

Summary
Pseudomonas aeruginosa is an opportunistic human pathogen whose survival is aided by forming communities known as biofilms, in which cells are encased in a self-produced matrix. We devised a mutant screen based on colony morphology to identify additional genes with previously unappreciated roles in biofilm formation. Our screen, which identified most known biofilm-related genes, also uncovered PA14_16550 and PA14_69700, deletions of which abrogated and augmented biofilm formation, respectively. We also identified ptsP, which encodes enzyme I of the nitrogen-regulated phosphotransferase (PTSNtr) system, as being important for cyclic-di-GMP production and for biofilm formation. Further experiments showed that biofilm formation is hindered in the absence of phosphotransfer through the PTSNtr, but only in the presence of enzyme II (PtsN), the putative regulatory module of the PTSNtr. These results implicate unphosphorylated PtsN as a negative regulator of biofilm formation and establish one of the first known roles of the PTSNtr in P. aeruginosa.
Keywords: colony morphology, biofilms, mutagenesis, PTSNtr, GAF domain, cyclic-di-GMP
Introduction
The Gram-negative bacterium Pseudomonas aeruginosa is an important human opportunistic pathogen (Driscoll et al., 2007), especially in nosocomial pneumonia (Richards et al., 2000). Its success as a pathogen is in part driven by its ability to form biofilms. In a biofilm, a community of bacterial cells is held together and protected from the surrounding environment by a self-produced extracellular matrix generally comprising polysaccharides, DNA, and proteins (Hall-Stoodley et al., 2004). Biofilms enhance the resistance of bacterial cells to environmental insults such as antibiotic treatment (Drenkard & Ausubel, 2002, Nguyen et al., 2011), making chronic infections difficult to treat.
The extracellular polysaccharide (EPS) component of the biofilm matrix, termed Pel in strain PA14, is crucial for the ability of P. aeruginosa to effectively form a biofilm (Friedman & Kolter, 2004a). The synthesis of Pel is regulated both transcriptionally and post-transcriptionally in response to various environmental signals by a complex regulatory network (Goodman et al., 2004, Hickman et al., 2005, Ventre et al., 2006). One signal is an imbalanced intracellular redox state; cells lacking a terminal electron acceptor upregulate Pel production, as the resultant colony wrinkling allows cells better access to atmospheric oxygen (Dietrich et al., 2013). As in many bacterial species (Boyd & O’Toole, 2012), the bacterial second messenger cyclic diguanylate (c-di-GMP) also has a central role in biofilm signaling in P. aeruginosa (Cotter & Stibitz, 2007). For example, the PelD protein directly binds c-di-GMP, and mutations that abolish c-di-GMP binding also prevent Pel production (Lee et al., 2007b). Meanwhile, mutations that increase the intracellular c-di-GMP concentration, for instance by increasing the production or activity of diguanylate cyclases or decreasing the activity of phosphodiesterases, generally promote biofilm formation while suppressing non-biofilm behaviors such as cell motility (Kulasakara et al., 2006). However, the existence of additional layers of biofilm regulation is evidenced by the absence of a strict correlation between intracellular c-di-GMP concentration and the degree of biofilm formation (Merritt et al., 2010).
Many of the key regulators of P. aeruginosa biofilm formation, including the Pel proteins, multiple diguanylate cyclases and phosphodiesterases, upstream regulators, and cellular adhesion proteins, were discovered using a now-classic crystal violet-based screen for submerged biofilm formation in liquid medium (O’Toole & Kolter, 1998a, O’Toole & Kolter, 1998b). An alternative assay for biofilm formation relies on the visual inspection of bacterial colony morphology on solid biofilm-inducing media. Such an approach has proven useful for uncovering biofilm-related genes in bacterial species that do not form submerged biofilms in liquid culture, such as Bacillus subtilis (Branda et al., 2006, Chu et al., 2006, Kearns et al., 2005, McLoonn et al., 2011). In this approach, the appearance and degree of colony wrinkling is used as a proxy for biofilm formation, as the final morphology of a colony biofilm is an endpoint that reflects matrix production. Moreover, Congo red binding allows for quantification of the exopolysaccharide content of a homogenized colony. Different degrees of wrinkling by P. aeruginosa colonies have long been reported, and in fact the “rugose small-colony variant” phenotype that commonly appears in chronic human infections is associated with hyper-wrinkled colony morphology and elevated intracellular c-di-GMP concentration (Starkey et al., 2009). Colony wrinkling and Congo red binding have been also been effectively used to monitor biofilm formation by P. aeruginosa (Friedman & Kolter, 2004b). However, colony wrinkling has not been used in a systematic screen to identify genes that affect P. aeruginosa biofilm formation.
Here, we report a colony morphology-based screen in P. aeruginosa for mutants altered in biofilm formation. The screen robustly identified known and unknown genes with different roles in biofilm formation, highlighting its effectiveness as well as its value as a tool for further discovery. We also describe three genes, identified using this screen, with distinct roles in biofilm formation that had not previously been appreciated.
Results and Discussion
A colony morphology-based screen for genes with roles in biofilm formation
As a means for discovering previously unrecognized genes that act within the biofilm regulatory network of P. aeruginosa, we employed a visual screen of colony morphology on agar plates (Fig 1A). We thus sought to find mutants with diminished or enhanced colony wrinkling. To facilitate the discovery of such mutant morphologies, we constructed a amrZ deletion mutant of strain PA14 for use as the parental strain in the screen, as it overproduces the Pel matrix exopolysaccharide and exhibits wrinkled colony morphology (Jones et al., 2014) that we found easy to distinguish from flat or hyper-wrinkled morphologies. AmrZ, a transcriptional repressor, controls switching between an alginate-producing mucoid state and a Pel-producing biofilm state, such that the amrZ mutant is locked in the biofilm-producing state. An additional advantage of using an amrZ deletion strain was that it exhibits an approximately twofold elevated c-di-GMP level relative to the wild type (Jones et al., 2014). Elevated c-di-GMP is a phenotype that is associated both with stronger biofilm formation and with chronic infection (Starkey et al., 2009) and so our screen would also be expected to uncover genes that are required for such high c-di-GMP levels or augmented biofilm formation. In the case of the amrZ mutant, derepression of a gene that encodes a diguanylate cyclase enzyme, termed adcA, is thought to be largely responsible for the resulting rise in c-di-GMP levels and hence for enhanced biofilm formation (Jones et al., 2014).
Figure 1. A visual colony-morphology screen for biofilm-related genes in P. aeruginosa.
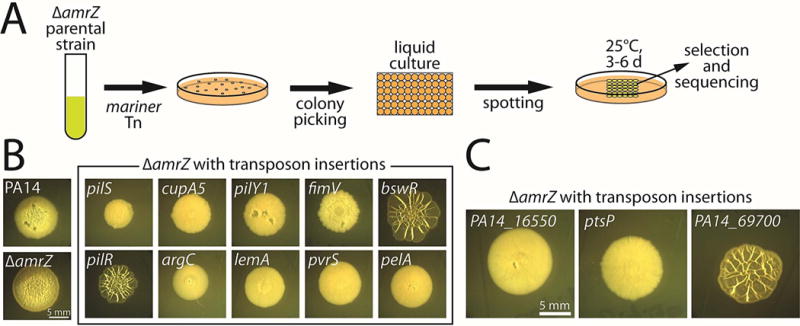
A. Schematic of the colony-morphology screen. The amrZ-deleted parental strain was mutagenized with a Mariner-based transposon, and individual mutant colonies were then grown overnight in static liquid culture before being spotted in an array on M6301-1% agar plates and grown at 25°C for 4–6 days. Visual inspection of the colonies was used to select promising candidates for subsequent sequencing to identify the position of transposon insertion. B. Colony morphologies (4 d) of mutants identified in the screen with known roles in biofilm formation. C. Colony morphologies (4 d) of mutants identified in the screen with no previously described role in biofilm formation.
