

Abstract
Cohesin is a large ring-shaped protein complex, conserved from yeast to human, which participates in most DNA transactions that take place in the nucleus. It mediates sister chromatid cohesion, which is essential for chromosome segregation and homologous recombination (HR)-mediated DNA repair. Together with architectural proteins and transcriptional regulators, such as CTCF and Mediator, respectively, it contributes to genome organization at different scales and thereby affects transcription, DNA replication, and locus rearrangement. Although cohesin is essential for cell viability, partial loss of function can affect these processes differently in distinct cell types. Mutations in genes encoding cohesin subunits and regulators of the complex have been identified in several cancers. Understanding the functional significance of these alterations may have relevant implications for patient classification, risk prediction, and choice of treatment. Moreover, identification of vulnerabilities in cancer cells harboring cohesin mutations may provide new therapeutic opportunities and guide the design of personalized treatments.
Cohesin has many roles related to genome organization and is often mutated in cancer. Changes in chromatin accessibility and transcription are the main effects of cohesin dysfunction, at least in some cell types.
Cohesin is one of the three structural maintenance of chromosomes (SMC) complexes that exist in eukaryotic cells. The other two are condensin and the Smc5/6 complex. They are all composed of an SMC heterodimer and additional non-SMC subunits arranged in a characteristic domain architecture (Haering and Gruber 2016). Remarkably, bacteria and archea also possess SMC complexes, although in this case the SMC proteins homodimerize. In all three kingdoms of life, the functions of SMC complexes are critical for genome organization, chromosome duplication, and segregation. In particular, cohesin was initially identified for its role in sister chromatid cohesion (Guacci et al. 1997; Michaelis et al. 1997; Losada et al. 1998), a requirement for proper chromosome segregation in mitosis and meiosis, as well as for homologous recombination (HR)-mediated DNA repair (Nasmyth and Haering 2009). In addition, cohesin is currently recognized as a major player in higher-order chromatin structure together with the CCCTC-binding factor (CTCF) (Phillips-Cremins et al. 2013; Mizuguchi et al. 2014). How the same complex can perform all of these different functions is far from understood. Germline mutations in cohesin and its regulators are at the origin of human developmental syndromes collectively known as cohesinopathies, the most prevalent of which is Cornelia de Lange syndrome (CdLS) (Horsfield et al. 2012). Recent sequencing efforts of cancer genomes have revealed the presence of somatic mutations in cohesin in several cancer types. Understanding how cohesin works and how it is regulated will likely help us recognize the contribution of these mutations to tumor initiation and progression.
THE BASIC BIOLOGY OF COHESIN
Composition and Architecture
Cohesin is a ring-shaped complex that consists of Smc1, Smc3, Rad21, and SA (see Table 1 for nomenclature). SMCs are 1000–1500 amino-acid-long proteins that contain two coiled-coil stretches separated by a flexible globular domain called “hinge.” When folded at this domain, the amino and carboxyl termini of the protein are brought in proximity to create an ATPase head domain (hd; Fig. 1). Smc1 and Smc3 interact stably through their hinges and on the other end are bridged by the Rad21 subunit (Haering et al. 2002, 2004; Gligoris et al. 2014; Huis in ‘t Veld et al. 2014). The central region of Rad21 binds the fourth subunit of cohesin, SA (Haering et al. 2002; Orgil et al. 2015). SA is composed of many homologous huntingtin, elongation factor 3, A subunit, and TOR (HEAT) repeats and likely serves as an interaction platform for cohesin-interacting proteins (Hara et al. 2014). Among these are two regulatory subunits associated with chromatin-bound cohesin complexes throughout the cell cycle, Pds5 and Wapl (Sumara et al. 2000; Losada et al. 2005; Gandhi et al. 2006; Kueng et al. 2006). There are also transient or position-specific interactors such as CTCF (Xiao et al. 2011) or the telomeric protein TRF1 (Canudas et al. 2007). In vertebrate somatic cells cohesin contains one of two SA subunits, SA1 or SA2 (Losada et al. 2000; Sumara et al. 2000; Remeseiro et al. 2012a). Additional meiosis-specific versions of all cohesin subunits exist except Smc3 (in red in Table 1).
