cortex, hippocampus, and thalamus (Supp. Info. Fig. S1A). Next, in the samples of the dorsal part of the cortex (which included the primary sensory and motor cortices), we analyzed by Western blot the levels of the following major myelin proteins: myelin basic protein (MBP), PLP1, 2',3'‐cyclic‐nucleotide 3'‐phosphodiesterase (CNP), myelin oligodendrocyte glycoprotein (MOG), and myelin oligodendrocyte basic protein (MOBP). This analysis was performed with juvenile (P15), adolescent (P30), and mature adult (P90) mice. Lower levels of all MBP isoforms, as well as CNP, were observed in St8sia2 −/− mice at all ages examined (Fig. 1). MOG was reduced on P30 and P90, whereas the levels of PLP1 and MOBP were significantly lower in mature St8sia2 −/− mice (P90). No alterations were seen in younger mice. In contrast, in 4‐month‐old (M4) St8sia4 −/− mice, the levels of PLP1 were unchanged (Fig. S1B in Supp. Info.). These results demonstrated that ST8SIA2 deficiency but not ST8SIA4 deficiency reduced the levels of myelin‐specific proteins in the brain.
Figure 1.
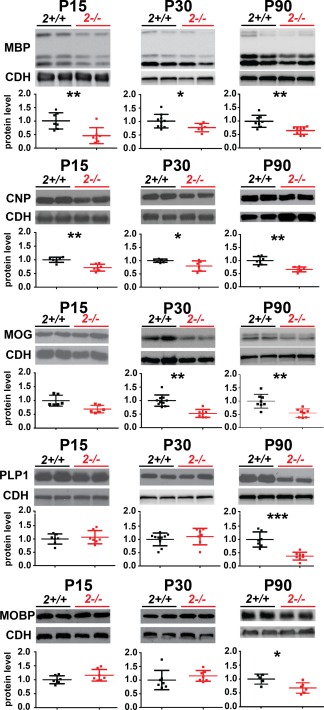
Myelin protein levels in the cortex in wildtype and St8sia2 −/− mice. Representative Western blots and densitometric analyses of MBP, CNP, MOG, PLP1, and MOBP levels in the cortex in St8sia2+/+ and St8sia2 −/− mice on P15, P30, and P90. The band intensity was normalized to pan‐Cadherin (CDH). Each dot represents one animal. The mean for the control is set as 1. The data are expressed as mean ± SD. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
Myelin Onset Is Not Delayed in St8sia2−/− Mice
Low content of myelin proteins may result from a delay in myelination onset. To test this, we compared the progression of brain myelination in St8sia2 −/− pups vs. wildtype pups. We stained brain slices from neonatal (P5) and juvenile (P15) mice for MBP or used Black Gold II to stain myelin. On P5, MBP immunoreactivity was visible in the medulla, pons, and hindbrain basal plate (Fig. 2A). By P15, only fibers in the superficial layers of the cortex were not stained (Fig. 2B). No difference was found in spatial myelination patterns between St8sia2 −/− and wildtype mice on either P5 or P15. The histological staining also revealed the normal progression of myelination in St8sia2 −/− mice on P15 (Fig. 2C). However, MBP staining on P15 revealed a nonsignificant tendency toward a decrease in intensity in specimens from St8sia2 −/− mice compared with controls, consistent with the Western blot results. These results indicated that the time‐course and developmental pattern of myelination were unaltered in St8sia2 −/− mice, although the content of the specific myelin protein MBP was lower even before the whole brain was myelinated.
Figure 2.
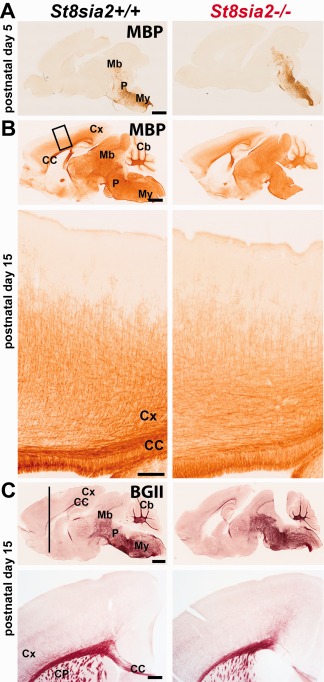
Progression of brain myelination in wildtype and St8sia2 −/− mice during postnatal development. A, B: Immunohistochemical staining for MBP on sagittal brain sections on P5 and P15. C: Histological Black Gold II staining of brain sections on P15. Representative sections of one of four brains are shown for each genotype. Scale bar: 1000 μm in upper panels and 100 μm in bottom panels. Cb, cerebellum; CC, corpus callosum; CP, caudate putamen; Cx, cortex; Mb, midbrain; My, medulla; P, pons. [Color figure can be viewed at wileyonlinelibrary.com]
Myelin Content Is Low in the Brain in St8sia2−/− Adult Mice
To determine whether myelin deficits in St8sia2 −/− mice progress with age, we stained brain slices from adolescent (P30), mature adult (P90), and older adult (M8) mice with Black Gold II (Fig. 3A,B). The densitometric analysis across all cortical layers revealed no significant difference between St8sia2 −/− and wildtype mice on P30 (Fig. 3A, upper panel) but lower myelin content in St8sia2 −/− mice on P90 and at M8 (Fig. 3A, middle and lower panels). In the corpus callosum on P90, the myelin content was also lower in St8sia2 −/− mice (Fig. 3B), and similar results were obtained for other brain structures, including the hippocampus and striatum (data no shown). In contrast, myelin content in St8sia4 −/− mice at M4 was unchanged (Fig. 3C). In summary, ST8SIA2 deficiency led to progressive myelin deficits with age.
Figure 3.
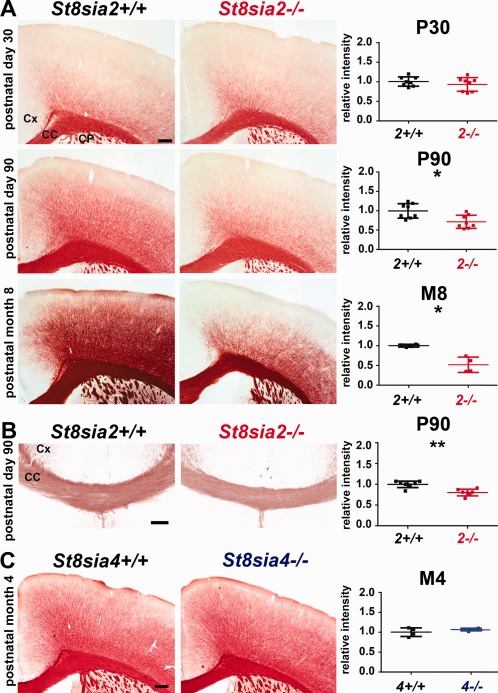
Histological evaluation of myelin content in the brains in adult wildtype, St8sia2 −/−, and St8sia4 −/− mice. A, B: Black Gold II staining and densitometric analysis in the cortex and corpus callosum in St8sia2+/+ and St8sia2 −/− mice on P30 and P90 and at M8. C: Black Gold II staining and densitometric analysis in the cortex in St8sia4+/+ and St8sia4 −/− mice at M4. Each dot represents one animal. The mean for the control is set as 1. Scale bar: 250 μm in A and C, and 100 μm in B. The data are expressed as mean ± SD. CC, corpus callosum, CP, caudate putamen; Cx, cortex. *P ≤ 0.05; **P ≤ 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
Axons Are Altered in the Cortex in St8sia2−/− Mice
Polysialic acid‐deficient mice display hypoplasia in major brain axon tracts (Hildebrandt et al., 2009; Kröcher et al., 2015; Schiff et al., 2011; Weinhold et al., 2005). To determine whether axonal agenesis occurs in the cortex in St8sia2 −/− mice, which could cause the observed decrease in myelin content, we stained P90 brain sections for major axonal protein neurofilaments (NEFs). Compared with wildtypes, the NEF staining intensity in the upper cortical layers appeared to be weaker in St8sia2 −/− mice (Fig. 4A), but the densitometric analysis did not reveal a significant difference. However, the Western blot analysis of the levels of NEF in the cortex on P15, P30, and P90 revealed significant reductions of the levels of all NEF isoforms (light, medium, and heavy [NEF‐L, NEF‐M, and NEF‐H]) in the cortex in St8sia2 −/− mice at all ages examined (Fig. 4B). These results were consistent with compromised axon development.