Both previously characterized and unrecognized genes are identified by visual colony screening
We subjected the parental PA14 amrZ mutant strain to mutagenesis with a Mariner-based transposon (Kulasakara et al., 2006) and arrayed the resulting mutants on agar plates for visual inspection (Fig. 1A). We then verified the enhanced or diminished colony wrinkling phenotypes of the selected candidates and performed sequencing to identify the transposon insertion sites. We screened approximately 5,000 mutants and identified 119 candidates (Supplementary Table S1), representing a hit rate of approximately 2.4%. We found that the screen yielded genes spanning many of the known biofilm-related pathways (Fig. 1B; Supplementary Table S1). The number of independent hits per gene varied widely, ranging from genes that were only recovered once or twice to more-frequently recovered genes such as pelA (5 insertions) and pilY1 (12 insertions, the most for any identified gene). Among many others, we identified bifA, which encodes a c-di-GMP-degrading phosphodiesterase (Kuchma et al., 2007); pilR, pilS, pilY1, fimV, and cupA5, which encode surface-attachment proteins or their regulators (Alm et al., 1996, Hobbs et al., 1993, Ishimoto & Lory, 1992, Semmler et al., 2000, Vallet et al., 2001, Wehbi et al., 2011); hpd, phzA, and pqsL, which encode or regulate quorum-sensing proteins and virulence factors (Ahuja et al., 2008, Lepine et al., 2004, Yoon et al., 2007); bswR, which is involved in small RNA regulation (Wang et al., 2014); argC and argG, which are involved in arginine metabolism (Haas et al., 1977), and pelA, pelC, and gacS, which are involved in regulating or producing the exopolysaccharide matrix (Friedman & Kolter, 2004a, Parkins et al., 2001). Some genes with known roles in biofilm formation, such as the diguanylate cyclase-encoding genes sadC (Merritt et al., 2007) and roeA (Merritt et al., 2010) and the regulatory protein-encoding gene wspF (Hickman et al., 2005), were not detected by the screen. While genes whose main roles are in surface attachment might be missed by a colony morphology-based screen, we primarily attribute such omissions to the screen not yet being close to saturation. Indeed, in a pilot screen using a similar parental background, we uncovered roeA. In sum, these findings demonstrate the ability of a colony morphology-based P. aeruginosa screen to uncover biofilm-related genes in an accurate, efficient, and unbiased manner.
In addition to the known genes uncovered by our screen, we also identified genes with no previously known roles in biofilm formation (Fig. 1C). These candidate biofilm genes included PA14_16550 (orthologous to PA3699; hereafter 16550), which encodes a TetR-type DNA repressor; PA14_69700 (orthologous to PA5279; hereafter 69700), which encodes an uncharacterized GAF-domain protein; and ptsP, which encodes enzyme I of the nitrogen-regulated phosphotransferase (PTSNtr) system. Insertions in these genes were recovered once, twice, and twice, respectively. Transposon disruption of 16550 and ptsP strongly diminished colony wrinkling, whereas the insertion in 69700 enhanced wrinkled biofilm morphology (Fig. 1C).
Deletion mutations of candidate genes perturb biofilm formation
To confirm that the altered colony morphologies of the mutants with insertions in 16550, 69700, and ptsP were a consequence of the inactivation of the respective genes, we constructed markerless, in-frame deletions of each gene in the parental amrZ mutant strain. Qualitative visual inspection of colony morphology confirmed that deletions of 16550 and of ptsP led to biofilm attenuation and that deletion of 69700 resulted in biofilm augmentation (Fig. 2A). The respective effects of the 16550 and ptsP deletions were each reversed by complementation of the deleted gene at the attB locus. The 69700 gene is located in the middle of an operon, and efforts to complement the deletion mutation have so far been unsuccessful. However, the original insertion mutant and the deletion mutant exhibit indistinguishable phenotypes, increasing our confidence that 69700 is indeed the relevant locus.
Figure 2. Three genes with previously unknown roles in biofilm formation.
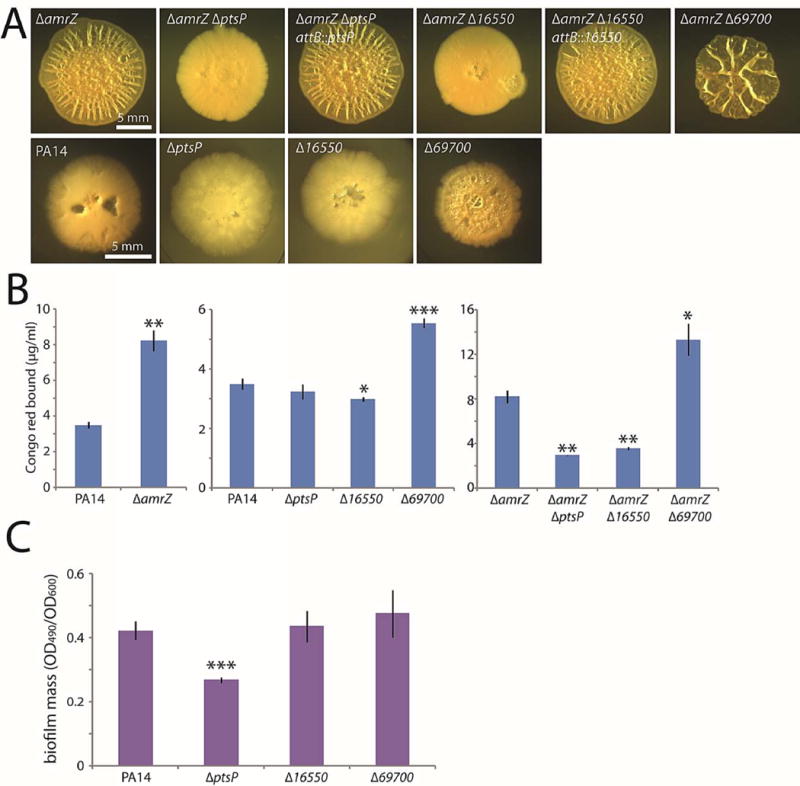
A. Colony morphologies (4–5 d) of strains bearing markerless in-frame deletions of the three new genes identified in the screen. In the amrZ deletion background, deletions were complemented at the ectopic attB locus. B. Congo red binding by the indicated mutant colonies. Mean values of at least 3 replicates are shown; error bars indicate ± 1 standard deviation. *, p < 0.05; **, p < 0.01; ***, p < 0.001 relative to the leftmost strain in each graph. C. Crystal violet binding by the indicated mutants after 24 h of static culture. Mean values of at least 4 replicates are shown; error bars indicate ± 1 standard deviation. ***, p < 0.001 relative to PA14.
The qualitative colony morphologies were supported by quantitative biofilm measurements using Congo red binding (Fig. 2B). As expected, amrZ mutant colonies bound significantly more Congo red than the wild type (Fig. 2B, left graph). Our visual observations in the amrZ mutant background were confirmed by the Congo red data, as significant variations from the parental strain were apparent in the deletion mutants (Fig. 2B, right graph).
Biofilm formation is impaired by mutations of 16550 and ptsP and enhanced by 69700 mutation in otherwise wild-type cells
A key question was whether the deletion mutations of the genes we identified in our screen would exert a biofilm phenotype independently of the amrZ mutation. The deletion in 69700, which increased biofilm formation in an amrZ mutant, did likewise in wild-type cells, visibly enhancing colony wrinkling (Fig. 2A) and increasing Congo red binding (Fig. 2B, center graph). Because wild-type PA14 colonies are relatively flat, it was difficult to visually distinguish the 16550 and ptsP mutations from the wild type by visual inspection (Fig. 2A). However, the 16550 mutant colonies bound slightly but detectably less Congo red than did the wild type (Fig. 2B, center graph). The ptsP mutant exhibited no significant difference from the wild type in Congo red binding (Fig. 2B, center graph), but it did show a clear defect in biofilm mass as judged by crystal violet staining (Fig. 2C).
Mutations in 16550 and ptsP lower c-di-GMP levels
Because c-di-GMP is an important second messenger in biofilm formation, we next examined the effects of our candidate genes on cellular levels of the dinucleotide. We first asked whether a deletion in 16550, which reduced colony wrinkling, correspondingly lowered c-di-GMP levels in the cells of a biofilm colony. For comparison, and as expected, the amrZ mutation elevated c-di-GMP levels roughly twofold (Fig. 3A). Interestingly, the 16550 deletion on its own caused a modest but significant increase in c-di-GMP (Fig. 3B). However, the 16550 mutation in combination with the amrZ mutation caused a significant decrease in c-di-GMP, lowering it to approximately wild-type levels (Fig. 3B). One possible interpretation of this result is that the 16550 protein, which encodes a TetR-type repressor, has multiple targets within the biofilm signaling network. For example, 16550 might be important for the transcription both of a pel gene and of a diguanylate cyclase-encoding gene that is upregulated in an amrZ mutant. In this case, deletion of 16550 would invariably lower matrix production by virtue of its pel regulation. However, it would only lower cyclic-di-GMP levels in the amrZ mutant.
