

Abstract
Most indigenous citrus varieties are assumed to be natural hybrids, but their parentage has so far been determined in only a few cases because of their wide genetic diversity and the low transferability of DNA markers. Here we infer the parentage of indigenous citrus varieties using simple sequence repeat and indel markers developed from various citrus genome sequence resources. Parentage tests with 122 known hybrids using the selected DNA markers certify their transferability among those hybrids. Identity tests confirm that most variant strains are selected mutants, but we find four types of kunenbo (Citrus nobilis) and three types of tachibana (Citrus tachibana) for which we suggest different origins. Structure analysis with DNA markers that are in Hardy–Weinberg equilibrium deduce three basic taxa coinciding with the current understanding of citrus ancestors. Genotyping analysis of 101 indigenous citrus varieties with 123 selected DNA markers infers the parentages of 22 indigenous citrus varieties including Satsuma, Temple, and iyo, and single parents of 45 indigenous citrus varieties, including kunenbo, C. ichangensis, and Ichang lemon by allele-sharing and parentage tests. Genotyping analysis of chloroplast and mitochondrial genomes using 11 DNA markers classifies their cytoplasmic genotypes into 18 categories and deduces the combination of seed and pollen parents. Likelihood ratio analysis verifies the inferred parentages with significant scores. The reconstructed genealogy identifies 12 types of varieties consisting of Kishu, kunenbo, yuzu, koji, sour orange, dancy, kobeni mikan, sweet orange, tachibana, Cleopatra, willowleaf mandarin, and pummelo, which have played pivotal roles in the occurrence of these indigenous varieties. The inferred parentage of the indigenous varieties confirms their hybrid origins, as found by recent studies.
Introduction
The genus Citrus L. (Family Rutaceae, subfamily Aurantiodeae) covers a wide range of edible and commercial varieties, including sweet orange, lemon, lime, grapefruit, and mandarins such as Clementine, Satsuma, King, and ponkan [1–4]. The production of major citrus varieties in tropical to sub-tropical and temperate zones exceeds 90 million tons, and the citrus industry occupies a significant position not only in the fruit industry but also in global agriculture [5,6]. In addition to the worldwide production of these major citrus varieties, numerous indigenous citrus varieties have also been produced in specific regions, and consumed locally [2,7]. Wide genetic diversity observed in Citrus, however, has made it difficult for taxonomists to draw a clear picture of their classification. Furthermore, mutants have occasionally been selected from limb sports or nucellar seedlings, and these constitute large variant strains [2,8–10]. Understanding how these modern citrus varieties arose from the ancestral basic species would bring us important insights for future citrus breeding.
Many botanists and taxonomists have proposed various approaches for the classification of a wide range of citrus varieties. Among them, two systems proposed by Swingle [11] and Tanaka [7,12] have been used in many studies. These two systems presume that most indigenous and commercial varieties arose from hybridization of ancestral ones, but differ in the way they treat indigenous varieties and cultivated varieties. Swingle primarily classified indigenous varieties rather than the cultivated varieties, placing two subgenera Papeda and Citrus in the genus Citrus [11]. The subgenus Papeda consists of section Papeda with four species, and section Papedocitrus with two species. He classified ten species in the subgenus Citrus, and regarded most cultivated varieties as natural hybrids of these indigenous species. He assigned most mandarin varieties to the scientific name Citrus reticulata, classified tachibana separately as C. tachibana, and also classified grapefruit, which arose from a chance seedling [2,9], separately as C. paradisi. In contrast, Tanaka stressed the importance of both indigenous varieties and cultivated varieties, and classified them equally as a species. He primarily placed two subgenera (Archicitrus and Metacitrus) in genus Citrus. The subgenus Archicitrus consists of five sections (Papeda, Limonellus, Citrophorum, Cephalocitrus and Aurantium) with 111 species, including grapefruit as C. paradisi. The subgenus Metacitrus consists of three sections (Osmocitrus, Acrumen and Pseudofortunella) with 48 species [12]. According to Tanaka’s system, individual mandarin varieties and tachibana were classified as a species with individual scientific names, and C. reticulata was assigned to the ponkan mandarin. Tanaka classified 145 citrus species in 22 different categories [12]. Since then, he has added several indigenous varieties to his classification system, and he released the ultimate list consisting of 159 species in 1969 [13]. Swingle considered C. ichangensis as a species of subgenus Papeda, and did not assign a scientific name to yuzu because he regarded it as a natural hybrid of C. ichangensis. In contrast, Tanaka classified C. ichangensis in subgenus Metacitrus section Osmocitrus, and classified yuzu to subgenus Metacitrus section Euosmocitrus as C. junos [12].
By the 1970s, various studies had been launched to classify citrus varieties using biochemical markers. In 1975, Scora published a novel paper based on his own chemotaxonomical study of citrus together with a survey of past literature [14]. He postulated three hypothetical taxa, mandarin (C. reticulata), citron (C. medica) and pummelo (C. maxima, formerly C. grandis), as the ancestors, and proposed that modern citrus varieties arose from repeated hybridization of these ancestors. In 1976, Barrett and Rhodes examined correlations among 22 indigenous varieties based on similarities for 146 traits, then estimated their affinities according to their deduced distance [15]. Similar chemotaxonomical studies gradually revealed the phylogenies of citrus varieties [16–21]. When DNA marker technology became available, taxonomical studies attempted classification of citrus using various DNA markers such as RAPD [22–26], RFLP [27], AFLP [28,29], ISSR [29–31] and SRAP [8,32]. Nicolosi and colleagues deduced a citrus phylogeny according to the genotypes of nuclear and chloroplast markers, and demonstrated that the origins of citrus varieties proposed by Scora [14] and Barrett and Rhodes [15] were acceptable [33,34]. Since then, the origins of some citrus varieties have gradually been revealed, and new classifications have been proposed [35,36]. Nowadays, codominant precision simple sequence repeat (SSR) or single nucleotide polymorphism (SNP) markers have been developed and used in most studies (see the reviews [34,37–40]). In addition, the chloroplast genome sequence of sweet orange has been released [41], and genome sequences of major citrus varieties are now public [42,43]. These genome sequence resources enable the design of precision DNA markers, and have revealed the parentage of Clementine, grapefruit, sweet orange, and limes and lemons [43–48]. However, the parentage of most indigenous varieties has not yet been determined.
Identifying the combination of seed parent and pollen parent is another important issue to be solved in parentage analysis. Many studies have revealed the phylogeny of citrus varieties by evaluating polymorphisms in the chloroplast or mitochondrial genome, or both [33,47,49–57]. However, some of these studies have only evaluated local citrus varieties [51,52], or limited numbers of varieties in the genus Citrus [50,57,58]. Next generation sequencing (NGS) technology has become commonplace, and it has been applied to the genotyping of citrus chloroplast genomes [56], but it is still a costly and time-consuming approach. Simple but reproducible and low-cost technologies that reveal sufficient polymorphisms are needed for the parentage analysis of a wide range of citrus varieties.
DNA marker analysis has been used in forensic genetics for inferring parentage or paternity, and identifying missing persons from their remains [59,60]. These techniques have also been used to infer sibships of wild populations [61–64], and are anticipated to be able to reveal unknown genealogy among indigenous citrus varieties. Two basic approaches have been adopted for parentage estimation with DNA marker analysis [64]. The first uses allele-sharing tests that estimate the number of alleles shared between two individuals at codominant DNA markers according to the Mendelian rules of inheritance. These tests estimate the probability of parentage from the proportion of DNA markers with shared alleles, and can also eliminate unrelated individuals. The discriminatory power of the test is proportional to the number of loci evaluated and the polymorphism of each DNA marker. However, these tests are susceptible to genotyping errors, and may give false positive or negative results [64]. Another approach is a likelihood ratio analysis, which compares the probabilities of alternate hypotheses for the parentage of two individuals (e.g., whether they are parent and offspring or unrelated) then estimates an odds score between these two hypotheses [62–64]. This is a widely used technique for examining proposed paternity or parentage and also to identify individuals [59,60,65]. The likelihood ratio analysis estimates the probability of the proposed parentage according to the likelihood of alleged parents and child, then compares it with a null relation between them deduced from the allele frequency within the population. The logarithm of likelihood ratio odds (LOD score) is often used to indicate the estimated score, but the number of DNA markers used for the evaluation and their allele frequency in the population influence the score [64]. Genotyping errors can also influence the score, and it is thus difficult to demonstrate a clear threshold for discrimination [63]. These two methods each have pros and cons; therefore, an approach that first excludes unrelated individuals using an allele-sharing test, then examines the probability of the proposed parentage using likelihood ratio analysis, will be a simple but effective way to infer parentage in a given population.
Because genotyping error severely affects the reliability of both methods, detecting such error and evaluating parentage with error-free DNA markers is a prerequisite for reliability. In the genotyping analysis of citrus varieties, however, wide genetic diversity among natural varieties reduces the transferability of DNA markers, resulting in false genotypes [44,46,64,66]. Selected somatic mutants could also be a drawback because some of them, but not all, have mutations in their genotype that make it difficult to estimate their identity.
The objective of the present study is to infer parentage among various citrus varieties using DNA marker analysis, and verify the inferred parentage statistically. We have attempted 1) to develop sufficient DNA markers for parentage analysis and eliminate erroneous DNA markers by examining them with a large enough set of known hybrid varieties, 2) to estimate genetic structures of indigenous varieties using these certified DNA markers, 3) to determine the cytosolic genotypes of individual varieties by evaluating chloroplast and mitochondrial genomes with DNA marker analysis, 4) to infer parentage among indigenous citrus varieties and verify it using a likelihood ratio approach.
Materials and Methods
Plant materials
We selected 371 citrus accessions consisting of 208 indigenous varieties, 78 hybrid varieties, and 85 selected strains (Table 1 and S1 Table). The indigenous varieties are from the collections of the Institute of Fruit Tree and Tea Science, NARO (NIFTS) that have been maintained at the Okitsu Citrus Research Division in Shizuoka prefecture, Japan. These varieties were selected from major mandarins (C. reticulata, C. tangerina, C. unshiu, C. clementina, C. kinokuni, C. tachibana, C. nobilis), pummelos (C. maxima and its hybrids), lemon (C. limon), sweet orange (C. sinensis), yuzu (C. junos), ichanchii (C. ichangensis) and their assumed natural hybrids. Sixteen varieties included variant selections to evaluate their genetic identity: four Clementines, two varieties classified to C. tangerine hort. ex Tanaka (Dancy and Obeni mikan), three grapefruits, five hyuganatsu, two iyos, 16 Kishus, 10 kunenbos, four ponkans, 12 pummelos, 21 Satsumas, two shiikuwashas, five sour oranges, 20 sweet oranges, 12 tachibanas, four tankans, and two willowleaf mandarins, respectively. Among them, kunenbo included both C. nobilis Lour. (King) and C. nobilis Lour. var. kunep Tanaka. Hybrid varieties used in this study are from the collections of NIFTS. Forty-five of them were developed by NIFTS, 11 by UC Riverside, 10 by the USDA, and the other 12 varieties were developed by seven other institutes or by farmers. We also used 85 strains that were selections from various crosses in NIFTS.
Table 1. Summary of citrus samples used in this study.
| Category | Scientific names | Samples | Genotyped samples | Representative samples | ||
|---|---|---|---|---|---|---|
| Swingle's system | Tanaka's system | |||||
| Indigenous varieties | 208 | 269 | 101 | |||
| Clementine | C. reticulata Blanco | C. clementina hort. ex Tanaka | 4 | 4 | 1 | |
| Dancy | C. reticulata Blanco | C. tangerina hort. ex Tanaka | 2 | 2 | 1 | |
| Grapefruit | C. paradisi Macf. | C. paradisi Macf. | 3 | 3 | 1 | |
| Hyuganatsu | C. sinensis (L.) Osbeck | C. tamurana hort. ex Tanaka | 5 | 6 | 1 | |
| Iyo | C. sinensis (L.) Osbeck | C. iyo hort. ex Tanaka | 2 | 2 | 1 | |
| Kishu | C. reticulata Blanco | C. kinokuni hort. ex Tanaka | 16 | 21 | 1 | |
| Kunenbo 1) | C. reticulata Blanco | C. nobilis Lour. var. kunep Tanaka | 10 | 13 | 4 | |
| Natsudaidai | C. paradisi Macf. | C. natsudaidai Hayata | 4 | 5 | 1 | |
| Ponkan | C. reticulata Blanco | C. reticulata Blanco | 4 | 5 | 1 | |
| Pummelo | C. grandis Osbeck 2) | C. grandis Osbeck 2) | 12 | 14 | 12 | |
| Satsuma | C. reticulata Blanco | C. unshiu Marcov. | 21 | 33 | 1 | |
| Shiikuwasha | C. indica | C. depressa Hayata | 2 | 3 | 2 | |
| Sour orange | C. aurantium L. | C. aurantium L. | 5 | 6 | 1 | |
| Sweet orange | C. sinensis (L.) Osbeck | C. sinensis (L.) Osbeck | 20 | 22 | 1 | |
| Tachibana | C. tachibana Makino | C. tachibana (Makino) Tanaka | 12 | 13 | 3 | |
| Tankan | C. sinensis (L.) Osbeck | C. tankan Hayata | 4 | 4 | 1 | |
| Willowleaf mandarin | C. reticulata Blanco | C. deliciosa Ten. | 2 | 2 | 2 | |
| Others | 80 | 111 | 66 | |||
| Hybrid varieties | C.spp | C.spp | 78 | 83 | 75 | |
| Selected strains | C.spp | C.spp | 85 | 90 | 85 | |
| Total | 371 | 442 | 261 | |||
1) Kunenbo (C. nobilis Lour. var. kunep Tanaka) includes King mandarin (C. nobilis Lour.)
2) Now classified as C. maxima Merr.
DNA extraction
Fully matured leaves were collected from each sample in the field at Okitsu, Shizuoka, then provided for DNA extraction using a modified protocol with a Nucleon Phytopure kit (GE Healthcare Life Science, NJ, USA) [67]. For certain varieties, several samples were collected from different trees. These were used as biological replicates to confirm the reproducibility of genotyping (RA in S1 Table). DNA concentration of the prepared DNA samples was determined using a Qubit Assay kit (ThermoFisher Scientific, Tokyo, Japan). UV absorbance analysis was used to confirm sample quality (A260/A280 > 1.8, and A260/A230 > 2.0), and gel electrophoresis analysis to verify the size and integrity of the extracted DNA samples.
Citrus sequence resources for DNA marker design
Nucleotide sequences of expressed genes of citrus were obtained from public cDNA sequence databases dbEST (http://www.ncbi.nlm.nih.gov/dbEST/), RefSeq (http://www.ncbi.nlm.nih.gov/refseq/) and HarvEST (http://harvest.ucr.edu/) [68]. Citrus genome sequence resources in public databases, including BAC end sequences of Clementine [69] and Satsuma [70,71], and whole genome shotgun sequences of sweet orange ‘Ridge Pineapple’ in the trace file repository of Sanger reads (ftp://ftp.ncbi.nlm.nih.gov/pub/TraceDB/citrus_sinensis/), were also used for DNA marker design. Preliminary evaluation of the quality and length of each of these data sets was carried out using pregap4 [72], then a consensus sequence set was obtained for each set with Mira assembler [73] to reduce redundancy.
NGS analysis of citrus varieties
NGS analysis of citrus varieties for mining SSR and indel regions was performed with a HiSeq 2000 sequencing system (Illumina, CA, USA) in paired-end mode [67]. Quality-checked NGS reads were mapped to the haploid Clementine reference sequence v.0.9 or v.1.0 [43] using BWA [74]. Candidate SSR or indel regions in the re-sequenced data were scored and identified using SAMtools and BCFtools [75], or using mreps [76].
DNA marker design for genotyping nuclear genomes
SSR regions of each sequence were mined using mreps [76], then candidate regions with motif length between two and six nucleotides were selected. The identified candidate regions found in expressed genes or genomic sequences were used for oligonucleotide primer design with PerlPrimer [77] or Primer3 [78]. Previously reported SSR markers designed from BAC end sequences [46], or from EST sequences [79,80] were also used in this study.
DNA marker design for genotyping organelle genomes
SSR markers for detecting polymorphisms in the chloroplast genome were designed from the chloroplast genome sequence of sweet orange ‘Ridge Pineapple’ (accession No. DQ864733) [41] by searching candidate SSR regions using mreps [76] as described in the previous section. Oligonucleotide primer sets for citrus mitochondrial genomes [53], and universal primer sets for the chloroplast genomes of dicotyledonous angiosperms [81] were also used for genotyping organelle genomes.
Genotyping analysis
All genotyping analysis of nuclear or organelle genomes followed the multiplexed and multicolored post-labeling method in single tube with BStag reported by Shimizu and Yano [82]. Post-labeling of the PCR product with BStag is a simple but inexpensive method that does not require large alteration of the PCR program, and it reduces the total cost of analysis significantly. One of the six standard BStag sequences or an additional BStag sequence (F9TCC: 5'-CTAGTATCAGGACTCC-3') was added at the 5' end of the designed forward primer. A short ‘pigtail’ sequence was added at the 5' end of the reverse primer in order to suppress stuttering of the detected peak [83]. For each genotyping analysis, four oligonucleotide primer sets that were individually attached to different BStag sequences were mixed with the corresponding fluorescently labeled BStag primers. A typical PCR program for the amplification and post-labeling of the target region of the nuclear genome was: initial denaturation at 94°C for 3 min; 32 cycles of target amplification (20 s at 94°C followed by 35 s at 52–65°C); then three post-labeling cycles (20 s at 94°C followed by 10 s at 49°C and 5 s at 72°C); and final extension at 72°C for 10 min then terminated at 4°C. Each DNA marker was labeled separately with one of four different fluorescent dyes in a single tube at the labeling step. The reaction mixture was diluted twofold with water after the PCR. Then, a 0.4-μL aliquot of the diluted mixture was mixed with 0.1 μL GeneScan 600 LIZ® dye Size Standard (ThermoFisher Scientific, Tokyo, Japan) and adjusted to be 10 μL with deionized formamide, and then heat denatured at 95°C for 4 min. Electrophoresis of the labeled product was carried out on an ABI 3130xl DNA sequencer (ThermoFisher Scientific, Tokyo, Japan) with 36 cm length capillary using the standard program. Genotypes of each DNA marker/sample were called using GeneMapper 4.0 software (ThermoFisher Scientific, Tokyo, Japan).
Parentage test and identity test
Parentage was confirmed for assumed parent–offspring triads by considering the inheritance of each allele from parents to offspring according to the Mendelian rule. Any DNA markers showing discrepancies in known hybrids were excluded from the analysis. The evaluation was carried out using a function of GUGS (General Utilities for Genotyping Study) software (Shimizu, T. in preparation). The identity test is a simple exact match test of each genotype to others for all combinations. If a pair of samples coincided with each other for the genotypes of all of the DNA markers, they were treated as identical. In this study, we counted the number of DNA markers that did not agree between any given pair of samples.
Statistical evaluation of the genotype data
Observed heterozygosity (Ho), expected heterozygosity (He, equivalent to the unbiased estimator of gene diversity given by equation 8.4 of Nei [84]), number of unique alleles, and polymorphic information content (PIC, representing the probability of distinguishing a marker allele derived from either one of the parents [85]) were calculated using the frequency analysis function of Cervus [62] and confirmed with GUGS. The probability of match (PM), representing the probability that an unrelated individual happens to have the same genotype to others [60] is given by:
| (1) |
Here, pk is the observed frequency of each unique genotype k in the population, and m is the number of unique genotypes at a given nuclear locus. The gene diversity (GD) of a single allelic organelle genotype at a given locus was evaluated by
| (2) |
(equation 8.1 of Nei [84]). Here, xi is the observed frequency of the ith single allele in the population, and m is the number of alleles at an organelle locus. This parameter (Nei’s GD) is an equivalent of the expected heterozygosity for diploid organisms. The values of the unique genotypes, PM and GD, were obtained using a function of GUGS. Wright’s fixation index (Fw) was obtained by the equation Fw = (He−Ho)/He (equation 12.9 of Nei and Kumar [86]).
All statistical evaluations of the normal distribution (Shapiro–Wilk test) and one-way ANOVA (Kruskal–Wallis test) were conducted with the stats package of R (version 3.1.3, https://www.r-project.org/) in the Rstudio environment (version 0.99.893, https://www.rstudio.com/). Tests for equal variance and stochastic equality of two samples were conducted according to Brown–Forsythe test and Brunner–Munzel test using functions levene.test and brunner.munzel.test in the lawstat package [87]. The p-value adjustment for multiple samples was carried out by Benjamini–Hochberg (BH) correction with the p.adjust function of R. F-statistics for population analysis (FIT, FIS) [86,88,89] were estimated for each sample category or individual DNA marker using R packages hierfstat [90] and pegas [91] in combination with adegenet [92]. Additionally, Hedrick’s G''ST [88], which is an equivalent of FST extended to multiallelic DNA markers, was estimated globally or pairwise using the mmod package of R [93] in combination with adegenet [92].
Evaluation of Hardy–Weinberg equilibrium
An exact test of Hardy–Weinberg proportions for multiallelic genotype data was estimated with a Markov Chain Monte Carlo (MCMC) simulation method developed by Guo and Thompson [94], that was implemented as a function of Arlequin (version 3.5.2.2) [95]. The genotype data file used as input for Arlequin was formatted with CONVERT software [96] with no prior inferred population structure. We continued the MCMC simulation runs 10 times each for 1,000,000 iterations in both initial burn-in and de-memorization steps, and then the average of the estimated p-values was provided for evaluation.
Factorial analysis and phylogenetic evaluation
Principal coordinate analysis (PCoA) and phylogenetic analysis of the obtained genotype data were carried out with DARWin (version 6.0.13) [97,98]. A dissimilarity matrix was obtained from the genotypes of each sample pair using a simple matching method (nuclear genotypes) or from modalities by Rogers and Tanimoto’s coefficient (organelle genotypes). The PCoA analysis assumed two to six axes (typically five), and data for the first two axes were used to draw a scatter plot. A consensus phylogenetic tree was inferred from the bootstrapped dissimilarity matrices obtained from 30,000 iterations for the nuclear genotype data or 5,000 iterations for the organelle genotype data using the weighted neighbor-joining method [99], then obtained consensus trees.
Structure analysis
Structure analysis for the inference of the basic taxa and their proportions was carried out using STRUCTURE [100]. The genotype data for the 101 representative indigenous varieties obtained with the 123 selected DNA markers were formatted using CONVERT software [96] with no prior inferred population structure. Missing data were treated as lost (assigned ‘-9’ for the genotype data). The analysis assumed the admixture model for ancestry and that allele frequencies were correlated. In the estimation of the number of basic taxa (K), we varied K stepwise from two to ten, then evaluated the probability ten times for each K with 100,000 iterations of the initial burn-in and 1,000,000 MCMC runs. The inferred proportions of the K populations, and the estimated lnPr(X|K), mean lnP(K) and its variance were used to obtain stdev LnP(K), L'(K) and |L''(K)|, then ΔK was estimated as the mean of (|L''(K)| / stdev LnP(K)), following Evanno et al [101]. We used the Structure Harvester web service [102] at http://taylor0.biology.ucla.edu/structureHarvester/ for this purpose. The inferred proportions of the K basic taxa were deduced individually from the output of Structure Harvester using the Greedy algorithm of CLUMPP [103]. We compared the full search and random input order running modes of CLUMPP, and also changed the running period for the permutation analysis from 1,000 to 1,000,000, but all results were identical. We therefore used the simulation results from CLUMPP run in Greedy mode with 100,000 permutation runs. The bar plot of inferred proportions was drawn with MS Excel.
Allele-sharing test and stochastic verification of inferred parentage
Possible parent-to-offspring relationships between varieties were examined using an allele-sharing test. The test evaluates the ratio of the number of DNA markers that share at least one allele between two varieties to the total number of DNA markers. Any pair of varieties in which nearly all DNA markers shared an allele between the two varieties was selected as a candidate parent–offspring pair. When two varieties were assumed to be the parents of a particular offspring variety, the parentage of the assumed triad was examined using the parentage test.
The probability of the inferred dyad or triad being true single parent-to-offspring or parents-to-offspring combinations was examined by likelihood ratio analysis according to Marshal et al and Jones and Ardren [62,63]. In this analysis, the probabilities of two hypotheses (H1 and H2) are compared. Assume P(G|H1) is the probability of observing a particular pair of genotypes G under the hypothesis H1, and P(G|H2) is the probability of G under the hypothesis H2. The evaluated P(G|H1) relative to the evaluated P(G|H2) will give a likelihood ratio L(H1, H2 | G) that the G will be observed under the two hypotheses H1 and H2:
| (3) |
In the parentage test, H1 presumes that a particular variety is an offspring of the alleged parent or parents, and H2 presumes that it is not an offspring of the alleged parents but a chance seedling that has arisen from a given population. The likelihood ratio L represents the probability that the offspring was obtained from the alleged parent(s) rather than being a chance seedling.
For the stochastic evaluation of the parentage test, let gS, gP and gO represent the genotypes of the alleged seed parent, alleged pollen parent and offspring, respectively, at a DNA marker. The likelihood ratio that the alleged parents are the true parents of the given offspring variety was estimated according to Eq (3) from Jones and Ardren [63]:
| (4) |
Here, the numerator T(gO|gS,gP) is the transition probability of gO given gS and gP. This probability was estimated from the allele frequencies and a genotype combination according to Table 1 of Marshall et al [62]. The denominator P(gB) is the frequency of the offspring’s genotype in a particular population obtained according to Table 2 of Marshall et al [62]. The value L is the likelihood ratio that the parentage of this triad is correct compared to the offspring obtained its genotype from an unknown hybrid combination.
Table 2. Summary of DNA markers used in this study.
| Type/source | Evaluated | Selected | (%) | Certified | (%) | Reference | |
|---|---|---|---|---|---|---|---|
| Genomic SSR/INDEL | 154 | 104 | 67.5% | 58 | 37.7% | This study | |
| EST/cDNA SSR | 201 | 110 | 54.7% | 87 | 43.3% | This study | |
| Ollitrault, F et al. 2010 | 79 | 6 | 7.6% | 6 | 7.6% | 1) | |
| Chen, C. et al. 2008 | 106 | 19 | 17.9% | 12 | 11.3% | 2) | |
| Chen, C. et al. 2006 | 56 | 7 | 12.5% | 6 | 10.7% | 3) | |
| Total | 596 | 246 | 41.3% | 169 | 28.4% | ||
1) Ollitrault, F et al. (2010) Am. J. Bot. e124-e129.
2) Chen, C et al. (2008) Tree Genet. Genom. 4:1–10.
3) Chen, C et al. (2006) Theor Appl Genet. 112:1248–1257.