Figure 4.
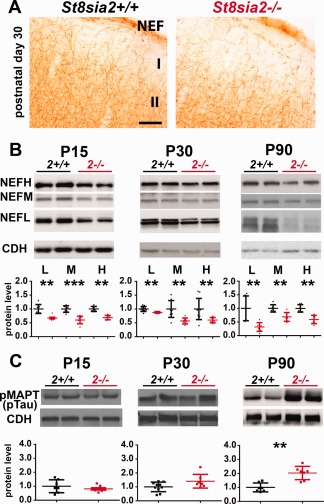
The levels of Neurofilament and the axonal degeneration marker pMAPT in the cortex in wildtype and St8sia2 −/− mice. A: Immunohistochemical staining for NEF on coronal brain sections on P30. n = 4. B: Representative Western blots and (C) densitometric analyses of NEF isoforms and pMAPT (pTAU) in the cortex in St8sia2+/+ and St8sia2 −/− mice on P15, P30, and P90. The band intensity was normalized to pan‐Cadherin (CDH). Each dot represents one animal. The mean for the control is set as 1. The data are expressed as mean ± SD. I, molecular layer of cortex; II, external granular layer of cortex. **P ≤ 0.01; ***P ≤ 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
Phosphorylated microtubule‐associated protein tau (pMAPT) is a marker of degenerating axons (Evans et al., 2000). To determine whether axons in St8sia2 −/− mice degenerate with age, which might explain the age‐related decline of myelin, we examined the phosphorylation status of MAPT at Ser396 and Ser404. On P15 and P30, the levels of pMAPT in St8sia2 −/− mice were not different from wildtype mice (Fig. 4C). However, on P90, the level of pMAPT was significantly higher in St8sia2 −/− mice, suggesting age‐related axonal degeneration.
Axons Become Smaller, the Myelin Sheath Thins, and Nerve Fibers Degenerate with Age in the Corpus Callosum in St8sia2−/− Mice
To determine whether only axons or both axons and myelin deteriorate in St8sia2 −/− mice, we examined the white matter ultrastructure in the corpus callosum on P90 and at M8 by TEM (Fig. 5A). In sections from St8sia2 −/− mice, we observed dark degeneration of some axons on P90 and white matter lesions at M8. We then performed a morphometric analysis of the TEM images. The cross‐sectional areas of axons were calculated, and the circularity of axons was assessed for each genotype and age. At both of the investigated time‐points, axons in the corpus callosum were significantly thinner in St8sia2 −/− mice, and their shape was less regular than in wildtype mice (Fig. 5B, left and middle panels). Furthermore, the g‐ratio (the ratio of axon diameter to axon plus myelin diameter), a reliable measure of axonal myelination (Chomiak and Hu, 2009), was significantly higher in St8sia2 −/− mice (Fig. 5B, right panel), indicating thinner myelin sheaths in these mice. The difference was more pronounced in M8 mice than in P90 mice, indicating progressive demyelination of axons in knockout mice. A regression analysis of the g‐ratio and axon size measurements was performed e. The g‐ratios increased with axon size in both wildtype and St8sia2 −/− mice, reflected by the trend lines, and the slopes of the trend lines were similar (Fig. 5C). To determine whether small‐ or large‐diameter axons displayed myelin thinning in St8sia2 −/− mice we compared regression coefficients between knockout and wildtype mice, but we did not observe a statistically significant difference (p = 0.59 for P90 mice, p = 0.11 for M8 mice). Thus, regardless of axonal caliber, the myelin sheaths were thinner in St8sia2 −/− mice. This suggested that the thinning of axons and the thinning of myelin in St8sia2 −/− mice developed independently. Finally, TEM revealed the premature degeneration of nerve fibers, which worsened with age.
Figure 5.
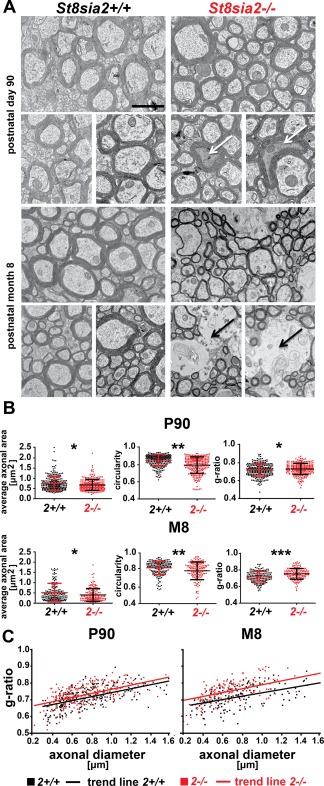
Ultrastructure of white matter in the corpus callosum in adult wildtype and St8sia2 −/− mice. A: Electron micrographs of axons in the corpus callosum in St8sia2+/+ and St8sia2 −/− mice on P90 and at M8. White arrows point to dark degeneration. Black arrows point to white matter lesions. Scale bar = 1,000 nm. B: Scatter diagram of axonal area, circularity, and g‐ratio values in the corpus callosum on P90 and at M8. Three photomicrographs of the corpus callosum from each mouse were used to analyze the area and circularity of axons. n = 5 for P90; n = 4 for M8. The analysis was performed on the area of the corpus callosum that lies beneath the primary motor cortex (1.5 mm posterior to bregma to −0.3 mm anterior to bregma; lateral, ∼1.0 to 1.5 mm). A total of 250 axons were analyzed for each genotype on P90. A total of 150 axons were analyzed for each genotype on M8. C: Scatter diagrams of g‐ratios as a function of axon diameters in the corpus callosum in St8sia2+/+ and St8sia2 −/− mice on P90 (left) and at M8 (right). The data are expressed as mean ± SD *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.[Color figure can be viewed at wileyonlinelibrary.com]
The Number of Oligodendroglia Decreases in the Cortex and Corpus Callosum in St8sia2−/− Mice
The myelin deficits that were observed in St8sia2 −/− mice might be attributable to a decrease in the number of oligodendroglia. To test this possibility, we analyzed the level of OLIG2, an oligodendrocyte lineage‐specific transcription factor (Li and Richardson 2015), in cortical extracts from wildtype and St8sia2 −/− mice. At all three time points (P15, P30, and P90), the levels of OLIG2 were significantly lower in extracts from St8sia2 −/− mice compared with wildtype animals (Fig. 6A). To clarify whether this difference was caused by fewer OLIG2+ cells or lower expression levels of OLIG2 in knockouts, OLIG2+ cells were evaluated. In St8sia2 −/− mice, the density of OLIG2+ cells significantly decreased in both the cortex (Fig. 6C) and corpus callosum (Fig. 6D). These results suggested that deficits in oligodendrocyte development might have caused the aberrant myelin maintenance in St8sia2 −/− mice.
Figure 6.

