

Abstract
This study describes the design, production, and testing of functionalized variants of a recombinant protein‐based polymer that forms nanofibrillar hydrogels with self‐healing properties. With a view to bone tissue engineering applications, we equipped these variants with N‐terminal extensions containing either (1) integrin‐binding (RGD) or (2) less commonly studied proteoglycan‐binding (KRSR) cell‐adhesive motifs. The polymers were efficiently produced as secreted proteins using the yeast Pichia pastoris and were essentially monodisperse. The pH‐responsive protein‐based polymers are soluble at low pH and self‐assemble into supramolecular fibrils and hydrogels at physiological pH. By mixing functionalized and nonfunctionalized proteins in different ratios, and adjusting pH, hydrogel scaffolds with the same protein concentration but varying content of the two types of cell‐adhesive motifs were readily obtained. The scaffolds were used for the two‐dimensional culture of MG‐63 osteoblastic cells. RGD domains had a slightly stronger effect than KRSR domains on adhesion, activity, and spreading. However, scaffolds featuring both functional domains revealed a clear synergistic effect on cell metabolic activity and spreading, and provided the highest final degree of cell confluency. The mixed functionalized hydrogels presented here thus allowed to tailor the osteoblastic cell response, offering prospects for their further development as scaffolds for bone regeneration. © 2016 Wiley Periodicals, Inc. J Biomed Mater Res Part A: 104A: 3082–3092, 2016.
Keywords: KRSR cell‐adhesive domain, Pichia pastoris, protein‐based polymers, self‐assembled hydrogel, tissue engineering
INTRODUCTION
In natural tissues, cells are surrounded by the extracellular matrix (ECM), which ensures the mechanical support and signaling necessary for regulation of cell metabolism and functions such as migration, proliferation, differentiation, and apoptosis.1, 2, 3, 4 The ECM can be seen as a complex hydrogel containing multiple functionalities and bioactive groups at different length scales,5 which synergistically influence cell response. Mimicry of the properties and structure of the ECM remains a key objective in biomaterial design,2, 6, 7 which is why hydrogels are considered particularly promising candidates for use as scaffolds in tissue engineering.3, 8, 9, 10
As highly hydrated polymers, hydrogels are usually biocompatible, noncytotoxic, well permeable to oxygen and nutrients, and often biodegradable.5, 10 Hydrogels based on natural proteins and carbohydrates of the ECM, as well as a variety of synthetic polymers, have been investigated as biomaterials.5 Despite their generally weak mechanical strength, hydrogels have not only shown promise in soft tissue engineering but also in bone regeneration applications.11 In applications not requiring gap filling, the hydrogel must primarily facilitate delivery of cells and/or release of growth factors.11 Coating of surgical implants with hydrogels that provide biological cues, growth factors, or antibiotics can promote osseointegration.12 Composite hydrogels complexed with ceramics or other additives have been devised to improve the mechanical properties,13 although no study has yet applied hydrogels in a load‐bearing bone defect model.11 Injectable hydrogels, that is, hydrogels that form in situ by chemical or physical crosslinking, are also of interest. They conform to the shape of the defect, and allow delivery of cells by mixing cells and polymer solution prior to crosslinking.5 Self‐healing hydrogels are especially suitable for this purpose, in that they can recover from strain‐induced damage during application to the desired tissue,14 or afterwards. In the design of biomaterial scaffolds as matrices for anchor‐dependent cells such as osteoblasts, recognition sites for cell adhesion are crucial. The most commonly used peptide to improve cell adhesion is the Arg‐Gly‐Asp (RGD) motif derived from ECM proteins such as bone sialoproteins, vitronectin, and fibronectin.15 RGD‐mediated adhesion is based on the interaction with multiple integrins and promotes adhesion of different cell types. Osteoblasts also attach through another mechanism, less explored in the literature,16, 17 which involves the interaction between heparan sulfate proteoglycans on the cell membrane and heparin‐binding sites in the ECM. Based on sequences found in bone adhesive proteins, Cardin and Weintraub16 proposed that the amino acid sequence basic‐basic‐nonbasic‐basic, in terms of protonation, binds heparan sulfate. One such sequence, Lys‐Arg‐Ser‐Arg (KRSR), selectively improved adhesion of osteoblasts on model surfaces.17, 18, 19, 20
In recent years, a few studies used combinations of both RGD and KRSR adhesive motifs, in an effort to possibly create conditions more similar to the environment of the multifunctional native ECM.21, 22, 23, 24, 25, 26, 27 However, these studies involved the post‐production modification of scaffold surfaces with chemically synthesized RGD and KRSR peptides. In the present study, we report hydrogels self‐assembled from genetically engineered proteins that contain these cell‐adhesive motifs. Genetically engineered protein‐based polymers used as scaffolds represent an emerging technology in the field of tissue engineering.2, 28, 29, 30, 31, 32, 33, 34 Because amino acid sequence and molecular weight are directly defined by the genetic design, these polymers offer exquisite control over the chemistry, and desired bioactive modules can be incorporated without chemical modification.2, 28 Protein‐based polymers are in principle monodisperse, possible biological complications during production aside, which is difficult to achieve using conventional chemistry. These properties allow the generation of precisely defined, tailored materials. To our knowledge, this report presents the first recombinant protein‐based hydrogel containing KRSR domains.
The hydrogel framework is based on the genetically engineered pH‐responsive polymer C2 C2 developed by our group.35, 36 At pH 6, or above, this protein self‐assembles into stable, physical nanofibrillar hydrogels, thereby resembling nanostructural elements of the ECM. C2 C2 hydrogels are furthermore noncytotoxic and possess self‐healing properties.37 The material could thus potentially be used for example as an injectable scaffold, forming hydrogels in situ under physiological conditions, without use of chemical crosslinkers. We describe here the high‐yield biotechnological production of functionalized variants of this protein‐based polymer, and analyze the potential of the resulting fibrillar hydrogels for osteoblast cell culture. By mixing functionalized proteins, containing either RGD or KRSR motifs, and non‐functionalized proteins in different ratios, hydrogel scaffolds were conveniently prepared with precisely controlled overall concentration of the two motifs. We show that this tailoring allowed to control osteoblastic cell adhesion, activity, spreading, and formation of a confluent layer.
MATERIALS AND METHODS
Construction of recombinant strains
The production strain for triblock copolymer C2 C2 has been described by us previously.36 Two variants of the corresponding gene were designed; one encoding N‐terminal RGD‐motifs (BRGDC2 C2), and another encoding N‐terminal KRSR‐motifs (BKRSRC2 C2). These genes and corresponding strains were constructed as follows.