In a similar manner, another likelihood ratio for the alleged single parent to an offspring was estimated according to Eq (2) of Jones and Ardren [63], or Eq (5) of Marshall et al [62]:
| (5) |
Here, the numerator T(gO|gS) is the transition probability of gO given gS, estimated from their allele frequencies and genotype combination according to Brenner [104] or Table 2 of Marshall et al [62]. In most parentage analyses of wild plant populations, it is unknown which variety is the seed parent or the pollen parent. Thus, a particular alleged parent sample without any prior supporting information was assigned to either gS or gP arbitrarily. The probability of obtaining a particular genotype in a population was estimated from the allele frequencies at a given DNA marker, as x2 for homozygous genotype, or 2xy for a heterozygous genotype, where x and y are the allele frequencies in a population. The obtained value L is the ratio of the likelihood that this is a parent–offspring dyad to the likelihood that the offspring is from some unknown hybrid combination. All DNA markers used in the parentage test were presumed to be at Hardy–Weinberg equilibrium (HWE) in the given population. The LOD score (the natural logarithm of the likelihood ratio, LR) for the set of genotypes at multiple DNA markers is given by the product of LR:
| (6) |
where LRm is a likelihood ratio for a triad or dyad at the mth DNA marker. Any DNA markers that showed discrepancies in the parentage test or allele-sharing test were excluded from LOD score estimation. The required cross trial index (RCI) was obtained by:
| (7) |
Here, N is the number of individuals with unique genotype in the proposed population, fk is the expected frequency of a particular genotype at the kth DNA marker estimated from the allele frequencies of the two alleles in the population (equation 7.4 in Nei [84]), and m is the total number of DNA markers used for the evaluation. Single parent–offspring probability (SPP) is not a likelihood ratio value but a cumulative probability between two particular individuals assuming that one is the alleged parent of a particular offspring variety without prior information on the other parent. The SPP value for the particular offspring (gO) and the alleged parent (gP) is obtained from the transition probability T(gO|gP) of gO given gP in a similar manner to that described above by:
| (8) |
where m is the total number of DNA markers used for the evaluation. These tests, frequency analyses and probability estimations were carried out using functions of GUGS software. The inferred genealogy was drawn as a family tree manually, or using Helium [105].
Results
Development and evaluation of DNA markers for nuclear genotyping of citrus
DNA sequences of citrus expressed genes from cloned cDNA, EST, and RefSeq in public sequence database repositories or the harvEST citrus database were used for DNA marker design. Preliminary clustering analysis of EST sequences with a sequence assembler reduced duplication in these data sets, and yielded 98,869 consensus sequences from 582,270 EST sequences. Another clustering analysis of whole genome shotgun sequences of sweet orange ‘Ridge Pineapple’ yielded 381,909 consensus sequences from 866,700 reads, but 46,341 Clementine BAC end sequences were not used for assembly because of their low redundancy. SSR mining of these data sets with mreps [76] identified 143,825 candidate regions from the consensus EST sequences, 314,967 from the consensus sweet orange whole genome shotgun sequences, and 16,159 from the Clementine BAC end sequences. SSR mining of the Clementine haploid genome sequence [43] (https://www.citrusgenomedb.org/) also identified 310,413 candidate SSR regions for both v0.9 (release 165) and v1.0 (release 182) genomes. These candidate regions were verified with resequencing data obtained from NGS analysis of 15 citrus varieties (banpeiyu A004, Clementine A009, dancy A016, hyuganatsu A036 and A038, King A054, Kishu A066, ponkan A108, Satsuma A113 and A122, sweet orange A162, willowleaf (Mediterranean) mandarin A200, ‘Encore’ B014, ‘Harehime’ B017, and ‘Kiyomi’ tangor B031). Candidate SSR regions that were supported with more than 40× Illumina read coverage were selected for primer design by referring their motif size, repeat length, genome position, gene annotation, specificity and versatility among citrus varieties. We also identified indel regions by referring to resequencing data, and these were also used for primer design. Consequently, we designed SSR and indel markers (S2 Table lists DNA markers by type and gives their sources).
Verifying genotyping errors to select certified DNA markers
The genotypes of the DNA markers were preliminarily evaluated for peak height and peak height ratio, product size, and number of alleles in a small sample set consisting of Satsuma, sweet orange, Clementine, pummelos, lemon, ponkan and Kishu. Most of the evaluated primers successfully amplified PCR products, but a portion of them failed to amplify in particular varieties, or yielded multiple peaks in lemon. Consequently, 154 genomic SSR/indel markers and 201 EST/cDNA markers were primarily selected. The selected DNA markers were further examined for inconsistency using the parentage test with two hybrid varieties ‘Kiyomi’ tangor (Satsuma × sweet orange) and ‘Harumi’ (‘Kiyomi’ tangor × ponkan). Consequently, 104 genomic SSR/indel markers and 110 EST/cDNA SSR markers were selected for further evaluation (Table 2). Genomic SSR markers reported by Ollitrault et al [46] were also evaluated in a similar manner, and six valid markers were selected (Table 2). EST SSR markers reported by Chen et al [79,80] were also examined and 19 and seven SSR markers were selected.
Genotyping analyses of 371 plant samples (Table 1 and S1 Table) were conducted with the 246 selected SSR/indel markers and their genotype data were obtained (S3 Table). Genotyping error in these data was examined using the parentage test with 59 known hybrid varieties (Table 3, S1 Fig), and also with 63 of 85 selected strains. The hybrid varieties used for the test were developed from various crosses of Satsuma, Clementine, sweet orange, grapefruit, hassaku, ponkan, several pummelos, hyuganatsu, dancy, King, willowleaf mandarin, Kishu, and offspring of these varieties (Table 3). The parentage test using multiallelic DNA markers is strict to the combination; it will fail even when the correct triad is examined but their parents-to-child combination is incorrect (eg. AB × CD will give AC, but AC × CD will not give AB). Accordingly, this test examined not only erroneous DNA markers, but also incorrect hybrid combinations. For example, we found during the evaluation that ‘Fortune’ (Clementine × dancy) was discrepant to the reported parents [2]. Therefore, ‘Fortune’ was excluded from the reference varieties for the parentage test (the correct parentage of ‘Fortune’ will be discussed in a later section).
Table 3. Parentage of hybrid varieties with their organellar genotypes and the LOD scores used for error checking of DNA markers.
| # | Hybrid variety | Seed parent | Pollen parent | LOD 2) | RCI 3) | SPP 4) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Variety name | CT1) | ID | Variety name | CT 1) | ID | Variety name | CT1) | score | Seed | Pollen | ||
| 1 | B001 | Akemi | C12 | B031 | Kiyomi | C12 | B056 | Seminole | C04 | 91.3 | 172.3 | 51.8 | 48.1 |
| 2 | B003 | Aki Tangor | C12 | A125 | Satsuma | C12 | A162 | Sweet orange | C07 | 78.8 | 162.3 | 51.3 | 49.2 |
| 3 | B005 | Amaka | C12 | B031 | Kiyomi | C12 | B014 | Encore | C12 | 95.9 | 169.0 | 47.5 | 55.3 |
| 4 | B006 | Ariake | C07 | A162 | Sweet orange | C07 | A009 | Clementine | C12 | 108.0 | 185.3 | 49.2 | 50.9 |
| 5 | B007 | Asumi | C12 | B077 | Okitsu-46 | C12 | B021 | Harumi | C12 | 99.5 | 171.8 | 49.3 | 53.7 |
| 6 | B008 | Aurastar | C04 | B072 | H/FD-1 | C04 | A004 | Banpeiyu | C04 | — 5) | — 5) | 36.4 | 34.5 |
| 7 | B009 | Awa Orange | C05 | A036 | Hyuganatsu | C05 | A162 | Sweet orange | C07 | 72.9 | 175.2 | 46.8 | 45.4 |
| 8 | B010 | Benibae | C12 | B073 | HF9 | C12 | B014 | Encore | C12 | 100.9 | 161.1 | 54.3 | 55.5 |
| 9 | B011 | Benimadoka | C04 | A088 | Mato buntan | C04 | A032 | Hirado buntan | C04 | 210.0 | 256.9 | 45.0 | 44.9 |
| 10 | B014 | Encore | C12 | A054 | King | C12 | A201 | Willowleaf mandarin | C12 | 102.5 | 167.2 | 53.1 | 58.9 |
| 11 | B015 | Fairchild | C12 | A009 | Clementine | C12 | B045 | Orland | C04 | 84.0 | 156.2 | 56.3 | 50.1 |
| 12 | B017 | Harehime | C12 | B070 | E-647 | C12 | A125 | Satsuma | C12 | 103.3 | 179.5 | 49.6 | 46.4 |
| 13 | B018 | Hareyaka | C12 | B014 | Encore | C12 | A107 | Ponkan | C12 | 104.0 | 163.9 | 56.7 | 55.1 |
| 14 | B019 | Haruhi | C12 | B077 | Okitsu-46 | C12 | B009 | Awa Orange | C05 | 97.5 | 179.3 | 49.8 | 49.3 |
| 15 | B021 | Harumi | C12 | B031 | Kiyomi | C12 | A107 | Ponkan | C12 | 94.6 | 172.0 | 49.5 | 51.9 |
| 16 | B022 | Hayaka | C12 | A125 | Satsuma | C12 | A107 | Ponkan | C12 | 91.8 | 169.1 | 48.8 | 51.7 |
| 17 | B023 | Hayasaki | C04 | A088 | Mato buntan | C04 | A032 | Hirado buntan | C04 | 209.5 | 257.1 | 44.4 | 44.2 |
| 18 | B024 | Himekoharu | C12 | B031 | Kiyomi | C12 | A101 | Oogonkan | C05 | 81.4 | 171.5 | 49.0 | 47.0 |
| 19 | B026 | Honey | C12 | A054 | King | C12 | A201 | Willowleaf mandarin | C12 | 114.5 | 181.6 | 52.9 | 56.0 |
| 20 | B027 | Kanpei | C12 | B039 | Nishinokaori | C12 | A107 | Ponkan | C12 | 98.4 | 167.7 | 51.4 | 48.7 |
| 21 | B028 | Kara mandarin | C12 | A125 | Satsuma | C12 | A054 | King | C12 | 77.2 | 148.8 | 53.9 | 54.0 |
| 22 | B030 | Kinnow mandarin | C12 | A054 | King | C12 | A201 | Willowleaf mandarin | C12 | 104.2 | 171.6 | 52.3 | 57.5 |
| 23 | B031 | Kiyomi | C12 | A125 | Satsuma | C12 | A162 | Sweet orange | C07 | 81.4 | 169.2 | 47.3 | 48.4 |
| 24 | B033 | Lee | C12 | A009 | Clementine | C12 | B045 | Orland | C04 | 103.9 | 178.9 | 53.7 | 48.1 |
| 25 | B034 | May Pomelo | C04 | A028 | Hassaku | C04 | A032 | Hirado buntan | C04 | 139.7 | 216.0 | 41.9 | 50.2 |
| 26 | B035 | Mihaya | C12 | B066 | Tsunonozomi | C12 | B076 | No.1408 | C12 | 98.7 | 160.1 | 53.9 | 56.2 |
| 27 | B036 | Mihocore | C12 | A125 | Satsuma | C12 | B014 | Encore | C12 | 97.5 | 163.9 | 53.9 | 56.2 |
| 28 | B037 | Mineola | C04 | A024 | Grapefruit | C04 | A016 | Dancy tangerine | C12 | 90.2 | 164.0 | 47.5 | 52.1 |
| 29 | B038 | Nankou | C12 | A125 | Satsuma | C12 | A009 | Clementine | C12 | 104.8 | 175.7 | 49.7 | 48.9 |
| 30 | B039 | Nishinokaori | C12 | B031 | Kiyomi | C12 | A162 | Sweet orange | C07 | 94.8 | 186.5 | 47.9 | 44.6 |
| 31 | B040 | Nou 5 gou | C12 | B033 | Lee | C12 | A059 | Kishu | C12 | 113.9 | 175.5 | 53.0 | 54.0 |
| 32 | B041 | Nou 6 gou | C12 | A054 | King | C12 | A059 | Kishu | C12 | 106.3 | 174.4 | 53.6 | 53.0 |
| 33 | B042 | Nou 7 gou | C04 | B072 | H/FD-1 | C04 | A004 | Banpeiyu | C04 | — 5) | — 5) | 37.7 | 38.8 |
| 34 | B043 | Nova | C12 | A009 | Clementine | C12 | B045 | Orland | C04 | 119.1 | 193.3 | 51.2 | 46.3 |
| 35 | B045 | Orland | C04 | A024 | Grapefruit | C04 | A016 | Dancy tangerine | C12 | 101.1 | 174.0 | 46.5 | 49.9 |
| 36 | B047 | Osceola | C12 | A009 | Clementine | C12 | B045 | Orland | C04 | 100.5 | 176.9 | 53.9 | 48.7 |
| 37 | B048 | Page | C04 | B037 | Mineola | C04 | A009 | Clementine | C12 | 110.7 | 180.3 | 49.3 | 53.5 |
| 38 | B051 | Robinson | C12 | A009 | Clementine | C12 | B045 | Orland | C04 | 104.2 | 180.5 | 54.5 | 47.6 |
| 39 | B052 | Saga Mandarin | C12 | A125 | Satsuma | C12 | B015 | Fairchild | C12 | 93.8 | 166.1 | 52.7 | 50.7 |
| 40 | B054 | Seihou | C12 | B031 | Kiyomi | C12 | B037 | Mineola | C04 | 98.0 | 180.7 | 50.3 | 49.4 |
| 41 | B055 | Seinannohikari | C12 | B071 | EnOw21 | C12 | B069 | Youkou | C12 | 92.7 | 160.1 | 54.5 | 52.7 |
| 42 | B056 | Seminole | C04 | A024 | Grapefruit | C04 | A016 | Dancy tangerine | C12 | 93.3 | 171.6 | 46.3 | 51.1 |
| 43 | B057 | Setomi | C12 | B031 | Kiyomi | C12 | A107 | Ponkan | C12 | 107.5 | 185.5 | 47.7 | 48.4 |
| 44 | B058 | Shiranuhi | C12 | B031 | Kiyomi | C12 | A107 | Ponkan | C12 | 82.4 | 161.9 | 52.8 | 49.5 |
| 45 | B059 | Southern Yellow | C05 | B063 | Tanikawa Buntan | C05 | A059 | Kishu | C12 | 104.8 | 185.1 | 46.1 | 42.0 |
| 46 | B060 | Summer Fresh | C04 | A028 | Hassaku | C04 | A098 | Natsudaidai | C04 | 94.0 | 191.0 | 45.6 | 42.9 |
| 47 | B061 | Sweet Spring | C12 | A125 | Satsuma | C12 | A028 | Hassaku | C04 | 69.3 | 155.5 | 52.3 | 48.1 |
| 48 | B062 | Tamami | C12 | B031 | Kiyomi | C12 | B067 | Willking | C12 | 109.6 | 174.5 | 49.9 | 53.9 |
| 49 | B064 | Tsunokagayaki | C12 | B074 | KyOw14 | C12 | B014 | Encore | C12 | 94.7 | 164.2 | 52.5 | 52.3 |
| 50 | B065 | Tsunokaori | C12 | B031 | Kiyomi | C12 | A125 | Satsuma | C12 | 84.9 | 159.5 | 52.0 | 45.9 |
| 51 | B066 | Tsunonozomi | C12 | B031 | Kiyomi | C12 | B014 | Encore | C12 | 94.1 | 165.4 | 50.4 | 53.3 |
| 52 | B068 | Yellow Pummelo | C04 | A028 | Hassaku | C04 | A032 | Hirado buntan | C04 | 108.5 | 183.5 | 47.5 | 52.6 |
| 53 | B069 | Youkou | C12 | B031 | Kiyomi | C12 | A107 | Ponkan | C12 | 77.2 | 159.1 | 50.3 | 52.4 |
| 54 | B070 | E-647 | C12 | B031 | Kiyomi | C12 | B047 | Osceola | C12 | 111.8 | 183.7 | 49.3 | 49.9 |
| 55 | B071 | EnOw21 | C12 | B014 | Encore | C12 | A125 | Satsuma | C12 | 97.2 | 165.5 | 52.5 | 52.2 |
| 56 | B073 | HF9 | C12 | A125 | Satsuma | C12 | A162 | Sweet orange | C07 | 72.9 | 165.8 | 48.4 | 46.3 |
| 57 | B074 | KyOw14 | C12 | B031 | Kiyomi | C12 | A125 | Satsuma | C12 | 91.7 | 170.1 | 51.3 | 50.6 |
| 58 | B075 | KyOw21 | C12 | B031 | Kiyomi | C12 | A125 | Satsuma | C12 | 96.9 | 174.3 | 51.4 | 48.8 |
| 59 | B077 | Okitsu-46 | C12 | B061 | Sweet Spring | C12 | A162 | Sweet orange | C07 | 76.9 | 173.0 | 46.8 | 47.0 |
1) Category of organellar genotype (see Table 9).
2) LOD: See the section ‘Stochastic evaluation of inferred parentage’.
3) RCI: required cross trial index obtained by Eq 7.
4) SPP: Single parent–offspring probability for seed parent or pollen parent obtained by Eq 8.
5) Score or value was indeterminate due to lack of allele in H/FD-1.
All DNA markers were also evaluated for error with 64 selected strains.
The parentage test confirmed that 176 DNA markers were consistent on all hybrid varieties, and 182 DNA markers were consistent on all of the selected strains (S4 Table). However, 31 DNA markers showed discrepancies in more than two hybrid varieties, and 24 in more than two selected strains. Thirteen DNA markers failed to give an amplified product in just one hybrid variety, and 27 in just one selected strain. Most of these failures were due to simple technical error, and they were ignored in the following analysis. However, four DNA markers failed to amplify in the hybrid varieties and two in the selected strains (S4 Table). These DNA markers were assumed to contain a null allele, and they were excluded. Accordingly, the parentage test selected 58 certified genomic markers and 87 certified EST/cDNA markers. A similar evaluation for the published SSR markers also selected 6 certified SSR markers from the Clementine BAC end sequence [46], and 12 and 6 certified SSR markers from the EST sequence [79,80]. These 169 certified markers (166 SSR and 3 indel markers) are indicated by asterisks in S2, S3, S4, S6 and S7 Tables.
Most of the varieties used for the error check were offspring of mandarin, sweet orange or pummelo. Therefore, the selected DNA markers were expected to show less discrepant genotypes for those varieties or their offspring. On the contrary, lemon, yuzu, sour orange and citron were less frequently used as breeding parents for the hybrids. Consequently, the selected DNA markers could show discrepant genotypes for parentage analysis when these varieties or their offspring were used.
The 246 selected DNA markers were also used to construct a linkage map for two cross populations (Shimizu, T. in preparation). As a result, 225 of the selected DNA markers, including 154 certified markers, were mapped to one of the maps, or to both as a single locus (S2 Table gives the mapped linkage group in the ‘LG’ column). The mapped linkage groups of all DNA markers present in both maps agreed with each other. Among the mapped markers, 16 exactly matched the positions of other DNA markers on the two maps, and they were regarded as duplicate markers. These duplicate markers are indicated with double asterisks in S2, S3, S4, S6 and S7 Tables, and were excluded from the statistical evaluation. According to this selection and validation process, 153 certified DNA markers (150 SSR markers and 3 indel markers) that were consistent with 148 crosses, and uniquely mapped to a linkage group as a single locus without duplication, were finally selected.
Genetic identity of indigenous varieties
All citrus samples were examined for their genetic identity to each other using the 169 certified DNA markers. The number of mismatched genotypes between all combinations of two indigenous varieties was scored using the identity test, and then summarized (S5 Table). Considerable numbers of mismatches were confirmed between most unrelated varieties or strains. None of the hybrid varieties except ‘Kuchinotsu-41’ (B032) and ‘Sagakashi 34’ (B053) coincide with any other varieties or strains. ‘Kuchinotsu-41’ is an autotetraploid selection of hyuganatsu and its genotypes completely agreed with those of hyuganatsu (Table 4, S5 Table). Likewise, ‘Sagakashi 34’ was confirmed to be a nucellar selection of ‘Shiranuhi’. The genotypes of each selected strain showed no coincidence with other varieties or strains, and these strains were confirmed to have been selected from diverse crosses. Twelve pummelo varieties (C. maxima or C. maxima hybrid) did not agree in their genotypes with the others; therefore we conclude that these pummelo varieties were not mutant selections.
Table 4. List of varieties that hold identical genotypes to the representative variety.
| Variety | (Sub type) | No. | Representative variety | ND | Identical varieties | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variety/strain name | Scientitific name | No. | Variety name | Strain name | MM | ND | ||||
| Andoukan | A001 | (stock strain) | C. maxima (hybrid) | 0 | A056 | Kinukawa | (stock strain) | 2 | 2 | |
| Clementine | A009 | (stock strain) | C. clementina hort. ex Tanaka | 0 | A010 | Clementine | A peau fin | 0 | 0 | |
| A011 | Clementine | Caffin | 0 | 0 | ||||||
| A012 | Clementine | de nules | 0 | 0 | ||||||
| Dancy | A016 | (stock strain) | C. tangerina hort. ex Tanaka | 0 | A017 | Dancy tangerine | Obenimikan | 0 | 0 | |
| Girimikan | A023 | (stock strain) | C. tardiva hort. ex Shirai | 0 | A180 | Tajima mikan | (stock strain) | 0 | 0 | |
| Grapefruit | A024 | Marsh | C. paradisi Macfad. | 0 | A025 | Grapefruit | Red blush | 0 | 0 | |
| A026 | Grapefruit | Triumph | 0 | 0 | ||||||
| Henka mikan | A030 | (stock strain) | C. pseudo-aurantium hort. ex Yu.Tanaka | 0 | A094 | Nansho daidai | (stock strain) | 0 | 0 | |
| Hiroshimanatsubuntan | A033 | (stock strain) | C. hiroshimana hort. ex Yu.Tanaka | 0 | A181 | Takumanatsukunenbo | 0 | 0 | ||
| Hyuganatsu | A036 | (stock strain) | C. tamurana hort. ex Tanaka | 0 | A037 | Hyuganatsu | Ihara 1 | 0 | 0 | |
| A038 | Hyuganatsu | Muroto konatsu | 0 | 0 | ||||||
| A039 | Hyuganatsu | Orange hyuga | 1 | 0 | ||||||
| A040 | Hyuganatsu | Shoukakukei hyuganatsu | 0 | 0 | ||||||
| B032 | 'Kuchinotsu-41' | Hyuganatasu NC | 0 | 0 | ||||||
| Iyo | A044 | Ootani Iyo | C. iyo hort. ex Tanaka | 0 | A043 | Iyo | Miyauchi Iyo | 0 | 0 | |
| Kishu | A059 | Kishu | C. kinokuni hort. ex Tanaka | 0 | A057 | Kishu | Hira Kishu | 0 | 0 | |
| A058 | Kishu | Hisago komikan | 1 | 1 | ||||||
| A060 | Kishu | Kishu mikan | 0 | 0 | ||||||
| A061 | Kishu | Kishu mikan Ihara Ichijoji | 0 | 0 | ||||||
| A062 | Kishu | Komikan Fukuyama (Kinkou pearl) | 0 | 0 | ||||||
| A063 | Kishu | Komikan Kawachi | 0 | 0 | ||||||
| A064 | Kishu | Komikan Tensui | 0 | 0 | ||||||
| A065 | Kishu | Kouda mikan | 0 | 0 | ||||||
| A066 | Kishu | Mukaku Kishu (seedless Kishu) | 0 | 0 | ||||||
| A067 | Kishu | Nanfengmiju | 0 | 0 | ||||||
| A068 | Kishu | Ozaki komikan | 0 | 0 | ||||||
| A069 | Kishu | Sakurajima komikan Matsuura | 0 | 0 | ||||||
| A070 | Kishu | Sakurajima komikan senbatsu 1gou | 0 | 0 | ||||||
| A071 | Kishu | Sakurajima komikan Shirahama | 0 | 0 | ||||||
| A072 | Kishu | Taka mikan | 0 | 0 | ||||||
| Kizu | A073 | (stock strain) | C. kizu hort. ex Yu.Tanaka | 0 | A029 | Hebesu | (stock strain) | 0 | 0 | |
| Koji | A076 | (stock strain) | C. leiocarpa hort. ex Tanaka | 0 | A077 | Komikan 2009–130 | (stock strain) | 0 | 0 | |
| A189 | Toukan | (stock strain) | 0 | 0 | ||||||
| Kunenbo | Kunenbo A | A081 | (stock strain) | C. nobilis Lour. var. kunep Tanaka | 0 | A007 | Bendiguangju | (Honchi kokitsu) | 0 | 0 |
| A083 | Kunenbo | Kagoshima 0027 | 1 | 1 | ||||||
| A084 | Kunenbo | Kamikoshikijima | 1 | 1 | ||||||
| A190 | Tookunin | (stock strain) | 2 | 2 | ||||||
| A193 | Twukkunin | (stock strain) | 0 | 0 | ||||||
| A194 | Twukunihu | (stock strain) | 0 | 0 | ||||||
| A195 | Twuukuribu | (stock strain) | 0 | 0 | ||||||
| Kunenbo B | A082 | Kagoshima 0007 | C. nobilis Lour. var. kunep Tanaka | 0 | (none) | — | ||||
| Twukkuni | A192 | (stock strain) | C. nobilis Lour. var. kunep Tanaka | 1 | (none) | — | ||||
| King | A054 | (stock strain) | C. nobilis Lour. | 0 | (none) | — | ||||
| Natsudaidai | A098 | Kawano | C. natsudaidai Hayata | 0 | A096 | Natsudaidai | (stock strain) | 0 | 0 | |
| A097 | Natsudaidai | Beniamanatsu | 0 | 0 | ||||||
| A099 | Natsudaidai | Tachibana orange | 0 | 0 | ||||||
| Ponkan | A107 | Oota | C. reticulata Blanco | 0 | A105 | Ponkan | Ihara ponkan | 0 | 0 | |
| A106 | Ponkan | Morita ponkan | 0 | 0 | ||||||
| A108 | Ponkan | Yoshida ponkan | 0 | 0 | ||||||
| Rokugatsumikan | A111 | (stock strain) | C. rokugatsu hort. ex Yu.Tanaka | 0 | A020 | Fukushukan | (stock strain) | 0 | 0 | |
| Satsuma | A125 | Okitsu Wase | C. unshiu Marcov. | A113 | Satsuma | Aoshima unshu | 0 | 0 | ||
| A114 | Satsuma | Dobashi beni | 1 | 0 | ||||||
| A115 | Satsuma | Haraguchi wase | 0 | 0 | ||||||
| A116 | Satsuma | Imamura unshu | 0 | 0 | ||||||
| A117 | Satsuma | Iwasaki wase | 0 | 0 | ||||||
| A118 | Satsuma | Juman unshu | 0 | 0 | ||||||
| A119 | Satsuma | Jutaro unshu NC | 0 | 0 | ||||||
| A120 | Satsuma | Kinokuni unshu | 0 | 0 | ||||||
| A121 | Satsuma | Kuno unshu | 0 | 0 | ||||||
| A122 | Satsuma | Miyagawa wase | 0 | 0 | ||||||
| A123 | Satsuma | Nagahashi unshu NC | 0 | 0 | ||||||
| A124 | Satsuma | Niu unshu | 0 | 0 | ||||||
| A126 | Satsuma | Original tree | 0 | 0 | ||||||
| A127 | Satsuma | Otsu-4 (NC) | 0 | 0 | ||||||
| A128 | Satsuma | Shirakawa unshu | 0 | 0 | ||||||
| A129 | Satsuma | Sugiyama unshu | 0 | 0 | ||||||
| A130 | Satsuma | Suruga beni | 0 | 0 | ||||||
| A131 | Satsuma | Ueno wase | 0 | 0 | ||||||
| A132 | Satsuma | Yamada unshu NC | 0 | 0 | ||||||
| A133 | Satsuma | Yamashita beni | 0 | 0 | ||||||
| Satsuma Kikoku | A134 | (stock strain) | C. spp | 0 | A078 | Konejime | (stock strain) | 0 | 0 | |
| Sour orange | A141 | (stock strain) | C. aurantium L. var. crispa Yu.Tanaka | 0 | A139 | Sour orange | Bouquet de fleurs | 0 | 0 | |
| A140 | Sour orange | Chaozhouchen | 0 | 0 | ||||||
| A142 | Sour orange | Kaiseito | 2 | 2 | ||||||
| A143 | Sour orange | Zadaidai | 0 | 0 | ||||||
| A093 | Myrttle leaf orange | Chinott | 1 | 0 | ||||||
| Sweet orange | A162 | Trovita | C. sinensis (L.) Osbeck | 0 | A148 | Sweet orange | Cadenera | 0 | 0 | |
| A149 | Sweet orange | Cara cara | 3 | 2 | ||||||
| A150 | Sweet orange | Crescent | 0 | 0 | ||||||
| A151 | Sweet orange | Hamlin | 0 | 0 | ||||||
| A152 | Sweet orange | Jincheng | 0 | 0 | ||||||
| A153 | Sweet orange | Joppa | 0 | 0 | ||||||
| A154 | Sweet orange | Mediterranean sweet orange | 0 | 0 | ||||||
| A155 | Sweet orange | Moro NC | 1 | 0 | ||||||
| A156 | Sweet orange | Parson Brown | 0 | 0 | ||||||
| A157 | Sweet orange | Pineapple | 0 | 0 | ||||||
| A158 | Sweet orange | Santa Catarina | 0 | 0 | ||||||
| A159 | Sweet orange | Seike navel | 0 | 0 | ||||||
| A160 | Sweet orange | Shamouti | 0 | 0 | ||||||
| A161 | Sweet orange | Tongzigan | 0 | 0 | ||||||
| A163 | Sweet orange | Washington navel | 0 | 0 | ||||||
| A164 | Sweet orange | Valencia | 0 | 0 | ||||||
| A165 | Sweet orange | Wuyuecheng | 0 | 0 | ||||||
| A166 | Sweet orange | Xuegan | 0 | 0 | ||||||
| A167 | Sweet orange | Yinzigan | 1 | 0 | ||||||
| Tachibana | Tachibana A | A172 | Heda 1 | C. tachibana (Makino) Tanaka | 1 | A168 | Tachibana | (stock strain) | 0 | 1 |
| A173 | Tachibana | Heda 2 | 0 | 1 | ||||||
| A178 | Tachibana | Okitsu | 0 | 1 | ||||||
| Tachibana B | A175 | Ishinami No.1 | C. tachibana (Makino) Tanaka | 0 | A169 | Tachibana | Anettaishijou | 0 | 0 | |
| A171 | Tachibana | Hananoiwaya | 0 | 0 | ||||||
| A176 | Tachibana | Ishinami No.2 | 0 | 0 | ||||||
| A177 | Tachibana | Oodomari OP-2 | 0 | 0 | ||||||
| A179 | Tachibana | Reizanji | 0 | 0 | ||||||
| Tachibana C | A174 | Ishinami Minka | C. tachibana (Makino) Tanaka | 0 | (none) | |||||
| Tankan | A183 | Taishun | C. tankan Hayata | 0 | A182 | Tankan | (stock strain) | 0 | 0 | |
| A184 | Tankan | Tarumizu 1 | 0 | 0 | ||||||
| A185 | Tankan | T-132 | 0 | 0 | ||||||
| Tosa buntan | A191 | (stock strain) | C. maxima (L.) Merr. | 0 | A102 | Ootachibana | (stock strain) | 0 | 0 | |
| Ujukitsu | A197 | (stock strain) | C. ujukitsu hort. ex Tanaka | 0 | A034 | Houraikan | (stock strain) | 0 | 0 | |
| 'Shiranuhi' | B058 | (stock strain) | (Hybrid) | 0 | B053 | 'Sagakashi 34' | (stock strain) | 0 | 0 | |
NC: Nucellar seedling
ND: Number of failed genotypes
MM: Mismatched genotypes
Any pair of samples that showed fewer than four mismatches were assumed identical. This threshold was determined empirically. According to this criterion, all genotypes of the four Clementine strains (A009 to A012) were identical, and they were confirmed as selected somatic mutants (Table 4, S5 Table). In the same way, genotypes of two C. tangerina varieties (A016: dancy and A017: obeni mikan), three grapefruit strains (A024–A026), two iyo strains (A043 and A044), four natsudaidai strains (A096–A099), four ponkan strains (A105–A108) and four tankan strains (A182–A185) agreed exactly among themselves, and were revealed to be somatic mutants. Except for one mismatch observed in the strain Hisago komikan (A058), the genotypes of 16 strains of Kishu (A057–A072) agreed with each other exactly, and they were confirmed as somatic mutants. Interestingly, these 15 strains of Kishu were collected in Japan, but a Chinese strain nanfengmiju (A067) exactly matched Kishu. The identity tests of hyuganatsu, Satsuma and sweet orange demonstrated slight mismatches within them. These mismatches were attributed to accidental technical failure. However, biological replication of Cara cara (A149) and the nucellar seedling selection Moro (A155) confirmed that their discrepancies were reproducible. These observations confirmed that the mutation of SSR markers is not frequent but a rare event, and unlikely to alter many genotypes of a strain from the original. We therefore concluded that all evaluated strains of hyuganatsu, Satsuma and sweet orange were somatic mutants. The identity test of five strains of sour orange (A141: stock strain of sour orange, A139: bouquet de fleurs, A140: chaozhouchen, A142: kaiseito, and A143: za daidai) confirmed them to be somatic mutants of sour orange. The examined genotype of myrtle-leaf orange Chinott (A093: C. myrtifolia Raf.) was discrepant in one DNA marker (NSX23) but otherwise identical to sour orange, and it was consequently confirmed to be another somatic mutant of sour orange. Though significant differences are widely recognized in their fruit shape, tree architecture and leaf size, such discrepancies between sour orange strains and myrtle-leaf orange strains were confirmed to be natural variations within sour orange.