Expression of the pan‐oligodendroglia marker OLIG2 in the cortex and corpus callosum in wildtype and St8sia2 −/− mice. A: Representative Western blots and densitometric analyses of OLIG2 levels in the cortex in St8sia2+/+ and St8sia2 −/− mice on P15, P30, and P90. The band intensity was normalized to pan‐Cadherin (CDH). Each dot represents one animal. The mean for the control is set as 1. The data are expressed as mean ± SD. B: Immunohistochemical staining for OLIG2 on coronal brain sections and (C, D) analysis of the number of OLIG2+ cells in the cortex and corpus callosum on P30 St8sia2+/+ and St8sia2 −/− mice. Scale bar = 100 μm. n = 5. The data are expressed as mean ± SD. *P ≤ 0.05; **P ≤ 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
The Percentage of Undifferentiated Oligodendrocytes in the Corpus Callosum Is High in St8sia2−/− Mice
Myelin integrity requires the proper formation of white matter and its subsequent maintenance, and these two depend on the proper differentiation of myelinating cells (Takebayashi and Ikenaka, 2015). We hypothesized that the differentiation of OPCs in St8sia2 −/− mice is disturbed. To identify the population of immature oligodendroglia, we first analyzed the expression of PDGFRα and TCF7L2 in OLIG2+ cells. PDGFRα is a marker of OPCs (Baracskay et al., 2007), and TCF7L2 is expressed in OPCs and premyelinating oligodendrocytes (Guo et al., 2015; Zhao et al., 2016). We found that the percentages of both OLIG2+/PDGFRα+ and OLIG2+/TCF7L2+ cells in the corpus callosum on P15 were almost two times higher in St8sia2 −/− mice compared with control mice (Fig. 7A,B,D). The translocation of OLIG1 protein from the nucleus (OLIG1nuc) to the cytosol (OLIG1cyt) highly correlated with the maturation of oligodendrocytes (Niu et al., 2012). Therefore, we also investigated the number of undifferentiated oligodendroglia by analyzing the percentage of OLIG2+/OLIG1nuc cells in the corpus callosum. The quantification revealed an approximately two‐fold higher percentage of OLIG2+/OLIG1nuc cells in St8sia2 −/− mice compared with control mice. These results suggested substantial impairment in oligodendrocyte differentiation in St8sia2 −/− mice.
Figure 7.
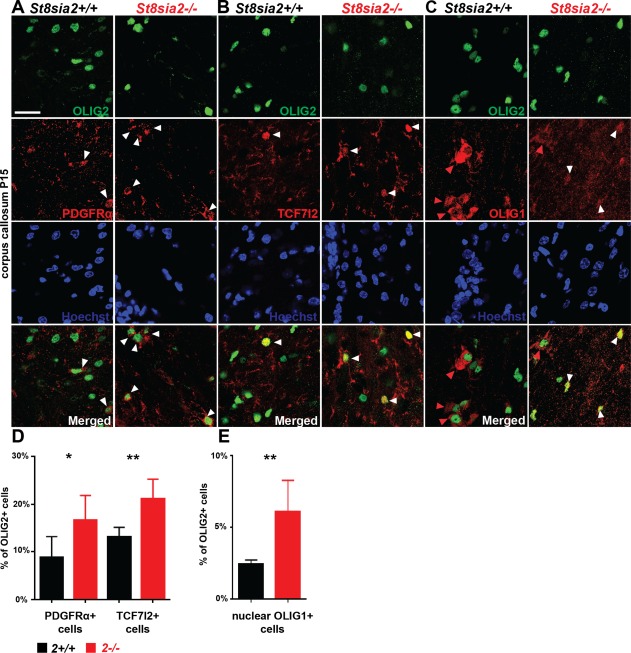
Expression of differentiation markers in oligodendroglial in the corpus callosum in juvenile wildtype and St8sia2 −/− mice. A−C: Representative photographs of the corpus callosum on coronal brain sections of St8sia2+/+ and St8sia2 −/− mice on P15. Slices were stained for OLIG2 (green) and PDGFRα (red, A), TCF7l2 (red, B) or OLIG1 (red, C). Cell nuclei were counterstained with Hoechst (blue). White arrowheads point to PDGFRα+ cells, TCF7l2+ cells, or cell with nuclear localization of OLIG1. Red arrowheads point to cell with cytosolic localization of OLIG1. Scale bar = 25 µm. D: Quantification of OLIG2+/PDGFRα+ cells (left panel) and OLIG2+/TCF7l2+ cells (right panel). E: Quantification of OLIG2+ cells expressing nuclear OLIG1. n = 6. The data are expressed as mean ± SD. *P ≤ 0.05; **P ≤ 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
St8sia2−/− OPCs Exhibit Lower Differentiation Potential
Possible defects in the differentiation of OPCs in St8sia2 −/− mice were studied in vitro. Oligodendrocyte precursor cells were obtained from mixed glial cultures that were isolated from the cortex in mice on P2. The cells were cultured for 48 h in media that were supplemented with either PDGF‐AA/bFGF (maintenance conditions) or T3 (differentiation conditions) to estimate spontaneous and induced differentiation, respectively (Billon et al., 2002).
Oligodendrocyte precursor cells express ST8SIA2 and ST8SIA4 (Werneburg et al., 2015a). To determine whether the OPCs that were derived from St8sia2 −/− mice were polysialylation‐deficient, we analyzed polySia expression in cultures that were grown under maintenance conditions. Immunostaining and Western blot revealed a clear reduction of polySia expression in St8sia2 −/− OPCs (Fig. 8A–C).
Figure 8.
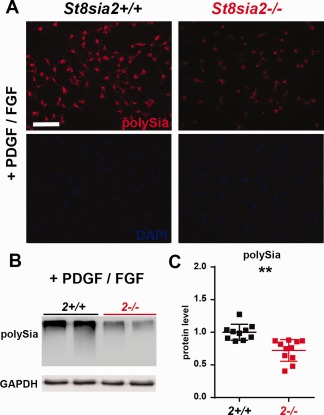
Levels of polySia in wildtype and St8sia2 −/− OPCs in vitro. A: Immunofluorescence analysis of polySia in OPCs cells in vitro. The cells were labeled with anti‐polySia antibody (red), and the nuclei were counterstained with DAPI. Representative photographs of St8sia2+/+ and St8sia2 −/− OPCs from one of three experiments are shown. Scale bar = 50 μm. B: Representative Western blots and (C) densitometric analyses of polySia levels in protein extracts from OPCs cultured in medium supplemented with PDGF‐AA/bFGF. The band intensity was normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). Each dot represents one independent OPC culture. The mean for the control is set as 1. The data are expressed as mean ± SD. **P ≤ 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
We then examined the capacity of wildtype and St8sia2 −/− OPCs to differentiate in a model of spontaneous and induced differentiation. In cultures that were grown in PDGF‐AA/bFGF medium, we assessed the number of MBP+ cells (i.e., mature oligodendrocytes). Few of these cells differentiated into oligodendrocytes in wildtype OPCs under these conditions. In St8sia2 −/− cultures, however, nearly 4‐times fewer MBP+ cells were found compared with wildtype cultures (Fig. 9A,B), suggesting a defect in differentiation. In the cultures that were grown in T3 medium, the mRNA levels of Olig2 and the mature oligodendrocyte markers Mbp, Plp1, and Mag were assessed by quantitative PCR (qPCR). Olig2 levels were unaltered, whereas Mbp and Plp1 levels were significantly lower in samples from St8sia2 −/− cultures than from control cultures, and Mag showed a tendency toward lower levels of expression (Fig. 9C). These results indicated that St8sia2 −/− OPCs had a low differentiation capability in vitro compared with wildtype OPCs.