Table 1.
Nomenclature of cohesin subunits and regulators
| Category | Yeast (Saccharomyces cerevisiae) | Mouse/human |
|---|---|---|
| Cohesin | Smc1 | Smc1α (SMC1A) Smc1β (SMC1B) |
| Smc3 | Smc3 (SMC3) | |
| Scc1 Rec8 | Rad21 (RAD21) Rad21L (RAD21L1) Rec8 (REC8) | |
| Scc3 | SA1 (STAG1) SA2 (STAG2) SA3 (STAG3) | |
| Associated factors | Pds5 | Pds5A (PDS5A) Pds5B (PDS5B) |
| Wapl/Rad61 | Wapl (WAPL) | |
| – | Sororin (CDCA5) | |
| Loader | Scc2 | Nipbl (NIPBL) |
| Scc4 | Mau2 (MAU2) | |
| CoAT | Eco1 | Esco1 (ESCO1) Esco2 (ESCO2) |
| CoDAC | Hos1 | Hdac8 (HDAC8) |
| Mitotic regulators | Sgo1 | Sgo1 (SGOL1) |
| Separase/Esp1 | Separase (ESPL1) | |
| Securin/Pds1 | Securin (PTTG1) |
Meiosis-specific variants are shown in red. For mouse/human, the name of the gene appears in parentheses. CoAT, cohesin acetyl transferase; CoDAC, cohesin deacetylase.
Figure 1.
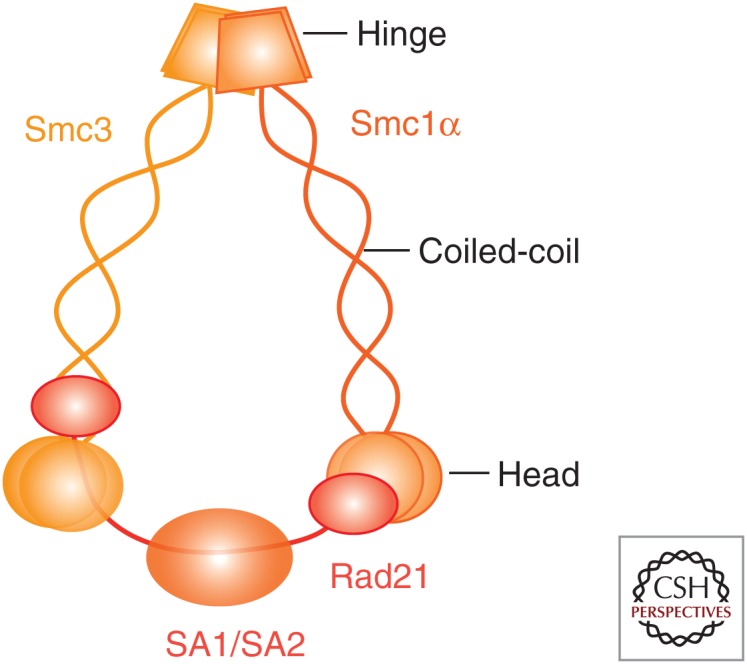
Cohesin composition and architecture. When the Smc1 and Smc3 proteins are folded at their flexible hinge domains, the NTP-binding motif and the DA box present at their amino- and carboxy-terminal globular domains come together to form a functional ATPase. Smc1 and Smc3 interact through their hinges, whereas the kleisin subunit Rad21 bridges their head domains and associates with SA. The outer diameter of the resulting ring-shaped complex is estimated at ∼50 nm and could hold two 10-nm chromatin fibers.