Figure 3. Deletions of 16550 and ptsP reduce cellular c-di-GMP levels.
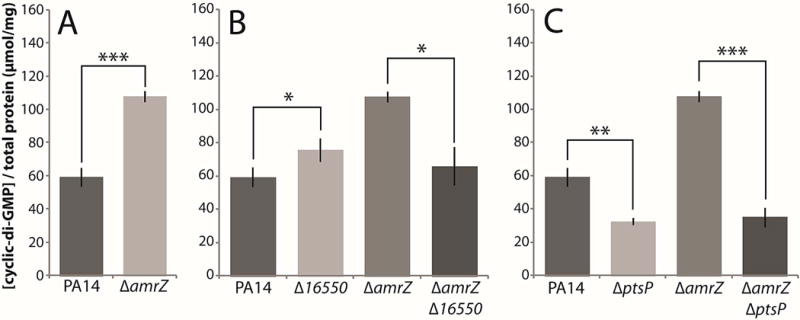
Cellular c-di-GMP was extracted from homogenized colonies and measured by HPLC. A. c-di-GMP levels in the parental PA14 and amrZ deletion strains. B. c-di-GMP levels in 16550 mutants. C. c-di-GMP levels in ptsP mutants. Mean values of at least 3 replicates are shown; error bars indicate ± 1 standard deviation. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Next, we examined the effect of the ptsP deletion. It caused a pronounced decrease in c-di-GMP levels both in a wild-type background and in a double mutant with the amrZ mutation. (Fig. 3C). Hence, the ptsP gene product appeared to have an important role in cellular c-di-GMP production under both wild-type and elevated-biofilm conditions.
The effects of 16550 and ptsP mutations are not mediated by AdcA
Because the amrZ mutation is known to cause derepression of adcA, which encodes a diguanylate cyclase (Jones et al., 2014), we hypothesized that the 16550 and ptsP deletions might be inhibiting adcA expression. As a first test, we constructed an adcA deletion to confirm its role in elevated biofilm formation. Surprisingly, when we examined the colony phenotype of an amrZ adcA double mutant strain, we observed no discernible change in colony morphology (Fig. S1A), suggesting that, in strain PA14 under our experimental conditions, AdcA plays only a minor role in the elevated-biofilm phenotype of the amrZ mutant. When we then analyzed the expression of adcA, it was indeed elevated in an amrZ mutant, but this derepression was not reversed by the 16550 mutation, and the ptsP mutation even caused a further elevation in expression (Fig. S1B). Collectively, these results appear to eliminate the hypothesis that the 16550 and ptsP mutations exert their effects by lowering the levels of the AdcA diguanylate cyclase. They also indicate that, in PA14, the inactivation of AmrZ provokes c-di-GMP synthesis by other pathway(s) in addition to adcA derepression.
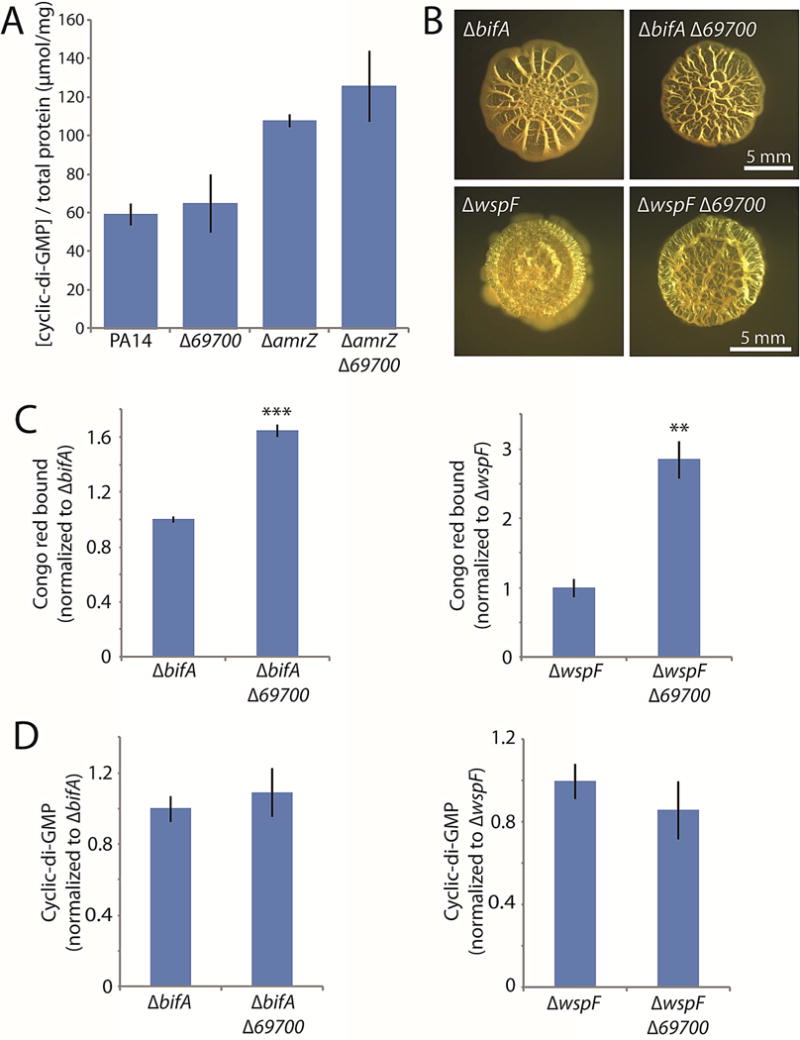
Deletions of 16550 and ptsP inhibit hyper-biofilm formation by bifA and wspF mutants
We next asked whether 16550 and ptsP mutations would impair the hyper-biofilm phenotypes of other mutants (in addition to amrZ) that exhibit elevated c-di-GMP levels. We therefore introduced 16550 and ptsP mutations into mutants lacking BifA, a phosphodiesterase that degrades cellular c-di-GMP (Kuchma et al., 2007), and WspF, a regulatory protein that inhibits the synthesis of the diguanylate cyclase WspR (Hickman et al., 2005). Both the 16550 and ptsP mutations substantially decreased colony wrinkling in the bifA and wspF mutants, with the 16550 mutant having a slightly stronger effect (Fig. 4A). Accordingly, Congo red binding assays showed substantial decreases from the bifA (Fig. 4B) and wspF (Fig. 4C) mutant strains. In addition, mutations of our newly identified biofilm genes significantly decreased cellular c-di-GMP concentrations in the bifA mutant background (Fig. 4D). We conclude that 16550 and ptsP mutations exert their negative effects on the hyper-biofilm phenotype of the bifA mutant by reversing the elevated levels of c-di-GMP. Interestingly, in the case of the wspF mutant, only the ptsP mutation significantly lowered the elevated levels of c-di-GMP (Fig. 4E). This result suggests that the 16550 mutation may have a distinct effect in the wspF mutant background that is not related to c-di-GMP production or clearance.
Figure 4. Deletions of 16550 and ptsP reduce biofilm formation by bifA and wspF mutants.