Figure 9.
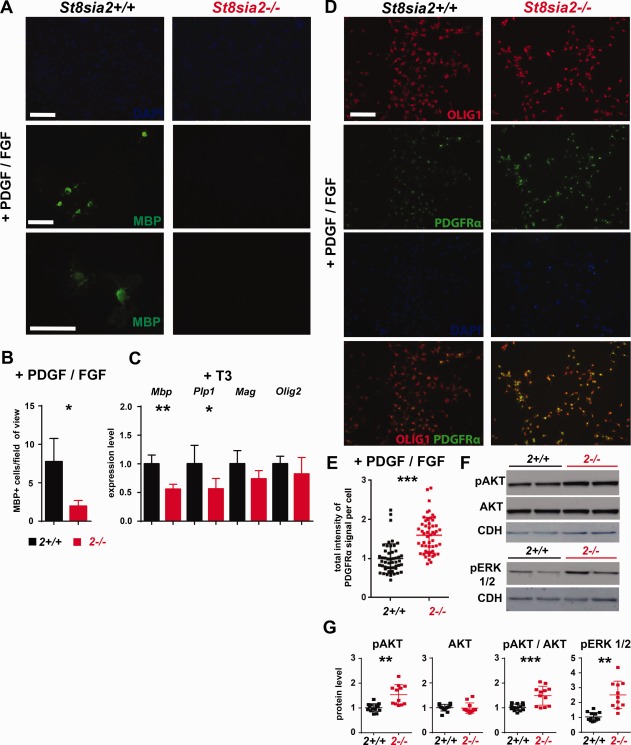
Expression of differentiation markers in wildtype and St8sia2 −/− oligodendroglia in vitro during spontaneous and induced differentiation. A: The spontaneous differentiation of St8sia2+/+ and St8sia2 −/− OPCs under maintenance conditions (PDGF‐AA/bFGF) was analyzed using immunofluorescence. The cells were labeled with anti‐MBP antibody (green), and the nuclei were counterstained with DAPI. Representative photographs of St8sia2+/+ and St8sia2 −/− OPCs from one of three experiments are shown. Scale bar = 50 μm. B: The number of MBP+ cells from 51 fields of view for each genotype were calculated for three independent OPC cultures. The quantification data are expressed as mean ± SD. n = 6. (C) Real‐time PCR analysis of Mbp, Plp1, Mag, and Olig2 expression in St8sia2+/+ and St8sia2 −/− OPC cultures grown in medium supplemented with 3,3',5‐triiodo‐L‐thyronine (T3; differentiation conditions). The results are expressed relative to the level of Gapdh expression. The mean for the control is set as 1 for each transcript. The data are expressed as mean ± SD. n = 7. D: Immunofluorescence analysis of PDGFRα and OLIG1 in OPCs under maintenance conditions in medium supplemented with PDGF‐AA/bFGF. The cells were labeled with anti‐OLIG1 antibody (red) and anti‐PDGFRα antibody (green). Nuclei were counterstained with DAPI (blue). Representative photographs of St8sia2+/+ and St8sia2 −/− OPCs from one of six experiments are shown. Scale bar = 50 μm. E: Quantification of the intensity of the PDGFRα signal per cell. Each dot represents a single cell (n = 50). The mean for the control is set as 1. The data are expressed as mean ± SD. F: Representative Western blots and (G) densitometric analyses of pAKT, AKT, and pERK1/2 levels in protein extracts from OPCs cultured under maintenance conditions (PDGF‐AA/bFGF). The band intensity was normalized to GAPDH. Each dot represents one independent OPC culture. The mean for the control for each protein is set as 1. The data are expressed as mean ± SD. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
To explore the possible mechanism that underlies the lower propensity for differentiation of St8sia2 −/− OPCs, we assessed the expression of PDGFRα. PDGF is a major factor for maintaining the undifferentiated state of OPCs (Barateiro and Fernandes 2014). The cultures were grown under maintenance conditions and stained, in parallel, for PDGFRα and the oligodendrocyte lineage marker OLIG1 (Dai et al., 2015). Although the percentage of PDGFRα+ cells within the OLIG1+ population was similar in St8sia2 −/− and wildtype cultures, the intensity of PDGFRα staining was significantly stronger in St8sia2 −/− cells (Fig. 9D,E). To study the possible consequences of this increase in PDGFRα levels, we evaluated the activity of the signaling pathways that are downstream from PDGFRα, AKT and extracellular signal‐regulated kinase (ERK) (Alessi et al., 1996), in St8sia2 −/− and wildtype cells using Western blot. The levels of the phosphorylated forms of both of these kinases were higher in extracts from St8sia2 −/− cultures than from wildtype cultures (Fig. 9F,G), indicating the overactivity of PDGFRα signaling in St8sia2 −/− OPCs, which might have caused the reduction of the differentiation potential of these cells.
Discussion
This study provides evidence that ST8SIA2 is linked to oligodendrocyte dysfunction, demyelination, and axon degeneration in the brain. We found that the loss of ST8SIA2 led to impairments in both axons and myelin sheaths in the cortex and corpus callosum in adult mice. Alterations in myelin content were also detected in the hippocampus, striatum, and thalamus, indicating that this phenotype is not limited to specific brain regions. The myelination defects were specific to ST8SIA2 deficiency. St8sia4 knockout had no effect on the developmental formation of myelin (Koutsoudaki et al., 2010) or myelin maintenance (present study). We also found that oligodendrocyte differentiation was compromised in St8sia2 −/− mice in a cell‐autonomous manner.
The onset of myelination in the brain in St8sia2 −/− mice was fairly normal at the histological level at early ages (P15 and P30), despite reductions of NEF immunoreactivity and protein levels that possibly reflected a defective axon outgrowth during development. Consistently, deficits in PLP and MOBP, markers of myelin, were not detected before P90, implying that the generation and assembly of compact myelin are undisturbed. Nevertheless, lower levels of CNP and MBP proteins were observed in juvenile mice on P15. The deficits in myelin proteins in St8sia2 −/− mice occurred in the order in which the individual proteins are expressed during the transition from late OPCs to myelinating oligodendrocytes (Fulton et al., 2010). CNP and MBP are synthetized already in premyelinating oligodendrocytes. Therefore, decreases in these proteins in St8sia2 −/− mice on P15 (i.e., when the amount of myelin is still low) might reflect the lower number of differentiating oligodendrocytes in these mice, rather than hypomyelination. In contrast, deficits in MOBP and PLP1 that did not occur in St8sia2 −/− mice until P90, as mentioned above, are indicative of demyelination. In agreement with this conclusion, the thinning of myelin was apparent on P90 and further deteriorated at M8, as observed by TEM and histological staining.
Because simultaneously to demyelination axon degeneration occurred, reflected by an increase in tau protein phosphorylation on P90 and axon shape irregularities (observed by TEM), it is possible that axonal dysfunctions, in addition to oligodendroglia dysfunction, contributed to degeneration of myelin in St8sia2 −/− mice at later ages, or myelin deficits compromised axon integrity. However, myelinated axons are negative for polySia (Charles et al., 2000). Therefore, it is very unlikely that myelin and axonal pathologies were initiated by the loss of axonal polySia.