Cohesin Interaction with DNA
Evidence from a number of in vivo and in vitro studies supports a model in which cohesin entraps the chromatin fiber within its ring structure (Gruber et al. 2003; Haering et al. 2008). Cohesin loading occurs in G1 and requires ATP hydrolysis and a heterodimeric complex composed of Nipbl and Mau2 (Fig. 2) (Arumugam et al. 2003; Weitzer et al. 2003; Gillespie and Hirano 2004; Watrin et al. 2006). In vitro, loading can occur in the absence of the loader, albeit very inefficiently (Murayama and Uhlmann 2014). After loading, cohesin binding to chromatin is dynamic and unloading mediated by Wapl occurs throughout the cell cycle (Gerlich et al. 2006; Bernard et al. 2008). Some evidence supports the idea that DNA enters and exits the cohesin ring through different interfaces or “gates” (Nasmyth 2011; Buheitel and Stemmann 2013; Eichinger et al. 2013). The entry gate requires dissociation of the Smc1 and Smc3 hinges (Gruber et al. 2006), whereas the exit gate would be located in the interface formed by the coiled coil emerging from the Smc3 hd and two α helices in the amino terminus of Rad21 (Gligoris et al. 2014; Huis in ‘t Veld et al. 2014). The opening/closure of this second gate is regulated by the ATPase activity of the SMCs, by DNA sensing through two lysines present in the Smc3 hd (K105 and K106 in human Smc3), and by Pds5-Wapl (Murayama and Uhlmann 2015).
Figure 2.

Cohesin and its regulators throughout the cell cycle. Cohesin is loaded on chromatin by Nipbl-Mau2 throughout the cell cycle, starting in early G1. Pds5 and Wapl associate with chromatin-bound cohesin and promote its unloading. Cohesin complexes may be encircling a single chromatin fiber or two fibers at the base of a chromatin loop that brings distal regions in proximity. During S phase, acetylation of Smc3 by CoATs Esco1/2 and Sororin recruitment, both facilitated by Pds5A/B (not depicted), results in cohesion establishment. A fraction of cohesin remains dynamic even after DNA replication (not depicted). Whether cohesin at the base of chromatin loops is also involved in tethering sister chromatids is not known. In prophase, most cohesin dissociates from chromatin in a process that requires Wapl and phosphorylation of cohesin and Sororin. Sgo1 and its partner PP2A prevent the dissociation of a population of cohesin, enriched at centromeres. This population is removed at the onset of anaphase when Securin (not depicted) is destroyed and the active Separase cleaves Rad21.
Cohesin Distribution
Chromatin immunoprecipitation (ChIP) studies provide a genome-wide view of cohesin distribution. In yeast, cohesin accumulates in a 50-kb region around centromeres and at sites of convergent transcription (Glynn et al. 2004; Lengronne et al. 2004). Because there is little colocalization of cohesin and its loader, it has been proposed that once topologically entrapping chromatin, the cohesin ring can slide away from the loading site (Hu et al. 2011; Ocampo-Hafalla and Uhlmann 2011). In contrast, cohesin and its loader do colocalize at sites of active transcription in Drosophila (Misulovin et al. 2008). Comparison of cohesin distribution in two different mouse tissues also reveals a correlation between active transcription and the presence of cohesin (Cuadrado et al. 2015). Importantly, cohesin accumulates at CTCF binding sites along the human and mouse genomes, but only when CTCF is present (Parelho et al. 2008; Rubio et al. 2008; Wendt et al. 2008; Remeseiro et al. 2012b). Whether cohesin is loaded at these CTCF sites or is loaded elsewhere and then slides to reach them is not known. The number of Nipbl positions identified by ChIP is 5–10 times lower that the number of cohesin or CTCF sites, and cohesin and its loader colocalize only at a subset of sites near active genes (Kagey et al. 2010) but not at CTCF sites (Zuin et al. 2014b). Considering the data from different model organisms, it is possible that cohesin is loaded mainly at sites of active transcription and then moves along the genome until it finds an obstacle, such as CTCF or another chromatin-binding protein with which it interacts. This ability of cohesin to slide along DNA may be consistent with “loop-extrusion” models recently proposed to explain how cohesin and CTCF contribute to the formation of chromatin loops (de Wit et al. 2015; Nichols and Corces 2015; Sanborn et al. 2015).