The identity test also revealed unforeseen relationships between particular varieties. A possible pummelo hybrid variety andoukan (A001) coincided in its genotypes, except for two with missing data, with those of kinukawa (A056: C. glaberrima hort. ex Tanaka), which was thought to be a chance seedling of pummelo. A mandarin variety girimikan (A023: C. tardiva hort. ex Shirai) exactly matched in genotypes with those of Tajima mikan (A180: C. spp). Such identical relationships were also revealed between henka mikan (A030: C. pseudo-aurantium hort. ex Yu.Tanaka) and nansho daidai (A094: C. taiwanica Tanaka et Shimada), Hiroshimanatsubuntan (A033: C. hiroshimana hort. ex Yu.Tanaka) and Takumanatsukunenbo (A181: C. spp), kizu (A073: C. kizu hort. ex Yu.Tanaka) and hebesu (A029: C. junos hybrid), rokugatsumikan (A111: C. rokugatsu hort. ex Yu.Tanaka) and fukushukan (A020: C. spp), Satsuma kikoku (A134: C. spp) and konejime (A078: C. junos hybrid), and Tosa buntan (A191: C. maxima Merr.) and Ootachibana (A102: C. otachibana hort. ex Yu.Tanaka). Koji (A076: C. leiocarpa hort. ex Tanaka) matched in its genotypes with two varieties: komikan 2009–130 (A077: C. spp), which was a collection of NIFTS found in the Southwest Islands of Japan; and toukan (A189: C. spp). Ujukitsu (A197: C. ujukitsu hort. ex Tanaka) and horaikan (A034: C. ujukitsu hort. ex Tanaka) were presumed to be synonymous with each other, and this study confirmed the assumption with evidence. Tanaka described kizu (A073) and mochiyu (A091) as synonyms from different localities [12], but the identity test revealed that they arose from different origins. With these observations, we selected one of the natural variations from each set of identical genotypes, and regarded them as representatives of each genotype in the subsequent analysis.
Genetic variation in the indigenous varieties
In contrast to the genetic identity found among the strains of various indigenous citrus varieties, variations in the strains of kunenbo and tachibana were identified (Table 4 and S5 Table). One kunenbo strain (A081: C. nobilis Lour. var. kunep Tanaka) agreed in its genotypes with six other strains (A083: kunenbo Kagoshima 0027, A084: kunenbo Kamikoshikijima, A190: tookunin, A193: twukkunin, A194: twukunihu, and A195: twuukuribu) that were classified to the same scientific name. Although three of them (A083, A084, A190) showed one or two mismatches to kunenbo, these were attributed to technical failure (S5 Table). Furthermore, bendi guangju (A007: C. spp, also known as honchi kokitsu in Japanese) exactly agreed in its genotypes with kunenbo (A081) (S5 Table). Two C. nobilis strains kunenbo Kagoshima 007 (A082: C. nobilis Lour. var. kunep Tanaka) and twukkuni (A192: C. nobilis Lour. var. kunep Tanaka) revealed 133 and 115 mismatches to kunenbo (A081) among the 169 DNA markers used, and kunenbo Kagoshima 007 (A082) disagreed with twukkuni (A192) for 100 markers (S5 Table). Additionally, King (A054: C. nobilis Lour.) revealed 99, 110 and 107 mismatches to kunenbo (A081), kunenbo Kagoshima 007 (A082) and twukkuni (A192), respectively (S5 Table). Although twukkuni (A192) contained one missing marker, these four varieties are obviously derived from different origins considering the frequency of mismatches among them. Accordingly, we selected these four unique genotypes as the representative varieties of C. nobilis, and tentatively assigned ‘kunenbo-A’ to kunenbo (A081), ‘kunenbo-B’ to kunenbo Kagoshima 007 (A082), ‘twukkuni’ to twukkuni (A192), and ‘King’ to King (A054) in subsequent study.
Similar genetic variations were also found among the strains of tachibana (C. tachibana (Makino) Tanaka). One tachibana strain, Heda 1 (A172), agreed in its genotype with three others (A168: tachibana stock strain, A173: Heda 2 and A178: Okitsu), but large discrepancies were found for tachibana Ishinami No.1 (A175) and tachibana ishinami minka (A174), with 45 and 72 mismatches (S5 Table). Tachibana ishinami No.1 (A175) and tachibana ishinami minka (A174) disagreed at 72 DNA markers (S5 Table). Tachibana ishinami minka (A176) agreed only with itself. However, tachibana ishinami No.1 (A175) agreed in genotype with five other tachibana strains (A169: anettaishijou, A171: hananoiwaya, A176: ishinami No.2, A177: Oodomari OP-2 and A179: Reizanji). On the basis of these observations, we selected these three unique genotypes as the representative varieties of tachibana, and tentatively assigned tachibana-A to Heda 1 (A172), tachibana-B to ishinami No.1 (A175), and tachibana-C to ishinami minka (A174) in subsequent study. Likewise, two shiikuwasha strains (A135 stock strain, and A136 Ogimikugani) disagreed at 44 DNA markers (S5 Table), and are therefore regarded as different strains of C. depressa Hayata.
According to these observations revealed by the genetic identity test, we selected 101 representative indigenous varieties that have unique genotypes. Kobayashi mikan (A074) was excluded from the representatives because it is a chimera and often gave three alleles. We also selected 75 representatives from 78 hybrid varieties by excluding one nucellar selection (B053: ‘Sagakashi 34’), one triploid variety (B046: ‘Oroblanco’), and one tetraploid variety (B032: ‘Kuchinotsu-41’). All 85 selected strains were selected as representatives since their genotypes were unique and did not overlap with others. Consequently, 261 unique representative varieties or strains were selected (Table 1). These are indicated by asterisks in S1, S3, S5 and S9 Tables.
Statistical evaluation of genetic characteristics
The 261 selected representative varieties or strains in the three sample categories were evaluated for seven genetic parameters: number of unique genotypes (Ng); number of unique alleles (Na); observed heterozygosity (Ho); expected heterozygosity (He); polymorphic information content (PIC); match probability (PM); and Wright’s fixation index (Fw); using the 169 certified DNA markers. Table 5 shows a summary of each parameter for the three sample categories (indigenous varieties, hybrid varieties and selected strains). S6 Table gives all the data for these seven parameters and the number of valid samples obtained with both certified and non-certified DNA markers.
Table 5. Summary of genetic characteristics and deduced population structure.
A. Genetic characteristics for three sample categories.
| Feature | Sample | N50 | N25 | N75 | Mean | S-W P | B-F P | K-W P | Sig | Pairs | 95% CI | Padj | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ng | IV | 9.0 | 6.0 | 17.0 | 12.0 | 1.5.E-10 | 2.05E-15 | 1.30E-11 | A | IV-HV | 0.267 | - | 0.381 | 4.94E-09 |
| HV | 6.0 | 4.0 | 10.0 | 7.1 | 1.8.E-09 | B | IV-SL | 0.241 | - | 0.352 | 8.98E-12 | |||
| SL | 6.0 | 3.0 | 9.0 | 6.4 | 1.3.E-08 | B | HV-SL | 0.404 | - | 0.527 | 0.268 | |||
| ALL | 10.0 | 6.0 | 18.0 | 13.4 | 8.4.E-11 | — | — | — | — | — | — | |||
| Na | IV | 5.0 | 3.0 | 7.0 | 5.6 | 2.5.E-09 | 7.05E-07 | 2.20E-09 | A | IV-HV | 0.291 | - | 0.406 | 5.38E-07 |
| HV | 3.5 | 3.0 | 5.0 | 4.1 | 3.7.E-09 | B | IV-SL | 0.263 | - | 0.374 | 1.29E-09 | |||
| SL | 3.0 | 2.0 | 5.0 | 3.8 | 1.2.E-08 | B | HV-SL | 0.403 | - | 0.524 | 0.231 | |||
| ALL | 5.0 | 4.0 | 8.0 | 5.9 | 3.4.E-09 | — | — | — | — | — | — | |||
| Ho | IV | 0.567 | 0.455 | 0.703 | 0.567 | 1.0.E-03 | 0.4994 | 0.005462 | A | IV-HV | 0.350 | - | 0.472 | 7.99E-03 |
| HV | 0.507 | 0.373 | 0.653 | 0.500 | 1.6.E-03 | B | IV-SL | 0.353 | - | 0.474 | 7.99E-03 | |||
| SL | 0.529 | 0.388 | 0.647 | 0.504 | 5.5.E-04 | B | HV-SL | 0.443 | - | 0.567 | 0.868 | |||
| ALL | 0.548 | 0.425 | 0.659 | 0.534 | 6.1.E-04 | — | — | — | — | — | — | |||
| He | IV | 0.567 | 0.478 | 0.716 | 0.570 | 2.4.E-05 | 0.8272 | 2.43E-09 | A | IV-HV | 0.295 | - | 0.412 | 2.06E-06 |
| HV | 0.499 | 0.870 | 0.602 | 0.484 | 3.5.E-04 | B | IV-SL | 0.255 | - | 0.368 | 6.24E-10 | |||
| SL | 0.489 | 0.361 | 0.572 | 0.461 | 1.8.E-04 | B | HV-SL | 0.397 | - | 0.520 | 0.159 | |||
| ALL | 0.529 | 0.439 | 0.643 | 0.527 | 3.4.E-05 | — | — | — | — | — | — | |||
| PIC | IV | 0.495 | 0.385 | 0.668 | 0.512 | 1.1.E-02 | 0.03945 | 1.08E-09 | A | IV-HV | 0.290 | - | 0.406 | 7.98E-07 |
| HV | 0.394 | 0.324 | 0.541 | 0.421 | 1.0.E-02 | B | IV-SL | 0.253 | - | 0.366 | 3.81E-10 | |||
| SL | 0.375 | 0.318 | 0.506 | 0.397 | 2.6.E-03 | B | HV-SL | 0.396 | - | 0.519 | 0.1793 | |||
| ALL | 0.460 | 0.366 | 0.589 | 0.468 | 1.8.E-03 | — | — | — | — | — | — | |||
| PM | IV | 0.261 | 0.132 | 0.361 | 0.279 | 7.8.E-10 | 0.8338 | 2.89E-13 | A | IV-HV | 0.623 | - | 0.737 | 2.34E-09 |
| HV | 0.364 | 0.232 | 0.471 | 0.381 | 1.5.E-07 | B | IV-SL | 0.672 | - | 0.780 | 1.47E-14 | |||
| SL | 0.403 | 0.277 | 0.489 | 0.408 | 3.4.E-08 | B | HV-SL | 0.488 | - | 0.611 | 0.117 | |||
| ALL | 0.307 | 0.184 | 0.404 | 0.323 | 2.4.E-09 | — | — | — | — | — | — | |||
| FW | IV | 0.004 | -0.052 | 0.075 | 0.008 | 0.01428 | 8.94E-05 | 2.23E-13 | A | IV-HV | 0.298 | - | 0.420 | 1.21E-05 |
| HV | -0.055 | -0.106 | 0.004 | -0.019 | 1.2E-10 | B | IV-SL | 0.213 | - | 0.324 | 4.32E-14 | |||
| SL | 0.090 | -0.169 | 0.000 | -0.083 | 5.9.E-03 | C | HV-SL | 0.325 | - | 0.446 | 2.29E-04 | |||
| ALL | -0.021 | -0.067 | -0.043 | -0.005 | 4.4.E-04 | — | — | — | — | — | — | |||
Average value of genetic characteristics with standard deviation for each sample category obtained with the 169 certified DNA markers. IV: 101 indigenous varieties, HV: 78 hybrid varieties, SL: 85 selected strains, ALL: 261 representative samples of these three categories.
Ng: number of unique genotypes, Na: number of unique alleles, Ho: observed heterozygosity, He: expected heterozygosity, PIC: polymorphic information content, PM: probability of match, Fw: fixation index.
N50, N25, N75: 50th, 25th and 75th percentile values.
S-W P: p-value of the normal distribution for each sample estimated by Shapiro–Wilk test.
B-F P: p-value of homogeneity of variance among the three sample categories estimated by Brown–Forsythe test.
K-W P: p-value of one-way ANOVA among the three sample categories estimated by Kruskal–Wallis test.
Sig: different letters represent significance between them at p < .01.
95% CI and Padj: The adjusted 95% confidence interval and p-value for each sample pair by Brunner–Munzel test. The p-value was adjusted by Benjamini–Hochberg (BH) correction for multiple sample comparison.
Pairs: combinations for the pairwise Brunner–Munzel test or G''ST analysis.
Statistical evaluation of the data did not confirm the normal distribution of these data even after several data conversion methods were applied, and equal variance was not confirmed for Ng, Na and Fw. Consequently, we adopted nonparametric analysis methods for the evaluation of these data. The medians (N50) of Ng, Na, Ho, He, PIC, PM and Fw for the indigenous varieties were 9.0, 5.0, 0.567, 0.567, 0.495, 0.261 and 0.04, respectively. The median and mean values of Ng, Na, Ho and He were higher than those reported by Curk et al [47], confirming that they were sufficiently polymorphic for the following genetic analysis. The observed Ho value demonstrated the heterozygous nature of the indigenous citrus varieties, and the He value demonstrated wide genetic diversity among them. The observed heterozygosity was high enough to use these DNA markers for the genetic mapping of crossed citrus populations. The observed high PIC value and low PM value confirmed their discriminatory power and indicates a low chance of misidentification of plant samples when using them.
The N50 and N75 values of Ng for the hybrid varieties (6.0 and 10.0) and the selected strains (6.0 and 9.0) were lower than for the indigenous varieties (9.0 and 12.0, Table 5). The observed differences between these were considered significant (p < .01), but not between the hybrid varieties and the selected strains (p > .05). Likewise, the Na values for the hybrid varieties and the selected strains were significantly lower than that of the indigenous varieties (p < .01), but the difference between the hybrid varieties and the selected strains was not obvious (p > .05). These decreases in Ng and Na strongly suggest that certain genotypes or alleles have been selected during the breeding program. These selected allele sets could be beneficial for citrus breeding. The differences in Ng and Na were not significant between the hybrid varieties and the selected strains, suggesting that the usefulness of the selected alleles in breeding continues in these offspring.
Similarly, the Ho, He and PIC values were significantly decreased in the hybrid varieties and the selected strains compared with the indigenous varieties (p < .01), but it was not obvious between the hybrid varieties and the selected strains. The observed decrease in PIC value coincided with the loss of alleles in the hybrid varieties and the selected strains. Though significant decreases were observed in Ho and He, these values in the hybrid varieties and the selected strains remained high, confirming their higher heterozygosity. On the contrary, PM was 0.261 for the indigenous varieties but was increased to 0.364 and 0.403 in the hybrid varieties and the selected strains, respectively. The observed increase in PM coinciding with the loss of alleles resulted in an increase in the probability that unrelated individuals show the same genotype.
The estimated fixation index (Fw) was not consistent among the three sample categories (p < .01). The Fw value for the indigenous varieties (0.004) suggested inbreeding within them. However, it was decreased for the hybrid varieties (-0.005) and this decrease is considered to have been achieved through artificial outcrossing. Interestingly, the Fw value was increased again in the selected strains (0.090). This increase is consistent with consanguineous mating among the indigenous varieties and the hybrid varieties during development of the selected strains, resulting in the loss of alleles.
The FIT value [86,88,89] suggests that inbreeding of all citrus samples within the three sample categories is not obvious (Table 6). The FIS value estimates that the inbreeding of individual varieties or lines in each sample category is not significant. However, the global G''ST value [88] was as high as 0.0703, suggesting substantial inbreeding in each of the three sample categories. The within-population inbreeding between each sample category is demonstrated by the increase of G''ST value between the indigenous varieties and the hybrid varieties (0.08637) or the selected strains (0.11925) (Table 6). In contrast, the increase was not significant between the hybrid varieties and the selected strains (0.00253). The deduced inbreeding within the hybrid varieties and the selected strains coincide well with the decrease in genotypes (Ng), alleles (Na), observed heterozygosity (Ho), expected heterozygosity (He) and PIC values, and also the increase in match probability (PM) among them (Table 5). These observations support the initial hypothesis that the indigenous varieties used in this study are high in genetic diversity but that hybrid varieties have selected particular alleles from the indigenous varieties, and fewer alleles are maintained in the selected strains. The decreased variation in alleles would increase the probability of sharing the same allele by crossing, as suggested by the increase in match probability, and results in an increase in inbreeding in the hybrid varieties and the selected strains by frequent use of particular varieties as breeding parents.
Table 6. Deduced F statistics and G''ST values among three sample categories.
| Statistics | Value | Pairs | G''ST |
|---|---|---|---|
| FIT | 0.0002 | IV—HV | 0.08637 |
| G''ST | 0.0703 | IV—SL | 0.11925 |
| FIS | -0.0384 | HV—SL | 0.00253 |
Refer Table 5 for the symbols.
Evaluation of genetic disequilibrium
Prior to estimating the population structure and analyzing the parentage of the indigenous varieties, which assume Hardy–Weinberg equilibrium (HWE) [100,106], we tested for HWE in the certified markers. Because SSR markers are highly polymorphic, we applied a Markov Chain Monte Carlo (MCMC) simulation method [94,107]. This method was implemented in Arlequin [95], and estimated the p-value for individual DNA markers, but it showed a slight variation in separate analyses because of its simulation principle. Accordingly, we tested for HWE in the 169 certified DNA markers ten times, and 31 DNA markers were considered not to satisfy HWE in the indigenous variety samples according to their average p-value (p < .05). These 31 DNA markers, along with the duplicated markers, were excluded in the following analysis, and 123 representative DNA markers that were confirmed to satisfy genetic consistency, singularity in the genome, and HWE in the indigenous varieties, were selected and provided to the following analysis.
Factorial analysis and phylogenetic evaluation based on nuclear genotypes
The population structure of the 101 representative indigenous varieties that excluded all identical genotype plant samples was examined with the 123 representative genomic DNA markers by principal coordinate analysis using DARWin [97,98]. The number of assumed axes was changed from two to six, but the values of the first two coordinates did not change. Therefore, the values of two major coordinates from five assumed coordinates were used to draw a scatter plot (Fig 1). These two coordinates explain about 39.1% of the total variation among the indigenous varieties. Five mandarin varieties (Kishu, dancy, willowleaf mandarin, sokitsu and kobeni mikan) are located together in the lower right region of the plot. Six pummelo varieties (banpeiyu, Egami buntan, Hirado buntan, Mato buntan, pummelo white type and uchimurasaki) are located on the left side of the plot. These five mandarins and six pummelos are located on opposite sides of the abscissa, and are considered to represent mandarin (C. reticulata) and pummelo (C. maxima), respectively. Meanwhile, three varieties (lemon, Mexican lime and ichanchii) that represent C. medica or C. ichangensis are located at the top center of the plot (Fig 1). The positions of these major citrus varieties, C. reticulata, C. maxima and C. medica or C. ichangensis, in the plot are reciprocal to each other, and constitute representative apexes in the plot. Though the absolute positions of these three basic groups are different, their triangular relationship is similar to previous reports [15,45], and confirms that these major citrus variety groups are well separated on this plot with the selected DNA markers.
Fig 1. Principal coordinate analysis plot of 101 citrus indigenous varieties using genomic DNA markers.

The plot was produced from the dissimilarity index deduced from 169 genomic DNA markers.
C. medica and C. ichangensis are located close together in the upper apex of the scatter plot. Preliminary evaluation of the DNA markers found that a considerable portion of the evaluated DNA markers yielded three or more PCR products, or failed on amplification, for the citrus varieties of C. medica. These defective DNA markers were eliminated in this study, but this may have suppressed the separation between C. medica and C. ichangensis. Satsuma and sweet orange are located between pummelos and mandarins but closer to mandarins, suggesting the contribution of mandarin and pummelo for their occurrence. Yuzu is close to C. ichangensis, agreeing with its proposed origin as a chance seedling of C. ichangensis as suggested by Swingle [11]. Likewise, sour orange is located close to the middle among these three apexes, and it is considered an offspring of C. maxima, C. reticulata and C. medica. Other indigenous citrus varieties are located anywhere between these three apexes on the scatter plot. However, their distribution is not discrete but continuous, and there is no clear isolated or aggregated structure. These observations suggest a complex admixture history for the occurrence of these varieties.