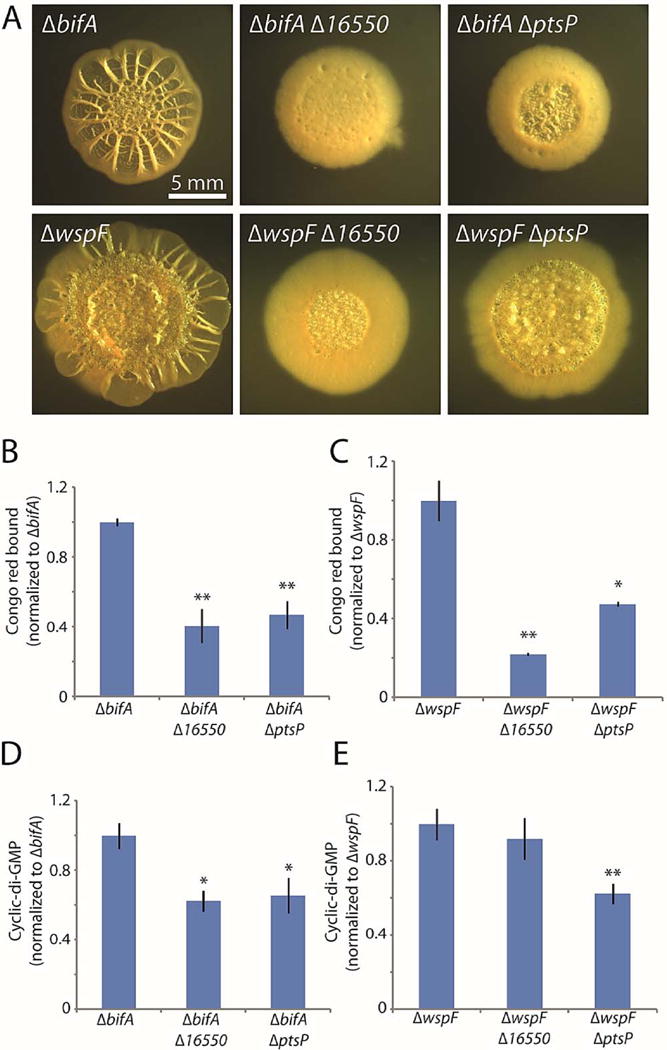
A. Colony morphologies (4 d for bifA, 6 d for wspF) of strains bearing markerless bifA or wspF deletions alone or in combination with markerless in-frame deletions of the three new genes identified in the screen. B and C. Congo red binding by the indicated colonies. D and E. c-di-GMP levels in the indicated colonies. For panels B–E, the mean values of at least 3 replicates are shown; error bars indicate ± 1 standard deviation. *, p < 0.05; **, p < 0.01 relative to the single mutants shown at the left of each graph.
The role of PtsP in biofilm formation is independent of its GAF domain
We next turned our attention to PtsP, which is best-characterized among our three novel biofilm regulators and also the one with the most robust phentypes. PtsP is Enzyme I of the PTSNtr phosphorelay (EINtr), which transfers phosphate groups from phosphoenolpyruvate (PEP), via Npr (encoded by ptsO), to Enzyme II (EIINtr, encoded by ptsN) (Fig. 5A) (Powell et al., 1995, Rabus et al., 1999, Zimmer et al., 2008). The PTSNtr system (reviewed in (Pfluger-Grau & Gorke, 2010)) is paralogous to the canonical sugar PTS pathway that phosphorylates imported sugars to ensure their retention within the cell. However, unlike the EIIs of the canonical pathway, which transfer phosphate groups to sugars, EIINpr has no known phosphorylation substrates; indeed, it lacks a second outward-facing His residue that is present in the sugar EII (van Montfort & Dijkstra, 1998). Instead, the PTSNtr is thought to exert its effects through protein-protein interactions. For example, in Escherichia coli, EIINtr interacts with TrkA and KdpD to regulate intracellular K+ concentration (Lee et al., 2007a, Lüttmann et al., 2009). Additionally, unphosphorylated Npr (PtsO) interacts with LpxD in E. coli to decrease lipid A synthesis (Kim et al., 2011). The phosphorylation state of the E. coli PTSNtr system appears to depend on the cellular glutamine/α-ketoglutarate ratio; a high ratio indicates nitrogen sufficiency and inhibits phosphotransfer, whereas nitrogen starvation stimulates the PTSNtr (Lee et al., 2013). The responsiveness of the PTSNtr to glutamine and α-ketoglutarate appears to require the GAF domain of PtsP, as its deletion in E. coli preserved phosphotransfer but abolished small-molecule regulation (Lee et al., 2013).
Figure 5. The GAF domain of PtsP is dispensable for its role in biofilm formation.
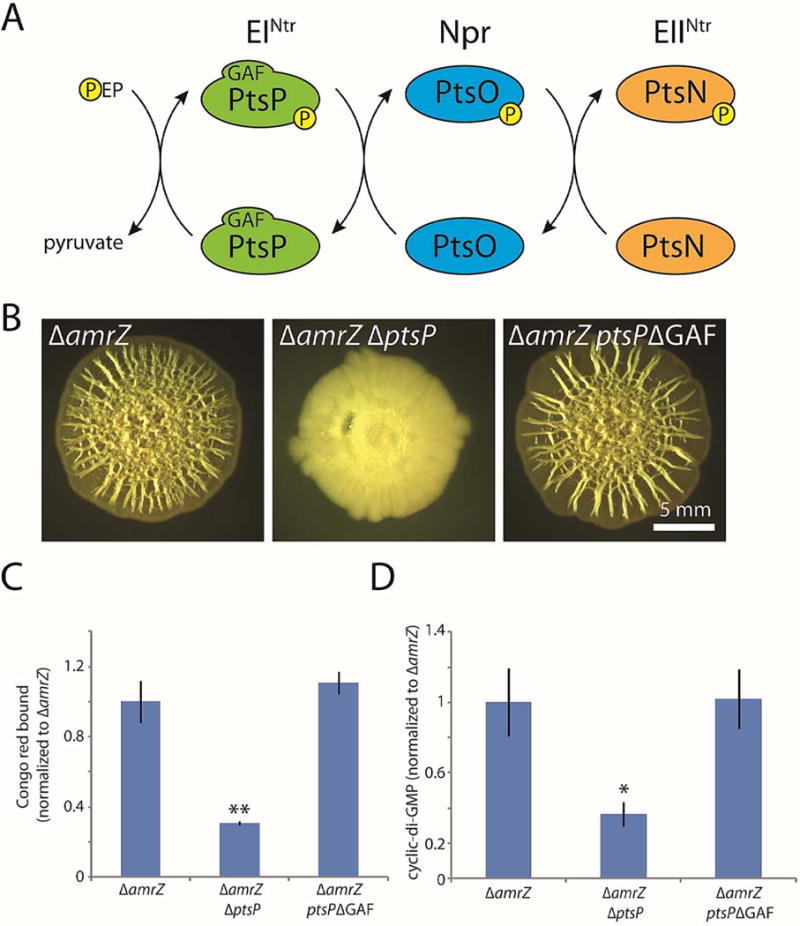
A. Schematic of the PTSNtr phosphorylation cascade. Phosphate is transferred by EINtr (encoded by ptsP) from phosphoenolpyruvate (PEP) to Npr (encoded by ptsO). Npr in turn transfers the phosphate to EIINtr (encoded by ptsN), which is thought to directly exert regulatory activity. B. Colony morphologies (6 d) of the indicated strains, including a strain in which the native copy of ptsP has been markerlessly replaced with a copy deleted for nucleotides 49–492, which encode the 148-residue GAF domain of EINtr. C. Congo red binding by the indicated colonies. Mean values of at least 3 replicates are shown; error bars indicate ± 1 standard deviation. **, p < 0.01 relative to amrZ. D. c-di-GMP levels in the indicated colonies. Mean values of at least 3 replicates are shown; error bars indicate ± 1 standard deviation. *, p < 0.05 relative to amrZ.
We therefore asked whether the GAF domain of PtsP was required for its role in biofilm formation, reasoning that it might be involved in sensing in c-di-GMP. GAF domains are well-known for allosterically binding cyclic mononucleotides to regulate enzyme activity. Thus, it was possible that PtsP might be activated by c-di-GMP under biofilm-forming conditions to further stimulate c-di-GMP synthesis and biofilm formation. To test this possibility, we replaced the native chromosomal copy of ptsP with a version containing a markerless, in-frame deletion of the GAF domain-encoding sequence. We used the amrZ mutant background to facilitate the detection of any phenotypic differences. In contrast to the pronounced defect in biofilm formation caused by removal of the entire gene, the wrinkled colony morphology of the GAF domain mutant was neither altered (Fig. 5B) nor quantitatively different (Fig. 5C) from the amrZ mutant parental strain. Moreover, cellular c-di-GMP was unchanged by the GAF deletion (Fig. 5D). This result indicates that the GAF domain is dispensable for the role of PtsP in biofilm formation.