Oligodendrocytes are required for myelin formation and turnover (Young et al., 2013). Additionally, oligodendroglia provide trophic support to axons independently of myelin itself (Lappe‐Siefke et al., 2003; Nave 2010a; Nave 2010b; Smith et al., 2013). In St8sia2 −/− mice the total number of OLIG2+ cells was low, whereas the percentage of OLIG2+/PDGFRα+, OLIG2+/TCF7L2+ and OLIG2+/nuclear OLIG1 cells (i.e., undifferentiated cells) was about twice higher compared with wildtype mice. This suggested an inhibition of oligodendrocyte differentiation, which was further corroborated in vitro. We propose that myelin deficits in St8sia2 −/− mice are at least partially caused by dysfunctions in oligodendroglia that result from an impairment of OPC differentiation. Furthermore, given that a low number of OLIG2+ cells was evident before the first signs of axon degeneration (i.e., an increase in tau protein phosphorylation and shape irregularity on P90), the deficits in oligodendroglia may have caused axonal alterations and subsequent degeneration in ST8SIA2‐deficient mice. However, additional experiments (e.g., oligodendroglial‐specific knockout of St8sia2) are required to test these hypotheses.
We found that St8sia2 −/− OPCs have a lower capacity to differentiate in vitro than wildtype cells, suggesting a cell‐autonomous role for ST8SIA2 in oligodendrocyte differentiation. Our results reveal previously unknown function of polySia and ST8SIA2 in differentiation of OPCs. We can speculate about the way in which the decrease in polySia in OPCs affects the differentiation of these cells in St8sia2 −/− mice. PolySia may interfere with ligand binding and consequently alter the activation of receptors that are involved in the proliferation or differentiation of OPCs, such as PDGFRα (Hill et al., 2013), fibroblast growth factor receptors (FGFRs) (Furusho et al., 2012), or receptor tyrosine‐protein kinase ERBB4 (Chen et al., 2006; Roy et al., 2007). This seems possible because we found increases in PDGFRα levels and activity of its downstream components AKT and ERK in St8sia2 −/− OPCs. Moreover, the polySia carriers NCAM1 and CADM1 are known to interact with FGFRs in neurons (Kiryushko et al., 2006) and ERBB4 in astrocytes (Ojeda et al., 2008; Sandau et al., 2011), respectively.
How do the current findings relate to previous reports on the role of polySia in oligodendrocyte development and myelination? The level of polySia was reported to decrease during the differentiation of myelinating oligodendrocytes (Franceschini et al., 2004) and on axons before myelin is deposited (Charles et al., 2000). Gain‐of‐function experiments in vitro and in vivo showed that polySia interferes with the maturation of oligodendrocytes (Decker et al., 2002; Fewou et al., 2007; Franceschini et al., 2004; Prolo et al., 2009) and myelin wrapping (Charles et al., 2000; Fewou et al., 2007; Franceschini et al., 2004; Prolo et al., 2009). Thus, polySia appears to act as a negative regulator of OPC differentiation and myelination. However, the results presented herein suggest that polySia may also play a positive role in myelination by promoting OPC differentiation. In addition, other in vitro studies have shown that polySia plays a positive regulatory role in the migration of oligodendroglia (Czepiel et al., 2014; Wang et al., 1994; Zhang et al., 2004). It was also demonstrated that attenuation of polySia by ablation of either St8sia2 or St8sia4 interferes with cortical interneuron migration, resulting in low number of interneurons in the adult brain (Angata et al., 2007; Kröcher et al., 2014; Wang et al., 1994). It raises the possibility that migration of OPCs is impaired in St8sia2 −/− mice; however, it was not examined in the present study. Interestingly, in both polySia‐deficient St8sia2 −/− mice and mice with elevated levels of polySia that were caused by deficiency in the lysosomal sialic acid transporter sialin or ectopic expression of St8sia4 under the Plp promotor (Fewou et al., 2007; Prolo et al., 2009), the number of mature oligodendrocytes and myelin content decreased. The fact that these three mouse lines exhibit similar deficits that result from opposite impairments in polysialylation implicates polySia in various aspects of oligodendrocyte development, myelin formation, and myelin maintenance. Altogether, these results support the notion that the precise regulation of polysialylation by the activity of ST8SIA2 and ST8SIA4 is required for the orchestration of oligodendrocyte development and myelination.
The myelin deficits and axon degeneration that were observed herein expand the list of similarities between St8sia2 −/− mice and schizophrenia patients (Kröcher et al., 2015) who present white matter changes and the disintegration of nerve fibers (Du et al., 2013; Holleran et al., 2014; Mighdoll et al., 2015; Najjar and Pearlman 2015; Wheeler and Voineskos 2014). Post‐mortem morphometric examinations of 40 schizophrenia brains revealed myelin thinning, axon deformation, dark degeneration, and white matter lesions (Uranova et al., 2011). ST8SIA2‐deficient mice also exhibit all of these alterations. The causes of these pathologies in schizophrenia are unclear. One presumption is that they are consequences of oligodendroglial deficits because variations in the genes that encode oligodendrocyte and myelin proteins, such as OLIG2, MBP, PLP1, MOG, MOBP, CNP, and MAG, have been associated with schizophrenia (reviewed in (Roussos and Haroutunian 2014), and lower levels of expression of these proteins were observed in schizophrenia brains post‐mortem (Hakak et al., 2001; Mauney et al., 2015; Tkachev et al., 2003). Moreover, a recent study reported a decrease in the density of OLIG2+ cells and an increase in PDGFRA expression in the prefrontal cortex (Mauney et al., 2015). Highly reminiscent of these findings in schizophrenia, the levels of all the investigated myelin proteins in this study were reduced in the cortex in adult St8sia2 −/− mice, and St8sia2 −/− OPCs exhibited an increase in the level of PDFGRα in vitro. Therefore, polysialylation disturbances that are caused by ST8SIA2 dysregulation may be one of many possible causes of deficits in oligodendroglia in schizophrenia patients. However, future genetic and neuroimaging studies are necessary to determine whether schizophrenia‐associated variants of ST8SIA2 are linked to white matter changes and nerve fiber alterations in schizophrenia patients. If these links are indeed established, then St8sia2 −/− mice may be a suitable model for studying the neuropathological mechanisms associated with schizophrenia.
Conclusions
This study revealed a new role for polySia in oligodendrocyte differentiation and myelin maintenance, and suggests a potential mechanism that underlies the white matter abnormalities that are observed in schizophrenia patients. The striking similarities in nerve fiber abnormalities and oligodendroglia‐related deficits between schizophrenia patients and St8sia2 −/− mice suggest that white matter pathology in at least some schizophrenia patients might be associated with dysfunctions in ST8SIA2 and polysialylation.
Supporting information
Supporting Information
Supporting Information Figure S1
Supporting Information Table S1
Acknowledgment
The authors want to express our special gratitude to Prof. Rita Gerardy‐Schahn from Hannover Medical School for inspiring discussions. Miriam Schiff and Tim Kröcher are acknowledged for their help with sample collection at Hannover Medical School. They thank Dr. Janusz Debski and Prof. Michal Dadlez for help with the mass spectrometry analysis, which was performed in the Core Facility for Mass Spectrometry at the Institute of Biochemistry and Biophysics in Warsaw using the LTQ‐Orbitrap Velos mass spectrometer (Thermo Scientific). They thank Andrzej Szczepankiewicz, MSc, and Prof. Grzegorz Wilczynski for their help with preparing the TEM samples and valuable advice.
The electron microscopy photos were taken with a JEM 1400 electron microscope (JEOL, Japan, 2008) equipped with an energy‐dispersive full‐range X‐ray microanalysis system (EDS INCA Energy TEM, Oxford Instruments, United Kingdom), tomographic holder, and high‐resolution digital camera (CCD MORADA, SiS‐Olympus, Germany) in the Laboratory of Electron Microscopy, Nencki Institute of Experimental Biology, Warsaw, Poland.
Contributor Information
Marta Barbara Wisniewska, Email: m.wisniewska@uw.edu.pl.