A double‐stranded adapter BRGD (short for cell‐binding block with RGD motifs) with HindIII and BsaI overhangs was prepared by annealing of oligonucleotides
5′‐AGCTTGAATTCGGTAGAGGAGATTCTCCTGGTGGATCCGGTGGAGGTTCTGGAGGTGGATCTGGTGGAAGAGGTGACTCACCAGGTCTCG‐3′ and
5′‐GCACCGAGACCTGGTGAGTCACCTCTTCCACCAGATCCACCTCCAGAACCTCCACCGGATCCACCAGGAGAATCTCCTCTACCGAATTCA‐3′. This adapter encodes two GRGDSP motifs separated by a (GGSG)3 flexible spacer. Likewise a double‐stranded adapter BKRSR (short for cell‐binding block with KRSR motifs) encoding three repeats of the sequence PKRSRGGG was prepared by annealing of oligonucleotides
5′‐AGCTTGAATTCGGTCCAAAGAGATCTAGAGGAGGTGGACCTAAACGTTCCAGAGGTGGAGGTCCAAAGAGGTCTAGAGGAGGTGGTCTCG‐3′ and
5′‐GCACCGAGACCACCTCCTCTAGACCTCTTTGGACCTCCACCTCTGGAACGTTTAGGTCCACCTCCTCTAGATCTCTTTGGACCGAATTCA‐3′. These adapters were inserted into the HindIII/BsaI sites of vector pMTL23‐C2 C2,36 resulting in vectors pMTL23‐BRGDC2 C2 and pMTL23‐BKRSRC2 C2, respectively. These were then digested with EcoRI/NotI, and the released inserts were cloned into the EcoRI/NotI sites of Pichia pastoris expression vector pPIC9 (Thermo Fisher Scientific). The vectors pPIC9‐BRGDC2 C2 and pPIC9‐BKRSRC2 C2 thus obtained were linearized with SalI and used to transform P. pastoris GS115 by electroporation as described previously.38
Fermentation and protein purification
Methanol fed‐batch fermentations of the BRGDC2 C2 and BKRSRC2 C2 strains at 30°C and pH 3 were performed in 2.5‐L Bioflo 3000 bioreactors (New Brunswick Scientific) as described previously for C2 C2. 37 At the end of fermentation, the cells were separated from the broth by centrifugation for 10 min at 10,000 × g (RT) in an SLA‐3000 rotor (Sorvall), and the supernatant was microfiltered.
Proteins were purified from the supernatant as previously described for C2 C2,37 with the exception that the acetone precipitation step was omitted. Shortly, proteins were selectively precipitated twice at 45% ammonium sulfate saturation and subsequently dialyzed, filter sterilized (0.2 µm), and lyophilized.
SDS‐PAGE, N‐terminal protein sequencing, amino acid analysis, and carbohydrate determination
The NuPAGE Novex system (Thermo Fisher Scientific) was used for SDS‐PAGE, with 10% Bis‐Tris gels, MES SDS running buffer, and SeeBlue Plus2 prestained molecular mass markers. Gels were stained with Coomassie SimplyBlue SafeStain (Thermo Fisher Scientific).
Purified proteins in solution were N‐terminally sequenced via Edman degradation by Midwest Analytical (St. Louis, MO). Amino acid composition analysis after acid hydrolysis was performed by Ansynth Service B.V. (Roosendaal, The Netherlands). Protein purity was estimated by linear least‐squares fitting to the obtained amino acid composition of (1) the theoretical composition of the respective pure protein and (2) the composition determined for host‐derived proteins present in the medium.
Because polysaccharides are the major nonprotein contaminant in the extracellular medium of P. pastoris cultures, the carbohydrate content in the purified protein was determined using a phenol–sulfuric acid assay as previously described.37 Proteins contained <2% (w/w) of polysaccharides.
Mass spectrometry
Matrix‐assisted laser desorption/ionization time‐of‐flight (MALDI‐TOF) mass spectrometry was performed using an ultrafleXtreme mass spectrometer (Bruker). Samples were prepared by the dried droplet method on a 600 µm AnchorChip target (Bruker), using 5 mg/mL 2,5‐dihydroxyacetophenone, 1.5 mg/mL diammonium hydrogen citrate, 25% (v/v) ethanol, and 1% (v/v) trifluoroacetic acid as matrix. Spectra were derived from ten 500‐shot (1,000 Hz) acquisitions taken at nonoverlapping locations across the sample. Measurements were made in the positive linear mode, with ion source 1, 25.0 kV; ion source 2, 23.3 kV; lens, 6.5 kV; pulsed ion extraction, 680 ns. Protein Calibration Standard II (Bruker) was used for external calibration.
Atomic force microscopy
Protein samples of (A) pure C2 C2, and (B) BKRSRC2 C/BRGDC2 C2 mixed in a 1:1 ratio were dissolved in 10 mM HCl (final pH 2 to 3). The pH of the ∼2 mL samples was adjusted to 7.4 ± 0.2 by addition of 0.1 M NaOH, using a pH meter with a micro electrode (Mettler Toledo InLab® Micro). Milli‐Q was then added to obtain protein solutions between 0.05% and 0.1% (w/v). After 24 h of incubation at room temperature, solutions were pipetted onto silica wafers and left for 20 min to allow fibril deposition. Wafers were subsequently rinsed with Milli‐Q to remove salts. The samples were dried by removing the bulk of the solution with a tissue and/or using a mild stream of nitrogen. The dry samples were analyzed with a Nanoscope V (Veeco), in Scan Asyst mode, using a nonconductive silicon nitride probe (NP‐10, Bruker) with a spring constant of 0.32 N/m. Images were recorded at a frequency of 0.997 Hz and processed with NanoScope Analysis 1.20 software (Veeco).
Rheology
Solutions of the purified proteins (A) C2 C2, (B) BRGDC2 C2, and (C) BKRSRC2 C2 were prepared in 10 mM HCl and vortexed for 1.5 h using a multi‐tube holder. The pH of the ∼2 mL samples was adjusted to 7.4 ± 0.2 by addition of 0.1M NaOH, using a pH meter with a micro electrode (Mettler Toledo InLab® Micro). PBS (phosphate buffered saline, pH 7.4, Sigma‐Aldrich) was then added to reach a 2% (w/v) protein concentration. Solutions were loaded immediately into the rheometer (Physica MCR 501 Rheometer, Anton Paar) with a Couette CC10/T200 geometry (bob diameter 10.002 mm, cup diameter 10.845 mm). Each sample was placed in the measuring cell on top of 500 µL of perfluorinated fluid (Galden® HT‐70, Solvay Specialty Polymers) to decrease the sample volume, and covered with paraffin oil to avoid evaporation. Gel formation was analyzed by recording storage moduli over time under sinusoidal deformation (frequency (f) = 1 Hz and strain (γ) = 1%). After 15 h, gels were broken (f = 1 Hz and logarithmically increasing deformation γ = 0.1 to 100%). Self‐healing behavior was then monitored by recording storage moduli under sinusoidal deformation as before. All measurements were performed at 37°C.
Preparation of scaffolds for cell culture
Freeze‐dried proteins were separately dissolved at 4% (w/v) in 10 mM HCl for at least 2 h by vortexing with a multi‐tube holder. Solutions were then mixed to obtain 1 mL samples of the required protein compositions. To allow simultaneous preparation of multiple scaffolds without pH measurement, one volume of 0.1 M NaOH was added to adjust the protein concentration to 2% (w/v) and to reach a pH above neutral in all samples. This ensures proper gel formation across samples independent of the precise pH, as self‐assembly occurs at pH 6 or above.36 The protein solutions were immediately transferred to 48‐well cell culture plates (250 µL per well) and left for 2 h at room temperature to allow full setting of the gels. These 2 h are a conservative approach, as gels already form (crossover point of storage modulus and loss modulus) within minutes (Supporting Information Figure 1). To ensure a pH value of 7.4 for cell seeding, the gels were further incubated in 0.5 mL of PBS for 3 h and then in 0.5 mL of cell culture medium (α‐MEM, 22571, Thermo Fisher Scientific) with 10% (v/v) fetal bovine serum (FBS) for 15 h (with one medium change after 12 h).