PtsN is required for biofilm inhibition in the absence of upstream phosphotransfer
As PtsP is the most upstream enzyme in the PTSNtr phosphorelay (Fig. 5A), we examined (again using the amrZ mutant background to facilitate the detection of phenotypes) whether the effect of a ptsP deletion involved one or more of the remaining members of the PTSNtr system. We considered two possibilities. The first was that the role of PtsP in biofilm formation is independent of the PTSNtr system, whereas the second was that the phenotype of a ptsP deletion is caused by disrupted phosphate flow through the PTSNtr system. To distinguish these possibilities, we deleted ptsO, which encodes the Npr enzyme downstream of PtsP (Fig. 5A). In the first case, ptsO deletion would be expected to have little or no effect on biofilm formation. However, in the second case, ptsO deletion would also block phosphate flow through the PTSNtr and thus phenocopy the ptsP deletion. In fact, the phenotype of a ptsO null mutant resembled that of the ptsP mutant, exhibiting severely inhibited colony wrinkling and significantly less Congo red binding than the amrZ deletion alone (Fig. 6A–B). This finding suggested that strong biofilm formation requires phosphate flow through the PTSNtr system. It also confirmed that, as in E. coli (Lee et al., 2013), the GAF domain deletion of PtsP did not interfere with its phosphotransfer function, as removal of the PtsP GAF domain (in contrast to removing PtsO) had no effect on colony morphology as we have seen (Fig. 5B).
Figure 6. Unphosphorylated EIINtr (PtsN) is responsible for the biofilm-suppressing effect of PTSNtr mutants.
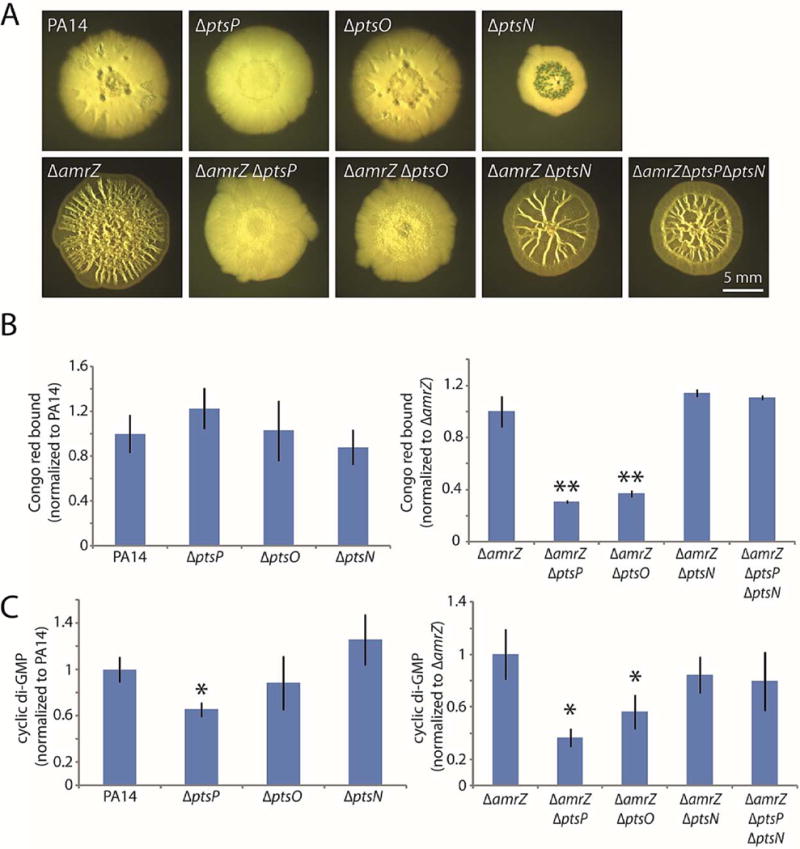
A. Colony morphologies (6 d) of the indicated strains with combinations of PTSNtr deletions. B. Congo red binding by the indicated colonies. Mean values of at least 3 replicates are shown; error bars indicate ± 1 standard deviation. **, p < 0.01 relative to amrZ. C. c-di-GMP levels in the indicated colonies. Mean values of at least 3 replicates are shown; error bars indicate ± 1 standard deviation. *, p < 0.05 relative to amrZ.
The phenotypic similarity of the ptsP and ptsO mutants prompted us to focus on PtsN, the EIINtr that is the final recipient of phosphates in the PTSNtr (Fig. 5A). Our results again raised two possibilities. First, phosphorylated PtsN might have a stimulatory effect on c-di-GMP levels and hence biofilm formation. In this case, deleting ptsN would phenocopy a ptsP or ptsO deletion, as it would eliminate PtsN function. Alternatively, unphosphorylated PtsN might have an inhibitory effect on biofilm formation. In this case, we would expect that deleting ptsN would have little or no effect on biofilm formation but that it would bypass the phenotype of a ptsP deletion by eliminating the effect of unphosphorylated PtsN. When we constructed an amrZ ptsN double mutant, we found that it exhibited wrinkled colony morphology and Congo red binding that did not significantly differ from those of the amrZ mutant strain (Fig. 6A–B), suggesting that PtsN is not required to stimulate biofilm development. Moreover, adding the ptsN deletion to an amrZ ptsP double mutant reversed the biofilm-inhibition phenotype of the ptsP mutation (Fig. 6A–B), arguing that the biofilm inhibition displayed by the ptsP and ptsO mutant strains is mediated by unphosphorylated PtsN. When we examined cellular c-di-GMP levels in these same strains, we observed that the ptsN deletion alone had no effect on c-di-GMP but that it rescued the lowered c-di-GMP levels of the ptsP deletion (Fig. 6C). Therefore, the PTSNtr system as a whole has an important role in biofilm formation. Phosphate flow through the PTSNtr ensures that PtsN remains phosphorylated, thus permitting elevation of c-di-GMP levels and strong biofilm formation. However, if the phosphorelay is blocked, unphosphorylated PtsN acts as an inhibitor of biofilm formation through a currently unknown mechanism.
69700 deletion enhances biofilm formation but does not increase c-di-GMP levels
Our screen also uncovered 69700, deletion of which yielded an elevated-biofilm phenotype (Fig. 2). We thus suspected that it might increase c-di-GMP levels. However, the 69700 deletion only slightly increased c-di-GMP levels, not reaching statistical significance in either the wild-type or amrZ mutant backgrounds (Fig. 7A). When we constructed double deletions of 69700 with wspF or bifA, we also observed further enhancement in colony wrinkling in both backgrounds (Fig. 7B). Congo red binding confirmed that the 69700 mutation significantly increased biofilm formation in these backgrounds (Fig. 7C). However, when we analyzed the c-di-GMP levels of these double mutants, they again showed only a slight increase (Fig. 7D). Collectively, these results suggest that the substantial enhancement of colony morphology in a 69700 mutant is not primarily mediated through c-di-GMP signaling. Therefore, in contrast to 16550 and PtsP, the normal function of the 69700 protein appears to be as a negative, c-di-GMP-independent regulator of biofilm formation, as its deletion in all tested backgrounds enhanced biofilm formation, even under conditions in which biofilm signaling was already substantially upregulated.
Figure 7. Deletion of 69700 increases biofilm formation but not cellular c-di-GMP.
A. c-di-GMP levels in the indicated 69700 mutants. B. Colony morphologies (4 d) of strains bearing markerless bifA or wspF deletions alone or in combination with 69700 deletions. C. Congo red binding by the indicated colonies. D. c-di-GMP levels in the indicated colonies. For panels C and D, the mean values of at least 3 replicates are shown; error bars indicate ± 1 standard deviation. **, p < 0.01; ***, p < 0.001 relative to the single mutants shown at the left of each graph.
Perspectives and outlook
The present work demonstrates the value of colony morphology for the discovery of previously unrecognized genes involved in biofilm formation, as we identified three such genes. Of special interest was the ptsP gene, whose robust phenotype allowed us to show that the P. aeruginosa PTSNtr has a role in c-di-GMP production and biofilm formation—the first established function of the PTSNtr in this species. We also identified 16550 as being needed for efficient biofilm formation. The 16550 gene encodes a TetR-type repressor whose PAO1 homolog (PA3699) binds to the promoter region of the gene encoding the LasR quorum-sensing regulator (Longo et al., 2013). However, whereas PA3699 repressed lasR transcription when overexpressed from a plasmid, its inactivation had no detectable effect on lasR (Longo et al., 2013), leading us to conclude that the biofilm-related role of the 16550 protein is distinct from a role in lasR repression. The third newly identified gene, 69700 (PA5279 in PAO1), is notable because its product inhibits biofilm formation and because it contains a crystallographically validated GAF domain of unknown function (PDB 3E98). GAF domains are generally considered small ligand-binding domains, but the functions of most GAF domains are not well-understood (Heikaus et al., 2009). Our initial results indicate that 69700 does not act via c-di-GMP signaling, implying that it impedes biofilm formation via some other yet-to-be elucidated pathway. Not only have the discovery of ptsP, 16550, and 69700 raised important unanswered questions about the circuitry governing biofilm formation, but our screen has not yet reached saturation and we anticipate that additional genes involved in biofilm formation remain to be discovered.