A phylogenetic tree of the 101 representative indigenous varieties based on 123 representative genomic DNA markers was constructed using the neighbor-joining method [99] from bootstrap analysis. We prepared the consensus tree from 30,000 bootstrap trials, but runs with 5,000 or 10,000 MCMC iterations produced identical trees. The tree classifies the 101 indigenous citrus varieties into three major clusters (Fig 2). Cluster I consists of 11 varieties including the three varieties (lemon, Mexican lime and ichanchii) that constitute the upper central apex in Fig 1. Sour orange and the offspring of lemon were classified into this cluster, and C. medica and C. ichangensis with their varieties are thus considered to be classified in this cluster. Cluster II consists of 30 varieties including the five pummelo varieties that constitute the left apex in the PCoA plot (Fig 1). All pummelo offspring varieties are found in this cluster; therefore, this cluster is considered to represent C. maxima and its offspring. Cluster III consists of 60 varieties, and the six mandarin varieties that constitute the remaining apex in Fig 1 are found in this cluster. However, various varieties that are located at diverse positions in the PCoA plot (Fig 1), for example Temple, ujukitsu, henka mikan, mochiyu, jabara, kizu, kabosu, sudachi, which are regarded as natural hybrids [7,11], are also found in this cluster. Consequently, this cluster is considered to represent C. reticulata and its offspring varieties. All representative varieties are classified into different clades, and no consolidated clade structure reminiscent of the PCoA plot is obvious in the tree.
Fig 2. Phylogenetic tree of 101 citrus indigenous varieties estimated from genomic DNA markers.

The tree was produced using the neighbor-joining method from genetic distances by the simple matching method. Node labels show bootstrap support values larger than 20.
Inferring basic taxa and their proportions in individuals of the indigenous varieties
The proportions of basic taxa for 101 indigenous varieties were inferred by a model-based clustering method with a Bayesian MCMC approach according to Pritchard et al [108]. The deduced number of basic taxa (K) was obtained from the ΔK value according to Evanno et al [102] by varying K from two to ten (Table 7). The magnitude plot of ΔK against K shows a large single peak at K = 3 (Fig 3). The delta K values for K = 4 to 9 were close to zero (Table 7). Changing the iteration period for initial burn-in (50,000 or 100,000) and MCMC runs (500,000 or 1,000,000) in the simulations gave the same result with a single large peak at K = 3 (data not shown). Another large peak at K = 5 appeared when all genotype data that deviated from HWE were included in the structure analysis (data not shown). However, this disappeared when the genotype data that deviated from HWE were removed. Therefore, that peak was considered to be spuriously caused by disequilibrium in particular DNA markers, and discarded. The deduced K value conforms to the current understanding that the basic citrus ancestral taxa consist of C. medica, C. maxima and C. reticulata [14,15,35,109].
Table 7. Summary of ΔK estimation for Structure analysis.
| K | Reps | Mean LnP(K) | Stdev LnP(K) | Ln'(K) | |Ln''(K)| | Delta K |
|---|---|---|---|---|---|---|
| 2 | 10 | -22738.00 | 0.49 | — | — | — |
| 3 | 10 | -21305.92 | 1.07 | 1432.08 | 681.49 | 637.78 |
| 4 | 10 | -20555.33 | 73.18 | 750.59 | 209.44 | 2.86 |
| 5 | 10 | -20014.18 | 182.62 | 541.15 | 2388.97 | 13.08 |
| 6 | 10 | -21862.00 | 3550.42 | -1847.82 | 4291.36 | 1.21 |
| 7 | 10 | -19418.46 | 259.15 | 2443.54 | 5009.38 | 19.33 |
| 8 | 10 | -21984.30 | 8041.19 | -2565.84 | 37.24 | 0.00 |
| 9 | 10 | -24587.38 | 4307.95 | -2603.08 | 5124.23 | 1.19 |
| 10 | 10 | -22066.23 | 5352.53 | 2521.15 | — | — |
The magnitude of ΔK from STRUCTURE analysis was calculated according to Evanno et al (2005) for 101 indigenous varieties with 123 DNA markers. The prior K value was varied from 2 to 10, with 10 simulation runs for each K. Simulations for each K used 100,000 iterations for initial burn-in, and 1,000,000 iterations for the subsequent MCMC estimation.
Fig 3. Magnitude of ΔK deduced from structure analysis as a function of K.

Simulation runs were performed for K = 2 to 10 with 10 iterations for each K, and the mean values of ΔK are plotted against K.
The inferred admixture population (Q) plot of the 101 indigenous varieties at K = 3 demonstrates that several varieties mostly consisted of one of the three populations (Fig 4, S8 Table). The inferred admixture proportion of the first population was over 90% for seven pummelo varieties (banpeiyu, Egami buntan, Hirado buntan, Mato buntan, suisho buntan, pummelo white type and uchimurasaki), and these were regarded as a taxon representing pummelo (C. maxima). Likewise, eight varieties (hanayu, ichanchii, Kourai Tachibana, lemon, limonia, Mexican line, rokugatsumikan and yuzu) had proportions of more than 80% for the second population, and this group was regarded as a taxon representing citron (C. medica). Sixteen varieties (bendizao, Clementine, Cleopatra, dancy, Hickson, Kishu, kobeni mikan, genshokan, Murcott, sokitsu, sunki, ponkitsu, youpiju, ponkan, willowleaf mandarin and Mediterranean mandarin) had proportions of more than 90% for the third population, and were regarded as a taxon representing mandarin (C. reticulata). The deduced proportions of these three basic taxa in individual varieties will be discussed in the following section.
Fig 4. Inferred admixture populations (Q) of 101 indigenous citrus varieties for a K = 3 population model in STRUCTURE.

Three clusters correspond to the deduced basic taxa at K = 3 (pummelo, citron and mandarin).
Development and evaluation of DNA markers for genotyping chloroplast and mitochondrial genomes
For the purpose of categorizing citrus varieties according to their cytoplasmic organellar genotypes (referred to as ‘cytotypes’ hereafter), genotypes of both chloroplast and mitochondria genomes were evaluated using DNA markers for each genome. Phylogenetic analyses of citrus varieties based on chloroplast genome polymorphisms have been reported for trnL–trnF and trnT–trnL sequences [50], rbcL–ORF106, psaA–trnS, trnH–trnK and trnD–trnT intergenic regions [57], trnL–trnF sequences [51], trnL–trnF intergenic regions [52], nine chloroplast genomic intergenic regions [110], matK gene sequences [55], and trnS–trnG, rps16, rpl16, atpB–rbcL and accD–psaI sequences [111]. However, our preliminary attempts at genotyping chloroplast genomes with previously reported DNA markers showed occasional failures or less amplification on particular varieties (data not shown). Therefore, we designed new SSR markers for the chloroplast genome by referring to the chloroplast genome sequence of sweet orange ‘Ridge Pineapple’ [41]. SSR mining of the sweet orange genome using mreps [76] identified 94 candidate regions. The forward and reverse primers of four SSR markers in the short single copy region and nine SSR markers in the large single copy region were designed to anchor at two adjacent genes to amplify an SSR found in an untranslated region between these genes. Preliminary evaluation selected two SSR markers for the short single copy region (CSS03: ndhE–ndhG and CSS04: ndhD–psaC) and two SSR markers for the large single copy region (CSL01: psbA–trnK and CSL09: rpl16–rps3) (Table 8). These regions have not been evaluated in citrus, or in other plant species. However, they were confirmed to be stable and versatile in a wide range of citrus varieties.
Table 8. SSR markers for the genotyping of organelle genomes.
| Chloroplast (this study1) | ||||||||
| Name | Target region | SSR From | To | Size | Program | F/R | BStag | Sequence |
| CSS03 | ndhE-ndhG | 122,864 | 122,878 | 292 | 54/28 | F | F9GTC | ctagtatgaggacgtcTCATTAACCAACTCCGTACCA |
| R | - | gtttcttGGCGCGTCAATAACAAATCT | ||||||
| CSS04 | ndhD-psaC | 121,941 | 121,955 | 211 | 56/28 | F | F9GCC | ctagtattaggacgccGTGGTAAAGACAAGATACACTTGG |
| R | - | gtttcttATGGCATGAAACAACCCGA | ||||||
| CSL01 | psbA-trnK | 1,743 | 1,775 | 355 | 60/28 | F | F9GAC | ctagtatcaggacgacCAGTGCTAGTTATCCAGTTACAGA |
| R | - | gtttcttCGGGCAACCCATTCTTATTATT | ||||||
| CSL09 | rpl16-rps3 | 86,713 | 86,730 | 276 | 54/32 | F | F9TAC | ctagtatcaggactacCGCACACTAAGCATAGCAAT |
| R | - | gtttcttCCTCTACAAACCATTGGAGCTA | ||||||
| Chloroplast (Weising and Gardner, 19912) | ||||||||
| Name | Target region | SSR From | To | Size | Program | BStag | Sequence | |
| ccmp2.2 | 5' to trnS | 8,609 | 189 | 54/28 | F | F9GAC | ctagtatcaggacgacATCGTACCGAGGGTTCGAAT | |
| R | - | GATCCAGGGCGTAATCCCG *) | ||||||
| ccmp6.2 | ORF77-ORF82 intergenic | 45,119 | 103 | 60/28 | F | F9GCC | ctagtattaggacgccCGATGCATATGTAGAAAGCC | |
| R | - | CATTACGTGCGACTATCTCT | ||||||
| ccmp7.2 | atp-B-rbcL intergenic | 57,339 | 133 | 52/36 | F | F9CCG | ctagtattaggacccgACATCATTATTGTATACTCTTTC | |
| R | - | CAACAGATAAAACTGTCAAG | ||||||
| ccmp10.2 | rpl2-rps19 intergenic | 86,694 | 103 | 54/32 | F | F9AGG | ctagtattaggacaggTTTTTTTTTAGTGAACGTGTCA | |
| R | - | TTCGCCGCCGTACTAAATAG | ||||||
| Mitochondria (Froelicher, et al. 20113) | ||||||||
| Name | Target region | SSR From | To | Size | Program | BStag | Sequence | |
| rrn5/rrn18-1 | rrn5-rrn18 | 161,129 | 161,383 | 273 | 56/28 | F | F9GTC | ctagtatgaggacgtcGGGTGAAGTCGTAACAAGGT |
| R | - | GAGGTCGGAATGGGATCGGG | ||||||
| nad2/4-3 | nad2-nad4-3 | 330,666 | 331,566 | 239 | 52/28 | F | F9CCG | ctagtattaggacccgGACCTTCACCTCAAATCA |
| R | - | TTCAGATAACACGCACC | ||||||
| nad7/1-2 | nad7-nad1-2 | 132,914 | 133,077 | 163 | 52/28 | F | F9AGG | ctagtattaggacaggGGAACATAGCATAGGG |
| R | - | TTTGATATAGGCTCGCT | ||||||
1 SSR markers developed in this study with reference to the sweet orange chloroplast genome (DQ864733).
2 SSR markers for chloroplast genomes by Weising and Gardner, 1991. The underlined nucleotides represent the modifications from their original sequences.
*) Forward and reverse primers were swapped from the original report.
3 DNA markers for genotyping mitochondrial genomes by Froelicher et al 2011.
All these DNA markers were designed for the single tube post labeling protocol with BStag. F/R: Forward/reverse primers.
We also evaluated the availability of recently published universal SSR markers for chloroplast genomes [81] and citrus mitochondrial genomes [53]. Preliminary evaluation of the 10 SSR markers for chloroplast genomes [81] failed to amplify or yielded a low amplification product with some citrus varieties. The primer sequences of these SSR markers were modified with reference to the sweet orange chloroplast genome [41], and four redesigned SSR markers (ccmp2.2, ccmp6.2, ccmp7.2 and ccmp10.2) were selected that show stable and consistent genotypes for a wide range of samples. Likewise, we evaluated 15 citrus DNA markers for mitochondria [53], but nine of them yielded amplified products too long for fragment analysis and were excluded. Three (rrn5/rrn18-1, nad2/4-3, and nad7/1-2) were selected according to their product size and stability on various citrus samples. Consequently, we selected 11 SSR markers, eight for chloroplast genomes and three for mitochondrial genomes.
Genetic characteristics estimated using organellar DNA markers
Genotyping analysis of 371 plant samples in three sample categories using the 11 selected organelle SSR markers yielded a single product in each sample, with no failure to amplify (S9 Table). The observed product sizes of these SSR markers almost agreed with previous reports [53,81]. All plant samples that were assumed to be somatic mutants on the basis of their nuclear genotypes (Table 4) revealed identical cytotypes.
The 11 organelle SSR markers evaluated each produced two to eight alleles among the samples and a total of 43 alleles were identified (Table 9). Their product sizes ranged from 129 to 370 bp (Table 9). The average number of alleles for the four chloroplast DNA markers from Weising was 3.8 [81], and that for the three mitochondrial DNA markers from Froelicher was 2.3 [53]. In contrast, the average number of alleles for the chloroplast DNA markers developed in this study was as high as 5.3. The genetic diversity (Nei’s GD) of the indigenous varieties ranged from 0.040 to 0.765 (Table 9). The median number of genotypes for all samples was 3.0 and the median GD was 0.477 (Table 10), and this SSR marker set was confirmed to be polymorphic enough to classify the cytotypes of a wide range of citrus samples. Previous studies classified the cytotypes of sweet orange (C. sinensis) and pummelo (C. maxima) into the same category [33,47,50,53,54,57], but this study separated them into different categories. Curk and colleagues classified citrus varieties into six categories with three chloroplast DNA markers and three mitochondrial DNA markers [47]. Their study classified sunki (C. sunki) and Cleopatra (C. reshni) into the same group as wild mandarin (C. reticulata) and limonia (C. limonia), and also classified Ichang lemon (C. sp.) into the group of C. maxima. In this study, those varieties were classified into independent groups, confirming the usefulness of the four chloroplast DNA markers developed in this study for fine separation and parentage analysis of citrus varieties.
Table 9. Organellar genotype characteristics of plant samples observed using SSR markers.
| Summary of identified genotypes for three sample categories | ||||||||||||||
| Markers | CSS03 | CSS04 | CSL01 | CSL09 | ccmp2.2 | ccmp6.2 | ccmp7.2 | ccmp10.2 | rrn5/rrn18-1 | nad2/4-3 | nad7/1-2 | Total | ||
| Indigenous varieties | 2 | 4 | 7 | 8 | 3 | 8 | 2 | 2 | 2 | 3 | 2 | 43 | ||
| Hybrid varieties | 1 | 3 | 3 | 2 | 1 | 3 | 1 | 1 | 2 | 2 | 2 | 21 | ||
| Selected lines | 1 | 3 | 3 | 3 | 1 | 3 | 1 | 1 | 2 | 2 | 2 | 22 | ||
| All samples | 2 | 4 | 7 | 8 | 3 | 8 | 2 | 2 | 2 | 3 | 2 | 43 | ||
| Size distribution of the amplified product (bp) * | ||||||||||||||
| Markers | CSS03 | CSS04 | CSL01 | CSL09 | ccmp2.2 | ccmp6.2 | ccmp7.2 | ccmp10.2 | rrn5/rrn18-1 | nad2/4-3 | nad7/1-2 | Total | ||
| Max | 335 | 237 | 379 | 311 | 224 | 149 | 159 | 130 | 274 | 269 | 172 | 379 | ||
| Min | 315 | 233 | 370 | 298 | 210 | 139 | 148 | 129 | 269 | 253 | 164 | 129 | ||
| Genetic diversity of each SSR markers estimated by Nei's GD | ||||||||||||||
| Markers | CSS03 | CSS04 | CSL01 | CSL09 | ccmp2.2 | ccmp6.2 | ccmp7.2 | ccmp10.2 | rrn5/rrn18-1 | nad2/4-3 | nad7/1-2 | Average | ||
| Indigenous varieties | 0.0776 | 0.6673 | 0.7651 | 0.6467 | 0.0398 | 0.6402 | 0.0776 | 0.0776 | 0.4321 | 0.5236 | 0.4321 | 0.3981 | ||
| Hybrid varieties | 0.0000 | 0.4193 | 0.4193 | 0.3930 | 0.0000 | 0.4154 | 0.0000 | 0.0000 | 0.3930 | 0.3930 | 0.3930 | 0.2569 | ||
| Selected lines | 0.0000 | 0.2570 | 0.2570 | 0.2510 | 0.0000 | 0.2372 | 0.0000 | 0.0000 | 0.2478 | 0.2478 | 0.2478 | 0.1587 | ||
| All samples | 0.0304 | 0.5736 | 0.6087 | 0.5658 | 0.0154 | 0.5300 | 0.0304 | 0.0304 | 0.4770 | 0.5218 | 0.4770 | 0.3509 | ||
*) Product size include BStag sequence (16 bp).
Table 10. Differences in genetic characteristics of organellar genotype among three sample categories.
| Feature | Sample | N50 | N25 | N75 | Mean | S-W P | B-F P | K-W P | Sig | Pair | 95% CI | Padj | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ng | IV | 3.0 | 2.0 | 5.5 | 3.9 | 0.00167 | 0.04179 | 0.04813 | a | IV-SV | 0.033 | - | 0.429 | 0.031 |
| HV | 2.0 | 1.0 | 2.5 | 1.9 | 0.01659 | b | IV-SL | 0.045 | - | 0.475 | 0.046 | |||
| SL | 2.0 | 1.0 | 3.0 | 2.0 | 0.00805 | b | SV-SL | 0.275 | - | 0.783 | 0.815 | |||
| ALL | 3.0 | 2.0 | 5.5 | 3.9 | 8.4.E-11 | — | — | — | — | — | — | |||
| GD | IV | 0.4320 | 0.0780 | 0.6418 | 0.3982 | 0.07757 | 0.1605 | 0.01515 | a | IV-SV | 0.019 | - | 0.467 | 0.041 |
| HV | 0.3930 | 0.0000 | 0.3985 | 0.2568 | 0.00093 | b | IV-SL | 0.000 | - | 0.447 | 0.041 | |||
| SL | 0.2425 | 0.0000 | 0.2487 | 0.1588 | 0.00082 | b | SV-SL | 0.024 | - | 0.546 | 0.098 | |||
| ALL | 0.4770 | 0.0300 | 0.5390 | 0.3509 | 0.00562 | — | — | — | — | — | — | |||
IV: indigenous varieties, HV: hybrid varieties, SL: selected strains, ALL: 261.
Refer Table 5 for other symbols.
The cytotypes obtained were categorized into 18 classes (C01 to C18) by the nonredundant allele set of 11 SSR markers (Table 11). Two SSR markers for the chloroplast genome (ccmp7.2 and ccmp10.2) showed identical genotype patterns, as did two SSR markers for the mitochondrial genome (rrn5/rrn18-1 and nad7/1-2) (Table 11). Each of the 18 classes consisted of a unique and non-redundant genotype set, and is referred to by its representative variety (Table 11). All representative plant samples of the three sample categories were classified into one of these 18 cytotype classes (Table 12 and S9 Table). The 18 proposed cytotypes agreed with the hybrid varieties that were used to evaluate genotyping error (Table 3), and showed no discrepancies. Therefore, we conclude that these classes demonstrate genuine cytotypes.
Table 11. The classes of citrus cytoplasmic genotype (cytotype) according to the nonredundant alleles of eight chloroplast and three mitochondrial SSR markers.
| Class | Cytotype | Chloroplast markers | Mitochondria markers | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CSS03 | CSS04 | CSL01 | CSL09 | ccmp2.2 | ccmp6.2 | ccmp7.2 | ccmp10.2 | rrn5/rrn18-1 | nad2/4-3 | nad7/1-2 | ||
| C01 | C. ichangensis type | 315 | 237 | 371 | 306 | 224 | 149 | 159 | 129 | 274 | 261 | 172 |
| C02 | Mexican lime type | 315 | 233 | 379 | 305 | 217 | 148 | 148 | 130 | 274 | 261 | 172 |
| C03 | Limonia type | 315 | 233 | 370 | 304 | 210 | 141 | 148 | 130 | 269 | 261 | 164 |
| C04 | pummelo type | 315 | 234 | 379 | 298 | 210 | 143 | 148 | 130 | 274 | 269 | 172 |
| C05 | Hyuganatsu type | 315 | 233 | 377 | 298 | 210 | 144 | 148 | 130 | 274 | 269 | 172 |
| C06 | Lemon type | 315 | 233 | 377 | 305 | 210 | 141 | 148 | 130 | 274 | 269 | 172 |
| C07 | Sweet orange type | 315 | 233 | 377 | 298 | 210 | 143 | 148 | 130 | 274 | 269 | 172 |
| C08 | Satsumakikoku type | 315 | 233 | 377 | 299 | 210 | 143 | 148 | 130 | 274 | 269 | 172 |
| C09 | Yuzu type | 315 | 233 | 371 | 311 | 210 | 139 | 159 | 129 | 274 | 261 | 172 |
| C10 | Ichang lemon type | 315 | 233 | 377 | 298 | 210 | 145 | 148 | 130 | 274 | 269 | 172 |
| C11 | Kunenbo_B type | 335 | 233 | 374 | 310 | 210 | 141 | 148 | 130 | 269 | 253 | 164 |
| C12 | Mandarin type | 315 | 235 | 372 | 303 | 210 | 141 | 148 | 130 | 269 | 253 | 164 |
| C13 | Sunki type | 315 | 235 | 370 | 305 | 210 | 141 | 148 | 130 | 269 | 261 | 164 |
| C14 | Tachibana type | 335 | 233 | 374 | 310 | 210 | 142 | 148 | 130 | 269 | 253 | 164 |
| C15 | Tachibana C type | 315 | 234 | 371 | 305 | 210 | 141 | 148 | 130 | 269 | 261 | 164 |
| C16 | Ogimikugani type | 335 | 234 | 374 | 310 | 210 | 142 | 148 | 130 | 269 | 253 | 164 |
| C17 | Cleopatra type | 315 | 234 | 370 | 304 | 210 | 141 | 148 | 130 | 269 | 261 | 164 |
| C18 | Koji type | 315 | 234 | 378 | 304 | 210 | 142 | 148 | 130 | 274 | 269 | 172 |
Table 12. Summary of all plant samples and representative samples according to the organelle genotype classes.
| Class | Type | Swingle's system (1943) | Tanaka's system (1961) | All plant samples | Representative samples | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| IV | HV | SL | IV | HV | SL | |||||
| C01 | C. ichangensis type | C. ichangensis | C. ichangensis Swingle | 1 | 0 | 0 | 1 | 0 | 0 | |
| C02 | Mexican lime type | C. aurantifolia | Citrus aurantifolia(Christm.) Swingle | 1 | 0 | 0 | 1 | 0 | 0 | |
| C03 | Limonia type | C. limon | C. limonia Osbeck | 1 | 0 | 0 | 1 | 0 | 0 | |
| C04 | pummelo type | C. grandis, C. paradisi | C. grandis, C. paradisi | 43 | 16 | 8 | 33 | 15 | 8 | |
| C05 | Hyuganatsu type | C. sinensis,C. paradisi | C. grandisC. aurantium L. | 9 | 5 | 3 | 5 | 4 | 3 | |
| C06 | Lemon type | C. limon, C. aurantium | C. limon Burm.f., C. aurantium L. | 12 | 0 | 1 | 5 | 0 | 1 | |
| C07 | Sweet orange type | C. limon, C. sinensi, C. nobilis hybrid | C. limon, C. sinensis, C. nobilis, C. reticulata | 44 | 1 | 0 | 16 | 1 | 0 | |
| C08 | Satsumakikoku type | C. aurantium | C. aurantium L. | 2 | 0 | 0 | 1 | 0 | 0 | |
| C09 | Yuzu type | (C. ichangensis hybrid) | C. junos | 3 | 0 | 0 | 3 | 0 | 0 | |
| C10 | Ichang lemon type | (C. ichangensis hybrid) | C. wilsonii Tanaka | 1 | 0 | 0 | 1 | 0 | 0 | |
| C11 | Kunenbo B type | C. reticulata hybrid | C. nobilis Lour. | 1 | 0 | 0 | 1 | 0 | 0 | |
| C12 | Mandarin type | C. reticulata | C. nobilis, C. reticulata, C. unshiu | 63 | 56 | 73 | 21 | 55 | 73 | |
| C13 | Sunki type | C.reticulata var. austera | C. sunki Hort. ex Tanaka | 7 | 0 | 0 | 4 | 0 | 0 | |
| C14 | Tachibana type | C. tachibana | C. tachibana | 11 | 0 | 0 | 2 | 0 | 0 | |
| C15 | Tachibana C type | C. tachibana | C. tachibana | 1 | 0 | 0 | 1 | 0 | 0 | |
| C16 | Ogimikugani type | C. reticulata hybrid | C. depressa Hayata | 1 | 0 | 0 | 1 | 0 | 0 | |
| C17 | Cleopatra type | C.reticulata var. austera | C. reshni Hort. ex Tanaka | 1 | 0 | 0 | 1 | 0 | 0 | |
| C18 | Koji type | (C. ichangensis hybrid), (C. indica hybrid) | C. leiocarpa, C.sudachi Hort. ex Shirai | 6 | 0 | 0 | 3 | 0 | 0 | |
| Total | 208 | 78 | 85 | 101 | 75 | 85 | ||||
IV: indigenous varieties, HV: hybrid varieties, SL: selected strains (see Table 1).
Among these cytotypes, C04 (pummelo type) was dominant in the indigenous varieties, followed by C12 (mandarin type) and C07 (sweet orange type) (Table 12). In contrast, nine indigenous varieties were exclusively classified with their own cytotypes (C01: C. ichangensis type, C02: Mexican lime type, C03: limonia type, C08: Satsumakikoku type, C10: Ichang lemon type, C11: kunenbo B type, C15: tachibana C type, C16: Ogimikugani type and C17: Cleopatra type). Two kunenbo varieties (A081: kunenbo A and A194: twukkuni) shared the same cytotype C07 (sweet orange type), in accord with the report by Yamamoto and colleagues [49]. However, two other kunenbo varieties were revealed to have different cytotypes (C12: mandarin type for A054: King and C11: kunenbo B type for A082: kunenbo B). Likewise, the cytotypes of two tachibana varieties (A174: tachibana A and A177: tachibana B) were identical (C14: tachibana type), but another tachibana (A176: tachibana C) had its own unique cytotype C15 and we refer to it as tachibana C type (S9 Table). One shiikuwasha variety (A136: shiikuwasha) shared same cytotype C13 with sunki and others, but another shiikuwasha variety (A137: shiikuwasha Ogimikugani) had its own unique cytotype C16 (S9 Table). These observations confirm their divergent origins as suggested by their nuclear genotypes (Table 4).
Other cytotypes (C05, C06, C09 and C18) were shared among three to five varieties. The cytotype C05 (hyuganatsu type) was shared among hyuganatsu, kawabata, lemonade, oogonkan and tengu (S9 Table). Similarly, bergamot, lemon, Hiroshimanatsubuntan, rokugatsumikan and sour oranges shared cytotype C06 (lemon type). Hanayu, Kourai tachibana and yuzu shared cytotype C09 (yuzu type). Cytotype C18 (koji type) was shared among girimikan, koji and sudachi. These observations enabled us to estimate their origin and possible hybrid combinations, and this is discussed in the following section.