Jacek Kuznicki, Email: jacek.kuznicki@iimcb.gov.pl.
References
- Ahrendsen JT, Macklin W. 2013. Signaling mechanisms regulating myelination in the central nervous system. Neurosci Bull 29:199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrens J, Foadi N, Eberhardt A, Haeseler G, Dengler R, Leffler A, Mühlenhoff M, Gerardy‐Schahn R, Leuwer M. 2011. Defective polysialylation and sialylation induce opposite effects on gating of the skeletal Na+ channel NaV1.4 in Chinese hamster ovary cells. Pharmacology 87:311–317. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. 1996. Mechanism of activation of protein kinase B by insulin and IGF‐1. Embo J 15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Angata K, Huckaby V, Ranscht B, Terskikh A, Marth JD, Fukuda M. 2007. Polysialic acid‐directed migration and differentiation of neural precursors are essential for mouse brain development. Mol Cell Biol 27:6659–6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angata K, Long JM, Bukalo O, Lee W, Dityatev A, Wynshaw‐Boris A, Schachner M, Fukuda M, Marth JD. 2004. Sialyltransferase ST8Sia‐II assembles a subset of polysialic acid that directs hippocampal axonal targeting and promotes fear behavior. J Biol Chem 279:32603–32613. [DOI] [PubMed] [Google Scholar]
- Arai M, Yamada K, Toyota T, Obata N, Haga S, Yoshida Y, Nakamura K, Minabe Y, Ujike H Sora I, et al. 2006. Association between polymorphisms in the promoter region of the sialyltransferase 8B (SIAT8B) gene and schizophrenia. Biol Psychiatry 59:652–659. [DOI] [PubMed] [Google Scholar]
- Bakhti M, Snaidero N, Schneider D, Aggarwal S, Möbius W, Janshoff A, Eckhardt M, Nave KA, Simons M. 2013. Loss of electrostatic cell‐surface repulsion mediates myelin membrane adhesion and compaction in the central nervous system. Proc Natl Acad Sci USA 110:3143–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baracskay KL, Kidd GJ, Miller RH, Trapp BD. 2007. NG2‐positive cells generate A2B5‐positive oligodendrocyte precursor cells. Glia 55:1001–1010. [DOI] [PubMed] [Google Scholar]
- Barateiro A, Fernandes A. 2014. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim Biophys Acta 1843:1917–1929. [DOI] [PubMed] [Google Scholar]
- Barbeau D, Liang JJ, Robitalille Y, Quirion R, Srivastava LK. 1995. Decreased expression of the embryonic form of the neural cell adhesion molecule in schizophrenic brains. Proc Natl Acad Sci USA 92:2785–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billon N, Jolicoeur C, Tokumoto Y, Vennström B, Raff M. 2002. Normal timing of oligodendrocyte development depends on thyroid hormone receptor alpha 1 (TRalpha1). Embo J 21:6452–6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calandreau L, Márquez C, Bisaz R, Fantin M, Sandi C. 2010. Differential impact of polysialyltransferase ST8SiaII and ST8SiaIV knockout on social interaction and aggression. Genes Brain Behav 9:958–967. [DOI] [PubMed] [Google Scholar]
- Charles P, Hernandez MP, Stankoff B, Aigrot MS, Colin C, Rougon G, Zalc B, Lubetzki C. 2000. Negative regulation of central nervous system myelination by polysialylated‐neural cell adhesion molecule. Proc Natl Acad Sci USA 97:7585–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Velardez MO, Warot X, Yu ZX, Miller SJ, Cros D, Corfas G. 2006. Neuregulin 1‐erbB signaling is necessary for normal myelination and sensory function. J Neurosci 26:3079–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomiak T, Hu B. 2009. What is the optimal value of the g‐ratio for myelinated fibers in the rat CNS? A theoretical approach. PLoS One 4:e7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czepiel M, Leicher L, Becker K, Boddeke E, Copray S. 2014. Overexpression of polysialylated neural cell adhesion molecule improves the migration capacity of induced pluripotent stem cell‐derived oligodendrocyte precursors. Stem Cells Transl Med 3:1100–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Bercury KK, Ahrendsen JT, Macklin WB. 2015. Olig1 function is required for oligodendrocyte differentiation in the mouse brain. J Neurosci 35:4386–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker L, Durbec P, Rougon G, Baron‐Van Evercooren A. 2002. Loss of polysialic residues accelerates CNS neural precursor differentiation in pathological conditions. Mol Cell Neurosci 19:225–238. [DOI] [PubMed] [Google Scholar]
- Du F, Cooper AJ, Thida T, Shinn AK, Cohen BM, Ongür D. 2013. Myelin and axon abnormalities in schizophrenia measured with magnetic resonance imaging techniques. Biol Psychiatry 74:451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt M, Bukalo O, Chazal G, Wang L, Goridis C, Schachner M, Gerardy‐Schahn R, Cremer H, Dityatev A. 2000. Mice deficient in the polysialyltransferase ST8SiaIV/PST‐1 allow discrimination of the roles of neural cell adhesion molecule protein and polysialic acid in neural development and synaptic plasticity. J Neurosci 20:5234–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers K, Werneburg S, Schertzinger A, Abeln M, Schiff M, Scharenberg MA, Burkhardt H, Mühlenhoff M, Hildebrandt H. 2011. Polysialic acid controls NCAM signals at cell‐cell contacts to regulate focal adhesion independent from FGF receptor activity. J Cell Sci 124:3279–3291. [DOI] [PubMed] [Google Scholar]
- Evans DB, Rank KB, Bhattacharya K, Thomsen DR, Gurney ME, Sharma SK. 2000. Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase II inhibits tau's ability to promote microtubule assembly. J Biol Chem 275:24977–24983. [DOI] [PubMed] [Google Scholar]
- Fewou SN, Ramakrishnan H, Büssow H, Gieselmann V, Eckhardt M. 2007. Down‐regulation of polysialic acid is required for efficient myelin formation. J Biol Chem 282:16700–16711. [DOI] [PubMed] [Google Scholar]
- Franceschini I, Vitry S, Padilla F, Casanova P, Tham TN, Fukuda M, Rougon G, Durbec P, Dubois‐Dalcq M. 2004. Migrating and myelinating potential of neural precursors engineered to overexpress PSA‐NCAM. Mol Cell Neurosci 27:151–162. [DOI] [PubMed] [Google Scholar]
- Fulton D, Paez PM, Campagnoni AT. 2010. The multiple roles of myelin protein genes during the development of the oligodendrocyte. ASN Neuro 2:e00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusho M, Dupree JL, Nave KA, Bansal R. 2012. Fibroblast growth factor receptor signaling in oligodendrocytes regulates myelin sheath thickness. J Neurosci 32:6631–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galuska SP, Rollenhagen M, Kaup M, Eggers K, Oltmann‐Norden I, Schiff M, Hartmann M, Weinhold B, Hildebrandt H Geyer R, et al. 2010. Synaptic cell adhesion molecule SynCAM 1 is a target for polysialylation in postnatal mouse brain. Proc Natl Acad Sci USA 107:10250–10255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G Zuchero JB, et al. 2014. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344:1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilabert‐Juan J, Nacher J, Sanjuán J, Moltó MD. 2013. Sex‐specific association of the ST8SIAII gene with schizophrenia in a Spanish population. Psychiatry Res 210:1293–1295. [DOI] [PubMed] [Google Scholar]
- Gilabert‐Juan J, Varea E, Guirado R, Blasco‐Ibáñez JM, Crespo C, Nácher J. 2012. Alterations in the expression of PSA‐NCAM and synaptic proteins in the dorsolateral prefrontal cortex of psychiatric disorder patients. Neurosci Lett 530:97–102. [DOI] [PubMed] [Google Scholar]