Experimental Procedures
Strains and growth conditions
Escherichia coli SM10 and Pseudomonas aeruginosa PA14 were grown in Luria-Bertani (LB) broth (10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) or on LB agar plates fortified with 1.5% Bacto agar at 37°C. When appropriate, 25 μg/ml irgasan (to specifically select for P. aeruginosa) plus 75 μg/ml tetracycline, 25 μg/ml irgasan plus 75 μg/ml gentamicin, 25 μg/ml tetracycline, or 20 μg/ml gentamicin was added to liquid or solid medium. The strains used in this work are listed in Table 1 and Supplementary Table S2. Markerless deletions were generated using the pEXG2 vector with counterselection on LB plates containing 6% sucrose and were screened by colony PCR for the presence of deletions. Complementation and reporter strains were constructed by integration of the mini-CTX-1 vector at the neutral chromosomal attB locus. Plasmids, primer sequences, and modes of strain construction are listed in the Supplementary Information.
Table 1.
Pseudomonas aeruginosa strains used in this study.
| Strain | Genotype or description | Source or reference |
|---|---|---|
| PA14 (MTC1) | Laboratory wild-type strain of P. aeruginosa | Laboratory stock; Stephen Lory, Harvard Medical School |
| MTC590 | PA14 ΔamrZ | This study |
| MTC1240 | PA14 ΔamrZ Tn69700 isolate 1, GentR | This study |
| MTC1241 | PA14 ΔamrZ Tn16550, GentR | This study |
| MTC1281 | PA14 ΔamrZ TnptsP, GentR | This study |
| MTC1284 | PA14 ΔamrZ Tn69700 isolate 2, GentR | This study |
| MTC1381 | PA14 ΔamrZ Δ16550 | This study |
| MTC1387 | PA14 ΔamrZ ΔptsP | This study |
| MTC1398 | PA14 ΔamrZ Δ69700 | This study |
| MTC1448 | PA14 Δ16550 | This study |
| MTC1450 | PA14 ΔptsP | This study |
| MTC1512 | PA14 Δ69700 | This study |
| MTC1521 | PA14 ΔbifA | This study |
| MTC1522 | PA14 ΔbifA Δ16550 | This study |
| MTC1523 | PA14 ΔbifA Δ69700 | This study |
| MTC1525 | PA14 ΔbifA ΔptsP | This study |
| MTC1533 | PA14 attB∷CTX-1-PadcA -lux, TetR | This study |
| MTC1535 | PA14 ΔamrZ attB∷CTX-1-PadcA -lux | This study |
| MTC1537 | PA14 ΔamrZ Δ16550 attB∷CTX-1-PadcA -lux, TetR | This study |
| MTC1539 | PA14 ΔamrZ Δ69700 attB∷CTX-1-PadcA -lux, TetR | This study |
| MTC1541 | PA14 ΔamrZ ΔptsP attB∷CTX-1-PadcA -lux, TetR | This study |
| MTC1543 | PA14 ΔamrZ ΔptsP CTX-1-ptsP, TetR | This study |
| MTC1549 | PA14 ΔamrZ Δ16550 CTX-1-16550, TetR | This study |
| MTC1562 | PA14 ΔamrZ ΔadcA | This study |
| MTC1603 | PA14 ΔamrZ ΔptsO | This study |
| MTC1605 | PA14 ΔamrZ ptsPΔGAF | This study |
| MTC1649 | PA14 ΔptsN | This study |
| MTC1651 | PA14 ΔamrZ ΔptsN | This study |
| MTC1666 | PA14 ΔamrZ ΔptsN ΔptsP | This study |
| MTC1709 | PA14 ΔwspF | This study |
| MTC1711 | PA14 ΔwspF ΔptsP | This study |
| MTC1713 | PA14 ΔptsO | This study |
| MTC1714 | PA14 ΔwspF Δ16550 | This study |
| MTC1717 | PA14 ΔwspF Δ69700 | This study |
P. aeruginosa biofilm studies were conducted using liquid or solid (1% agar) M6301 or M6301R medium. M6301 medium is composed of 100 μM KH2PO4, 15.14 mM (NH4)2SO4, and 0.36 μM FeSO4·H2O (pH-balanced to 7.0 using 10 M KOH) (Cabeen, 2014); after autoclaving and before use, 0.5% glycerol, 1 mM MgSO4, and 0.2% casamino acids (BD Bacto, USA) were added.
Transposon mutagenesis and biofilm colony morphology screen
P. aeruginosa PA14 ΔamrZ (MTC590) and E. coli SM10 pBT24 (MTC33) were grown as separate liquid cultures in 3 ml of LB for 6–8 h at 37°C in a tube roller. An aliquot of MTC33 (50 μl) was spot-dried on LB agar plates, and 100 μl of MTC590 was spot-dried atop the dried MTC33. The plates were incubated overnight at 37°C. The following day, the cells were harvested and resuspended in 500 μl sterile phosphate-buffered saline (PBS; 20 mM potassium phosphate, 150 mM NaCl) and plated for single colonies on LB agar containing irgasan and gentamicin. Individual single colonies were picked and used to inoculate LB (150 μl/well) in 96-well plates (Costar, Fisher Scientific, USA). The 96-well plates were incubated overnight at 37°C. Each plate contained 2 cultures each of PA14 and MTC590 as reference samples.
Cells from the 96-well plate cultures were deposited with a 6-by-8 48-pin array onto M6301 agar plates and grown at 25°C. At days 4 and 6, colonies exhibiting a biofilm morphology that differed from that of MTC590 were imaged and streaked for single colonies on LB agar with gentamicin. These colonies were then grown in 3 ml LB culture with gentamicin, and aliquots were archived in frozen glycerol (25% final concentration) stocks. A 2-μl sample of each culture was then spotted on M6301 agar to verify the initially observed colony morphologies. Colonies that exhibited reproducible colony morphologies were subjected to sequencing with the primer BT20TnMSeq following two rounds of colony PCR-based amplification with GoTaq Green (Promega, USA), RubyTaq (Affymetrix, USA), or Quick-Load Taq (New England Biolabs, USA) polymerase. Primers Rnd1-ARB1, Rnd1-ARB2, Rnd1-ARB3, and Rnd1-TnM20 were used for the first round of amplification. Primers Rnd2-ARB and Rnd2-TnM20 (Kulasekara et al., 2005) were used for the second round of amplification.
Biofilm assays
Plates containing 40 ml of M6301 with 1% agar were poured fresh for each experiment and allowed to harden for 6–7 hours. P. aeruginosa cultures grown at 37°C for 6–8 h in 3 ml LB were back-diluted to an OD600 of 1.0, and 2 μl of the diluted cultures were spotted on M6301 agar plates. The plates were incubated right-side-up at 25°C and were typically photographed after 4, 5, or 6 days, as indicated.
For further analysis, colonies were harvested from the agar surface and thoroughly resuspended by vortexing in 1 ml sterile PBS. Aliquots (100 μl) were removed from the resuspension to measure the optical density at 600 nm (OD600) using a Genesys 20 spectrophotometer (Thermo Scientific, USA). The remaining volume was pelleted in a microcentrifuge at 13,000 g for 3 minutes at room temperature, and the supernatant was discarded. The pellets were resuspended in 1 ml of 40 μg/ml Congo red and were incubated with vigorous shaking on a vortex platform for at least 90 minutes at room temperature. The samples were again pelleted at 13,000 g for 3 minutes at room temperature, and the OD of the supernatants was measured at 490 nm. A standard curve was constructed by measuring the OD490 of Congo red at 40, 20, 10, 5, 2, 1, and 0.5 μg/ml. 1X PBS was used as a blank and as the diluent for the standard curve.