Cohesion Establishment and Dissolution
Cohesion establishment is coupled to DNA replication and requires acetylation of the two aforementioned lysine residues in the Smc3 hd by cohesin acetyltransferases (CoATs) and Sororin recruitment (Fig. 2) (Rolef Ben-Shahar et al. 2008; Unal et al. 2008; Zhang et al. 2008). The functional links between Smc3 acetylation, ATP hydrolysis, and DNA entrapment are not clear yet (Heidinger-Pauli et al. 2010b; Ladurner et al. 2014; Camdere et al. 2015). There are two CoATs in mammalian cells, Esco1 and Esco2, with partially redundant functions (Hou and Zou 2005; Whelan et al. 2012; Minamino et al. 2015; Rahman et al. 2015). Pds5 proteins, which exist in two versions in vertebrate cells, Pds5A and Pds5B, are also required for cohesion establishment in yeast and mouse cells (Vaur et al. 2012; Carretero et al. 2013; Chan et al. 2013). As a result of establishment, Sororin displaces Wapl from its Pds5-interaction site and thereby counteracts its unloading activity (Nishiyama et al. 2010; Ouyang et al. 2016). In this way, a fraction of cohesin complexes tethering the two sister chromatids become stably bound to chromatin and maintain cohesion until mitosis (Gerlich et al. 2006; Schmitz et al. 2007). Whether a single complex embraces the two sister chromatids or two complexes are required, each one embracing a sister, is still a matter of debate (Eng et al. 2015).
At the time of chromosome segregation, cohesin dissociates from DNA in two steps, in prophase and anaphase (Fig. 2) (Losada et al. 1998; Waizenegger et al. 2000). The prophase pathway requires SA phosphorylation by Plk1 and release of Sororin, after phosphorylation by Cdk1 and Aurora B, to restore Wapl unloading activity (Losada et al. 2002; Sumara et al. 2002; Dreier et al. 2011; Nishiyama et al. 2013). Shugoshin (Sgo1) and its partner, the protein phosphatase 2A (PP2A), prevent cohesin release around centromeres (McGuinness et al. 2005). Sgo1 outcompetes the binding of Wapl to SA-Rad21 (Hara et al. 2014), whereas PP2A counteracts Sororin dissociation (Liu et al. 2013b). Pericentromeric cohesin remains on chromatin and is essential to hold the sister chromatids together until all the chromosomes establish proper attachments to opposite spindle poles (Toyoda and Yanagida 2006; Liu et al. 2013a). Once this task is completed, activation of the anaphase-promoting complex (APC/C) leads to degradation of Securin and activation of Separase (Shindo et al. 2012). The protease cleaves the kleisin subunit of chromatin-bound cohesin and sister chromatid separation ensues (Hauf et al. 2001). In yeast, all chromatin-bound cohesin is released from chromatin in anaphase by this cleavage pathway (Uhlmann et al. 2000). Acetylated cohesin removed from chromosomes in prophase and anaphase is deacetylated by a cohesin deacetylase (CoDAC), Hdac8 in human cells and Hos1 in yeast (Beckouet et al. 2010; Borges et al. 2010; Deardorff et al. 2012).
Cohesin Functions
Our knowledge of cohesin functions comes from studies in many different experimental systems, most notably yeast and human cells, but also Drosophila, zebrafish, and Xenopus egg extracts. Mouse models carrying knockout alleles for genes encoding cohesin subunits or their regulators have also been generated and characterized to different extents (Table 2). As mentioned above, cohesin was first recognized as a mediator of sister chromatid cohesion (Fig. 3, left). During mitosis, cohesion contributes to the proper orientation of sister kinetochores (Sakuno et al. 2009) and prevents the premature separation of sister chromatids under the pulling forces of spindle microtubules, whereas chromosomes try to align at the metaphase plate (Daum et al. 2011). In the absence of cohesin, chromosome missegregation is commonly observed (Sonoda et al. 2001; Vass et al. 2003; Toyoda and Yanagida 2006; Barber et al. 2008; Solomon et al. 2013; Covo et al. 2014). Cohesin is also important for HR-driven DNA repair (Sjogren and Nasmyth 2001; Schmitz et al. 2007; Heidinger-Pauli et al. 2010a; Xu et al. 2010; Wu et al. 2012). Cohesin promotes usage of the sister chromatid as a template for faithful repair while preventing both damage-induced recombination between homologs (Covo et al. 2010) and end joining of distal double-strand breaks (DSB) (Gelot et al. 2015). A most intriguing function of cohesin is cohesion between mother and daughter centrioles, and its regulation bears similarities with that of sister chromatid cohesion (Wang et al. 2008; Beauchene et al. 2010; Schockel et al. 2011; Mohr et al. 2015).