Cell pre‐culture
Cryo‐preserved MG‐63 cells (ATCC® CRL‐1427™; LGC Standards GmbH), passage 90, were cultured for 6 days in proliferation medium (α‐MEM, 22571, Thermo Fisher Scientific), supplemented with 10% (v/v) FBS, at 37°C, 95% relative humidity and 5% CO2. Prior to seeding, cells were washed twice with PBS, enzymatically detached for 5 min with trypsin‐EDTA (0.25% (w/v) trypsin, 0.02% (w/v) EDTA), and resuspended at 36,000 cells/mL in osteogenic medium. The medium was composed of α‐MEM supplemented with 10% (v/v) FBS, 100 U/mL penicillin and 10 µg/mL streptomycin (Thermo Fisher Scientific), 50 mg/L ascorbic acid (Sigma‐Aldrich), 10 nM dexamethasone (Sigma), 10 mM β‐glycerophosphate disodium salt hydrate (Sigma). Next, 500 µL of cell suspension was added per well (final seeding density: 20,000 cells/cm2). Scaffolds incubated in osteogenic medium without cells were included as controls. All samples were prepared in triplicate (n = 3). The entire study was run for 23 days in an incubator at 37°C, 95% relative humidity and 5% CO2. The culture medium was refreshed twice a week for all samples.
Cell adhesion
Cell adhesion was analyzed 4 h after cell seeding. The medium was removed from a dedicated set of wells to take out nonadherent cells. Gel surfaces were imaged by randomly taking light microscopy pictures (n = 3 per sample) using an inverted phase‐contrast microscope (Leica DMIL). Pictures were taken close to the center of the gels to ensure that the imaged area was relatively flat. The number of cells on the area of 2,048 μm × 1,536 μm was counted in manual mode using ImageJ 1.49o software. Cell adhesion was calculated as the number of cells per mm2.
Cell metabolic activity
To assess cell metabolic activity, an alamarBlue® (Thermo Fisher Scientific) assay was performed on a dedicated set of wells on days 1, 3, 10, and 23, following the protocol of the manufacturer. Shortly, the cell culture medium in the wells was exchanged for fresh medium containing 10% (v/v) of alamarBlue® reagent, followed by incubation at 37ºC for color development. In view of high cell activity on day 23, an incubation time of only 1.5 h was used for these samples, whereas for the earlier days 4 h was used. The medium containing the reagent was subsequently removed for measuring fluorescence (ex 560 nm, em 590 nm; FLx800 reader, Bio‐Tek Instruments), and all values were normalized to activity per hour. To compare cell activity on different scaffolds, the average fluorescence of the medium was calculated (n = 3).
Cell viability and spreading
To study cell viability, and to visualize changes over time in cell distribution and morphology (spreading), a LIVE/DEAD Viability/Cytotoxicity Kit (L3224, Thermo Fisher Scientific) was used on days 1 and 3, and only the calcein‐AM compound from the kit on days 10, and 23. Fluorescent staining of F‐actin and nuclei was performed on samples from days 1, 3, and 23 for further confirmation of cell distribution and spreading.
For the LIVE/DEAD assay, three samples of each gel type (n = 3) were washed twice with PBS and afterwards incubated in 0.5 mL of PBS with 2 mM calcein‐AM and, optionally, 4 mM ethidium homodimer, in the dark, for 30 min at 37°C. Next, scaffolds were washed twice with PBS and cells were visualized using a fluorescence microscope (Axio Imager Microscope Z1; Carl Zeiss Micro Imaging GmbH). Overlay images were prepared, with dead cells appearing in red (ex 517 nm, em 617 nm) and live cells in green (ex 488 nm, em 517 nm).
Samples for fluorescent staining of F‐actin and nuclei were prepared as follows. First, the cell culture medium was removed from the cell culture well‐plate. Subsequently, the scaffolds in the wells were washed gently with PBS, fixed for 1.5–4 h with 0.7 mL per well of 3.3% PFA (paraformaldehyde) in PBS, and then kept in 70% (v/v) ethanol until the day of imaging (for a maximum of 5 days). Prior to imaging the gels, ethanol was removed, scaffolds were washed twice in PBS, and cells were permeabilized for 20–30 min with 0.8 mL per well of 1% Triton X‐100 in PBS containing 1% FBS. Scaffolds were then incubated for 2 h with Alexa Fluor® 568 conjugated phalloidin (Thermo Fisher Scientific) for F‐actin staining and with DAPI (Thermo Fisher Scientific) for visualization of nuclei. Both reagents were diluted in PBS containing 1% FBS (1:200 and 1:2,500, respectively), and 0.5 mL of this mixture was added per well. After washing in PBS for 10 min, scaffolds were examined using an Axio Imager Microscope Z1 (Carl Zeiss Micro Imaging GmbH).
Statistical analysis
Statistical analysis was performed using SPSS Statistics software. Statistical differences were estimated using one‐way analysis of variance followed by an LSD post‐hoc test. Differences were considered significant at p < 0.05, if not stated differently. Results were reported as the mean ± standard deviation.
RESULTS
Polymer design
We previously reported the molecular design of the fibril‐forming protein C2 C2.35, 36 It features an (silk‐inspired) middle block consisting of 48 repeats of the octapeptide GAGAGAGH. Whereas at low pH the proteins are soluble owing to repulsion by the positively charged histidines, at neutral pH the charges are neutralized36 and the domains form β‐rolls or β‐sheets.39, 40, 41 These, in turn, form the fibril core through stacking, mediated by hydrophobic interactions. The block is flanked on both sides by hydrophilic C2 (random coil) blocks, each consisting of two 99 amino acid‐long unfolded domains in tandem, which form a corona surrounding the core and provide colloidal stability [Fig. 1(A)]. At higher protein concentrations (above several g/L), gels are formed, most likely as a result of the entanglement of long fibrils,42 and possibly the occurrence of weak physical crosslinks.36
Figure 1.
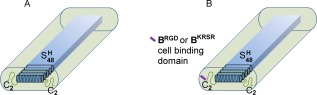
Tentative structure of fibrils at neutral pH (not to scale). A: unmodified C2 C2; B: functionalized variants. Representation based on the model proposed for the related polymer C2 C2.39
Here, we designed two functionalized variants of C2 C2 with N‐terminal cell‐binding extensions (B domains, for short). One variant, BRGDC2 C2, contains integrin‐binding RGD motifs,43 and the other, BKRSRC2 C2 contains proteoglycan‐binding KRSR motifs17 [Fig. 1(B)]. For the design, the following considerations were taken into account: (1) To potentially increase cell interaction, the design should preferably feature several copies of these motifs. (2) The length of the extensions should be limited, so as to minimize possible effects on the well‐characterized fibril‐forming properties of the C2 C2 core component. (3) The repeated cell‐binding motifs should be separated by suitable flexible linkers to allow sufficient conformational freedom.