As observed in the genotyping analysis of the nuclear genome (Table 5), a significant decrease in the number of genotypes and GD between the indigenous varieties and the hybrid varieties or the selected strains were also confirmed in the organelle genomes (Table 10). The observed decrease suggests the frequent use of specific cytotypes during cross breeding programs. Comparing the observed cytotype among three sample categories demonstrates that four out of 18 cytotypes have been selected during the breeding process (Table 12, Fig 5). The cytotype C12 (mandarin type) has been selected preferentially in the hybrid varieties and the selected strains from all cytotypes.
Fig 5. Organelle genome composition of the three sample categories.

Each pie chart shows the relative abundance of each cytotype within the three sample categories. C01: C. ichangensis type, C02: Mexican lime, C03: limonia type, C04: pummelo type, C05: hyuganatsu type, C06: lemon type, C07: sweet orange type, C08: Satsumakikoku type, C09: yuzu type, C10: Ichang lemon type, C11: kunenbo B type, C12: mandarin type, C13: sunki type, C14: tachibana type, C15: tachibana C type, C16: Ogimikugani type, C17: Cleopatra type, C18: koji type.
Factorial analysis and phylogenetic evaluation based on organellar genotypes
PCoA analysis of the 18 cytotypes clearly distinguishes them into three clusters (Fig 6). As with the PCoA analysis of the nuclear genome, the first two coordinates showed identical values when the number of assumed axes was changed from two to six, and the values of the first two of five assumed coordinates were used to draw the plot. These two coordinates explained about 55.6% of the total variation of 18 cytotypes. The three clusters observed were reminiscent of the three apexes in the PCoA plot from the nuclear genome (Fig 1). No cytotypes were classified in intermediate positions as observed in the nuclear PCoA plot (Fig 1). Thus, the classified cytotypes are considered to be a good measure to confirm parentage and the combination of seed parent and pollen parent. Among them, three cytotypes (C01, C02 and C09) are grouped in cluster I, corresponding to lime, yuzu and C. ichangensis. Seven cytotypes (C04, C05, C06, C07, C08, C10 and C18) are grouped in cluster II, which corresponds to pummelo and lemon. Eight cytotypes (C03, C11, C12, C13, C14, C15, C16, and C17) are grouped in cluster III, which corresponds to mandarin. Although the PCoA analysis of nuclear markers placed lemon (C. limon) with lime and C. ichangensis (Fig 1), it is classified in cluster II, which consists of the pummelo cytotype. Cytotypes C01 (C. ichangensis type) and C09 (yuzu type) are classified close together in cluster I, corresponding to the proposed relationship between them that was mentioned by Swingle [11] and Tanaka [12]. In cluster I, polymorphisms between C01 and C09 were observed in four chloroplast DNA markers (CSS04, CSL09, ccmp2.2m and ccmp6.2), and polymorphisms between C02 (Mexican lime type) and C09 in six chloroplast DNA markers (CSL01, CSL09, ccmp2.2, ccmp6.2, ccmp7.2 and ccmp10.2). However, no polymorphism was observed in mitochondrial markers within cluster I (Table 11). Consequently, distributions of these three cytotypes in cluster I represent polymorphism of chloroplast markers.
Fig 6. Principal coordinate analysis plot of 18 representative organelle genomes observed within 101 citrus indigenous varieties.

The plot was produced from modalities by Rogers and Tanimoto’s coefficient estimated from 11 organellar DNA markers. Three groups of genomes that cluster together are circled (I, II and III).
Five varieties in cluster II (hyuganatsu, Ichang lemon, koji, Satsumakikoku and sweet orange) are placed in intermediate positions among the three apexes in the nuclear PCoA plot (Fig 1), but their corresponding cytotypes are classified to the same pummelo and lemon cluster (Fig 6). Of those grouped in cluster II, C05 (hyuganatsu type) and C10 (Ichang lemon type) show just one polymorphism in ccmp6.2, and map to the same position. All of the cytotypes grouped in cluster II show polymorphism for the chloroplast markers, but no polymorphism was observed for the mitochondrial markers (Table 11). Six varieties in cluster III (Cleopatra, kunenbo-B, limonia, tachibana, tachibana-C, and shiikuwasha Ogimikugani) are placed in intermediate positions among the three apexes in the nuclear PCoA plot (Fig 1), but they are grouped in the same mandarin cluster (Fig 6). The cytotype of kunenbo-A (A081) is C07 (sweet orange type) and it falls in cluster II with pummelo, but the cytotype of kunenbo-B (C11) is in cluster III. These inconsistencies suggest different origins of their cytoplasmic genomes. All of the eight cytotypes in cluster III harbor unique mitochondrial alleles for rrn5/rrn18-1 and nad7/1-2 of length 269 and 164, respectively. Accordingly, we conclude that the first coordinate (horizontal axis) corresponds to the polymorphism of mitochondrial markers, and the second coordinate (vertical axis) corresponds to the polymorphism of chloroplast markers.
Of the eight cytotypes that are grouped in cluster III, four cytotypes (C03, C13, C15 and C17) and three cytotypes (C11, C14 and C16) are placed at opposite ends of the cluster along the second coordinate, with C12 (mandarin type) in the center (Fig 6). Interestingly, two cytotypes of tachibana (C14: tachibana type and C15: tachibana C type) are positioned at opposite ends of cluster III (Fig 6). The group of three cytotypes (C11, C14 and C16) harbored the 374 and 310 alleles for chloroplast CSL01 and CSL09, respectively, but the remaining five cytotypes harbored different alleles. Thus, these differences are considered to separate these groups in cluster III. Furthermore, the group of three cytotypes (C11, C14 and C16) and C12 harbored the 253 allele at the nad2/4-3 marker for mitochondria, which was not observed in other cytotypes (Table 11), suggesting that the mitochondria of these four cytotypes could be derived from the same origin. The phylogenetic tree estimated using the neighbor-joining method demonstrates the same three clusters (Fig 7).
Fig 7. Estimated phylogenetic tree of the 18 representative organelle genomes observed in 101 citrus indigenous varieties.

The tree was produced using the neighbor-joining method. The three main clades observed are indicated by different colors (I, II and III). Node labels show bootstrap support values.
Consequently, clusters I and II were revealed to harbor the same mitochondrial genotypes, and their differences were due to polymorphism in chloroplast genotypes. In contrast, cluster III was distinguished from clusters I and II by a polymorphism in mitochondrial genotypes. The observed isolation of two groups in cluster III was caused by polymorphisms in CSL01 and CSL09, and the observation that the mandarin type (C12) was placed between these groups could contribute to understanding the evolution of this cytotype.
Parentage analysis of parent–offspring triads in the indigenous varieties
Parentage was evaluated for all possible dyad combinations in the 101 indigenous varieties using the allele-sharing test with the 123 selected DNA markers confirmed to have Hardy–Weinberg proportions (Table 13). This test will succeed when a particular dyad shares at least one allele, but will fail whenever no allele is shared between them according to Mendel’s laws of inheritance [64]. The number of DNA markers not sharing any alleles was scored for each pairwise combination of the indigenous varieties using the allele-sharing test, and these scores are given in S10 Table. The test reveals that 74 varieties share all alleles with others, and 92 varieties share alleles with others when up to four mismatches are allowed (Table 13). Among those varieties, kaikoukan (A049), Kishu (A059), kunenbo-A (A081), sour orange (A141) and sweet orange (A162) were shown to share all alleles with more than five varieties without mismatch (S10 Table). Yuzu (A208) was shown to share alleles with 10 varieties when up to four mismatches were allowed.
Table 13. Summary of allele sharing test within the indigenous varieties for inferring parentage.
| Evaluated DNA markers | 123 | |
| Examined indigenous varieties | 101 | |
| (Combinations examined by the allele sharing test) | 10,201 | |
| Inferred parent-offspring dyads | ||
| No mismatch allowed | 74 | |
| (No shared alleles with others) | 27 | |
| Up to 4 mismatches allowed | 92 | |
| (No shared alleles with others) | 9 | |
Parent–offspring relationships were examined using the parentage test for all varieties that matched more than two other varieties. We allowed up to four mismatches among 123 DNA markers on the test in case of mutations or genotyping error (Table 13). The identities of seed parent and pollen parent were also deduced from their cytotypes (S9 Table). Any examined triads that disagreed on their parentage by more than five mismatches were rejected in this study. As an example, the parentage of ‘Fortune’ (B016) was inconsistent with the reported parentage (Clementine × dancy) [2], but the allele-sharing test proposed Clementine and Orland as the candidate parents with no mismatched DNA markers, and their cytotypes confirmed the true parentage with Clementine as seed parent and Orland as pollen parent (Table 14). In addition, the pollen parent of the hybrid variety ‘Haruka’ (B020), which was a selection of open pollinated hyuganatsu, was identified as natsudaidai. The cytotype of ‘Haruka’ agreed with the inferred parentage (Table 14).
Table 14. List of the inferred parentage in the indigenous varieties and orphan hybrid varieties.
| No. | Offspring variety | Inferred seed parent | Inferred pollen parent | Scores | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Variety | CT | ID | Variety | CT) | ID | Variety | CT | MM | Matched(%) | LOD 1)score | RCI 2) | |
| 1 | A001 | Andoukan | C04 | A049 | Kaikoukan | C04 | A059 | Kishu | C12 | 0 | 100.0% | 80.9 | 158.0 |
| 2 | A006 | Bergamot | C06 | A085 | Lemon | C06 | A141 | Sour orange | C06 | 4 | 96.7% | 125.9 | 221.7 |
| 3 | A009 | Clementine | C12 | A201 | Willowleaf mandarin | C12 | A162 | Sweet orange | C07 | 0 | 100.0% | 92.6 | 176.0 |
| 4 | A014 | Cravo | C12 | A201 | Willowleaf mandarin | C12 | A162 | Sweet orange | C07 | 0 | 100.0% | 90.7 | 168.5 |
| 5 | A019 | Fukure mikan | C12 | A059 | Kishu | C12 | A076 | Koji | C18 | 0 | 100.0% | 100.0 | 170.2 |
| 6 | A027 | Hanayu | C09 | A208 | Yuzu | C09 | A172 | Tachibana-A | C14 | 2 | 98.4% | 173.3 | 251.8 |
| 7 | A030 | Henka mikan | C07 | A081 | Kunenbo-A | C07 | A208 | Yuzu | C09 | 2 | 98.4% | 99.7 | 201.2 |
| 8 | A044 | Iyo | C04 | A049 | Kaikoukan | C04 | A016 | Dancy | C12 | 0 | 100.0% | 92.7 | 163.6 |
| 9 | A045 | Jabara | C07 | A081 | Kunenbo-A | C07 | A208 | Yuzu | C09 | 3 | 97.6% | 110.6 | 212.8 |
| 10 | A047 | Kabosu | C07 | A081 | Kunenbo-A | C07 | A208 | Yuzu | C09 | 1 | 99.2% | 101.4 | 202.8 |
| 11 | A052 | Keraji | C07 | A048 | Kabuchi | C07 | A081 | Kunenbo-A | C07 | 0 | 100.0% | 89.3 | 187.9 |
| 12 | A073 | Kizu | C07 | A081 | Kunenbo-A | C07 | A208 | Yuzu | C09 | 2 | 98.4% | 102.9 | 203.0 |
| 13 | A091 | Mochiyu | C07 | A081 | Kunenbo-A | C07 | A208 | Yuzu | C09 | 2 | 98.4% | 97.5 | 196.8 |
| 14 | A100 | Nidonari mikan | C12 | A059 | Kishu mandarin | C12 | A141 | Sour orange | C06 | 0 | 100.0% | 88.7 | 177.0 |
| 15 | A112 | Sanbokan | C04 | A049 | Kaikoukan | C04 | A059 | Kishu | C12 | 0 | 100.0% | 82.3 | 161.5 |
| 16 | A125 | Satsuma | C12 | A059 | Kishu | C12 | A081 | Kunenbo-A | C07 | 0 | 100.0% | 75.3 | 162.2 |
| 17 | A138 | Sokitsu | C12 | A059 | Kishu | C12 | A075 | Kobeni mikan | C12 | 0 | 100.0% | 127.1 | 178.6 |
| 18 | A147 | Suruga Yuko | C12 | A059 | Kishu | C12 | A076 | Koji | C18 | 0 | 100.0% | 114.6 | 189.0 |
| 19 | A186 | Temple | C12 | A201 | Willowleaf mandarin | C12 | A162 | Sweet orange | C07 | 4 | 96.7% | 81.9 | 160.4 |
| 20 | A188 | Tizon | C07 | A162 | Sweet orange | C07 | A013 | Cleopatra | C17 | 0 | 100.0% | 88.3 | 162.0 |
| 21 | A204 | Yatsushiro | C07 | A081 | Kunenbo-A | C07 | A059 | Kishu | C12 | 1 | 99.2% | 82.3 | 169.2 |
| 22 | A207 | Yuukunibu | C04 | A001 | Andoukan | C04 | A112 | Sanbokan | C04 | 1 | 99.2% | 74.1 | 164.5 |
| 23 | B016 | Fortune | C12 | A009 | Clementine | C12 | B045 | Orland | C04 | 0 | 100.0% | 98.9 | 174.7 |
| 24 | B020 | Haruka | C05 | A036 | Hyuganatsu | C05 | A096 | Natsudaidai | C04 | 0 | 100.0% | 65.5 | 171.8 |
Consistent with their cytotypes, Satsuma (A125) was inferred to be an offspring of Kishu (A059) as the seed parent, and kunenbo-A (A081) as the pollen parent (Table 14, Fig 8). All of the genotypes obtained not only from the 123 certified DNA markers but also the 169 passed DNA markers supported the parentage. Parentage analysis further identified Yatsushiro (A204) as another offspring of the same parents (Kishu and kunenbo-A), but their cross combinations were opposite to each other. Satsuma and Yatsushiro are therefore recognized as siblings (Table 14, Fig 8).
Fig 8. An inferred genealogy of citrus indigenous varieties, part 1.

The plot shows the pedigrees of Kishu, yuzu, lemon, sour orange and their inferred offspring. Codes in parentheses represent individual cytotypes, and the same color represents the same cytotype. Dashed boxes indicate postulated parents. Double-lined boxes correspond to key varieties in this plot.
In a similar fashion, Clementine was inferred to be an offspring of willowleaf mandarin × sweet orange as previously demonstrated [43,45]. Though Cravo (A014; Laranja Cravo) had been recognized as a variety of unknown origin [2], the parentage test proposed that it was an offspring of willowleaf mandarin × sweet orange. Furthermore, Temple (A186; C. temple Hort. ex Y. Tanaka) [2] was inferred to be another offspring of the same parents despite it showing four mismatches (Table 14, Fig 9A). Their cytotypes and the results of structure analysis supported these inferred parentages. Consequently, Clementine, Cravo and Temple are recognized as siblings from a willowleaf mandarin × sweet orange cross.
Fig 9. An inferred genealogy of citrus indigenous varieties, part 2.

A: The pedigree plot of sweet orange and its inferred offspring. B: The pedigree plot of three tachibanas and their inferred offspring with postulated parents. C: Various proposed pedigrees. Their codes, colors and lines are as described for Fig 8.
The allele-sharing test revealed close relationships of sour orange (A141) to six varieties: bergamot (A006), Hiroshimanatsubuntan (A033), kunenbo-B (A082), lemon (A085), nidonari mikan (A100), and rokugatsumikan (A111) (S10 Table). Among these varieties, nidonari mikan (C. nidonari Hort. ex Y. Tanaka) is an old mandarin variety of unknown origin, but it demonstrated no mismatch in any DNA marker with sour orange or Kishu (S10 Table). The parentage test inferred it to be an offspring of Kishu × sour orange with no mismatch, and their cytotypes proposed Kishu as the seed parent, and sour orange as the pollen parent. Likewise, the parentage test inferred bergamot (A006; C. bergamina Risso) to be an offspring of lemon (A085) × sour orange (Table 14). According to the cytotypes, bergamot (A006) is assumed to be a hybrid of lemon (A085) as the seed parent and sour orange (A141) as the pollen parent. Though four mismatches were observed in the score of bergamot, this is thought to result from residual genotyping error because lemon and its relatives were not provided for the initial verification of the DNA markers in sufficient numbers. Bergamot has been considered a natural hybrid of sour orange [2], and the inferred parentage agrees with the proposed one [45,47]. Very recently, Curk et al reported the identical combination for bergamot [47]. The parentage of the remaining four varieties (lemon, Hiroshimanatsubuntan, kunenbo-B and rokugatsumikan) will be examined in the next section.
Allele-sharing tests on Kishu revealed that at least 18 varieties could be kindred to it (S10 Table). Parentage tests on these inferred that both andoukan (A001) and sanbokan (A112) are offspring of kaikoukan (A049) as the seed parent and Kishu (A059) as the pollen parent without mismatches. Although one mismatch was observed, yuukunibu (A207) is inferred to be an offspring of these hybrid varieties (andoukan × sanbokan). Kaikoukan was inferred to be the seed parent of iyo (A044), crossed with dancy (A016) as the pollen parent. Consequently, both andoukan and sanbokan are revealed to be half-siblings of iyo. Sokitsu (A138) was inferred to be an offspring of Kishu × kobeni mikan (A075), but it is not possible to determine which is the seed parent and which is the pollen parent because of their identical cytotypes.
Five varieties (A030: henka mikan, A045: jabara, A047: kabosu, A073: kizu and A091: mochiyu) were inferred to be offspring of yuzu × kunenbo-A, though one to three mismatches were observed for them (Table 14). Likewise, hanayu (A027) was inferred to be a hybrid of yuzu as the seed parent and tachibana-A (A172) as the pollen parent with two mismatches. Given that control hybrids used to verify the DNA markers did not include sufficient numbers of yuzu or its relatives, the observed mismatches could be due to either unforeseen null alleles or mutations, or both.
Fukure mikan (A019) and Suruga yuko (A147) were thought to be mutant varieties of koji (A076) [112], but parentage analysis revealed that they are not mutants but offspring of Kishu × koji. Tanaka proposed a close relationship among koji, fukure mikan and tachibana [7] and this agrees with the inferred parentage. Tizon (A188; C. papillaris Blanco) [12] was inferred to be a hybrid of sweet orange × Cleopatra. Kabuchi (A048) shared all alleles with kunenbo-A (A081) and keraji (A052), and keraji also shared all alleles with kabuchi and kunenbo-A. The parentage test rejected kunenbo-A and keraji as the parents of kabuchi with 14 mismatches, but kunenbo-A and kabuchi were inferred to be the parents of keraji with no mismatch. Thus, kabuchi was inferred to be an offspring of kunenbo-A as seed parent and an unidentified variety, and keraji was inferred to be an offspring of kabuchi and kunenbo-A, but their combination was indeterminate. This inferred parentage suggests that keraji is a backcrossed offspring of kunenbo-A.
Despite these inferred parentages, most of the proposed parent–offspring combinations were rejected by significant discrepancies on the parentage test. Three varieties, Kawachi bankan (A051), ujukitsu (A197) and yuge hyoukan (A205) shared all alleles among them. Likewise, ujukitsu, Kishu (A059) and yuge hyoukan shared all alleles among them, and this was confirmed with Naruto (A095), Kishu and yuge hyoukan. These observed perfect matches suggested their parentage, but the parentage test rejected all combinations of them. We hypothesize that Naruto and ujukitsu are the offspring of Kishu with unknown parents. According to their cytotype, Kishu is thought to be their pollen parent. The unknown other parents of Naruto and ujukitsu should hold a pummelo-type cytotype. The result of structure analysis coincides with this hypothesis. In this fashion, yuge hyoukan was inferred to be the offspring of ujukitsu or Naruto and an unidentified variety with uncertain cytotype, but the parentage of Kawachi bankan remained uncertain. These assumed relationships are examined further using stochastic methods, as discussed in a later section.
Tankan has been considered a natural tangor [2,7], and the allele-sharing test revealed no mismatch to sweet orange, ponkitsu (A109) and genshokan (A022). Ponkitsu showed five and eight mismatches to genshokan and sweet orange, respectively. Likewise, genshokan showed 10 mismatches to sweet orange. Consequently, sweet orange and genshokan were proposed as the parents of tankan, but the parentage test rejected their parentage. Furthermore, the cytotypes of tankan, sweet orange and genshokan were sunki type (C13), sweet orange type (C07) and mandarin type (C12), respectively, and did not coincide with each other. Therefore, tankan is assumed to be an offspring of sweet orange and an unidentified variety with sunki-type cytotype. Ponkitsu is assumed to be an offspring of tankan since they share the same cytotype. The parentage of genshokan is unclear, but it could be an offspring of tankan.
Except for the inferred triads, kunenbo-A revealed no mismatch to asahikan, hassaku, hyoukan, kabuchi, kaikoukan, kawabata, kinkoji, Kishu and unzoki, and one mismatch to sweet orange. Because kunenbo-A and sweet orange share the same cytotype (C07; sweet orange type), all possible combinations of these were examined using the parentage test but rejected with 15–25 mismatches. Sweet orange shared the same cytotype with kunenbo-A, but showed allele mismatches when evaluated with the genotype data obtained from 169 certified DNA markers. None of the other indigenous varieties that have the sweet orange-type cytotype (Table 12) shared significant number of alleles with kunenbo-A, and they were thus rejected from the parentage test. In contrast, kunenbo-A did not show any mismatch to Kishu when evaluated with all of the DNA markers (data not shown). The deduced proportions of the three basic taxa in Kishu were 0.2%, 0.5% and 99.3%, and those of kunenbo-A were 35.2%, 0.4% and 64.2% for pummelo, citron and mandarin, respectively (S8 Table). The proportion of mandarin genome would decrease when Kishu is crossed with an unknown variety, as observed in kunenbo-A. On the contrary, it seems difficult to purge the entire pummelo genome portion in kunenbo-A by a single crossing event with an unknown variety to result in Kishu that has a minimum of pummelo genome. Consequently, kunenbo-A is thought to be an offspring of Kishu as pollen parent crossed with an unidentified variety that hold sweet orange-type cytotype as seed parent.
Suisho buntan (A145) shared all alleles with Tosa buntan (A191), and shared all except for one mismatch with Hirado buntan (A032). The parentage test rejected both of these as parents of suisho buntan with seven mismatches (data not shown). Suisho buntan has been selected from open pollinated pummelo, and banoukan and Tosa buntan were proposed to be the parents [112]. Although banoukan was not examined in this study, the allele-sharing test supports the parentage of Tosa buntan.
The inferred parentages agreed well with the population compositions deduced by structure analysis with K = 3 (Fig 4, S8 Table). The proportions of the assumed basic taxa coincided for those triads (Table 14). For example, the deduced proportions of the three basic taxa (P1: pummelo, P2: citron and P3: mandarin) in tizon (A188) were 16.6%, 1.0% and 82.4%, respectively (S8 Table), and these are close to the proportions (18.2%, 2.3% and 79.6%) estimated from the inferred parents sweet orange (36.1%, 0.5% and 63.4%) and Cleopatra (0.3%, 4.0% and 95.7%). The proposed proportions in sokitsu (A138) also agreed well with the inferred parents (Kishu × kobeni mikan). Meanwhile, measurable discrepancies in the proportions of the populations were observed occasionally. The estimated proportions in Kishu (0.2%, 0.5% and 99.3% for the three genomes) × kunenbo-A (35.2%, 0.4% and 64.2%) were 17.7%, 0.5% and 81.8%; however, the corresponding proportions estimated from the inferred offspring were 28.9%, 0.5% and 70.6% for Satsuma, and 24.3%, 0.5% and 75.2% for Yatsushiro. Interestingly, the deduced proportions in these offspring were not identical but fluctuated in these siblings. Similar discrepancies and fluctuation were also observed among other inferred full or half-siblings. Iyo was one of two inferred offspring of kaikoukan and Kishu, and the deduced proportions in iyo (40.9%, 0.3% and 58.8%) were close to those in kaikoukan × dancy (37.9%, 0.4% and 61.8%). In contrast, andoukan (49.6%, 0.4% and 50.0%) and sanbokan (48.6%, 0.7% and 50.7%) differed from the expected values of kaikoukan × Kishu (37.9%, 0.4% and 61.7%). Likewise, four offspring of kunenbo-A × yuzu (henka mikan, jabara, kabosu and mochiyu), two offspring of willowleaf mandarin × sweet orange (Clementine and Cravo), two siblings of Kishu × koji (fukure mikan and Suruga yuko) were consistent in their genome composition with those of the inferred parents but showed fluctuation between siblings. The deduced proportion of the pummelo genome in the four siblings of kunenbo-A × yuzu fluctuated widely, from 3.7% (henka mikan) to 20.5% (jabara), and the proportions of the two other genomes also fluctuated in a coordinated manner. Similar discrepancies were also observed in bergamot and hanayu, and were still evident at K = 4 (S2 Fig). These observed variations suggest that two alleles at particular heterozygous loci would have different effects on the estimation of the proportions of basic taxa, or that the lack of ‘pure’ citron or papeda varieties in this study might lead to underestimation of their contribution in the indigenous varieties.
Three types of tachibana and their relatives
We evaluated the mutual relationships between three types of tachibanas. The shared allele frequencies estimated using the allele-sharing test (S10 Table) were 93.5% (tachibana-A–B), 97.6% (tachibana-A–C) and 91.9% (tachibana-B–C). The observed shared allele frequencies were higher than those between the three types of tachibanas and other varieties; 66.2% (tachibana-A), 70.7% (tachibana-B), and 67.9% (tachibana-C). Hirai and colleagues reported similar genetic variation among wild tachibana collections using three isozymes [20]. The deduced proportions for the genomes of the three basic taxa for these types were similar to one another (Fig 4), and also suggested their hybrid origin as mandarin × citron. The cytotype of tachibana-A and B was the same (C14; tachibana type), but it was different in tachibana-C (C15; tachibana-C type). These observations suggest that these three types of tachibana might be siblings. Accordingly, a model was proposed in which these three types of tachibanas are offspring of two ancestors, one harboring tachibana-type cytotype (C14) and the other harboring tachibana-C type (C15) (Fig 9B). Because these cytotypes (C14 and C15) were not found in other varieties, those hypothetical ancestors may have been lost.
The allele-sharing test suggested that hanayu is an offspring of yuzu × tachibana-A as described above. Similarly, girimikan (A023) and hyuganatsu (A036) were proposed to be offspring of tachibana-B (A175), but their cytotypes were different to each other. Oogonkan (A101) was proposed to be the offspring of tachibana-C (Table 15). Although the seed parents of these three varieties were unidentified, cytotypes of hyuganatsu and oogonkan were identical (C05; hyuganatsu). Iwamasa has pointed out the close relationship among hyuganatsu, oogonkan and yuzu [112], and their unidentified seed parents could be siblings of yuzu. On the other hand, no previous studies have proposed the involvement of any of the tachibanas as the parents of hanayu, girimikan, hyuganatsu, or oogonkan.