Table 2.
Mouse models of cohesin subunits and regulators
| Targeted gene | References | Phenotype |
|---|---|---|
| SMC3 | White et al. 2013; Viny et al. 2015 | Embryonic lethality (prior to E14.5). Heterozygous animals have reduced body weight and higher mortality rates, and a subset showed a distinct craniofacial morphology (reminiscent of Cornelia de Lange syndrome [CdLS]). Deletion in hematopoietic compartment in adult mice results in rapid lethality. |
| RAD21 | Xu et al. 2010; Seitan et al. 2011 | Embryonic lethality (prior to E8.5). Heterozygous mouse embryo fibroblasts (MEFs) are defective in homologous recombination (HR)-mediated DNA repair. Heterozygous animals show increased sensitivity to irradiation, particularly in the gastrointestinal tract and the hematopoietic system. Deletion in thymocytes results in reduced differentiation efficiency and impairs TCRα locus rearrangement. |
| STAG1 | Remeseiro et al. 2012a,b | Embryonic lethality (from E12.5). Null MEFs show telomere cohesion defects leading to faulty replication and chromosome missegregation, altered transcription, and decreased colocalization of cohesin at CTCF sites and promoters. Heterozygous animals show increased incidence and earlier onset of cancer but are protected against acute carcinogenesis. |
| WAPL | Tedeschi et al. 2013 | Embryonic lethality. Heterozygous animals healthy. Conditional elimination in MEFs leads to aberrant retention of cohesin on chromatin, altered transcription, defects in cell-cycle progression, and chromosome segregation. |
| PDS5A | Zhang et al. 2009; Carretero et al. 2013 | Late embryonic lethality (from E12.5) or death soon after birth (depending on the allele) with cleft palate, skeletal patterning defects, growth retardation, congenital heart defects. Null MEFs proliferate slowly but show no chromosome missegregation. |
| PDS5B | Zhang et al. 2007; Carretero et al. 2013 | Late embryonic lethality (from E12.5) or death soon after birth (depending on the allele) with multiple congenital anomalies, including heart defects, cleft palate, fusion of the ribs, short limbs. Null MEFs show centromere cohesion defects and delocalization of the chromosomal passenger complex (CPC), chromosome missegregation, and aneuploidy. |
| NIPBL | Kawauchi et al. 2009; Remeseiro et al. 2013; Smith et al. 2014 | Embryonic lethality (prior to E9.5). Up to 80% of heterozygous animals die during the first weeks of life and display CdLS-like defects such as small size, craniofacial anomalies, heart defects, delayed bone maturation, and behavioral disturbances. Heterozygous MEFs show gene expression alterations but no cohesion defects. |
| MAU2 | Smith et al. 2014 | Embryonic lethality (prior to E9.5). Heterozygous animals are normal. |
| ESCO2 | Whelan et al. 2012 | Embryonic lethality (prior to E8). Conditional elimination in MEFs leads to defects in centromere cohesion and chromosome segregation. |
| HDAC8 | Haberland et al. 2009 | Homozygous mice show perinatal lethality with dramatic skull abnormalities. |
| SGO1 | Yamada et al. 2012 | Embryonic lethality. Heterozygous MEFs show chromosome missegregation and aneuploidy. Heterozygous animals viable but display increased susceptibility to colon and liver cancer induced by treatment with azoxymethane. |
| ESPL1 | Kumada et al. 2006; Wirth et al. 2006 | Embryonic lethality (E3.5). Conditional elimination in MEFs leads to proliferation defects and polyploidy, with multiple chromosomes connected at their centromeric regions. Depletion in bone marrow causes aplasia. |
| PTTG1 | Mei et al. 2001; Kumada et al. 2006 | Homozygous mice are viable but display testicular and splenic hypoplasia, thymic hyperplasia, and thrombocytopenia. Null MEFs grow slowly in culture and accumulate in G2. |
Figure 3.