In BRGDC2 C2, a flexible Gly/Ser‐rich sequence was chosen to separate the RGD motifs because this type of spacer is widely used as a constituent of engineered proteins produced in various hosts including P. pastoris.44, 45 A spacer length of 12 residues in the form of three GGSG repeats was used. Two RGD motifs were incorporated, where the specific motif used was GRGDSP from fibronectin.43 The N‐terminal extension relative to C2 C2 thus became: GRGDSPGGSGGGSGGGSGGRGDSP, which adds ∼3% to the length of the protein sequence. This sequence is N‐terminally preceded by the cloning‐derived sequence YVEF, as is the case in C2 C2.39
In the design of BKRSRC2 C2, poly‐glycine stretches were used as spacers between the KRSR motifs, in view of a study that reports osteoblast adhesion onto glass surfaces with C‐terminally immobilized KRSR, KRSRG3, or KRSRG6 peptides.17 However, as the authors found similar adhesion efficiency for all peptides tested, spacer length does not appear to be critical. Rather, because a tetravalent poly‐lysine peptide core with four attached KRSR peptides performed significantly better, the authors suggest peptide density may be more important. We, therefore, incorporated three KRSRG3 motifs, while maintaining the same small length of the N‐terminal extension, namely 24 residues, as used in BRGDC2 C2. Another critical design consideration was to prevent proteolysis of Lys‐Arg in the KRSR motifs by the P. pastoris homolog of Kex2 protease. This serine endoprotease was first identified in S. cerevisiae as the protease required for maturation of the α‐mating factor pheromone, via cleavage at the C‐terminal side of Lys‐Arg.46 As we have shown previously,38, 47 P. pastoris Kex2 can also digest particular mono‐arginyl sites, as governed to a large extent by the residues at positions −2 to −4 relative to the scissile bond. Under the assumption that P. pastoris and S. cerevisiae Kex2 have similar substrate specificity, the extent of putative cleavage at the C‐terminal side of the KRSR sequences is likely low, in view of the moderately unfavorable Ser at position −2 relative to the scissile bond.48 As for potential digestion of the canonical Lys‐Arg motif in KRSR, we reasoned it might be possible to minimize this by placing highly unfavorable residues at positions −4 and −3. We thus arrived at the following sequence design for the N‐terminal BKRSR extension: GPKRSRGGGPKRSRGGGPKRSRGG (again N‐terminally preceded by the cloning‐derived sequence YVEF, and C‐terminally followed by the C2 C2 sequence starting with Gly). For all three Lys‐Arg motifs in this sequence, position −4 relative to the scissile bond is occupied by Gly, and position −3 by Pro. Both amino acids rank as highly unfavorable in these positions, at least for S. cerevisiae Kex2.49
Protein synthesis and characterization
The polymers were produced as secreted proteins in methanol fed‐batch bioreactor cultures of P. pastoris, and purified as previously described.37 Based on the determined amino acid composition, the purity of BRGDC2 C2 was estimated at 99.8%, and that of BKRSRC2 C2 at 99.3%. The gravimetrically determined yields of the purified, desalted, and lyophilized material, expressed per volume of cell‐free fermentation broth, were 3.2 g/L for BRGDC2 C2 and 3.5 g/L for BKRSRC2 C2.
The functionalized proteins and unmodified C2 C2 were analyzed by SDS‐PAGE (Fig. 2). The main bands of all three proteins migrated in SDS‐PAGE at an apparent molecular weight of ∼105 kDa, although the calculated masses are ∼66 to ∼68 kDa. As described previously for C2 C2,36, 37 this discrepancy is due to the high content of hydrophilic and small amino acids in this protein. The faint upper band and low mass smear visible in all proteins are probably SDS‐PAGE artifacts, as has previously been shown for C2 C2.37 N‐terminal sequencing of BRGDC2 C2 showed the expected YVEFGRGD sequence without any secondary reactions, confirming that the protein was N‐terminally intact. Likewise, N‐terminal sequencing of BKRSRC2 C2 revealed mainly the expected YVEFGPKRSR sequence, which shows that most of the N‐terminal BKRSR extension was N‐terminally intact. However, besides spurious background signals, a minor secondary species, most likely corresponding to SRGG, was also observed in this sample. This sequence may have resulted from a limited extent of Kex2 cleavage at the C‐terminal side of the dibasic GPKR sequences. The molecular mass of the proteins was verified by MALDI‐TOF (Fig. 3). For both BRGDC2 C2 and BKRSRC2 C2, the observed masses of the main peaks (67,932 and 68,412 Da, respectively) correspond, within experimental error, to the calculated masses of the intact proteins (68,061 and 68,535 Da, respectively). The MALDI‐TOF spectra furthermore show that BRGDC2 C2 was fully monodisperse, and BKRSRC2 C2 nearly so. The slight shoulder to the left of the main peak in the spectrum of BKRSRC2 C2 (indicated by an arrow in Fig. 3) contains three peaks whose masses correspond to that of the protein after N‐terminal truncation following either the first, second, or third GPKR motif. This result is in agreement with the minor SRGG species observed in N‐terminal sequencing. Additionally, three minor peaks are present, with masses that could correspond to N‐terminal truncation following either the first, second, or third KRSR motif. Apparently, Kex2 cleavage occurred at the C‐terminal side of the dibasic GPKR and monobasic KRSR motifs, but only at a very low level. The shoulder to the right side of the main peak of both proteins (Fig. 3) corresponds to Glu‐Ala extended species in a small portion of the molecules, as observed previously for C2 C2,37 and results from incomplete processing of the α‐mating factor propeptide.38
Figure 2.
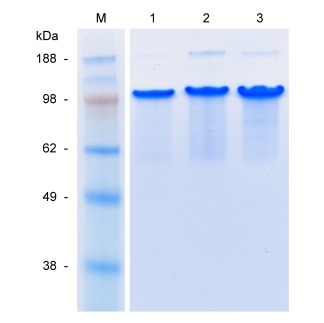
SDS–PAGE of purified proteins. Lane 1: C2 C2; lane 2: BRGDC2 C2; lane 3: BKRSRC2 C2; lane M: molecular weight marker. For all samples, ∼5 μg of protein was loaded.
Figure 3.
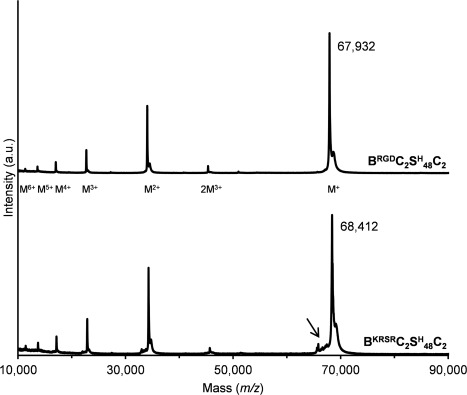
MALDI‐TOF of purified BRGDC2 C2 and BKRSRC2 C2. Molecular ions are indicated. The arrow points to a minor shoulder discussed in the text.