For P. aeruginosa biofilm assays conducted using a 96-well-plate format, stationary-phase LB liquid cultures grown for 6–8 h at 37°C were diluted 1:100 in M6301 or M6301R. The cultures were immediately plated in transparent Costar 96-well plates with lids (Fisher Scientific, USA) and were incubated at 25°C for 24 hours. The OD in each well was measured using a BioTek Synergy 2 plate reader (BioTek, USA) at 600 nm. The wells were then washed three times with dH2O and stained with 0.1% crystal violet. After a 20-minute incubation at room temperature, the crystal violet was removed and the wells were again washed three times with dH2O. After air-drying the plates overnight, the crystal violet was solubilized using 95% ethanol, and the OD600 was measured as described above. Biofilm biomass was calculated for each sample as the ratio of crystal violet staining to culture density.
Microscopy
Bacterial colonies were imaged with a Leica Wild M10 stereomicroscope with a Planapo 0.63X objective and a Diagnostic Instruments 18.2 Color Mosaic digital camera. Images were captured using Spot software v. 4.5 (Diagnostic Instruments).
Kinetic luciferase assay
Cultures of P. aeruginosa cells were grown in liquid LB shaking culture at 37°C to stationary phase and were then diluted to an OD600 of 1.0 in fresh LB. Aliquots (2 μl) of the resulting cultures were spotted on solid M6301, and plates were incubated at 25°C for 24, 72, or 120 hours. At the indicated time points, colonies were harvested in 1 ml sterile 1x PBS, homogenized for approximately 20 seconds, and plated in quadruplicate (200 μl each) in a 96-well polystyrene Costar plate (white with a clear bottom; Fisher Scientific, USA). Luciferase activity and OD were measured on a BioTek Synergy 2 plate reader (BioTek, USA). Luciferase luminescence was measured at a sensitivity setting of 200, and the culture OD was measured at 600 nm. In cases in which the culture OD exceeded the dynamic range of the reader, the analysis was repeated with two- or four-fold culture dilutions. Final luciferase activity values were calculated by normalizing luciferase luminescence to culture density.
Quantification of intracellular cyclic di-GMP
P. aeruginosa PA14 and derivative mutant strains were grown in 3 ml LB liquid shaking culture for 6–8 h at 37°C and spotted (2 μl each) at an OD600 of 1.0 on 40 ml M6301 agar as described above. After 4 d of growth at 25°C, the colonies were harvested for c-di-GMP extraction. We chose a 4-d time point after testing multiple incubation times, as it showed a clear distinction between our PA14 and ΔamrZ reference samples. Using a sterile spatula, the colonies were deposited into 5-ml Eppendorf™ tubes containing 1 ml sterile PBS and were homogenized for at least 20 seconds with an Argos pestle (Cole Parmer, USA). Cyclic di-GMP was extracted from triplicate biological samples and subjected to HPLC as previously described (Roy et al., 2013). The quantity of c-di-GMP in each sample (calculated from a standard curve prepared from commercially available pure c-di-GMP) was normalized to the total amount of protein from the same sample (calculated using the Bradford method with bovine serum albumin standards).
Supplementary Material
Acknowledgments
We are grateful to Sunia Trauger for technical help with the HPLC experiments. We also thank members of the Losick lab for helpful discussions. M.T.C. was a Merck Postdoctoral Fellow of the Jane Coffin Childs Memorial Fund for Medical Research. S.A.L. was supported in part by the Natural Health Research Institute and the Tracie Lawlor Trust for Cystic Fibrosis. Funding was provided by the NIH (GM18568, to R.L.).
References
- Ahuja EG, Janning P, Mentel M, Graebsch A, Breinbauer R, Hiller W, Costisella B, Thomashow LS, Mavrodi DV, Blankenfeldt W. PhzA/B catalyzes the formation of the tricycle in phenazine biosynthesis. J Am Chem Soc. 2008;130:17053–17061. doi: 10.1021/ja806325k. [DOI] [PubMed] [Google Scholar]
- Alm RA, Hallinan JP, Watson AA, Mattick JS. Fimbrial biogenesis genes of Pseudomonas aeruginosa: pilW and pilX increase the similarity of type 4 fimbriae to the GSP protein-secretion systems and pilY1 encodes a gonococcal PilC homologue. Mol Microbiol. 1996;22:161–173. doi: 10.1111/j.1365-2958.1996.tb02665.x. [DOI] [PubMed] [Google Scholar]
- Boyd CD, O’Toole GA. Second messenger regulation of biofilm formation: breakthroughs in understanding c-di-GMP effector systems. Annu Rev Cell Dev Biol. 2012;28:439–462. doi: 10.1146/annurev-cellbio-101011-155705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda SS, Chu F, Kearns DB, Losick R, Kolter R. A major protein component of the Bacillus subtilis biofilm matrix. Mol Microbiol. 2006;59:1229–1238. doi: 10.1111/j.1365-2958.2005.05020.x. [DOI] [PubMed] [Google Scholar]
- Cabeen MT. Stationary phase-specific virulence factor overproduction by a lasR mutant of Pseudomonas aeruginosa. PLoS One. 2014;9:e88743. doi: 10.1371/journal.pone.0088743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu F, Kearns DB, Branda SS, Kolter R, Losick R. Targets of the master regulator of biofilm formation in Bacillus subtilis. Mol Microbiol. 2006;59:1216–1228. doi: 10.1111/j.1365-2958.2005.05019.x. [DOI] [PubMed] [Google Scholar]
- Cotter PA, Stibitz S. c-di-GMP-mediated regulation of virulence and biofilm formation. Curr Opin Microbiol. 2007;10:17–23. doi: 10.1016/j.mib.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Dietrich LE, Okegbe C, Price-Whelan A, Sakhtah H, Hunter RC, Newman DK. Bacterial community morphogenesis is intimately linked to the intracellular redox state. J Bacteriol. 2013;195:1371–1380. doi: 10.1128/JB.02273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenkard E, Ausubel FM. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature. 2002;416:740–743. doi: 10.1038/416740a. [DOI] [PubMed] [Google Scholar]
- Driscoll JA, Brody SL, Kollef MH. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. 2007;67:351–368. doi: 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- Friedman L, Kolter R. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol Microbiol. 2004a;51:675–690. doi: 10.1046/j.1365-2958.2003.03877.x. [DOI] [PubMed] [Google Scholar]
- Friedman L, Kolter R. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J Bacteriol. 2004b;186:4457–4465. doi: 10.1128/JB.186.14.4457-4465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, Kulasekara B, Rietsch A, Boyd D, Smith RS, Lory S. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell. 2004;7:745–754. doi: 10.1016/j.devcel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Haas D, Holloway BW, Schambock A, Leisinger T. The genetic organization of arginine biosynthesis in Pseudomonas aeruginosa. Mol Gen Genet. 1977;154:7–22. doi: 10.1007/BF00265571. [DOI] [PubMed] [Google Scholar]
- Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- Heikaus CC, Pandit J, Klevit RE. Cyclic nucleotide binding GAF domains from phosphodiesterases: structural and mechanistic insights. Structure. 2009;17:1551–1557. doi: 10.1016/j.str.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman JW, Tifrea DF, Harwood CS. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci U S A. 2005;102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs M, Collie ES, Free PD, Livingston SP, Mattick JS. PilS and PilR, a two-component transcriptional regulatory system controlling expression of type 4 fimbriae in Pseudomonas aeruginosa. Mol Microbiol. 1993;7:669–682. doi: 10.1111/j.1365-2958.1993.tb01158.x. [DOI] [PubMed] [Google Scholar]
- Ishimoto KS, Lory S. Identification of pilR, which encodes a transcriptional activator of the Pseudomonas aeruginosa pilin gene. J Bacteriol. 1992;174:3514–3521. doi: 10.1128/jb.174.11.3514-3521.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CJ, Newsom D, Kelly B, Irie Y, Jennings LK, Xu B, Limoli DH, Harrison JJ, Parsek MR, White P, Wozniak DJ. ChIP-Seq and RNA-Seq reveal an AmrZ-mediated mechanism for cyclic di-GMP synthesis and biofilm development by Pseudomonas aeruginosa. PLoS Pathog. 2014;10:e1003984. doi: 10.1371/journal.ppat.1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns DB, Chu F, Branda SS, Kolter R, Losick R. A master regulator for biofilm formation by Bacillus subtilis. Mol Microbiol. 2005;55:739–749. doi: 10.1111/j.1365-2958.2004.04440.x. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Lee CR, Kim M, Peterkofsky A, Seok YJ. Dephosphorylated NPr of the nitrogen PTS regulates lipid A biosynthesis by direct interaction with LpxD. Biochem Biophys Res Commun. 2011;409:556–561. [Google Scholar]