Cohesin functions. Cohesin plays important roles in several cellular processes involving DNA. These roles rely on the ability of cohesin to hold two DNA strands in trans (the sister chromatids) or in cis (e.g., at the base of a chromatin loop). Accurate chromosome segregation in mitosis and meiosis, HR-mediated DNA repair, and restart and/or protection of stalled replication forks require sister chromatid cohesion (left). DNA looping mediated by cohesin in collaboration with CTCF, Mediator, or transcription factors, among others, likely provides a major organizational principle for the genome (right). This organization regulates transcription both globally, through generation of active/silent domains, and locally, facilitating interactions between enhancer and promoters required for gene activation or RNA Pol II pause release. It also facilitates coordinated origin firing at replication factories and recombination at loci such as IgH or TCRα. For simplicity, a single cohesin ring embracing the two DNA fibers is drawn, but alternative configurations are possible (Eng et al. 2015).
Cohesin-SA1 and cohesin-SA2 coexist in vertebrate cells and mediate cohesion at telomeres and centromeres, respectively (Canudas and Smith 2009; Remeseiro et al. 2012a). Telomeres are repeated regions prone to fork stalling (Sfeir et al. 2009). Cohesin-SA1 likely stabilizes stalled forks and facilitates their restart by HR, consistent with results in budding yeast (Tittel-Elmer et al. 2012). In SA1-null mouse embryo fibroblasts (MEFs), faulty telomere replication leads to chromosome missegregation (Remeseiro et al. 2012a). Cohesin-SA2 is preferentially recruited to laser-induced DNA damage sites in postreplicative human cells (Kong et al. 2014) but both complexes are loaded at double-strand breaks (DSBs) generated by a restriction enzyme (Caron et al. 2012). HR-mediated DNA repair in cells exposed to replication stress is also facilitated by both complexes (Remeseiro et al. 2012a).
Cohesin performs additional functions that do not require cohesion establishment (Fig. 3, right). These functions could be related to the ability of cohesin to tether chromatin fibers at the base of a chromatin loop to facilitate long-range interactions. Cohesin contributes to the spatial organization of the genome, together with CTCF. Recently developed chromosome conformation capture (3C)-related technologies together with improved microscopy and computational modeling offer the picture of a genome partitioned in “topological” domains that are conserved among cell types and even in evolution (Dixon et al. 2012). Within these domains, more local contacts allow or prevent communication between enhancers and promoters (Kagey et al. 2010; Phillips-Cremins et al. 2013; Dowen et al. 2014; Tang et al. 2015). Down-regulation of cohesin leads to a loss of contacts and deregulation of gene expression (Hadjur et al. 2009; Mishiro et al. 2009; Nativio et al. 2009; Seitan et al. 2013; Sofueva et al. 2013; Zuin et al. 2014a). Importantly, different loci display very different sensitivities to loss of cohesin (Ing-Simmons et al. 2015; Viny et al. 2015). In the pancreata of SA1 heterozygous mice, for instance, a twofold decrease in SA1 protein levels is sufficient to alter the chromatin architecture and the expression of the Reg gene cluster. The resulting down-regulation of Reg proteins, involved in inflammation, may contribute to the increased incidence of pancreatic cancer observed in SA1 heterozygous mice (Remeseiro et al. 2012a).
Cohesin depletion also increases RNA polymerase II pausing at cohesin binding genes in Drosophila, suggesting that it regulates its transition to elongation (Schaaf et al. 2013). In human cells, cohesin-SA1 is specifically involved in interactions with the super elongation complex (SEC) involved in mobilization of the paused polymerase (Izumi et al. 2015). Transcriptional control at the level of elongation is key for a number of developmental genes (Smith and Shilatifard 2013) and has been linked to pathogenesis in some leukemias (Lin et al. 2010).
In addition to transcription regulation, several lines of evidence support the idea that chromatin loops stabilized by cohesin organize DNA replication factories to promote efficient origin firing (Guillou et al. 2010), and facilitate V(D)J recombination (Degner et al. 2011) and T-cell receptor α locus rearrangement (Seitan et al. 2011).