Material characterization
AFM and rheology were performed to compare the fibril formation and mechanical properties of BRGDC2 C2 and BKRSRC2 C2 with those previously established for C2 C2. 36, 37
For AFM (Fig. 4), dilute solutions in 10 mM HCl were prepared of the unmodified C2 C2 protein, and of a 1:1 mixture of the functionalized proteins BRGDC2 C2 and BKRSRC2 C2. After raising the pH to a physiological level of 7.4 and 24 h of incubation, both samples showed self‐assembled fibrils in AFM with a length in the micrometer range, as described previously for C2 C2.36, 37 This indicates that the fibril forming capacity was not affected by functionalization of the molecules.
Figure 4.

AFM of 0.1% (w/v) protein solutions after 24 h of incubation at pH ∼7.4 and room temperature: A: C2 C2, and B: C/BRGDC2 C2 mixed in a 1:1 ratio.
A rheological study (Fig. 5) revealed that gel formation is as well relatively unaffected by the addition of functional domains to C2 C2. After adjusting the pH to a physiological level of 7.4, 2% (w/v) solutions of C2 C2, BRGDC2 C2, and BKRSRC2 C2 all developed storage moduli in the range of 500–1,500 Pa. The observed differences in the final storage modulus and in the kinetics of gel formation can be attributed to batch‐to‐batch variations. Also, minor differences in pH adjustment could play a role, in view of the known influence of pH on the storage modulus of C2 C2.36 All proteins achieved a storage modulus above 200 Pa already after 1–2 h, and reached a plateau after 5 h. Gelation, defined as the crossover point of storage modulus and loss modulus, occurred within minutes after adjusting pH (see Supporting Information Figure 1). As has been shown for C2 C2,36 also BRGDC2 C2 and BKRSRC2 C2 retained the ability to quickly recover after breaking, reaching storage moduli of 58–83% of the initial values.
Figure 5.

Rheometry of 2% gels of C2 C2, BRGDC2 C2, and BKRSRC2 C2. Development of storage moduli as a function of time during gel formation, and recovery after strain‐induced breakage of the gel at t ∼15 h.
Cell culture
Scaffolds for two‐dimensional cell culture studies were obtained by preparing 2% (w/v) gels with six different protein compositions, as listed in Table 1. This design allowed to determine the influence of the incorporated functional domains on cell behavior. By mixing BRGDC2 C2 and BKRSRC2 C2, hydrogels are obtained that possess both RGD and KRSR motifs. By mixing C2 C2 with either BRGDC2 C2 or BKRSRC2 C2, a reduced content of these cell‐adhesive domains is obtained.
Table 1.
Protein Composition of 2% (w/v) Cell Culture Scaffolds
| Scaffold | Relative Composition | ||||
|---|---|---|---|---|---|
|
C2
|
BRGDC2
|
BKRSRC2
|
|||
| 0R/0K | 100% | – | – | ||
| 50R | 50% | 50% | – | ||
| 100R | – | 100% | – | ||
| 50K | 50% | – | 50% | ||
| 100K | – | – | 100% | ||
| 50R/50K | – | 50% | 50% | ||
As we aim at developing materials for future use as bone cell scaffolds, and investigate the influence of KRSR as bone cell‐specific adhesive motifs, MG‐63 osteoblastic cells were used in the cell culture studies. The influence of the active domains on early cell response was studied by evaluating cell adhesion (Fig. 6). As compared to the unmodified 0R/0K gel, the number of adherent cells was significantly higher on the functionalized gels, except for 50K. The simultaneous presence of both RGD and KRSR did not have a synergistic effect on cell adhesion.
Figure 6.
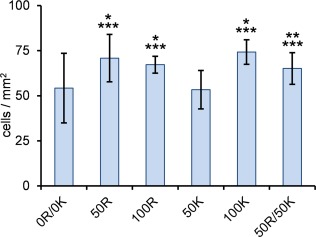
Number of cells adherent to the scaffold per mm2 after 4 h of incubation. * p < 0.05, compared to 0R/0K; ** p = 0.07 compared to 0R/0K; *** p < 0.05 compared to 50K; (n = 3).
The largest variation in the number of adherent MG‐63 cells was found in unmodified 0R/0K. Apparently, the distribution of cells on the surface of the scaffold was not uniform. In accordance with our previous study, where rat bone marrow stem cells (MSC) were grown on nonfunctionalized C2 C2,37 this suggests that scaffolds containing C2 C2 without adhesive domains are not particularly favorable for cell attachment. In contrast, the smallest variation was found for the 100R scaffold, which indicates a consistent positive effect from the RGD domains.
The effect of addition of RGD and KRSR domains on cell metabolic activity, as a measure of viability and possible proliferation, was tested with the alamarBlue® assay (Fig. 7). In this assay, the oxidized reagent resazurin is added to the culture medium, which after entering viable cells is reduced to resorufin by mitochondrial enzymes. This reduction can be observed as a change in color.50
Figure 7.
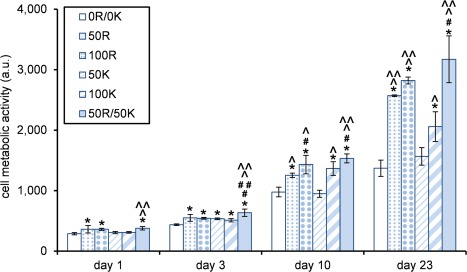
Cell metabolic activity as determined by the alamarBlue® assay. * Significantly higher compared to 0R/0K; # significantly higher compared to 50R; ## significantly higher compared to 100R; ^ significantly higher compared to 50K; ^ ^ significantly higher compared to 100K; (n = 3, p < 0.05).
Already after one day of cell culture, significant differences in cell activity were observed. Cells on the 50R, 100R, and 50R/50K scaffolds (i.e., scaffolds containing RGD domains) were metabolically more active in comparison with unmodified material. On day 23, 50R and 100R showed significantly more impact on cell activity than 50K and 100K, respectively, suggesting a stronger influence of RGD than of KRSR domains. Nonetheless, a synergistic effect of the scaffold modified with both functionalities (KRSR and RGD) became apparent from day 1 onward. With significantly higher metabolic activity of cells on 50R/50K compared to most of the other gel variants at each time point, there is a clear tendency for superior metabolic responses to our material when it contains both functionalities.
Cell survival was assessed after 1 and 3 days with a LIVE/DEAD assay. As visible in Figure 8, for all scaffold types, only a few individual cells were identified as dead (red‐stained nuclei), and the rest remained alive (green cells). For C2 C2, this result is in agreement with previous findings for scaffolds prepared from that protein.37 As expected, modification with RGD and/or KRSR motifs did not negatively affect the cytocompatibility of the gels.
Figure 8.