Table 15. Inferred parent-to-child combinations in the indigenous varieties.
| Offspring variety | Inferred seed parent | Inferred pollen parent | Scores | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # | ID | Variety | CT | ID | Variety *) | CT | ID | Variety *) | CT | MM | Matched (%) | LOD 1) score | RCI 2) | SPP 3) |
| 1 | A003 | Asahikan | C04 | (pummelo type) | C04 | A081 | Kunenbo-A | C07 | 0 | 100.0 | 6.6 | 173.4 | 41.2 | |
| 2 | A021 | Funadoko | C04 | (pummelo type) | C04 | A005 | Bendizao | C12 | 0 | 100.0 | 17.7 | 164.1 | 47.2 | |
| 3 | A023 | Girimikan | C18 | (Koji type) | C18 | A175 | Tachibana-B | C14 | 1 | 99.2 | 61.8 | 186.3 | 53.1 | |
| 4 | A024 | Grapefruit | C04 | (pummelo type) | C04 | A162 | Sweet orange | C07 | 0 | 100.0 | 14.5 | 177.9 | 42.0 | |
| 5 | A028 | Hassaku | C04 | (pummelo type) | C04 | A081 | Kunenbo-A | C07 | 0 | 100.0 | 16.2 | 176.6 | 42.6 | |
| 6 | A031 | Hickson | C12 | A107 | Ponkan | C12 | (Unidentified) | 0 | 100.0 | 50.9 | 179.6 | 51.7 | ||
| 7 | A033 | Hiroshimanatsubuntan | C06 | A141 | Sour orange | C06 | (Unidentified) | 0 | 100.0 | 41.7 | 187.3 | 47.6 | ||
| 8 | A035 | Hyoukan | C04 | (pummelo type) | C04 | A081 | Kunenbo-A | C07 | 0 | 100.0 | 4.0 | 169.9 | 42.1 | |
| 9 | A036 | Hyuganatsu | C05 | (Hyuganatsu type) | C05 | A175 | Tachibana-B | C14 | 0 | 100.0 | 34.0 | 188.4 | 44.5 | |
| 10 | A041 | Ichanchii (C.ichangensis) | C01 | (C. ichangensis type) | C01 | A085 | Lemon | C06 | 1 | 99.2 | 52.9 | 298.4 | 35.5 | |
| A085 | Lemon | C06 | (lemon type) | C06 | A041 | Ichanchii (C.ichangensis) | C01 | 1 | 99.2 | 52.9 | 278.5 | 37.3 | ||
| 11 | A042 | Ichang lemon | C10 | (Ichang lemon type) | C10 | A208 | Yuzu | C09 | 1 | 99.2 | 59.1 | 255.8 | 38.3 | |
| 12 | A046 | Jabon | C04 | (pummelo type) | C04 | A208 | Yuzu | C09 | 0 | 100.0 | 40.3 | 194.8 | 43.9 | |
| 13 | A048 | Kabuchi | C07 | A081 | Kunenbo-A | C07 | (Unidentified) | 0 | 100.0 | 7.6 | 189.8 | 42.6 | ||
| 14 | A049 | Kaikoukan | C04 | (pummelo type) | C04 | A081 | Kunenbo-A | C07 | 0 | 100.0 | 16.5 | 192.3 | 38.5 | |
| 15 | A050 | Kawabata | C05 | (Hyuganatsu type) | C05 | A081 | Kunenbo-A | C07 | 0 | 100.0 | 18.5 | 177.6 | 44.8 | |
| 16 | A051 | Kawachi bankan | C04 | A197 | Ujukitsu | C04 | (Unidentified) | 0 | 100.0 | 17.0 | 186.4 | 41.1 | ||
| A051 | Kawachi bankan | C04 | A205 | Yuge hyoukan | C04 | (Unidentified) | 0 | 100.0 | 34.9 | 186.4 | 45.6 | |||
| 17 | A076 | Koji | C18 | (Koji type) | C18 | A174 | Tachibana-C | C15 | 2 | 98.4 | 36.8 | 184.8 | 47.9 | |
| 18 | A055 | Kinkoji | C04 | (pummelo type) | C04 | A081 | Kunenbo-A | C07 | 0 | 100.0 | 19.0 | 187.4 | 41.0 | |
| 19 | A079 | Kourai Tachibana | C09 | A208 | Yuzu | C09 | (Unidentified) | 2 | 98.4 | 72.5 | 231.6 | 47.5 | ||
| 20 | A081 | Kunenbo-A | C07 | (Sweet orange type) | C07 | A059 | Kishu | C12 | 0 | 100.0 | 3.3 | 152.7 | 45.9 | |
| 21 | A082 | Kunenbo-B | C11 | (Kunenbo_B type) | C11 | A141 | Sour orange | C06 | 0 | 100.0 | 15.5 | 188.3 | 42.4 | |
| 22 | A085 | Lemon | C06 | A141 | Sour orange | C06 | (Unidentified) | 0 | 100.0 | 13.4 | 280.8 | 31.8 | ||
| A141 | Sour orange | C06 | A085 | Lemon | C06 | (Unidentified) | 0 | 100.0 | 13.4 | 180.6 | 42.2 | |||
| 23 | A090 | Meyer lemon | C07 | (sweet orange type) | C07 | A087 | Limonia | C03 | 1 | 99.2 | 113.1 | 266.7 | 43.5 | |
| 24 | A092 | Murcott | C12 | A054 | King | C12 | (Unidentified) | 0 | 100.0 | 40.8 | 177.0 | 50.8 | ||
| 25 | A098 | Natsudaidai | C04 | (pummelo type) | C04 | A059 | Kishu | C12 | 1 | 99.2 | -1.4 | 163.5 | 42.7 | |
| 26 | A095 | Naruto | C04 | (pummelo type) | C04 | A059 | Kishu | C12 | 0 | 100.0 | 1.3 | 169.9 | 40.0 | |
| 27 | A101 | Oogonkan | C05 | (Hyuganatsu type) | C05 | A174 | Tachibana-C | C15 | 0 | 100.0 | 34.6 | 180.9 | 47.7 | |
| 28 | A103 | Ootoukan | C04 | (pummelo type) | C04 | A005 | Bendizao | C12 | 0 | 100.0 | 22.0 | 178.1 | 43.7 | |
| 29 | A104 | Oukan | C04 | (pummelo type) | C04 | A059 | Kishu | C12 | 0 | 100.0 | 11.1 | 171.3 | 42.8 | |
| 30 | A107 | Ponkan | C12 | A016 | Dancy | C12 | (Unidentified) | 1 | 99.2 | 63.6 | 179.4 | 54.7 | ||
| 31 | A109 | Ponkitsu | C13 | A183 | Tankan | C13 | (Unidentified) | 0 | 100.0 | 61.4 | 190.5 | 52.1 | ||
| 32 | A111 | Rokugatsumikan | C06 | A141 | Sour orange | C06 | (Unidentified) | 0 | 100.0 | 17.0 | 208.5 | 40.4 | ||
| 33 | A135 | Shiikuwasha | C13 | (Sunki type) | C13 | A192 | Twukkuni | C07 | 1 | 99.2 | 63.6 | 196.0 | 52.0 | |
| 34 | A137 | Shunkokan | C04 | (pummelo type) | C04 | A204 | Yatsushiro | C07 | 0 | 100.0 | 19.8 | 167.6 | 47.8 | |
| 35 | A144 | Sudachi | C18 | (Koji type) | C18 | A208 | Yuzu | C09 | 3 | 97.6 | 67.2 | 210.1 | 47.8 | |
| 36 | A145 | Suisho buntan | C04 | A191 | Tosa buntan | C04 | (Unidentified) | 0 | 100.0 | 73.3 | 237.7 | 42.9 | ||
| 37 | A183 | Tankan | C13 | (Sunki type) | C13 | A162 | Sweet orange | C07 | 0 | 100.0 | 28.4 | 188.8 | 44.6 | |
| 38 | A187 | Tengu | C05 | (Hyuganatsu type) | C05 | A162 | Sweet orange | C07 | 0 | 100.0 | 14.5 | 167.8 | 43.2 | |
| 39 | A197 | Ujukitsu | C04 | (pummelo type) | C04 | A059 | Kishu | C12 | 0 | 100.0 | 9.3 | 161.8 | 46.4 | |
| A197 | Ujukitsu | C04 | A205 | Yuge hyoukan | C04 | (Unidentified) | 0 | 100.0 | 22.1 | 161.8 | 49.2 | |||
| 40 | A198 | Unzoki | C07 | A081 | Kunenbo-A | C07 | (Unidentified) | 0 | 100.0 | 6.3 | 188.8 | 42.0 | ||
| 41 | A199 | USSR Tangelo | C12 | A125 | Satsuma | C12 | (Unidentified) | 0 | 100.0 | 9.1 | 177.0 | 42.0 | ||
| 42 | A202 | Yamabuki | C04 | A049 | Kaikoukan | C04 | (Unidentified) | 0 | 100.0 | 38.3 | 166.3 | 50.2 | ||
| 43 | A203 | Yamamikan | C07 | A162 | Sweet orange | C07 | (Unidentified) | 0 | 100.0 | 15.3 | 181.0 | 42.4 | ||
| 44 | A205 | Yuge hyoukan | C04 | A197 | Ujukitsu | C04 | (Unidentified) | 0 | 100.0 | 22.1 | 179.0 | 44.4 | ||
| A205 | Yuge hyoukan | C04 | A095 | Naruto | C04 | (Unidentified) | 0 | 100.0 | 14.7 | 179.0 | 43.5 | |||
| 45 | A206 | Youpi ju (Yuhikitsu) | C12 | A200 | Willowleaf mandarin | C12 | (Unidentified) | 0 | 100.0 | 52.7 | 194.3 | 54.4 | ||
Parentage analysis of parent–offspring dyads in the indigenous varieties
The inferred triads were excluded from the proposed dyads, and the remaining dyads were further evaluated (Table 15). Unlike the parentage test, the allele-sharing test does not predict which variety is the parent and which is the offspring. Accordingly, parentage was estimated from cytotypes, asymmetry of parentage, the result of structure analysis, and past literatures.
The allele-sharing test found a close relationship of Kishu to oukan (A104) and natsudaidai (A098) (S10 Table). Oukan (C. suavissima Hort. ex Tanaka) is an old mandarin variety from China [7]. From the evidence obtained, it was suggested to be a hybrid of C. maxima and Kishu. Natsudaidai (C. natsudaidai Hayata) was a chance seedling in Yamaguchi prefecture [2,11], and the evidence suggests that this arose from hybridization between pummelo and Kishu.
Murcott (A092) and King (A054) shared all alleles and an identical cytotype (S9 and S10 Tables). This suggests that they could be a parent–offspring pair, but their deduced proportions of the ancestral populations were also similar (S8 Table), and it seemed difficult to determine which one would be the parent. According to Hodgson [2], Murcott was recognized as a tangor of unknown origin resulting from the breeding program of the USDA. Because King was frequently used in the USDA citrus breeding program [2], it is likely that Murcott was a selection of King. Nicolosi and colleagues also reported similarity between them [33]. Consequently, we postulate that Murcott is an offspring of King (Table 15).
Grapefruit has been regarded as a natural hybrid of sweet orange and pummelo [109,113], and recent molecular work supports this [34,43,45]. The allele-sharing test coincided with the current consensus on grapefruit, with no mismatches between sweet orange and grapefruit. The population structure analysis suggests the involvement of pummelo as the other parent, agreeing with the cytotype of grapefruit that is not the same as sweet orange but rather the pummelo type. Accordingly, grapefruit is assumed to be an offspring of sweet orange as the pollen parent with an unidentified variety harboring the pummelo cytotype. In addition, tengu (A187) and yamamikan (A203) are suggested their parentages with sweet orange (S11 Table). Because the cytotype of tengu (C. tengu Hort. ex Tanaka) [7,12] was hyuganatsu type (C05), sweet orange was assumed to be the pollen parent of tengu. Although the pollen parent of yamamikan (C. intermedia hort. ex Tanaka) is unidentified, the deduced genome proportions of the basic taxa suggest that yamamikan is a hybrid of mandarin and pummelo (Fig 4), and this agrees with the hypothesis of Tanaka [7]. Both tengu and yamamikan showed just one mismatch between them under the allele-sharing test, supporting the hypothesis that they are siblings of sweet orange.
Three mandarin varieties (Mediterranean mandarin, willowleaf mandarin and youpiju) are not mutant selections, but Mediterranean mandarin and willowleaf mandarin share 95.1% of alleles, suggesting a common ancestral origin. Mediterranean mandarin and youpiju shared all alleles including the excluded markers. They are old mandarin varieties of uncertain origin, and it has been suggested that they are kindred varieties. Furthermore, Hickson (A031) and ponkan (A107) shared all alleles, and these two varieties also shared significant numbers of alleles with willowleaf mandarin (S10 Table). Hickson was found as a sporting limb on ‘Ellendate’ tangor [2,114], but the deduced genome structure suggests that Hickson is almost a mandarin (Fig 4). These varieties share the same cytotype (C12; mandarin type) and their genome structures are quite similar to each other. Therefore, their parentage is indeterminate. We propose an alternative hypothesis that they are siblings.
Bendizao (A005) shared all alleles with two Japanese local varieties ootoukan (A103) and funadoko (A021), but their cytotypes were different to that of bendizao. Since the probability of selecting an identical genotype from two different varieties is negligible, the observed asymmetry confirms that both ootoukan and funadoko are the offspring of bendizao as pollen parent and an unidentified variety with pummelo-type organelle genomes. The deduced genome structure of basic taxa support their proposed kinship.
Similar asymmetric relationships were also found in sour orange and four varieties (A033: Hiroshimanatsubuntan, A082: kunenbo-B, A085: lemon, A111: rokugatsumikan) by the allele-sharing test (S10 Table). Since sour orange was inferred to be the parent of nidonari mikan and bergamot, those four varieties were also assumed to be either parents or offspring of sour orange. These varieties except for kunenbo-B share the same cytotype (C06; lemon type), but the allele-sharing test revealed that Hiroshimanatsubuntan, kunenbo-B and rokugatsumikan have 18, 20 and 15 mismatches with lemon, respectively (S10 Table). On the basis of these scores, sour orange is assumed to be an offspring of lemon, and Hiroshimanatsubuntan, kunenbo-B and rokugatsumikan are assumed to be offspring of sour orange. Their cytotypes suggest that sour orange is the seed parent of Hiroshimanatsubuntan and rokugatsumikan, but the pollen parent of kunenbo-B. The deduced genome structure of basic taxa agrees well with this inferred parentage. The inferred parentage also revealed that the two types of kunenbo (kunenbo-A and kunenbo-B) are different in origin.
It is interesting that ichanchii (C. ichangensis Swingle) shares all alleles with lemon but their cytotypes are different. Swingle considered it a unique variety related to papeda, and regarded yuzu as a chance seedling of C. ichangensis [11]. However, the allele-sharing test clearly refutes this proposal, with 31 out of 123 DNA markers not shared between yuzu and C. ichangensis. Because the cytotype of C. ichangensis was unique, but the lemon-type cytotype was found in 13 varieties (S9 Table), C. ichangensis could be an offspring of lemon with an unidentified seed parent whose cytotype should be identical to that of C. ichangensis. The position of C. ichangensis on the PCoA plot of the nuclear genome is close to lemon (Fig 1), as supported by the allele-sharing test. However, their cytotypes are different in eight out of 13 organelle DNA markers, and they are far apart in the organellar PCoA plot (Fig 6). These observations hypothesized that C. ichangensis could an offspring of an unidentified papeda × lemon, and yuzu might also be an offspring of this unidentified papeda.
As observed in the parentage analysis of the proposed triads, the allele-sharing test revealed a possible parent–offspring relationship of yuzu with Ichang lemon (A042), Kourai tachibana (A079), jabon (A046) and sudachi (A144). Swingle considered Ichang lemon (C. wilsonii Tanaka) to be a hybrid of C. ichangensis and C. maxima [11]. In contrast, Tanaka regarded it as an indigenous variety related to yuzu, and classified both C. ichangensis and C. wilsonii to subgenus Eucitrus [12]. Their inferred parentage in this study confirm that C. wilsonii is an offspring of yuzu as Tanaka stated [12]. However, there is no evidence to suggest kinship of C. ichangensis and yuzu, and direct parentage of C. ichangensis and C. wilsonii are consequently refuted. Their cytotypes also suggest no direct kinship between them (Table 15, Fig 8). Kourai tachibana (A079) was found in Yamaguchi prefecture [7] and initially classified as C. tachibana Tanaka, but later reclassified to C. nippokoreana Tanaka [7,12]. Although the allele-sharing test identified two mismatches between Kourai tachibana and yuzu (S10 Table), these mismatches were considered to be caused by unidentified genotyping error. They share the same cytotype (C09; yuzu type), suggesting that yuzu would be the seed parent of Kourai tachibana (S9 Table). The allele-sharing test did not identify the candidate pollen parent of Kourai tachibana but demonstrated fewer mismatches with the three types of tachibanas (11, 12 and 8 mismatches with tachibana-A, B and C, respectively) than with other varieties. The three types of tachibana and their proposed sibships suggest that there might be another sibling of tachibana, and it could be the pollen parent of Kourai tachibana.
The inferred relationships agreed that sudachi (C. sudachi Hort. ex Shirai) is a hybrid of yuzu [7]. The cytotype of sudachi was koji-type (C18), but neither koji nor any other variety with koji-type organelle genome was assumed to be the seed parent of sudachi. Jabon (A046) is an indigenous variety of unknown origin found in Hiroshima prefecture, Japan. ‘Jabon’ is a Japanese synonym of pummelo [7,12], but the morphological features of jabon are not reminiscent of a typical pummelo but suggest a hybrid of pummelo [115]. The inferred hybrid combination coincides with the observed features. Furthermore, koji is another indigenous variety in Japan and its cytotype (C18; koji type) is unique among the evaluated varieties. However, it shares all alleles except for two mismatches with tachibana-C (Table 15). The deduced genomic proportions of the basic taxa suggest that koji is a hybrid of mandarin (Fig 4). Furthermore, the cytotype of sudachi is identical to that of koji. These observations might imply that the unidentified parents of tachibana-C, koji and sudachi could be identical or very close to each other.
USSR tangelo (A199) is a germplasm collection of NIFTS of unknown origin, but it is recognized as a hybrid of Satsuma × pummelo [116]. The allele-sharing test confirmed it as an offspring of Satsuma as the seed parent (S10 Table). Its pollen parent was not identified but structure analysis suggests introgression of the pummelo genome (Fig 4). This is just one inferred offspring of Satsuma in the evaluated varieties. Satsuma possesses strong and stable male sterility, parthenocarpy, and apomixes [117], and these traits could make it difficult to obtain offspring of Satsuma. Yatsushiro (A204) was inferred to be an offspring of kunenbo-A × Kishu, and it was assumed to be the pollen parent of shunkokan (A137). Because the cytotype of shunkokan is pummelo type (C04), kunenbo-A is inferred to be its pollen grandparent. Shunkokan (C. shunkokan hort. ex Tanaka) was found in Wakayama prefecture in Japan, and Tanaka mentioned its resemblance to kunenbo and pummelo [7].
Despite these assumed parentages, several dyads remain uncertain. Dancy (A016) shares all alleles with ponkan (A107). Likewise, limonia (A087) and Meyer lemon (A090), and also shiikuwasha (A135) and the kunenbo variety twukkuni (A192) share all alleles except for one mismatch (S10 Table).
Thirty-eight of the 45 selected dyads revealed no mismatches between them. A single mismatch was observed in seven dyads, two and three mismatches were observed in two and one variety, respectively (Table 15). As observed in the parentage analysis of triads, kunenbo-A, sweet orange, sour orange, Kishu, yuzu, lemon and bendizao were found in eight, four, three, four, four, two and two dyads, respectively (Table 15). Four types of kunenbo (kunenbo-A, kunenbo-B, twukkuni and King) were found in these dyads. According to the score of the allele-sharing test and their cytotypes, seven varieties (asahikan, hassaku, hyoukan, kaikoukan, kawabata, kinkoji and unzoki) were assumed to be offspring of kunenbo-A and various unidentified varieties.
Stochastic evaluation of inferred parentage
While the allele-sharing test and the parentage test, coupled with the cytotypes, are an excellent approach to infer the parentage of uncertain varieties, these tests do not estimate the probability of the inferred parentage in the citrus population. In addition, these tests are susceptible to genotyping error or mutations, and could fail to estimate the correct combination. Although using the parentage test with known hybrid varieties or strains in this study eliminated all of the suspicious DNA markers, this does not guarantee the genotype to be perfect. As can be observed in Table 4, mutation is occasionally detected within mutant lines. Accordingly, the inferred parentage of triads or dyads was further examined by stochastic evaluation using a likelihood ratio approach. The likelihood ratio analysis is preferred over the parentage test because it can provide a basis for estimating the reliability of the inferred parentage. This approach has been used widely in forensic genetics in combination with Bayes’ theorem for missing person identification, paternity examination and kinship testing [59,60], and also in the field genetics [62–64]. A likelihood ratio represents the relative odds of two alternative hypotheses, and it has the advantage that it avoids postulating a posterior probability for the hypothesis. However, the likelihood ratio approach is implicitly premised on the minimum occurrence of relatives (full sib or half sib) in the given population [63]. Furthermore, it is known that this score is affected by the allele frequency in the population [60,118]. As observed in the previous section, a significant number of the indigenous varieties are thought to share kinship relations. Such a strained family structure might alter the LOD score due to uneven allele frequencies in populations.
Hence, we first evaluated the behavior of the LOD score with known hybrid triads (Table 3) using Eq 4. Their LOD scores show a wide range, from 69.3 (sweet spring) to 210.0 (benimadoka) (Table 3). In the case of the identification of a suspect from stains or remains, a likelihood ratio of 1,000 to 10,000 obtained from 10 to 15 (typically 13) STR markers is considered strong support for the prosecution hypothesis [60,118]. These values correspond to LOD scores of 6.9 to 9.2. Thus, the LOD scores of the known hybrid varieties obtained with 123 DNA markers were high enough to confirm their parentage (Table 3). Although all DNA markers used for the evaluation were confirmed to hold Hardy–Weinberg equilibrium, the indigenous varieties used in this study have been revealed to result from frequent and repeated crosses between several key varieties. Such complex population structure could unbalance the genotype frequencies of particular varieties, and could result in changes in their LOD scores. We therefore evaluated the whole genotype frequency of individuals in a population with a new score ‘required cross trial index’ (RCI). The RCI is a simple measure to estimate how many cross trials would be required to obtain a particular individual from the proposed population. This is a unitless logarithmic natural value, and observed differences in RCI value depend on allele frequency and abundance of sibs in the population. Higher RCI values mean there is less of an opportunity to select such an individual because of the lower allele frequency in the population of indigenous varieties, and vice versa. The observed RCI values of the known hybrid varieties range from 148.8 (kara mandarin) to 257.1 (Hayasaki) (Table 3). The structure analysis (Fig 4) suggests that these variations correlate with the less frequent occurrence of the pummelo genome, and the abundant occurrence of the mandarin genome within the indigenous varieties. On that premise, their LOD scores and RCI values show a high regression coefficient (r2 = 0.821), and the observed variations found in the LOD score were accordingly considered mostly due to allele abundance in the indigenous variety population. A similar influence of related individuals on frequency in the population was found in Marshall et al [62]. The estimated RCI values for the indigenous varieties showed a nonuniform distribution (S3 Fig) and the estimated p-value by the Shapiro–Wilk test was 2.07 × 10−10, supporting the hypothesis. Despite these constraints, the likelihood ratio analysis was considered valuable for the evaluation of confidence in the assumed triads even when applied to a small and structured population because it was avoidable by evaluating them with a sufficient number of DNA markers.
The LOD scores of the inferred parentage of indigenous varieties range from 75.3 (Satsuma) to 127.1 (sokitsu) (Table 14), and these values are comparable to those observed in the known hybrid varieties (Table 3). Therefore, we consider them sufficiently high enough to confirm the inferred combination. The observed LOD scores show a correlation to their RCI values (r2 = 0.711), as observed in the known hybrid varieties (Table 3). The observed LOD score of Satsuma was lower than others but the RCI value was proportionately low (Table 14). The inferred parents of Satsuma (Kishu and kunenbo-A) were also found in other parentage frequently as indicated by their low RCI values (161.5 for Kishu and 156.4 for kunenbo-A). This evidence confirms that frequent occurrence of sibship in the population depresses the LOD score of Satsuma. The LOD score of andoukan (80.9) was also considered to be depressed for similar reasons on the basis of its RCI value.
Likelihood ratio estimation of single parent and offspring dyads using Eq 5 was less informative than estimates from parent and offspring triads due to the lack of information of the second parent [65], resulting in lower LOD scores (Table 15). The observed LOD scores of most inferred single parent–offspring dyads in the indigenous varieties confirmed their relationships, but with a wide range from -1.4 (natsudaidai) to 113.1 (Meyer lemon) (Table 15). The likelihood ratio and LOD score were more susceptible to family structure than those observed in the triads [50]. The RCI values also showed large variation from 152.7 (kunenbo-A) to 298.4 (ichanchii) (Table 15). Meagher also pointed out a similar dependence although he evaluated the likelihood ratio in a different manner [61]. Because of these large variations, these measures were considered to be affected severely by the population structure of the indigenous varieties and not reliable for examining inferred parentage. Therefore, we verified the validity of the inferred dyads with another measure, ‘single parent–offspring probability’ (SPP). The SPP was a cumulative probability of two particular individuals being a single parent–offspring dyad, obtained from their transition probability (Eq 8). This score depends on the allele frequencies and combination of the single parent and offspring, but is less susceptible to family structure than the likelihood ratio. The score will increase according to the number of DNA markers for the analysis or the use of highly polymorphic DNA markers. In the estimation of the inferred single parent–offspring dyads, their obtained SPP values ranged from 35.5 to 54.7, and no large variation was observed (Table 15). The SPP values were comparable to those obtained from the known hybrid varieties (Table 3). Consequently, the inferred parentages were considered to be correct sufficiently.
The inferred parentage of several dyads was further examined with these scores. Direct parentage between ichanchii (C. ichangensis; A041) and lemon (A085) was suggested by the allele-sharing test, but their LOD scores were identical when either of them was assumed to be the parent and the other to the offspring. The SPP scores of these combinations were close, but slightly higher when ichanchii was assumed to be the parent of lemon. Structure analysis suggested a hybrid origin for ichanchii (Fig 4), they were insufficient to conclude the parentage between ichanchii and lemon (Table 15). The allele-sharing test suggested close kinship among Kawachi bankan (A051), yuge hyoukan (A205), ujukitsu (A197), Naruto (A095) and Kishu (A059), but their parentage relationships were not obvious. LOD and SPP scores suggested that Naruto was an offspring of Kishu with another unidentified parent harboring the pummelo-type cytotype (C04), and yuge hyoukan was inferred to be an offspring of Naruto and an unidentified parent. The parentage of ujukitsu and Kawachi bankan was not obvious, but they were assumed to be offspring of yuge hyoukan on the basis of their SPP scores (Fig 8). The possible parentage relationships between lemon and sour orange showed identical LOD scores, but the RCI score was higher when lemon was assumed to be the offspring of sour orange, and SPP score was higher when sour orange was assumed to be the offspring of lemon (Table 15). The result of structure analysis suggested admixture of the three basic taxa in sour orange, while lemon derived mostly from a single taxon. The PCoA analysis placed lemon close to one of the basic taxa; however, sour orange was placed in the middle of the three taxa. With this evidence, we infer that sour orange arose from a hybridization of lemon with an unidentified male variety.