COHESIN MUTATIONS IN CANCER
Recent pan-cancer studies have placed cohesin and its regulators among the networks most frequently mutated in cancer (Kandoth et al. 2013; Lawrence et al. 2014; Leiserson et al. 2015). Mutations in genes encoding cohesin subunits had been first reported in colorectal cancer after targeted sequencing of genes essential for chromosome segregation in yeast (Barber et al. 2008). A few years later, mutations in the gene encoding SA2, STAG2, were found in glioblastoma, Ewing sarcoma, and melanoma (Solomon et al. 2011). These two studies pointed to chromosome missegregation as the main contribution of cohesin dysfunction to tumorigenesis. However, sequencing of acute myeloid leukemia (AML) samples revealed the presence of recurrent mutations in STAG2, SMC3, RAD21, and SMC1A that were not associated with cytogenetic abnormalities, implying alternative pathological pathways (Welch et al. 2012). The correlation between STAG2 mutations and aneuploidy in bladder cancer was also unclear (Balbas-Martinez et al. 2013; Guo et al. 2013). In the next sections we will review these and other recent studies that provide evidence for the presence of cohesin mutations in cancer.
Cohesin Mutations in Myeloid Malignancies
Identification of Mutations in Cancer Cells
AML results from the aberrant proliferation and impaired differentiation of hematopoietic stem and progenitor cells. Reports using next-generation sequencing in AML samples appeared by 2012 and identified mutations in cohesin genes (Ding et al. 2012; Dolnik et al. 2012; Walter et al. 2012; Welch et al. 2012). According to The Cancer Genome Atlas (TCGA) Research Network (2013), AML genomes have fewer mutations than most adult cancers and, among them, 13% correspond to cohesin-related genes (Fig. 4A). Thol et al. (2014) performed targeted sequencing of genes encoding the five cohesin core subunits in samples from 389 AML patients and identified mutations in all of them (collectively in 6% of the cases). Most patients carrying cohesin mutations had a normal karyotype, supporting the hypothesis that they do not affect genome integrity. Importantly, cohesin mutations were found in myeloid malignancies other than AML (Kon et al. 2013). STAG2 and RAD21 were the most mutated cohesin genes (Fig. 4B). An even higher frequency of cohesin mutations (∼15%, most of them in STAG2) was found in MDS samples in another study (Haferlach et al. 2014). Even in the absence of cohesin mutations, low expression of cohesin components was detected in a significant fraction of myeloid malignancies (Thota et al. 2014). Mutations in additional components of the cohesin network including PDS5B, NIPBL, or ESCO2 were also identified in some studies (Fig. 4A,B).
Figure 4.

Cohesin mutations in myeloid malignancies and bladder cancer. Mutation matrix of cohesin subunits, associated factors (PDS5A, PDS5B, WAPL, CDC5A), loaders (NIPBL, MAU2), CoAT/CoDAC (ESCO1, ESCO2, HDAC8), mitotic regulators (SGOL1, ESPL1, PTTG1), and CTCF in AML (A), other myeloid malignancies (B), and bladder cancer (C). Nonsense, frameshift, and splice site mutations are grouped as truncating mutations (red), and missense and inframe indels are labeled as missense mutations (blue). (Data from Ding et al. 2012; Jan et al. 2012; Welch et al. 2012; Balbas-Martinez et al. 2013; Cancer Genome Atlas Research Network 2013, 2014; Guo et al. 2013; Kon et al. 2013; Yoshida et al. 2013; Lohr et al. 2014; Pellagatti et al. 2014; Thota et al. 2014.)