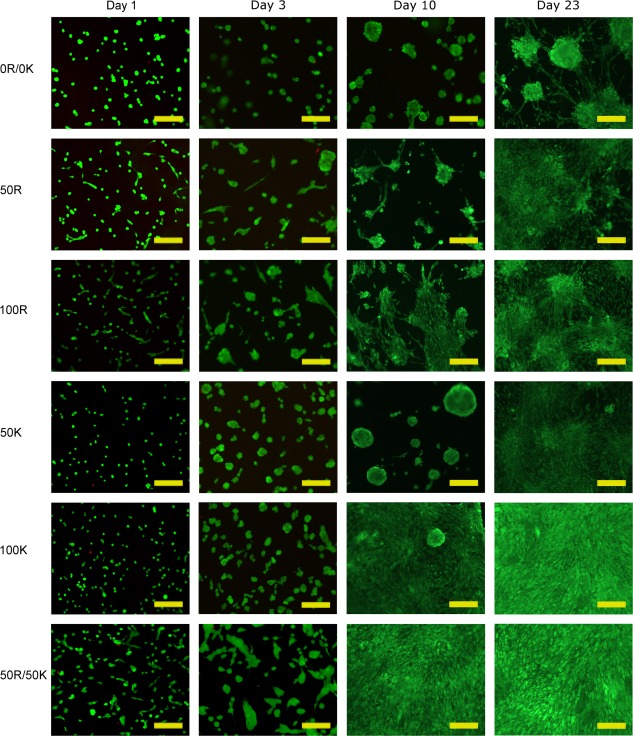
Cell survival, morphology, and distribution as monitored by the LIVE/DEAD assay. Viable cells show green fluorescence, whereas dead cells appear red (tested on days 1 and 3 only). Scale bar = 200 μm.
The LIVE/DEAD images (Fig. 8) also revealed changes over time in terms of cell morphology and cell distribution. Already on day 1, cells seeded on the scaffolds containing RGD motifs (50R, 100R, 50R/50K) started to show a more stretched morphology. On day 3, some cells with elongated shape were observed also on the gels exclusively containing KRSR domains (50K, 100K), although to a lesser extent than on scaffolds containing RGD. On day 3, the scaffold containing both RGD and KRSR motifs (50R/50K) yielded the highest degree of confluency and thus seemed most profitable for the cells. As of day 10, these samples showed a fully confluent layer. Also the 100K scaffold highly favored cell spreading and formation of a confluent layer on days 10 and 23. Interestingly, 50K scaffolds instead led to formation of clusters, and did not show cell spreading until day 23. A similar overall trend as apparent in Figure 8 was observed with fluorescent phalloidin/DAPI staining for the visualization of actin filaments and nuclei (Supporting Information Figure 2).
DISCUSSION
In previous work, we have shown that the C2 C2 protein‐based polymer can form self‐assembling, noncytotoxic fibrillar gels allowing to maintain viable cells for at least 21 days.37 However, the cells did not reveal a spread morphology over time, and did not proliferate to form a confluent layer. To further develop our material toward use as a scaffold in bone cell culture, we created in this study two variants of the protein with a genetic design that includes either integrin‐binding RGD motifs, or proteoglycan‐binding bone cell‐specific KRSR motifs.
The polymers were efficiently produced as secreted proteins in P. pastoris at up to 3.5 g/L of cell‐free broth. The possibility to obtain multiple grams of these polymers from benchtop bioreactors readily allows their use in preclinical research. The BRGDC2 C2 polymer produced was completely monodisperse. Also BKRSRC2 C2 was found to be nearly intact, apart from a very minor level of proteolytic cleavage in the KRSR motif C‐terminal of Lys‐Arg, and probably also C‐terminal of Ser‐Arg. As described in Results, proteolytic processing of these motifs in the N‐terminal random‐coil C2 block by the endogenous Kex2 protease of P. pastoris had been anticipated. The fact that this protease is responsible for processing at Lys‐Arg of the α‐mating factor prepro peptide, typically used to drive high‐level secretion of heterologous proteins in this host, only attests to the enzyme's processivity and/or abundance. In our sequence design, we therefore attempted to particularly minimize cleavage to the C‐terminal side of the canonical Lys‐Arg motif in KRSR by placing expectedly unfavorable residues at position −4 and −3, relative to the scissile bond. Because only a negligible level of degradation was observed, this approach was apparently remarkably effective. To our knowledge, this represents the first reported case where Kex2 cleavage of a dibasic Lys‐Arg motif in a recombinant protein was largely prevented by the careful design of the preceding sequence context. Importantly, the essentially monodisperse nature of the polymers adds to the level of control gained over the properties of the scaffolds produced from them.
By mixing nonfunctionalized C2 C2 with BRGDC2 C2 and/or BKRSRC2 C2 in a chosen ratio (Table 1), tailored fibrillar scaffolds for cell culture were obtained with different concentrations of the functional groups, independent of the total polymer concentration, and thus, in principle independent of properties such as nominal pore size and storage modulus. The storage moduli of gels prepared from either of the functionalized proteins were indeed in the same range as that of C2 C2, also in accordance with the observation that functionalized and nonfunctionalized proteins showed similar fibril‐forming properties in AFM. Apparently, the functional domains have little or no effect on the physico‐chemical characteristics of the proteins. This suggests that also other domains could be incorporated in the future, allowing convenient tuning of multiple functionalities by mixing. BRGDC2 C2 and BKRSRC2 C2 gels also retained the self‐healing characteristics of unmodified C2 C2.37 This may be a useful property for potential biomedical use, allowing recovery of the gels if inadvertently broken during in situ application or afterwards. While the mixing approach allows to instantaneously prepare hybrid scaffolds containing both RGD and KRSR domains, we do not claim that hybrid fibrils are certainly formed. Stochastically, however, this will most likely be the case, as each fibril consists of thousands of polymer molecules. To the cells it is probably not relevant whether both domains are present in the same or separate fibrils, as each cell is in contact with countless nanofibrils. In either case, there will likely be a certain optimal adhesive domain content, which can be reproduced at will by mixing the proteins in the appropriate ratio.
We previously observed that proliferation and mineralization of primary rat bone MSC were relatively low on nonfunctionalized C2 C2 in comparison with a collagen control, and anticipated that the cell response may be improved by functionalization of the polymer with cell‐adhesive motifs.37 Indeed, cell adhesion, metabolic activity, and spreading studied here using MG‐63 osteoblastic cells were all enhanced on the scaffolds containing either of the functional domains (KRSR or RGD). RGD domains had a slightly more pronounced influence than KRSR. Based on similar observations, Rezania et al.51 suggested that the principle binding domain is RGD, and that heparin‐binding sites are secondary.
A clear synergistic effect on cell metabolic activity and spreading was apparent from day 1 onwards when RGD and KRSR domains were simultaneously present in the scaffolds. Incorporating both integrin‐binding and heparin‐binding domains may create conditions more similar to the environment of the multifunctional native ECM and may thus be more profitable for the cells. Couchman and Woods suggested that integrins and proteoglycans strongly cooperate during regulation of cell adhesion and spreading.52
Interestingly, although cell activity and spreading in our study clearly benefited from the presence of both functional domains, no synergistic effect was seen for adhesion. Possibly, proteoglycan‐binding motifs influence the cell response only after initial adhesion and particularly play a role in mediating cell spreading.26 Similarly, cell adhesion, activity, and spreading in our study did not appear to have a simple causal relation to the degree of confluency. As mentioned above, RGD had a slightly more pronounced influence than KRSR on cell adhesion, activity, and spreading. However, the highest degree of confluency on days 10 and 23 was seen on scaffolds 100K and 50R/50K.