- Guo F, Lang J, Sohn J, Hammond E, Chang M, Pleasure D. 2015. Canonical Wnt signaling in the oligodendroglial lineage–puzzles remain. Glia 63:1671–1693. [DOI] [PubMed] [Google Scholar]
- Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V, Fienberg AA. 2001. Genome‐wide expression analysis reveals dysregulation of myelination‐related genes in chronic schizophrenia. Proc Natl Acad Sci USA 98:4746–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond MS, Sims C, Parameshwaran K, Suppiramaniam V, Schachner M, Dityatev A. 2006. Neural cell adhesion molecule‐associated polysialic acid inhibits NR2B‐containing N‐methyl‐D‐aspartate receptors and prevents glutamate‐induced cell death. J Biol Chem 281:34859–34869. [DOI] [PubMed] [Google Scholar]
- Hildebrandt H, Becker C, Mürau M, Gerardy‐Schahn R, Rahmann H. 1998. Heterogeneous expression of the polysialyltransferases ST8Sia II and ST8Sia IV during postnatal rat brain development. J Neurochem 71:2339–2348. [DOI] [PubMed] [Google Scholar]
- Hildebrandt H, Mühlenhoff M, Oltmann‐Norden I, Röckle I, Burkhardt H, Weinhold B, Gerardy‐Schahn R. 2009. Imbalance of neural cell adhesion molecule and polysialyltransferase alleles causes defective brain connectivity. Brain 132:2831–2838. [DOI] [PubMed] [Google Scholar]
- Hill RA, Patel KD, Medved J, Reiss AM, Nishiyama A. 2013. NG2 cells in white matter but not gray matter proliferate in response to PDGF. J Neurosci 33:14558–14566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleran L, Ahmed M, Anderson‐Schmidt H, McFarland J, Emsell L, Leemans A, Scanlon C, Dockery P, McCarthy P Barker GJ, et al. 2014. Altered interhemispheric and temporal lobe white matter microstructural organization in severe chronic schizophrenia. Neuropsychopharmacology 39:944–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski I, Mo Z, Zecevic N. 2007. Down‐regulation of the axonal polysialic acid‐neural cell adhesion molecule expression coincides with the onset of myelination in the human fetal forebrain. Neuroscience 149:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiermaier E, Moussion C, Veldkamp CT, Gerardy‐Schahn R, de Vries I, Williams LG, Chaffee GR, Phillips AJ, Freiberger F Imre R, et al. 2016. Polysialylation controls dendritic cell trafficking by regulating chemokine recognition. Science 351:186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryushko D, Korshunova I, Berezin V, Bock E. 2006. Neural cell adhesion molecule induces intracellular signaling via multiple mechanisms of Ca2+ homeostasis. Mol Biol Cell 17:2278–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochlamazashvili G, Senkov O, Grebenyuk S, Robinson C, Xiao MF, Stummeyer K, Gerardy‐Schahn R, Engel AK, Feig L Semyanov A, et al. 2010. Neural cell adhesion molecule‐associated polysialic acid regulates synaptic plasticity and learning by restraining the signaling through GluN2B‐containing NMDA receptors. J Neurosci 30:4171–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolkova K, Novitskaya V, Pedersen N, Berezin V, Bock E. 2000. Neural cell adhesion molecule‐stimulated neurite outgrowth depends on activation of protein kinase C and the Ras‐mitogen‐activated protein kinase pathway. J Neurosci 20:2238–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsoudaki PN, Hildebrandt H, Gudi V, Skripuletz T, Škuljec J, Stangel M. 2010. Remyelination after cuprizone induced demyelination is accelerated in mice deficient in the polysialic acid synthesizing enzyme St8siaIV. Neuroscience 171:235–244. [DOI] [PubMed] [Google Scholar]
- Kröcher T, Malinovskaja K, Jürgenson M, Aonurm‐Helm A, Zharkovskaya T, Kalda A, Röckle I, Schiff M, Weinhold B Gerardy‐Schahn R, et al. 2015. Schizophrenia‐like phenotype of polysialyltransferase ST8SIA2‐deficient mice. Brain Struct Funct 220:71–83. [DOI] [PubMed] [Google Scholar]
- Kröcher T, Röckle I, Diederichs U, Weinhold B, Burkhardt H, Yanagawa Y, Gerardy‐Schahn R, Hildebrandt H. 2014. A crucial role for polysialic acid in developmental interneuron migration and the establishment of interneuron densities in the mouse prefrontal cortex. Development 141:3022–3032. [DOI] [PubMed] [Google Scholar]
- Lappe‐Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. 2003. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33:366–374. [DOI] [PubMed] [Google Scholar]
- Li H, Richardson WD. 2015. Evolution of the CNS myelin gene regulatory program. Brain Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauney SA, Pietersen CY, Sonntag KC, Woo TU. 2015. Differentiation of oligodendrocyte precursors is impaired in the prefrontal cortex in schizophrenia. Schizophr Res 169:374–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maziade M, Roy MA, Chagnon YC, Cliche D, Fournier JP, Montgrain N, Dion C, Lavallée JC, Garneau Y Gingras N, et al. 2005. Shared and specific susceptibility loci for schizophrenia and bipolar disorder: A dense genome scan in Eastern Quebec families. Mol Psychiatry 10:486–499. [DOI] [PubMed] [Google Scholar]
- McAuley EZ, Scimone A, Tiwari Y, Agahi G, Mowry BJ, Holliday EG, Donald JA, Weickert CS, Mitchell PB Schofield PR, et al. 2012. Identification of sialyltransferase 8B as a generalized susceptibility gene for psychotic and mood disorders on chromosome 15q25‐26. PLoS One 7:e38172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mighdoll MI, Tao R, Kleinman JE, Hyde TM. 2015. Myelin, myelin‐related disorders, and psychosis. Schizophr Res 161:85–93. [DOI] [PubMed] [Google Scholar]
- Mitelman SA, Torosjan Y, Newmark RE, Schneiderman JS, Chu KW, Brickman AM, Haznedar MM, Hazlett EA, Tang CY Shihabuddin L, et al. 2007. Internal capsule, corpus callosum and long associative fibers in good and poor outcome schizophrenia: A diffusion tensor imaging survey. Schizophr Res 92:211–224. [DOI] [PubMed] [Google Scholar]
- Najjar S, Pearlman DM. 2015. Neuroinflammation and white matter pathology in schizophrenia: systematic review. Schizophr Res 161:102–112. [DOI] [PubMed] [Google Scholar]
- Nave KA. 2010a. Myelination and support of axonal integrity by glia. Nature 468:244–252. [DOI] [PubMed] [Google Scholar]
- Nave KA. 2010b. Myelination and the trophic support of long axons. Nat Rev Neurosci 11:275–283. [DOI] [PubMed] [Google Scholar]
- Niu J, Mei F, Wang L, Liu S, Tian Y, Mo W, Li H, Lu QR, Xiao L. 2012. Phosphorylated olig1 localizes to the cytosol of oligodendrocytes and promotes membrane expansion and maturation. Glia 60:1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara RW, Ryan SD, Colognato H, Kothary R. 2011. Derivation of enriched oligodendrocyte cultures and oligodendrocyte/neuron myelinating co‐cultures from post‐natal murine tissues. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda SR, Lomniczi A, Sandau US. 2008. Glial‐gonadotrophin hormone (GnRH) neurone interactions in the median eminence and the control of GnRH secretion. J Neuroendocrinol 20:732–742. [DOI] [PubMed] [Google Scholar]
- Oltmann‐Norden I, Galuska SP, Hildebrandt H, Geyer R, Gerardy‐Schahn R, Geyer H, Mühlenhoff M. 2008. Impact of the polysialyltransferases ST8SiaII and ST8SiaIV on polysialic acid synthesis during postnatal mouse brain development. J Biol Chem 283:1463–1471. [DOI] [PubMed] [Google Scholar]