Discussion
This study intended to infer the parentage of indigenous citrus varieties by the allele sharing test, parentage test, and likelihood ratio analysis. The identity test was used to identify mutant strains or synonymous varieties, and 101 representative varieties were selected. These selected representatives are valid as a core collection of citrus varieties. Similar approaches to infer parentage with DNA marker analysis have also been reported in pine [119], grape [120], and apple [121]. Genotypes of chloroplast and mitochondrial genomes were evaluated to estimate the combination of seed parent and pollen parent. Very recently, Curk et al revealed the parentage of lime, lemon and sour orange according to nuclear and organelle genome analysis [47]. However, this only revealed their parentage of a limited number of varieties, and the parentage of many indigenous varieties remained uncertain.
The allele-sharing test recognized the correct parentage of ‘Fortune’ (Table 14). The test also identified the unidentified parental variety of ‘Haruka’ (Table 14). Genotyping analysis of chloroplast and mitochondrial genomes enabled fine classification of the cytoplasmic genotype (cytotype) into 18 categories, and confirmed the inferred parentage of ‘Fortune’ and ‘Haruka’. Thus, the allele-sharing test with DNA markers, after eliminating erroneous ones by parentage test with 122 known hybrid triads (59 hybrid varieties and 63 selected strains), was confirmed to be a valid approach to infer parentage as described by Sieberts et al [122], and the deduced cytotype was sufficient to understand the combination of seed parent and pollen parent.
Consequently, the parentage of 22 indigenous varieties was inferred, and 12 of them revealed no mismatch in the parentage test (Table 14). Their cytotypes matched the inferred parentage entirely and contributed to determining the combination of these parents. LOD scores for all 22 varieties were sufficient to support the inferred parentage. The allele-sharing test also inferred 46 single parent–offspring parentages, and 36 of them showed no mismatches. The cytotypes of these inferred combinations were valuable to estimate whether the alleged single parent was the seed parent or the pollen parent. The reconstructed genealogy of the indigenous varieties was not tree-like, but reminiscent of a route map of a city with its ‘hub’ structure (Fig 8 and Fig 9).
Although the LOD score of the inferred parentage varied widely between varieties according to their RCI scores, it demonstrated that the inferred combination would be authoritative. The fixation index (Fw) of the indigenous varieties was not large enough and suggested that inbreeding did not affect the LOD score (Table 5). On the contrary, LOD showed a significant correlation to the RCI value, suggesting that uneven distribution of allele frequency affected the LOD score. Kunenbo-A, Kishu, yuzu, sweet orange and sour orange were found in 17, 13, 10, 8 or 5 inferred parentage combinations as parents, respectively (Tables 14 and 15). The frequent usage of particular varieties as parents would accumulate alleles specific to them in the given population and change allele frequencies, and this is considered to increase the difference in the LOD scores (S3 Fig). The influence of uneven distribution on the LOD score became prominent when inferring single parent–offspring dyads using Eq 5. Their deduced LOD scores showed large changes, and some of them had negative values (Table 15). The clear correlation between RCI and LOD scores was also observed in these varieties. The observed large influence of allele frequency on the LOD score strongly suggested that LOD score would not be a measure for inferring single parent–offspring parentage in citrus. To overcome the disadvantages of the LOD score, we proposed a simple SPP score to determine the inferred parentage. The SPP score was estimated from the genotypes and allele frequencies of the alleged single parent and offspring. The SPP scores for the known hybrid varieties support the parentage of the single parent in the known hybrids (Table 3). The SPP values of the inferred single parent–offspring were close to those of the known hybrids (Table 15).
Involvement of pummelo in the occurrence of the indigenous varieties
No pummelo varieties in this study were recognized as parents in triads (Table 14). However, the allele-sharing test with their cytotypes suggested five varieties (A003:asahikan, A028:hassaku, A035:hyoukan, A049:kaikoukan and A055:kinkoji) were hybrids of kunenbo-A and an unidentified variety or varieties harboring the pummelo-type cytotype. Likewise, three varieties (Naruto:A095, natsudaidai:A098 and oukan: A104) were inferred to be hybrids of Kishu (A059), shunkokan (A137) a hybrid of Yatsushiro (A204), jabon (A046) a hybrid of yuzu (A208), and two varieties (A021:funadoko and A103:ootoukan) hybrids of bendizao (A005), with unidentified varieties harboring the pummelo cytotype as the other parent in each case. Since pummelo is monoembryonic [2], its offspring should have different genotypes to the parent. We considered that the 12 evaluated pummelo varieties were not sufficient to find correct parentage, or some of them could be lost. However, these findings suggest that these unidentified pummelo varieties were cultivated close to kunenbo-A, Kishu, yuzu or bendizao in the past. Of those probable pummelo offspring inferred by the allele-sharing test, kaikoukan (A049) was inferred to be the parent of iyo (A044), and of sanbokan (A112) or andoukan (A001), with dancy (A016) and Kishu (A059) as pollen parents, respectively. Fukuba reported that 33 citrus varieties consisting of Kishu, Satsuma, Yatsushiro, kaikoukan, kunenbo, dancy, sour orange, sweet orange koji, ujukitsu, yuzu, citron and various pummelo varieties had been cultivated widely in the Wakayama region of Japan for a long time when his report was published in 1882 [123]. Similar records were found in old Japanese articles, suggesting that these varieties were selected in these regions.
The inferred roles of Kishu, kunenbo and yuzu in the occurrence of citrus varieties
The genetic identity test recognized four different types of kunenbo (C. nobilis Lour. var. kunep Tanaka; kunenbo-A, kunenbo-B, twukkuni and King) in the evaluated indigenous varieties (Table 4). These four types were inferred to be the parents of others, and kunenbo-B was inferred to be the offspring of sour orange (Table 14 and Table 15). Yamamoto et al reported that kunenbo is self-incompatible [124], and this would contribute to the many offspring of kunenbo-A. Interestingly, the allele-sharing test inferred that kunenbo-A was an offspring of Kishu (C. kinokuni hort. ex Tanaka) and an unidentified variety harboring the sweet orange-type cytotype. This observation revealed that both Satsuma and Yatsushiro would be BC1 selections of Kishu, and demonstrated the introgression of the Kishu genome into at least 30 varieties through kunenbo-A. Consequently, the inferred parentage revealed the pivotal role of Kishu in the occurrence of these indigenous varieties.
The allele-sharing test also inferred yuzu (C. junos Siebold ex Tanaka) to be the parent of 10 varieties, five of which were hybrids with kunenbo-A (Table 14). The involvement of yuzu and kunenbo-A in these varieties suggests that they have been cultivated together for a considerable period. With these observations, not only kunenbo-A, but also yuzu was regarded as another key variety for the occurrence of the indigenous varieties. Tanaka stated that C. nobilis must have played some important part in creating a new subsection Microacrumen in Japanese southern islands [12], and this evidence supports his proposal.
Three types of tachibana and their offspring
Three types of tachibana (C. tachibana (Makino) Tanaka) were inferred to be parents of hanayu, girimikan, hyuganatsu and ogonkan (aka ogonto), which are indigenous varieties in Japan [7,12]. The name ‘Tachibana’ appears in the historic Japanese article ‘Kojiki’ published in 712 A.D. Kaibara describes tachibana as having been cultivated in diverse regions of Japan in his book published in 1709 [125]. The allele-sharing test and structure analysis raise the hypothesis that the three types of tachibana found in this study arose from hybridization of the same unknown parents. Hirai et al reported isozyme polymorphism in tachibana accessions collected from various regions of Japan [20], and these results support this hypothesis. These tachibana varieties are presumed to have crossed with others close to them at various places in Japan.
Kourai tachibana (C. nippokoreana Tanaka) was found in Yamaguchi prefecture in Japan, and it was initially misidentified as tachibana [7]. Though these three types of tachibana were not inferred to be parents of Kourai tachibana, the allele-sharing test suggested that an unidentified type of tachibana not evaluated in this study might be its parent. Additionally, the allele-sharing test revealed that koji, another indigenous variety of Japan, shared the same parental variety with tachibana, suggesting their unidentified kinship. Tanaka stated that koji might have arisen from a cross between tachibana and fukure mikan [7]. Though koji is not an offspring of tachibana × fukure mikan, close kinship of koji and tachibana was suggested in this study.
Sour orange, lemons and Cleopatra
Sour orange (C. aurantium L.) was initially regarded as an offspring of citron (C. medica), mandarin (C. reticulata) and pummelo (C. maxima) [14–16]. Recent molecular studies suggest that regular lemon (C. limon (L.) Burm.f.) arose from hybridization between sour orange and mandarin [33,34,45,47,126]. The allele-sharing test inferred that bergamot arose from hybridization of lemon and sour orange as previously demonstrated [48]. Three varieties (Hiroshimanatsubuntan, kunenbo-B and rokugatsumikan) were also inferred to be hybrids of sour orange and unidentified varieties (Table 15, Fig 8). The allele-sharing test revealed a close relationship between sour orange and lemon as Curk et al reported [47]. However, all of the evidence from the allele-sharing test, PCoA, structure analysis and their cytotypes suggest that sour orange is an offspring of lemon (Fig 8). The deduced admixture of three basic taxa suggests that lemon might be a BC2 of citron and pummelo, and sour orange was deduced to be the offspring of lemon and the F1 of pummelo and mandarin. Another admixture analysis with K = 4 also demonstrated a similar result (S2 Fig). The reason for the discrepancy between the parentage of lemon and sour orange and that reported by Curk et al [47] is unclear, but genotyping error or any DNA markers that deviate from HWE might change allele frequencies in the population and could result in the opposite results.
Cleopatra was regarded as a variety of Indian origin [2]. Its cytotype was unique and no similar ones were found. Tizon (C. papillaris Blanco) was inferred to arise from hybridization of Cleopatra and sweet orange (Table 14). With these inferred parentages, lemon and sour orange, and also Cleopatra and sweet orange are considered to have been cultivated in the same regions for considerable durations. Future evaluation with this approach could identify the parent of sour orange or Cleopatra unless they have been lost.
Origins of C. ichangensis and other acid citrus varieties
Many studies based on DNA marker analysis of nuclear genomes have reported the unique position of C. ichangensis (ichanchii) in citrus taxonomy [33,34,55,57]. Swingle considered C. ichangensis as a variation of Papeda, and he classified it to subgenus Papeda section Papedocitrus [11]. He defined ‘Ichandarin’ as a hybrid of C. ichangensis × mandarin, and assumed yuzu as an Ichandarin [11]. Tanaka also recognized the similarity between C. ichangensis and yuzu, but he classified C. ichangensis to section Osmocitrus subsection Euosmocitrus by yuzu [12]. The allele-sharing test and parentage analysis in this study did not confirm direct parentage of C. ichangensis to yuzu as Swingle assumed, and their cytotypes unfortunately did not coincide. The allele-sharing test suggests the direct parentage between C. ichangensis and lemon, but the cytotype of C. ichangensis is unique and does not match that of lemon (Table 15). Because this study did not include sufficient number of papeda or citron as reference, the parentage of C. ichangensis and lemon was required further investigation. The origin of yuzu must be examined in detail with further evidence, but the similarity between yuzu and C. ichangensis suggests the unidentified parent of C. ichangensis as a primary candidate. Meanwhile, Swingle regarded Ichang lemon (C. wilsoni) as a probable hybrid of C. ichangensis and pummelo [11], but Tanaka classified it in the same section with yuzu (section Papedocitrus). The allele-sharing test revealed that the Ichang lemon is not an offspring of C. ichangensis but an offspring of yuzu as the pollen parent and an unidentified variety having the unique Ichang lemon cytotype as the seed parent (Table 15, Fig 8). In a similar fashion, Swingle regarded sudachi (C. sudachi hort. ex Shirai) as an Ichandarin [11], but Tanaka regarded sudachi, kizu and hanayu to be natural hybrids of yuzu and classified them in the same section [7,12]. The allele-sharing test inferred them to be hybrids of yuzu with various varieties (Table 14 and Table 15, Fig 8 and Fig 9), and confirmed the implications of Tanaka.
Origins of Satsuma and kunenbo
The allele-sharing test and parentage analysis inferred the parentages among Satsuma, Yatsushiro, kunenbo-A and Kishu. Many studies have pointed out similarities among Satsuma, kunenbo and Yatsushiro [12,13,123,127–130]. The close relationship between Satsuma and kunenbo has been observed in the DNA marker analysis [8,33,34]. Yatsushiro (C. yatsushiro hort. ex Yu. Tanaka) is an old but abandoned variety that has been produced in several regions as a substitute for Satsuma recently [123,127,128].
In 1709, Kaibara described 15 citrus varieties including ‘Unshukitsu’, which is an old name for Satsuma [125], but Tanaka considered it to represent Kishu [129]. The first document that is considered to describe Satsuma appeared in 1848 by Okamura [127]. In this document, he reported that Satsuma had been cultivated in many regions of Japan for several hundred years as a high quality seedless variety. George R. Hall introduced the first Satsuma trees to Florida from Japan in 1876 [2], indicating that Satsuma was already widely recognized for superior fruit characteristics by this time. Abe described nine and 15 local names for Kishu and Satsuma, respectively, in his book published in 1904 [128]. Together with these old documents and his own survey of old Satsuma trees in the Kyushu region, Tanaka proposed that Satsuma arose at Nagashima town in Kagoshima prefecture from the 15th to 16th centuries [129,130]. Kishu was a major citrus variety from the 12th to 18th centuries in Japan that was produced in wide regions including Kagoshima [127–130]. The origin of Kishu is not known. However, the occurrence of a Chinese biotype (nanfengmiju) agreed with recent speculation that it was transmitted from China to Japan in ancient times [12]. Kunenbo (C. nobilis Lour. var. kunep Tanaka) is not an indigenous variety of Japan but is regarded to have been transmitted from South China through Taiwan, Sakishima Islands and Ryukyu Islands to the Kyushu region around the 8th century [125,129,130]. The inferred parentage of kunenbo-A suggests that kunenbo is a hybrid of Kishu selected in ancient China or else, then propagated to many places. Therefore, it is likely that kunenbo was backcrossed to Kishu in the Kagoshima region of Japan several times and Satsuma and Yatsushiro were selected from their offspring.
Some characteristics of Satsuma contrast to those of its parents Kishu and kunenbo. For example, kunenbo is self-incompatible [124], but Kishu and Satsuma are not. Both kunenbo and Satsuma are polyembryonic but Kishu is monoembryonic [70]. Nakano et al isolated candidate genes involved in the polyembryony of Satsuma [131]. Likewise, Kishu shows no parthenocarpy (Shimizu, T., unpublished) but Satsuma is an exceptionally high and stable parthenocarpic variety [117]. Kotoda et al isolated two gibberellin 20-oxidase genes from Satsuma thought to be involved in parthenocarpy, and demonstrated their different biological functions [71]. Very recently, Kotoda et al isolated three gibberellin 2-oxidase genes of Satsuma involved in the degradation of bioactive gibberellic acid [132]. Furthermore, Satsuma is an entirely male sterile variety but Kishu and kunenbo are fertile varieties [117,133]. Goto et al recently revealed that male sterility of Satsuma is mostly caused by a decrease in pollen in the anther, and suggested the involvement of a nuclear gene to decrease pollen number [134]. The inferred parentage of Satsuma, Kishu and kunenbo with yuzu, sweet orange, sour orange, koji, tachibana and various pummelos is anticipated to enable a deep understanding of these traits of importance to the citrus industry. Another genotyping study with more than 1,000 certified SNP markers confirmed these inferred parentages (Shimizu, T. et al, in preparation). Whole genome sequence analysis and a comparative genomic approach for Satsuma, Kishu and kunenbo will reveal them at a molecular level.
In conclusion, the allele-sharing test and parentage test with the certified DNA markers inferred the parentage of 22 indigenous citrus varieties, and single parents of 46 indigenous citrus varieties. Genotyping analysis of chloroplast and mitochondrial genomes with 11 DNA markers classified cytotypes into 18 categories, and these were helpful in confirming the inferred parentages. Likelihood ratio analysis of triads verified the inferred parentages with significant scores. However, the scores of the triads were susceptible to the allele frequencies of particular varieties in a given population and showed large changes. Such susceptibility of the score became evident when it was applied to validate the parentage of single parents to offspring. Alternatively, a single parent–offspring probability (SPP) score was proposed to verify the inferred single parent to offspring parentage. The inferred parentage identified 12 types of varieties, consisting of Kishu, several types of kunenbo, yuzu, koji, sour orange, dancy, kobeni mikan, sweet orange, three types of tachibana, Cleopatra, willowleaf mandarin, and unidentified pummelo varieties, that were deeply involved in the occurrence of these indigenous varieties. The inferred parentage of the indigenous varieties confirmed their hybrid origins as stated by recent studies [11,12,14,15]. This study will also contribute to a reconsideration of their taxonomy.
Supporting Information
This chart was drawn using Helium software.
(PNG)
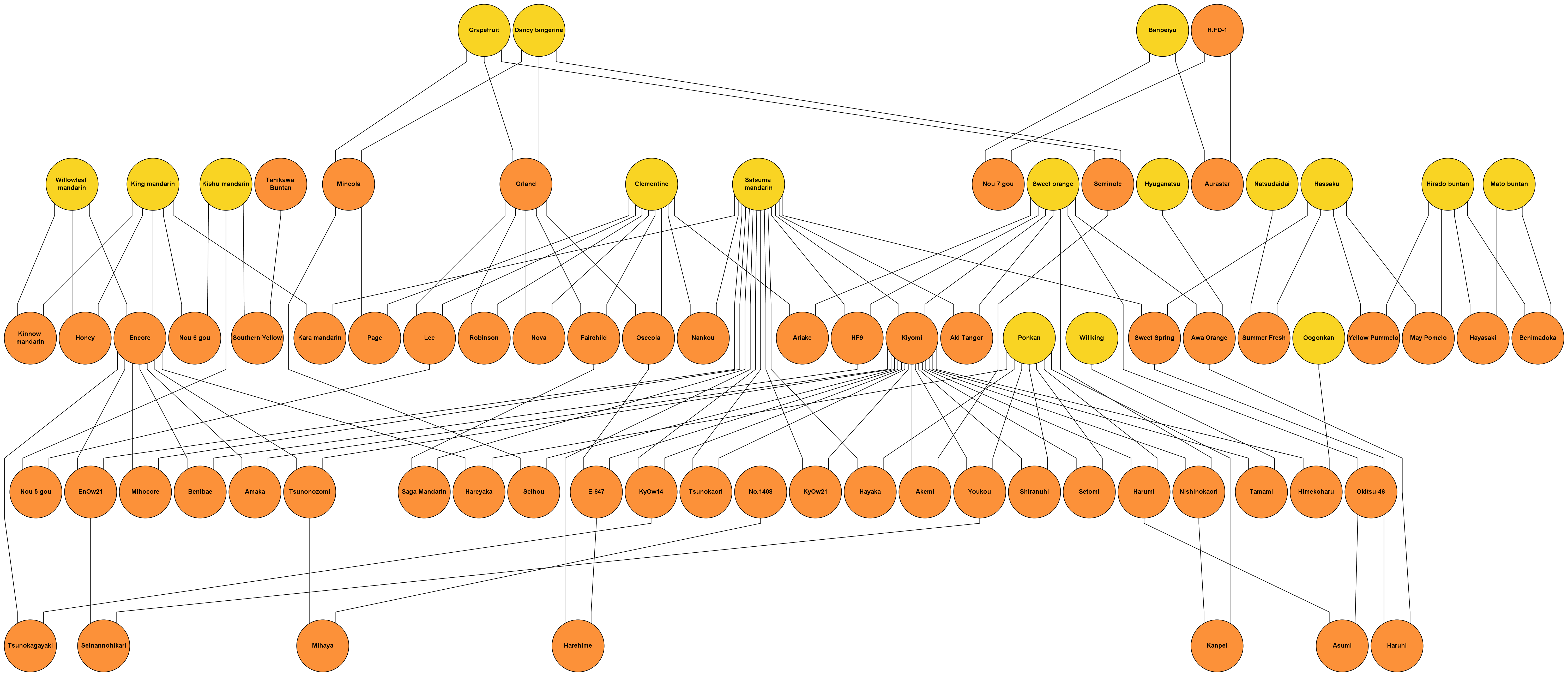{kind=link}
Three clusters correspond to the deduced basic taxa at K = 4 (pummelo, citron mandarin, and probable papeda).
(TIF)
(TIF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(XLS)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Acknowledgments
The authors thank Ms. Chiaki Hirano for her excellent technical assistance. We are also grateful to Dr. Junko Kaneyoshi for suggesting the origin of jabon, Dr. Akira Wakana of the Kyushu University and Dr. Hiroshi Yamagishi of the Kyoto Sangyo University for providing us valuable suggestions, and Dr. Paul D. Shaw of the James Hutton Institute, for providing us with a beta release of the Helium software. Computations were partially performed on the NIG supercomputer at ROIS National Institute of Genetics.
Data Availability
All relevant data are within the paper and its Supporting Information files. All nucleotide sequence data that were obtained by the authors had been deposited to the public database with accession numbers given in this paper. Accession numbers can be found in the file titled S1 Table.
Funding Statement
This work was supported by a grant for Research Project "Genomics for Agricultural Innovation NGB1006" from Japanese Ministry of Agriculture, Forestry and Fisheries for TS. A part of this work was also supported by KAKEN (No. 24405025) for AK.