CONCLUSIONS
We described the design and production of functionalized variants of the genetically engineered polymer C2 C2, and evaluated the performance of osteoblastic cells on self‐assembled fibrillar hydrogels formed by these proteins. Functionalization involved incorporation of either integrin‐binding (RGD) or bone cell‐specific heparin‐binding (KRSR) sequences into the genetic design. Both proteins, efficiently secreted by Pichia pastoris at g/L levels, were essentially monodisperse, and formed self‐supporting gels under physiological conditions. Control over the overall concentration of functional groups, independently of the total polymer concentration, was readily achieved by mixing functionalized and nonfunctionalized proteins in various ratios. Functionalization promoted attachment, metabolic activity, and spreading of osteoblastic cells. The combined cell culture studies suggest that the scaffold containing both bioactive domains was most profitable. Overall, these results hold promise for further development of the material toward bone tissue engineering applications.
Supporting information
Supporting Information
ACKNOWLEDGMENT
The authors thank Prof. E. Pamuła for the cooperation with AGH University of Science and Technology and C. J. Slingerland for assistance with AFM imaging.
How to cite this article: Włodarczyk-Biegun MK, Werten MWT, Posadowska U, Storm IM, de Wolf FA, van den Beucken JJJP, Leeuwenburgh SCG, Cohen Stuart MA, Kamperman M. 2016. Nanofibrillar hydrogel scaffolds from recombinant protein‐based polymers with integrin‐ and proteoglycan‐binding domains. J Biomed Mater Res Part A 2016:104A:3082–3092.
REFERENCES
- 1. Cai R, Nakamoto T, Kawazoe N, Chen G. Influence of stepwise chondrogenesis‐mimicking 3D extracellular matrix on chondrogenic differentiation of mesenchymal stem cells. Biomaterials 2015; 52:199–207. [DOI] [PubMed] [Google Scholar]
- 2. Sengupta D, Heilshorn SC. Protein‐engineered biomaterials: Highly tunable tissue engineering scaffolds. Tissue Eng. Part B Rev 2010; 16:285–293. [DOI] [PubMed] [Google Scholar]
- 3. Patterson J, Martino MM, Hubbell JA. Biomimetic materials in tissue engineering. Mater Today 2010; 13:14–22. [Google Scholar]
- 4. Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol 2011; 209:139–151. [DOI] [PubMed] [Google Scholar]
- 5. Jia X, Kiick KL. Hybrid multicomponent hydrogels for tissue engineering. Macromol Biosci 2009; 9:140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shin H, Jo S, Mikos AG. Biomimetic materials for tissue engineering. Biomaterials 2003; 24:4353–4364. [DOI] [PubMed] [Google Scholar]
- 7. Langer R, Tirrell DA. Designing materials for biology and medicine. Nature 2004; 428:487–492. [DOI] [PubMed] [Google Scholar]
- 8. Zhu J, Marchant RE. Design properties of hydrogel tissue‐engineering scaffolds. Expert Rev Med Devices 2011; 8:607–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geckil H, Xu F, Zhang X, Moon S, Demirci U. Engineering hydrogels as extracellular matrix mimics. Nanomedicine (Lond) 2010; 5:469–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drury JL, Mooney DJ. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003; 24:4337–4351. [DOI] [PubMed] [Google Scholar]
- 11. Gibbs DMR, Black CRM, Dawson JI, Oreffo ROC. A review of hydrogel use in fracture healing and bone regeneration. J Tissue Eng Regen Med 2016; 10:187–198. [DOI] [PubMed] [Google Scholar]
- 12. Agarwal R, García AJ. Biomaterial strategies for engineering implants for enhanced osseointegration and bone repair. Adv Drug Deliv Rev 2015; 94:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Short AR, Koralla D, Deshmukh A, Wissel B, Stocker B, Calhoun M, Dean D, Winter JO. Hydrogels that allow and facilitate bone repair, remodeling, and regeneration. J Mater Chem B 2015; 3:7818–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tseng TC, Tao L, Hsieh FY, Wei Y, Chiu IM, Hsu SH. An injectable, self‐healing hydrogel to repair the central nervous system. Adv Mater 2015; 27:3518–3524. [DOI] [PubMed] [Google Scholar]
- 15. Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol 1996; 12:697–715. [DOI] [PubMed] [Google Scholar]
- 16. Cardin AD, Weintraub HJR. Molecular modeling of protein‐glycosaminoglycan interactions. Arteriosclerosis 1989; 9:21–32. [DOI] [PubMed] [Google Scholar]
- 17. Dee KC, Andersen TT, Bizios R. Design and function of novel osteoblast‐adhesive peptides for chemical modification of biomaterials. J Biomed Mater Res 1998; 40:371–377. [DOI] [PubMed] [Google Scholar]
- 18. Dettin M, Conconi MT, Gambaretto R, Pasquato A, Folin M, Di Bello C, Parnigotto PP. Novel osteoblast‐adhesive peptides for dental/orthopedic biomaterials. J Biomed Mater Res 2002; 60:466–471. [DOI] [PubMed] [Google Scholar]
- 19. Hasenbein ME, Andersen TT, Bizios R. Micropatterned surfaces modified with select peptides promote exclusive interactions with osteoblasts. Biomaterials 2002; 23:3937–3942. [DOI] [PubMed] [Google Scholar]
- 20. Palchesko RN, Romeo JD, McGowan KA, Gawalt ES. Increased osteoblast adhesion on physically optimized KRSR modified calcium aluminate. J Biomed Mater Res Part A 2012; 100A:1229–1238. [DOI] [PubMed] [Google Scholar]
- 21. Schuler M, Hamilton DW, Kunzler TP, Sprecher CM, de Wild M, Brunette DM, Textor M, Tosatti SGP. Comparison of the response of cultured osteoblasts and osteoblasts outgrown from rat calvarial bone chips to nonfouling KRSR and FHRRIKA‐peptide modified rough titanium surfaces. J Biomed Mater Res Part B 2009; 91B:517–527. [DOI] [PubMed] [Google Scholar]
- 22. Shin JH, Lee JW, Jung JH, Cho DW, Lim G. Evaluation of cell proliferation and differentiation on a poly(propylene fumarate) 3D scaffold treated with functional peptides. J Mater Sci 2011; 46:5282–5287. [Google Scholar]
- 23. Heuts J, Salber J, Goldyn AM, Janser R, Moller M, Klee D. Bio‐functionalized star PEG‐coated PVDF surfaces for cytocompatibility‐improved implant components. J Biomed Mater Res Part A 2010; 92A:1538–1551. [DOI] [PubMed] [Google Scholar]
- 24. Broggini N, Tosatti S, Ferguson SJ, Schuler M, Textor M, Bornstein MM, Bosshardt DD, Buser D. Evaluation of chemically modified SLA implants (modSLA) biofunctionalized with integrin (RGD)‐ and heparin (KRSR)‐binding peptides. J Biomed Mater Res Part A 2012; 100A:703–711. [DOI] [PubMed] [Google Scholar]