- Piras F, Schiff M, Chiapponi C, Bossù P, Mühlenhoff M, Caltagirone C, Gerardy‐Schahn R, Hildebrandt H, Spalletta G. 2015. Brain structure, cognition and negative symptoms in schizophrenia are associated with serum levels of polysialic acid‐modified NCAM. Transl Psychiatry 5:e658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolo LM, Vogel H, Reimer RJ. 2009. The lysosomal sialic acid transporter sialin is required for normal CNS myelination. J Neurosci 29:15355–15365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen M, Kuckuck S, Ulm C, Hartmann M, Galuska SP, Geyer R, Geyer H, Mühlenhoff M. 2012. Polysialylation of the synaptic cell adhesion molecule 1 (SynCAM 1) depends exclusively on the polysialyltransferase ST8SiaII in vivo. J Biol Chem 287:35170–35180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos P, Haroutunian V. 2014. Schizophrenia: Susceptibility genes and oligodendroglial and myelin related abnormalities. Front Cell Neurosci 8:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy K, Murtie JC, El‐Khodor BF, Edgar N, Sardi SP, Hooks BM, Benoit‐Marand M, Chen C, Moore H O'Donnell P, et al. 2007. Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc Natl Acad Sci USA 104:8131–8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandau US, Mungenast AE, Alderman Z, Sardi SP, Fogel AI, Taylor B, Parent AS, Biederer T, Corfas G, Ojeda SR. 2011. SynCAM1, a synaptic adhesion molecule, is expressed in astrocytes and contributes to erbB4 receptor‐mediated control of female sexual development. Endocrinology 152:2364–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato C, Kitajima K. 2013. Impact of structural aberrancy of polysialic acid and its synthetic enzyme ST8SIA2 in schizophrenia. Front Cell Neurosci 7:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff M, Röckle I, Burkhardt H, Weinhold B, Hildebrandt H. 2011. Thalamocortical pathfinding defects precede degeneration of the reticular thalamic nucleus in polysialic acid‐deficient mice. J Neurosci 31:1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaar RL, Gerardy‐Schahn R, Hildebrandt H. 2014. Sialic acids in the brain: Gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol Rev 94:461–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenfaden R, Krauter A, Schertzinger F, Gerardy‐Schahn R, Hildebrandt H. 2003. Polysialic acid directs tumor cell growth by controlling heterophilic neural cell adhesion molecule interactions. Mol Cell Biol 23:5908–5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkov O, Tikhobrazova O, Dityatev A. 2012. PSA‐NCAM: Synaptic functions mediated by its interactions with proteoglycans and glutamate receptors. Int J Biochem Cell Biol 44:591–595. [DOI] [PubMed] [Google Scholar]
- Smith CM, Cooksey E, Duncan ID. 2013. Myelin loss does not lead to axonal degeneration in a long‐lived model of chronic demyelination. J Neurosci 33:2718–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW. 2003. Mapping cortical change across the human life span. Nat Neurosci 6:309–315. [DOI] [PubMed] [Google Scholar]
- Takebayashi H, Ikenaka K. 2015. Oligodendrocyte generation during mouse development. Glia 63:1350–1356. [DOI] [PubMed] [Google Scholar]
- Tao R, Li C, Zheng Y, Qin W, Zhang J, Li X, Xu Y, Shi YY, Feng G, He L. 2007. Positive association between SIAT8B and schizophrenia in the Chinese Han population. Schizophr Res 90:108–114. [DOI] [PubMed] [Google Scholar]
- Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB, Starkey M, Webster MJ, Yolken RH, Bahn S. 2003. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 362:798–805. [DOI] [PubMed] [Google Scholar]
- Uranova NA, Vikhreva OV, Rachmanova VI, Orlovskaya DD. 2011. Ultrastructural alterations of myelinated fibers and oligodendrocytes in the prefrontal cortex in schizophrenia: A postmortem morphometric study. Schizophr Res Treatment 2011:325789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaithianathan T, Matthias K, Bahr B, Schachner M, Suppiramaniam V, Dityatev A, Steinhaüser C. 2004. Neural cell adhesion molecule‐associated polysialic acid potentiates alpha‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid receptor currents. J Biol Chem 279:47975–47984. [DOI] [PubMed] [Google Scholar]
- Wake H, Lee PR, Fields RD. 2011. Control of local protein synthesis and initial events in myelination by action potentials. Science 333:1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Rougon G, Kiss JZ. 1994. Requirement of polysialic acid for the migration of the O‐2A glial progenitor cell from neurohypophyseal explants. J Neurosci 14:4446–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhold B, Seidenfaden R, Röckle I, Mühlenhoff M, Schertzinger F, Conzelmann S, Marth JD, Gerardy‐Schahn R, Hildebrandt H. 2005. Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J Biol Chem 280:42971–42977. [DOI] [PubMed] [Google Scholar]
- Werneburg S, Buettner FF, Mühlenhoff M, Hildebrandt H. 2015a. Polysialic acid modification of the synaptic cell adhesion molecule SynCAM 1 in human embryonic stem cell‐derived oligodendrocyte precursor cells. Stem Cell Res 14:339–346. [DOI] [PubMed] [Google Scholar]
- Werneburg S, Mühlenhoff M, Stangel M, Hildebrandt H. 2015b. Polysialic acid on SynCAM 1 in NG2 cells and on neuropilin‐2 in microglia is confined to intracellular pools that are rapidly depleted upon stimulation. Glia 63:1240–1255. [DOI] [PubMed] [Google Scholar]
- Wheeler AL, Voineskos AN. 2014. A review of structural neuroimaging in schizophrenia: From connectivity to connectomics. Front Hum Neurosci 8:653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R, Krämer‐Albers EM. 2014. Axon‐glia interaction and membrane traffic in myelin formation. Front Cell Neurosci 7:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobrock T, Kamer T, Roy A, Vogeley K, Schneider‐Axmann T, Wagner M, Maier W, Rietschel M, Schulze TG Scherk H, et al. 2008. Reduction of the internal capsule in families affected with schizophrenia. Biol Psychiatry 63:65–71. [DOI] [PubMed] [Google Scholar]
- Woodward ND, Karbasforoushan H, Heckers S. 2012. Thalamocortical dysconnectivity in schizophrenia. Am J Psychiatry 169:1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SY, Huh IS, Baek JH, Cho EY, Choi MJ, Ryu S, Kim JS, Park T, Ha K, Hong KS. 2015. Association between ST8SIA2 and the risk of schizophrenia and bipolar I disorder across diagnostic boundaries. PLoS One 10:e0139413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, Tohyama K, Richardson WD. 2013. Oligodendrocyte dynamics in the healthy adult CNS: Evidence for myelin remodeling. Neuron 77:873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Vutskits L, Calaora V, Durbec P, Kiss JZ. 2004. A role for the polysialic acid‐neural cell adhesion molecule in PDGF‐induced chemotaxis of oligodendrocyte precursor cells. J Cell Sci 117:93–103. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng Y, Liu L, Yu K, Zhang L, Wang H, He X, Wang J, Lu C Wu LN, et al. 2016. Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage‐specific partners propels oligodendroglial maturation. Nat Commun 7:10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information Figure S1
Supporting Information Table S1