References
- 1.Saunt J. Citrus Varieties of the World, 2nd Edition Norwich, England: Sinclair Intl Business Resources; 2000. [Google Scholar]
- 2.Hodgson RW. Horticultural varieties of Citrus In: Reuther Webber H.J., Batchelor L.D. W, editor. The Citrus Industry. California: University of California; 1967. pp. 431–591. [Google Scholar]
- 3.Ladaniya MS. Citrus Fruit. Academic Press; 2008. 10.1016/B978-012374130-1.50001-2 [DOI] [Google Scholar]
- 4.Rouse RE. Major citrus cultivars of the world as reported from selected countries. HortScience. 1988;23: 680–684. [Google Scholar]
- 5.Foreign Agricultural Service/USDA O of GA. Citrus: World Markets and Trade. 2016. Available: http://www.fas.usda.gov/data/citrus-world-markets-and-trade
- 6.Webber HJ. History and development of the citrus industy In: Reuther W., Webber H. J., Batchelor L. D, editor. The Citrus Industry I. Riverside: University of California; 1967. pp. 1–39. [Google Scholar]
- 7.Tanaka T. Citrologia. Sakai, Osaka: Citrologia supporting foundation; 1961. [Google Scholar]
- 8.Uzun A, Yesiloglu T. Genetic Diversity in Citrus In: Çalişkan M, editor. Genetic Diversity in plants. Intech; 2012. pp. 213–230. 10.5772/32885 [DOI] [Google Scholar]
- 9.Gmitter FG. Origin, Evolution, and Breeding of the Grapefruit. Plant Breeding Reviews. Oxford, UK: John Wiley & Sons, Inc.; 2010. pp. 345–363. 10.1002/9780470650059.ch10 [DOI] [Google Scholar]
- 10.Nishiura M. Citrus breeding through nucellar seedling selection. JARQ Japan Agric Res Q. 1967;2: 15–19. Available: http://www.jircas.affrc.go.jp/english/publication/jarq/02-1/02-1index.html [Google Scholar]
- 11.Swingle WT. The botany of Citrus and its wild relatives In: Reuther W., Webber H. J., Batchelor L. D, editor. The Citrus Industry. California: University of California; 1967. pp. 190–430. [Google Scholar]
- 12.Tanaka T. Species problem in citrus: a critical study of wild and cultivated units of citrus, based upon field studies in their native homes (Revisio Aurantiacearum IX). Tokyo: Japanese Society for the Promotion of Science; 1954. Available: http://www.worldcat.org/title/species-problem-in-citrus-a-critical-study-of-wild-and-cultivated-units-of-citrus-based-upon-field-studies-in-their-native-homes/oclc/5768249 [Google Scholar]
- 13.Tanaka T. Misunderstanding with regards Citrus classification and nomenclature. Bull Univ Osaka Prefect Ser B, Agric Biol. 1969;21: 139–145. Available: http://hdl.handle.net/10466/2982 [Google Scholar]
- 14.Scora RW. On the history and origin of Citrus. Bull Torrey Bot Club. 1975;102: 369–375. 10.2307/2484763 [DOI] [Google Scholar]
- 15.Barrett HC, Rhodes AM. A Numerical taxonomic study of affinity relationships in cultivated citrus and its close relatives. Syst Bot. 1976;1: 105–136. Available: http://www.jstor.org/stable/2418763 [Google Scholar]
- 16.Torres AM., Soost RK, Diedenhofen U. Leaf isozymes as genetic markers in citrus. Am J Bot. 1978;65: 869–881. Available: http://www.jstor.org/stable/2442183 [Google Scholar]
- 17.Potvin C, Bergeron Y, Simon J. A numerical taxonomic study of selected citrus species (Rutaceae) based on biochemical characters. Syst Bot. 1983;8: 127–133. Available: http://www.jstor.org/stable/2418689 [Google Scholar]
- 18.Handa T, Ishizawa Y, Oogaki C. Phylogenetic study of Fraction I protein in the genus Citrus and its close related genera. Japanese J Genet. 1986;61: 15–24. 10.1266/jjg.61.15 [DOI] [Google Scholar]
- 19.Hirai M, Kajiura I. Genetic analysis of leaf isozymes in citrus. Ikushugaku zasshi. 1987;37: 377–388. 10.1270/jsbbs1951.37.377 [DOI] [Google Scholar]
- 20.Hirai M, Mitsue S, Kita K, Kajiura I. A survey and isozyme analysis of wild mandarin, Tachibana (Citrus tachibana (Mak.) Tanaka) growing in Japan. J Japanese Soc Hortic Sci. 1990;59: 1–7. 10.2503/jjshs.59.1 [DOI] [Google Scholar]
- 21.Novelli V, Machado M, Lopes C. Isoenzymatic polymorphism in Citrus spp. and Poncirus trifoliata (L.) Raf.(Rutaceae). Genet Mol Biol. 2000;23: 163–168. Available: http://www.scielo.br/scielo.php?pid=S1415-47572000000100030&script=sci_arttext [Google Scholar]
- 22.Machado MA, Coletta Filho HD, Targon MLPN, Pompeu J. Genetic relationship of Mediterranean mandarins (Citrus deliciosa Tenore) using RAPD markers. Euphytica. 1995;92: 321–326. Available: 10.1007/BF00037115 [DOI] [Google Scholar]
- 23.Corazza-Nunes MJ, Machado MA, Nunes WMC, Cristofani M, Targon MLPN. Assessment of genetic variability in grapefruits (Citrus paradisi Macf.) and pummelos (C. maxima (Burm.) Merr.) using RAPD and SSR markers. Euphytica. 2002;126: 169–176. 10.1023/A:1016332030738 [DOI] [Google Scholar]
- 24.Elisiário P, Justo E, Leitão J. Identification of mandarin hybrids by isozyme and RAPD analysis. Sci Hortic (Amsterdam). 1999;81: 287–299. Available: http://www.sciencedirect.com/science/article/pii/S0304423899000138 [Google Scholar]
- 25.Natividade Targon Machado M.A., Coletta Filho H.D. and Cristofani M. MLP. Genetic polymorphism of sweet orange (Citrus sinensis [L.] Osbeck) varieties evaluated by random amplified polymorphic DNA. Acta Hortic. 2000;535: 51–54. Available: http://www.actahort.org/books/535/535_5.htm [Google Scholar]
- 26.Coletta Filho HD, Machado MA, Targon MLPN, Moreira MCPQDG, Pompeu J Jr. Analysis of the genetic diversity among mandarins (Citrus spp.) using RAPD markers. Euphytica. 1998;102: 133–139. 10.1023/A:1018300900275 [DOI] [Google Scholar]
- 27.Luro F, Lorieux M, Laigret F, Bove J, Ollitrault P. Genetic mapping of an intergeneric citrus hybrid using molecular markers. Fruits. 1994;49: 404–408. Available: http://cat.inist.fr/?aModele=afficheN&cpsidt=2895059 [Google Scholar]
- 28.Kepiro JL, Roose ML. AFLP markers closely linked to a major gene essential for nucellar embryony (apomixis) in Citrus maxima × Poncirus trifoliata. Tree Genet Genomes. 2009;6: 1–11. 10.1007/s11295-009-0223-z [DOI] [Google Scholar]
- 29.Bretó MP, Ruiz C, Pina J a, Asíns MJ. The diversification of Citrus clementina Hort. ex Tan., a vegetatively propagated crop species. Mol Phylogenet Evol. 2001;21: 285–93. 10.1006/mpev.2001.1008 [DOI] [PubMed] [Google Scholar]
- 30.Fang DQ, Roose ML. Identification of closely related citrus cultivars with inter-simple sequence repeat markers. TAG Theor Appl Genet. 1997;95: 408–417. 10.1007/s001220050577 [DOI] [Google Scholar]
- 31.Tripolitsiotis C, Nikoloudakis N, Linos A, Hagidimitriou M. Molecular characterization and analysis of the greek citrus germplasm. Not Bot Horti Agrobot Cluj-Napoca. 2013;41: 463–471. [Google Scholar]
- 32.Uzun A, Yesiloglu T, Aka-Kacar Y, Tuzcu O, Gulsen O. Genetic diversity and relationships within Citrus and related genera based on sequence related amplified polymorphism markers (SRAPs). Sci Hortic (Amsterdam). 2009;121: 306–312. 10.1016/j.scienta.2009.02.018 [DOI] [Google Scholar]
- 33.Nicolosi E, Deng ZN, Gentile A, La Malfa S, Continella G, Tribulato E. Citrus phylogeny and genetic origin of important species as investigated by molecular markers. TAG Theor Appl Genet. 2000;100: 1155–1166. 10.1007/s001220051419 [DOI] [Google Scholar]
- 34.Nicolosi E. Origin and taxonomy In: Khan IA, editor. Citrus genetics, breeding and biotechnology. Oxfordshire, UK: CABI, CAB International; 2007. pp. 19–43. [Google Scholar]
- 35.Mabberley DJ. Citrus (Rutaceae): A review of recent advances in etymology, systematics and medical applications. Blumea—Biodiversity, Evol Biogeogr Plants. 2004;49: 481–498. 10.3767/000651904X484432 [DOI] [Google Scholar]
- 36.Mabberley D. A classification for edible Citrus (Rutaceae). Telopea. 1997;7: 167–172. Available: https://www.rbgsyd.nsw.gov.au/__data/assets/pdf_file/0019/73216/Tel7Mab167.pdf [Google Scholar]
- 37.Khan IA. Citrus Genetics, Breeding and Biotechnology. Khan IA, editor. Nosworthy Way Wallingford: CAB International; 2007. [Google Scholar]
- 38.Talon M, Gmitter FG. Citrus genomics. Int J Plant Genomics. 2008;2008: 1–17. 10.1155/2008/528361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gmitter FG, Chen C, Machado M a., Souza AA, Ollitrault P, Froehlicher Y, et al. Citrus genomics. Tree Genet Genomes. 2012;8: 611–626. 10.1007/s11295-012-0499-2 [DOI] [Google Scholar]
- 40.Roose M, Close T. Genomics of citrus, a major fruit crop of tropical and subtropical regions Moore PH, Ming R, editors. Genomics of Tropical Crop Plants. Springer; New York; 2008. 10.1007/978-0-387-71219-2_8 [DOI] [Google Scholar]
- 41.Bausher MG, Singh ND, Lee S-B, Jansen RK, Daniell H. The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var “Ridge Pineapple”: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol. 2006;6: 21 10.1186/1471-2229-6-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Q, Chen L-L, Ruan X, Chen D, Zhu A, Chen C, et al. The draft genome of sweet orange (Citrus sinensis). Nat Genet. Nature Publishing Group; 2013;45: 59–66. 10.1038/ng.2472 [DOI] [PubMed] [Google Scholar]
- 43.Wu GA, Prochnik S, Jenkins J, Salse J, Hellsten U, Murat F, et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat Biotechnol. 2014;32: 656–662. 10.1038/nbt.2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luro FL, Costantino G, Terol J, Argout X, Allario T, Wincker P, et al. Transferability of the EST-SSRs developed on Nules clementine (Citrus clementina Hort ex Tan) to other Citrus species and their effectiveness for genetic mapping. BMC Genomics. 2008/06/19. 2008;9: 287 1471-2164-9-287 [pii] 10.1186/1471-2164-9-287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ollitrault P, Terol J, Garcia-Lor A, Bérard A, Chauveau A, Froelicher Y, et al. SNP mining in C. clementina BAC end sequences; transferability in the Citrus genus (Rutaceae), phylogenetic inferences and perspectives for genetic mapping. BMC Genomics. 2012;13: 13 10.1186/1471-2164-13-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ollitrault F, Terol J, Pina JA, Navarro L, Talon M, Ollitrault P. Development of SSR markers from Citrus clementina (Rutaceae) BAC end sequences and interspecific transferability in Citrus. Am J Bot. 2010;97: e124–9. 10.3732/ajb.1000280 [DOI] [PubMed] [Google Scholar]
- 47.Curk F, Ollitrault F, Garcia-Lor A, Luro F, Navarro L, Ollitrault P. Phylogenetic origin of limes and lemons revealed by cytoplasmic and nuclear markers. Ann Bot. 2016;117: 565–583. 10.1093/aob/mcw005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng Z, Gentile A, Nicolosi E, Continella G, Tribulato E. Parentage determination of some citrus hybrids by molecular markers. Proc Int Soc Citric. 1996;2: 84–854. [Google Scholar]
- 49.Yamamoto M, Kobayashi S, Nakamura Y, Yamada Y. Phylogenic relationships of Citrus revealed by RFLP analysis of mitochondrial and chloroplast DNA. Japanese J Breed. 1993;43: 355–365. 10.1270/jsbbs1951.43.355 [DOI] [Google Scholar]
- 50.de Araújo Edson Freitas, Queiroz LP de, Machado MA. What is Citrus? Taxonomic implications from a study of cp-DNA evolution in the tribe Citreae (Rutaceae subfamily Aurantioideae). Org Divers Evol. 2003;3: 55–62. 10.1078/1439-6092-00058 [DOI] [Google Scholar]
- 51.Jung Y-H, Kwon H-M, Kang S-H, Kang J-H, Kim S-C. Investigation of the phylogenetic relationships within the genus Citrus (Rutaceae) and related species in Korea using plastid trnL-trnF sequences. Sci Hortic (Amsterdam). 2005;104: 179–188. 10.1016/j.scienta.2004.08.008 [DOI] [Google Scholar]
- 52.Jena SN, Kumar S, Nair NK. Molecular phylogeny in Indian Citrus L. (Rutaceae) inferred through PCR-RFLP and trnL-trnF sequence data of chloroplast DNA. Sci Hortic (Amsterdam). 2009;119: 403–416. 10.1016/j.scienta.2008.08.030 [DOI] [Google Scholar]
- 53.Froelicher Y, Mouhaya W, Bassene J-B, Costantino G, Kamiri M, Luro F, et al. New universal mitochondrial PCR markers reveal new information on maternal citrus phylogeny. Tree Genet Genomes. 2011;7: 49–61. 10.1007/s11295-010-0314-x [DOI] [Google Scholar]
- 54.Yamamoto M, Tsuchimochi Y, Ninomiya T, Koga T, Kitajima A, Yamasaki A, et al. Diversity of chloroplast DNA in various mandarins (Citrus spp.) and other Citrus demonstrated by CAPS analysis. J Japanese Soc Hortic Sci. 2013;82: 106–113. [Google Scholar]
- 55.Penjor T, Yamamoto M, Uehara M, Ide M, Matsumoto N, Matsumoto R, et al. Phylogenetic relationships of citrus and its relatives based on matK gene sequences. PLoS One. 2013;8: e62574 10.1371/journal.pone.0062574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carbonell-Caballero J, Alonso R, Ibañez V, Terol J, Talon M, Dopazo J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol Biol Evol. 2015;32: 2015–35. 10.1093/molbev/msv082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abkenar AA, Isshiki S, Tashiro Y. Phylogenetic relationships in the “true citrus fruit trees” revealed by PCR-RFLP analysis of cpDNA. Sci Hortic (Amsterdam). 2004;102: 233–242. 10.1016/j.scienta.2004.01.003 [DOI] [Google Scholar]
- 58.Penjor T, Nagano Y, Mimura T, Matsumoto R, Yamamoto M. Exploration of local citrus genetic resources in Bhutan and their chloroplast DNA analysis. Hortic Res. 2014;13: 307–314. 10.2503/hrj.13.307 [DOI] [Google Scholar]
- 59.Brenner CH, Weir BS. Issues and strategies in the DNA identification of World Trade Center victims. Theor Popul Biol. 2003;63: 173–178. 10.1016/S0040-5809(03)00008-X [DOI] [PubMed] [Google Scholar]
- 60.Goodwin W, Linacre A, Hadi S. An introduction to forensic genetics. Journal of Chemical Information and Modeling. Wiley; 2011. [Google Scholar]
- 61.Meagher TR. Analysis of paternity within a natural population of Chamaelirium luteum. 1. Identification of most-likely male parents. Am Nat. 1986;128: 199–215. Available: http://www.jstor.org/stable/2461545 [Google Scholar]
- 62.Marshall TC, Slate J, Kruuk LEB, Pemberton JM. Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol. 1998;7: 639–655. 10.1046/j.1365-294x.1998.00374.x [DOI] [PubMed] [Google Scholar]
- 63.Jones AG, Ardren WR. Methods of parentage analysis in natural populations. Mol Ecol. 2003;12: 2511–2523. 10.1046/j.1365-294X.2003.01928.x [DOI] [PubMed] [Google Scholar]
- 64.Blouin MS. DNA-based methods for pedigree reconstruction and kinship analysis in natural populations. Trends Ecol Evol. 2003;18: 503–511. 10.1016/S0169-5347(03)00225-8 [DOI] [Google Scholar]
- 65.Brenner CH. Calculation of paternity index In: Walker R, editor. Inclusion probabilities in paternity testing. Arlington: American Association of Blood Banks; 1983. pp. 632–638. [Google Scholar]
- 66.Pemberton JM, Slate J, Bancroft DR, Barrett JA. Nonamplifying alleles at microsatellite loci: a caution for parentage and population studies. Mol Ecol. 1995;4: 249–252. 10.1111/j.1365-294X.1995.tb00214.x [DOI] [PubMed] [Google Scholar]
- 67.Shimizu T, Kaminuma E, Nonaka K, Yoshioka T, Goto S, Matsumoto T, et al. A genomic approach to selecting robust and versatile SNP sets from next-generation sequencing data for genome-wide association study in citrus cultivars. Acta Hortic. 2016;1135: 23–32. 10.17660/ActaHortic.2016.1135.4 [DOI] [Google Scholar]
- 68.Close TJ, Wanamaker S, Roose ML, Lyon M. HarvEST. Methods Mol Biol. 2007;406: 161–77. Available: http://www.ncbi.nlm.nih.gov/pubmed/18287692 [DOI] [PubMed] [Google Scholar]
- 69.Terol J, Naranjo MA, Ollitrault P, Talon M. Development of genomic resources for Citrus clementina: characterization of three deep-coverage BAC libraries and analysis of 46,000 BAC end sequences. BMC Genomics. 2008;9: 423 10.1186/1471-2164-9-423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakano M, Shimada T, Endo T, Fujii H, Nesumi H, Kita M, et al. Characterization of genomic sequence showing strong association with polyembryony among diverse Citrus species and cultivars, and its synteny with Vitis and Populus. Plant Sci. 2012;183: 131–42. 10.1016/j.plantsci.2011.08.002 [DOI] [PubMed] [Google Scholar]
- 71.Kotoda N, Matsuo S, Honda I, Yano K, Shimizu T. Isolation and functional analysis of two Gibberellin 20-oxidase genes from Satsuma mandarin (Citrus unshiu Marc.). Hortic J. 2015; 10.2503/hortj.MI-085 [DOI] [Google Scholar]
- 72.Staden R, Beal KF, Bonfield JK. The Staden package, 1998. Methods Mol Biol. 2000;132: 115–30. Available: http://www.ncbi.nlm.nih.gov/pubmed/10547834 [DOI] [PubMed] [Google Scholar]
- 73.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14: 1147–1159. 10.1101/gr.1917404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kolpakov R, Bana G, Kucherov G. mreps: efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res. 2003;31: 3672–3678. 10.1093/nar/gkg617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marshall OJ. PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics. 2004/04/10. 2004;20: 2471–2472. 10.1093/bioinformatics/bth254 bth254 [pii] [DOI] [PubMed] [Google Scholar]
- 78.Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers Bioinforma Methods Protoc Methods Mol Biol. Totowa: Humana Press; 2000; 365–386. [DOI] [PubMed] [Google Scholar]
- 79.Chen C, Zhou P, Choi YA, Huang S, Gmitter FG Jr. Mining and characterizing microsatellites from citrus ESTs. Theor Appl Genet. 2006/02/14. 2006;112: 1248–1257. 10.1007/s00122-006-0226-1 [DOI] [PubMed] [Google Scholar]
- 80.Chen C, Bowman KD, Choi Y a, Dang PM, Rao MN, Huang S, et al. EST-SSR genetic maps for Citrus sinensis and Poncirus trifoliata. Tree Genet Genomes. 2007;4: 1–10. 10.1007/s11295-007-0083-3 [DOI] [Google Scholar]
- 81.Weising K, Gardner RC. A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome. 1999;42: 9–19. Available: http://www.ncbi.nlm.nih.gov/pubmed/10207998 [PubMed] [Google Scholar]
- 82.Shimizu T, Yano K. A post-labeling method for multiplexed and multicolored genotyping analysis of SSR, indel and SNP markers in single tube with bar-coded split tag (BStag). BMC Res Notes. 2011;4: 161 10.1186/1756-0500-4-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brownstein MJ, Carpten JD, Smith JR. Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques. 1996;20: 1004–6, 1008–10. Available: http://www.ncbi.nlm.nih.gov/pubmed/8780871 [DOI] [PubMed] [Google Scholar]
- 84.Nei M. Molecular Evolutionary Genetics. Columbia University Press; 1987; 512. [Google Scholar]
- 85.Botstein D, White RL, Skolnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet. 1980;32: 314–331. [PMC free article] [PubMed] [Google Scholar]
- 86.Nei M, Kumar S. Molecular Evolution and Phylogenetics. New Nork, U.S.A., U.S.A.: Oxford University Press; 2000. Available: https://global.oup.com/academic/product/molecular-evolution-and-phylogenetics-9780195135855?cc=jp&lang=en [Google Scholar]
- 87.Hui W, Gel Y, Gastwirth J. lawstat: an R package for law, public policy and biostatistics. J Stat Softw. 2008;28: 1–26. Available: http://www.jstatsoft.org/v28/i03/paper27774042 [Google Scholar]
- 88.Meirmans PG, Hedrick PW. Assessing population structure: FST and related measures. Mol Ecol Resour. 2011;11: 5–18. 10.1111/j.1755-0998.2010.02927.x [DOI] [PubMed] [Google Scholar]
- 89.Ma L, Ji YJ, Zhang DX. Statistical measures of genetic differentiation of populations: Rationales, history and current states. Curr Zool. 2015;61: 886–896. 10.1093/czoolo/61.5.886 [DOI] [Google Scholar]
- 90.Goudet J. hierfstat, a package for r to compute and test hierarchical F-statistics. Mol Ecol Notes. 2005;5: 184–186. 10.1111/j.1471-8286.2004.00828.x [DOI] [Google Scholar]
- 91.Paradis E. pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics. 2010;26: 419–20. 10.1093/bioinformatics/btp696 [DOI] [PubMed] [Google Scholar]
- 92.Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24: 1403–1405. 10.1093/bioinformatics/btn129 [DOI] [PubMed] [Google Scholar]
- 93.Winter DJ. mmod: an R library for the calculation of population differentiation statistics. Mol Ecol Resour. 2012;12: 1158–1160. 10.1111/j.1755-0998.2012.03174.x [DOI] [PubMed] [Google Scholar]
- 94.Guo SW, Thompson EA. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48: 361–72. Available: http://www.ncbi.nlm.nih.gov/pubmed/1637966 [PubMed] [Google Scholar]
- 95.Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10: 564–7. 10.1111/j.1755-0998.2010.02847.x [DOI] [PubMed] [Google Scholar]
- 96.Glaubitz JC. CONVERT: A user-friendly program to reformat diploid genotypic data for commonly used population genetic software packages. Mol Ecol Notes. 2004;4: 309–310. 10.1111/j.1471-8286.2004.00597.x [DOI] [Google Scholar]
- 97.Xavier Perrier, Jacquemoud-Collet J-P. DARWin software. 2006. Available: http://darwin.cirad.fr/
- 98.Perrier X, Flori A, Bonnet F. Data analysis methods In: Hamon P, Seguin M, Perrier X, Glaszmann JC, editors. Genetic diversity of cultivated tropical plants. Montpellier: Enfield, Science Publishers; 2003. pp. 43–76. [Google Scholar]
- 99.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4: 406–25. Available: http://mbe.oxfordjournals.org/content/4/4/406.short [DOI] [PubMed] [Google Scholar]
- 100.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155: 945–59. Available: http://www.ncbi.nlm.nih.gov/pubmed/10835412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14: 2611–20. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- 102.Earl DA, VonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4: 359–361. 10.1007/s12686-011-9548-7 [DOI] [Google Scholar]
- 103.Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23: 1801–1806. 10.1093/bioinformatics/btm233 [DOI] [PubMed] [Google Scholar]
- 104.Brenner C. A note on paternity computation in cases lacking a mother. Transfusion. 1993;33: 51–54. 10.1046/j.1537-2995.1993.33193142310.x [DOI] [PubMed] [Google Scholar]
- 105.Shaw PD, Graham M, Kennedy J, Milne I, Marshall DF. Helium: visualization of large scale plant pedigrees. BMC Bioinformatics. 2014;15: 259 10.1186/1471-2105-15-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes. 2007;7: 574–578. 10.1111/j.1471-8286.2007.01758.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ayres KL, Balding DJ. Measuring departures from Hardy–Weinberg: a Markov chain Monte Carlo method for estimating the inbreeding coefficient. Heredity (Edinb). 1998;80: 769–777. 10.1046/j.1365-2540.1998.00360.x [DOI] [PubMed] [Google Scholar]
- 108.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155: 945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moore GA. Oranges and lemons: clues to the taxonomy of Citrus from molecular markers. Trends Genet. 2001;17: 536–540. 10.1016/S0168-9525(01)02442-8 [DOI] [PubMed] [Google Scholar]
- 110.Bayer RJ, Mabberley DJ, Morton C, Miller CH, Sharma IK, Pfeil BE, et al. A molecular phylogeny of the orange subfamily(Rutaceae: Aurantioideae) using nine cpDNA sequences. Am J Bot. 2009;96: 668–85. 10.3732/ajb.0800341 [DOI] [PubMed] [Google Scholar]
- 111.Yang X, Li H, Liang M, Xu Q, Chai L, Deng X. Genetic diversity and phylogenetic relationships of citron (Citrus medica L.) and its relatives in southwest China. Tree Genet Genomes. 2015;11: 129 10.1007/s11295-015-0955-x [DOI] [Google Scholar]
- 112.Iwamasa M. Varieties of citrus (Kankitsu no Hinshu). (Japanese). Shizuoka: Shizuoka Citrus Growers Association; 1976. [Google Scholar]
- 113.Gmitter FG. Origin, evolution and breeding of the grapefruit. Plant Breed Rev. 1995;13: 345–363. [Google Scholar]
- 114.Siebert T, Krueger R, Kahn T, Bash J, Vidalakis G. Descriptions of new varieties recently distributed from the Citrus Clonal Protection Program. Citrograph. 2010;Mar-Apr: 20–26. Available: http://www.citrusvariety.ucr.edu/citrus/documents/Siebert_etal_2010_CCPP_New_Varieties_CitrographMarchApril2010.pdf [Google Scholar]
- 115.Nakatani M. On the characteristics of “Jabon”, an acid citrus for vinegar (Japanese). Stud Citrol. 1989;15: 75–78. [Google Scholar]
- 116.Matsumoto R, Okudai N. The difference of cultivar with an increase in bitterness and bitter components flavanone neohesperidoside content in citrus fruit caused by freezing (In Japanese with English abstract). Bull Fruit Tree Res Stn Ser D. 1985;10: 1–10. [Google Scholar]
- 117.Tanaka T. A monograph of the Satsuma orange: with special reference to the occurrence of new varieties through bud variation. Mem Fac Sci Agric Taihoku Imp Univ. 1932;4: 1–698. Available: http://ci.nii.ac.jp/ncid/BA67366487 [Google Scholar]
- 118.Committee on Identifying the Needs of the Forensic Sciences Community, et al. Strengthening Forensic Science in the United States. Washington, D.C.: National Academies Press; 2009. July 10.17226/12589 [DOI] [Google Scholar]
- 119.Grattapaglia D, do Amaral Diener PS, dos Santos GA. Performance of microsatellites for parentage assignment following mass controlled pollination in a clonal seed orchard of loblolly pine (Pinus taeda L.). Tree Genet Genomes. 2014;10: 1631–1643. 10.1007/s11295-014-0784-3 [DOI] [Google Scholar]
- 120.Lacombe T, Boursiquot JM, Laucou V, Di Vecchi-Staraz M, Péros JP, This P. Large-scale parentage analysis in an extended set of grapevine cultivars (Vitis vinifera L.). Theor Appl Genet. 2013;126: 401–414. 10.1007/s00122-012-1988-2 [DOI] [PubMed] [Google Scholar]
- 121.Lassois L, Denancé C, Ravon E, Guyader A, Guisnel R, Hibrand-Saint-Oyant L, et al. Genetic diversity, population structure, parentage analysis, and construction of core collections in the French apple germplasm based on SSR markers. Plant Mol Biol Report. Plant Molecular Biology Reporter; 2016; 1–18. 10.1007/s11105-015-0966-7 [DOI] [Google Scholar]
- 122.Sieberts SK, Wijsman EM, Thompson EA. Relationship inference from trios of individuals, in the presence of typing error. Am J Hum Genet. 2002;70: 170–180. 10.1086/338444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fukuba H. Records of Citrus from Kii Province (Kishu Kankitsuroku, in Japanese). Tokyo, Japan: Ministry of Home Affairs, Department of Agriculture; 1882. Available: http://dl.ndl.go.jp/info:ndljp/pid/840099/48 [Google Scholar]
- 124.Yamamoto M, Kubo T, Tominaga S. Self-and cross-incompatibility of various citrus accessions. J Japan Soc Hort Sci. 2006;75: 372–378. Available: http://ir.kagoshima-u.ac.jp/handle/10232/18900 [Google Scholar]
- 125.Kaibara E. Yamato Honzo. 1709. Available: http://www.nakamura-u.ac.jp/library/kaibara/archive01/
- 126.Curk F, Ancillo G, Garcia-Lor A, Luro F, Perrier X, Jacquemoud-Collet J-P, et al. Next generation haplotyping to decipher nuclear genomic interspecific admixture in Citrus species: analysis of chromosome 2. BMC Genet. 2014;15: 152 10.1186/s12863-014-0152-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Okamura S. Keien Kiffu. 1848. 10.11501/2537133
- 128.Abe K. Citrus of Japan. 1904. Available: http://dl.ndl.go.jp/info:ndljp/pid/840260
- 129.Tanaka Y. Developmental history of citrus varieties (1). Kankitsu. 1941;8: 308–315. [Google Scholar]
- 130.Tanaka Y. Developmental history of citrus varieties (2). Kankitsu. 1942;9: 5–16. [Google Scholar]
- 131.Nakano M, Kigoshi K, Shimizu T, Endo T, Shimada T, Fujii H, et al. Characterization of genes associated with polyembryony and in vitro somatic embryogenesis in Citrus. Tree Genet Genomes. 2013;9: 795–803. 10.1007/s11295-013-0598-8 [DOI] [Google Scholar]
- 132.Kotoda N, Matsuo S, Honda I, Yano K, Shimizu T. Gibberellin 2-Oxidase Genes from Satsuma Mandarin (Citrus unshiu Marc.) Caused Late Flowering and Dwarfism in Transgenic Arabidopsis. Hortic J. 2017;85: 128–140. 10.2503/hortj.OKD-016 [DOI] [Google Scholar]
- 133.Nakano M, Nesumi H, Yoshioka T, Yoshida T. Segregation of plants with undeveloped anthers among hybrids derived from the seed parent, “Kiyomi” (Citrus unshiu × C. sinensis). J Japan Soc Hort Sci. 2001;70: 539–545. [Google Scholar]
- 134.Goto S, Yoshioka T, Ohta S, Kita M, Hamada H, Shimizu T. Segregation and heritability of male sterility in populations derived from progeny of Satsuma mandarin. PLoS One. 2016;11: e0162408 10.1371/journal.pone.0162408 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This chart was drawn using Helium software.
(PNG)
Three clusters correspond to the deduced basic taxa at K = 4 (pummelo, citron mandarin, and probable papeda).
(TIF)
(TIF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(XLS)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. All nucleotide sequence data that were obtained by the authors had been deposited to the public database with accession numbers given in this paper. Accession numbers can be found in the file titled S1 Table.