- 25. Bell BF, Schuler M, Tosatti S, Textor M, Schwartz Z, Boyan BD. Osteoblast response to titanium surfaces functionalized with extracellular matrix peptide biomimetics. Clin Oral Implant Res 2011; 22:865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sawyer AA, Hennessy KM, Bellis SL. The effect of adsorbed serum proteins, RGD and proteoglycan‐binding peptides on the adhesion of mesenchymal stem cells to hydroxyapatite. Biomaterials 2007; 28:383–392. [DOI] [PubMed] [Google Scholar]
- 27. Kim JW, Ki CS, Park YH, Kim HJ, Um IC. Effect of RGDS and KRSR peptides immobilized on silk fibroin nanofibrous mats for cell adhesion and proliferation. Macromol Res 2010; 18:442–448. [Google Scholar]
- 28. Romano NH, Sengupta D, Chung C, Heilshorn SC. Protein‐engineered biomaterials: Nanoscale mimics of the extracellular matrix. Biochim Biophys Acta 2011; 1810:339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qiu W, Huang Y, Teng W, Cohn CM, Cappello J, Wu X. Complete recombinant silk‐elastinlike protein‐based tissue scaffold. Biomacromolecules 2010; 11:3219–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gomes S, Leonor IB, Mano JF, Reis RL, Kaplan DL. Natural and genetically engineered proteins for tissue engineering. Prog Polym Sci 2012; 37:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Werkmeister JA, Ramshaw JAM. Recombinant protein scaffolds for tissue engineering. Biomed Mater 2012; 7:012002 [DOI] [PubMed] [Google Scholar]
- 32. Maskarinec SA, Tirrell DA. Protein engineering approaches to biomaterials design. Curr Opin Biotechnol 2005; 16:422–426. [DOI] [PubMed] [Google Scholar]
- 33. Nettles DL, Chilkoti A, Setton LA. Applications of elastin‐like polypeptides in tissue engineering. Adv Drug Deliv Rev 2010; 62:1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spiess K, Lammel A, Scheibel T. Recombinant spider silk proteins for applications in biomaterials. Macromol Biosci 2010; 10:998–1007. [DOI] [PubMed] [Google Scholar]
- 35. Yan Y, Martens AA, Besseling NAM, de Wolf FA, de Keizer A, Drechsler M, Stuart MAC. Nanoribbons self‐assembled from triblock peptide polymers and coordination polymers. Angew Chem Int Ed 2008; 47:4192–4195. [DOI] [PubMed] [Google Scholar]
- 36. Golinska MD, Wlodarczyk‐Biegun MK, Werten MWT, Cohen Stuart MA, de Wolf FA, de Vries R. Dilute self‐healing hydrogels of silk‐collagen‐like block copolypeptides at neutral pH. Biomacromolecules 2014; 15:699–706. [DOI] [PubMed] [Google Scholar]
- 37. Wlodarczyk‐Biegun MK, Werten MWT, de Wolf FA, van den Beucken JJJP, Leeuwenburgh SCG, Kamperman M, Cohen Stuart MA. Genetically engineered silk‐collagen‐like copolymer for biomedical applications: Production, characterization and evaluation of cellular response. Acta Biomater 2014; 10:3620–3629. [DOI] [PubMed] [Google Scholar]
- 38. Werten MWT, van den Bosch TJ, Wind RD, Mooibroek H, de Wolf FA. High‐yield secretion of recombinant gelatins by Pichia pastoris . Yeast 1999; 15:1087–1096. [DOI] [PubMed] [Google Scholar]
- 39. Martens AA, Portale G, Werten MWT, de Vries RJ, Eggink G, Cohen Stuart MA, de Wolf FA. Triblock protein copolymers forming supramolecular nanotapes and pH‐responsive gels. Macromolecules 2009; 42:1002–1009. [Google Scholar]
- 40. Razzokov J, Naderi S, van der Schoot P. Prediction of the structure of a silk‐like protein in oligomeric states using explicit and implicit solvent models. Soft Matter 2014; 10:5362–5374. [DOI] [PubMed] [Google Scholar]
- 41. Zhao B, Cohen Stuart MA, Hall CK. Dock 'n roll: Folding of a silk‐inspired polypeptide into an amyloid‐like beta solenoid. Soft Matter 2016; 12:3721–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raghavan SR, Douglas JF. The conundrum of gel formation by molecular nanofibers, wormlike micelles, and filamentous proteins: Gelation without cross‐links? Soft Matter 2012; 8:8539–8546. [Google Scholar]
- 43. Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984; 309:30–33. [DOI] [PubMed] [Google Scholar]
- 44. Whyteside G, Alcocer MJC, Kumita JR, Dobson CM, Lazarou M, Pleass RJ, Archer DB. Native‐state stability determines the extent of degradation relative to secretion of protein variants from Pichia pastoris . PLoS One 2011; 6:e22692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Niu B, Huang Y, Zhang S, Wang D, Xu H, Kong D, Qiao M. Expression and characterization of hydrophobin HGFI fused with the cell‐specific peptide TPS in Pichia pastoris . Protein Expr Purif 2012; 83:92–97. [DOI] [PubMed] [Google Scholar]
- 46. Julius D, Brake A, Blair L, Kunisawa R, Thorner J. Isolation of the putative structural gene for the lysine‐arginine‐cleaving endopeptidase required for processing of yeast prepro‐alpha‐factor. Cell 1984; 37:1075–1089. [DOI] [PubMed] [Google Scholar]
- 47. Werten MWT, de Wolf FA. Reduced proteolysis of secreted gelatin and Yps1‐mediated alpha‐factor leader processing in a Pichia pastoris kex2 disruptant. Appl Environ Microbiol 2005; 71:2310–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bevan A, Brenner C, Fuller RS. Quantitative assessment of enzyme specificity in vivo: P2 recognition by Kex2 protease defined in a genetic system. Proc Natl Acad Sci USA 1998; 95:10384–10389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Suzuki Y, Ikeda N, Kataoka E, Ohsuye K. Effect of amino acid substitution at the P(3) and P(4) subsites of fusion proteins on kex2 protease activity. Biotechnol Appl Biochem 2000; 32:53–60. [DOI] [PubMed] [Google Scholar]
- 50. Nociari MM, Shalev A, Benias P, Russo C. A novel one‐step, highly sensitive fluorometric assay to evaluate cell‐mediated cytotoxicity. J Immunol Methods 1998; 213:157–67. [DOI] [PubMed] [Google Scholar]
- 51. Rezania A, Healy KE. Biomimetic peptide surfaces that regulate adhesion, spreading, cytoskeletal organization, and mineralization of the matrix deposited by osteoblast‐like cells. Biotechnol Prog 1999; 15:19–32. [DOI] [PubMed] [Google Scholar]
- 52. Couchman JR, Woods A. Syndecan‐4 and integrins: Combinatorial signaling in cell adhesion. J Cell Sci 1999; 112:3415–3420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information