- Kuchma SL, Brothers KM, Merritt JH, Liberati NT, Ausubel FM, O’Toole GA. BifA, a cyclic-Di-GMP phosphodiesterase, inversely regulates biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J Bacteriol. 2007;189:8165–8178. doi: 10.1128/JB.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, Miyata S, Lee DG, Neely AN, Hyodo M, Hayakawa Y, Ausubel FM, Lory S. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′–5′)-cyclic-GMP in virulence. Proc Natl Acad Sci U S A. 2006;103:2839–2844. doi: 10.1073/pnas.0511090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulasekara HD, Ventre I, Kulasekara BR, Lazdunski A, Filloux A, Lory S. A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol Microbiol. 2005;55:368–380. doi: 10.1111/j.1365-2958.2004.04402.x. [DOI] [PubMed] [Google Scholar]
- Lee CR, Cho SH, Yoon MJ, Peterkofsky A, Seok YJ. Escherichia coli enzyme IIANtr regulates the K+ transporter TrkA. Proc Natl Acad Sci U S A. 2007a;104:4124–4129. doi: 10.1073/pnas.0609897104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CR, Park YH, Kim M, Kim YR, Park S, Peterkofsky A, Seok YJ. Reciprocal regulation of the autophosphorylation of enzyme INtr by glutamine and alpha-ketoglutarate in Escherichia coli. Mol Microbiol. 2013;88:473–485. doi: 10.1111/mmi.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VT, Matewish JM, Kessler JL, Hyodo M, Hayakawa Y, Lory S. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol Microbiol. 2007b;65:1474–1484. doi: 10.1111/j.1365-2958.2007.05879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepine F, Milot S, Deziel E, He J, Rahme LG. Electrospray/mass spectrometric identification and analysis of 4-hydroxy-2-alkylquinolines (HAQs) produced by Pseudomonas aeruginosa. J Am Soc Mass Spectrom. 2004;15:862–869. doi: 10.1016/j.jasms.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Longo F, Rampioni G, Bondi R, Imperi F, Fimia GM, Visca P, Zennaro E, Leoni L. A new transcriptional repressor of the Pseudomonas aeruginosa quorum sensing receptor gene lasR. PLoS One. 2013;8:e69554. doi: 10.1371/journal.pone.0069554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüttmann D, Heermann R, Zimmer B, Hillmann A, Rampp IS, Jung K, Görke B. Stimulation of the potassium sensor KdpD kinase activity by interaction with the phosphotransferase protein IIA(Ntr) in Escherichia coli. Mol Microbiol. 2009;72:978–994. doi: 10.1111/j.1365-2958.2009.06704.x. [DOI] [PubMed] [Google Scholar]
- McLoon AL, Guttenplan SB, Kearns DB, Kolter R, Losick R. Tracing the Domestication of a Biofilm-Forming Bacterium. J Bacteriol. 2011 doi: 10.1128/JB.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt JH, Brothers KM, Kuchma SL, O’Toole GA. SadC reciprocally influences biofilm formation and swarming motility via modulation of exopolysaccharide production and flagellar function. J Bacteriol. 2007;189:8154–8164. doi: 10.1128/JB.00585-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt JH, Ha DG, Cowles KN, Lu W, Morales DK, Rabinowitz J, Gitai Z, O’Toole GA. Specific control of Pseudomonas aeruginosa surface-associated behaviors by two c-di-GMP diguanylate cyclases. MBio. 2010;1 doi: 10.1128/mBio.00183-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, Britigan BE, Singh PK. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science. 2011;334:982–986. doi: 10.1126/science.1211037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998a;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- O’Toole GA, Kolter R. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol. 1998b;28:449–461. doi: 10.1046/j.1365-2958.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- Parkins MD, Ceri H, Storey DG. Pseudomonas aeruginosa GacA, a factor in multihost virulence, is also essential for biofilm formation. Mol Microbiol. 2001;40:1215–1226. doi: 10.1046/j.1365-2958.2001.02469.x. [DOI] [PubMed] [Google Scholar]
- Pfluger-Grau K, Gorke B. Regulatory roles of the bacterial nitrogen-related phosphotransferase system. Trends Microbiol. 2010;18:205–214. doi: 10.1016/j.tim.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Powell BS, Court DL, Inada T, Nakamura Y, Michotey V, Cui X, Reizer A, Saier MH, Jr, Reizer J. Novel proteins of the phosphotransferase system encoded within the rpoN operon of Escherichia coli. Enzyme IIANtr affects growth on organic nitrogen and the conditional lethality of an erats mutant. J Biol Chem. 1995;270:4822–4839. doi: 10.1074/jbc.270.9.4822. [DOI] [PubMed] [Google Scholar]
- Rabus R, Reizer J, Paulsen I, Saier MH., Jr Enzyme I(Ntr) from Escherichia coli. A novel enzyme of the phosphoenolpyruvate-dependent phosphotransferase system exhibiting strict specificity for its phosphoryl acceptor, NPr. J Biol Chem. 1999;274:26185–26191. doi: 10.1074/jbc.274.37.26185. [DOI] [PubMed] [Google Scholar]
- Richards MJ, Edwards JR, Culver DH, Gaynes RP. Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect Control Hosp Epidemiol. 2000;21:510–515. doi: 10.1086/501795. [DOI] [PubMed] [Google Scholar]
- Roy AB, Petrova OE, Sauer K. Extraction and Quantification of Cyclic Di-GMP from. Bio Protoc. 2013;3 doi: 10.21769/bioprotoc.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semmler AB, Whitchurch CB, Leech AJ, Mattick JS. Identification of a novel gene, fimV, involved in twitching motility in Pseudomonas aeruginosa. Microbiology. 2000;146(Pt 6):1321–1332. doi: 10.1099/00221287-146-6-1321. [DOI] [PubMed] [Google Scholar]
- Starkey M, Hickman JH, Ma L, Zhang N, Long SDe, Hinz A, Palacios S, Manoil C, Kirisits MJ, Starner TD, Wozniak DJ, Harwood CS, Parsek MR. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol. 2009;191:3492–3503. doi: 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet I, Olson JW, Lory S, Lazdunski A, Filloux A. The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Proc Natl Acad Sci U S A. 2001;98:6911–6916. doi: 10.1073/pnas.111551898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Montfort RL, Dijkstra BW. The functional importance of structural differences between the mannitol-specific IIAmannitol and the regulatory IIAnitrogen. Protein Sci. 1998;7:2210–2216. doi: 10.1002/pro.5560071019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventre I, Goodman AL, Vallet-Gely I, Vasseur P, Soscia C, Molin S, Bleves S, Lazdunski A, Lory S, Filloux A. Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. Proc Natl Acad Sci U S A. 2006;103:171–176. doi: 10.1073/pnas.0507407103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Ye F, Kumar V, Gao YG, Zhang LH. BswR controls bacterial motility and biofilm formation in Pseudomonas aeruginosa through modulation of the small RNA rsmZ. Nucleic Acids Res. 2014;42:4563–4576. doi: 10.1093/nar/gku106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehbi H, Portillo E, Harvey H, Shimkoff AE, Scheurwater EM, Howell PL, Burrows LL. The peptidoglycan-binding protein FimV promotes assembly of the Pseudomonas aeruginosa type IV pilus secretin. J Bacteriol. 2011;193:540–550. doi: 10.1128/JB.01048-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SS, Karabulut AC, Lipscomb JD, Hennigan RF, Lymar SV, Groce SL, Herr AB, Howell ML, Kiley PJ, Schurr MJ, Gaston B, Choi KH, Schweizer HP, Hassett DJ. Two-pronged survival strategy for the major cystic fibrosis pathogen, Pseudomonas aeruginosa, lacking the capacity to degrade nitric oxide during anaerobic respiration. EMBO J. 2007;26:3662–3672. doi: 10.1038/sj.emboj.7601787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer B, Hillmann A, Gorke B. Requirements for the phosphorylation of the Escherichia coli EIIANtr protein in vivo. FEMS Microbiol Lett. 2008;286:96–102. doi: 10.1111/j.1574-6968.2008.01262.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.