The prognostic impact of cohesin mutations in myeloid disorders is unclear, with studies reporting a positive (Kihara et al. 2014), negative (Thota et al. 2014), or no significant effect (Thol et al. 2014) on survival. The presence of these mutations in the major tumor populations points to their early origin during the neoplastic process (Kon et al. 2013; Thol et al. 2014). Interestingly, two patients analyzed by Kon et al. harbored each two independent subclones with different STAG2 mutations, which suggests that loss of STAG2 could confer a strong advantage to preexisting leukemic cells during clonal evolution. Analysis of clonal dynamics in another report revealed that cohesin mutations were not commonly present in the founder clone but rather promoted clonal expansion and transformation to more aggressive disease (Thota et al. 2014). Cohesin mutations often co-occurred with other mutations, most often in nucleophosmin (NPM1), epigenetic regulators such as ASXL1, TET2, or DNMT3A, or transcription factors like RUNX1 (Cancer Genome Atlas Research Network 2013; Walter et al. 2013; Kihara et al. 2014; Thol et al. 2014). Further support for a key role of cohesin mutations in malignant transformation came from the analysis of Down syndrome–related acute megakaryocitic leukemia (DS-AMKL) (Yoshida et al. 2013). Children with Down syndrome often suffer transient abnormal myelopoiesis (TAM) that, in some cases, evolves to DS-AMKL. Genomic profiling of TAM, DS-AMKL, and non-DS-AMKL samples revealed that TAM is caused by a GATA1 mutation and progression to DS-AMKL requires additional mutations. More than half of them were found in cohesin genes. Here again, most cases with mutated cohesin had normal karyotypes, except for constitutive trisomy 21.
In summary, sequencing studies have revealed a high prevalence of mutations in the genes encoding cohesin components in AML and other myeloid cancers (Fig. 4A,B). As expected from proteins working as part of the same complex, cohesin mutations are mutually exclusive. Although mutation rates differ among these studies, they typically account collectively for less than 10% of the cases. Cohesin mutations are usually heterozygous, with the exception of those present in the X-linked genes STAG2 and SMC1A in male patients. In female samples, mutations in STAG2 and SMC1A often reside in the active chromosome. While SMC1A, SMC3, and STAG1 mutations are often missense, those in STAG2 and RAD21 are usually truncating (i.e., frameshift, nonsense, or splice site mutations). No clear mutation hotspot has been identified in any of these genes, a characteristic of tumor suppressors. Importantly, no association of cohesin mutations and unstable karyotypes or aneuploidy has been reported. Thus, the contribution of cohesin dysfunction to development of myeloid malignancies is possibly not related to cohesion defects and genomic instability, and instead could be the result of altered transcription.
Functional Studies in Hematopoietic Cells
Cohesin mutations may reduce the levels of cohesin complexes in the cell or may alter their functionality. Understanding the contribution of these changes to tumorigenesis requires functional studies. Mazumdar et al. (2015) introduced a missense SMC1A mutant (SMC1A R711G) and a RAD21 truncation mutant (RAD21 Q592*), previously identified in AML (Cancer Genome Atlas Research Network 2013), in primary human hematopoietic stem and progenitor cells (HSPCs). No clear defects in proliferation or cell death were observed, but differentiation was impaired. The defects were restricted to the most immature populations of cord blood cells, consistent with the observation of cohesin mutations in the most immature forms of AML (Welch et al. 2012). Increased chromatin accessibility was observed in regions enriched for DNA-binding motifs of ERG, GATA2, and RUNX1, all transcription factors (TFs) involved in maintenance of the stem-cell program in HSPCs (Wilson et al. 2010). Knockdown of any of these factors reversed the differentiation block of cohesin mutants.
Two additional studies have explored the consequences of cohesin knockdown (kd) in hematopoiesis. Transgenic mice carrying inducible shRNAs against SA2, Smc1α, and Rad21 allowed ubiquitous and inducible cohesin kd in vivo in adult mice (Mullenders et al. 2015). Efficient reduction of cohesin levels, at least in the hematopoetic organs, was well tolerated, suggesting that a small fraction of cohesin complexes is sufficient to carry out its essential functions. Lineage skewing toward myeloid lineage commitment was observed in the spleen of cohesin-deficient mice, as well as in HSPCs, and gene expression changed accordingly. SA2 kd led to increased chromatin accessibility in regions enriched in the GATA motif. In the other study, mice carrying a conditional KO allele of SMC3 were used (Viny et al. 2015). Complete ablation of SMC3 in the hematopoietic compartment led to rapid lethality, whereas deletion of a single SMC3 allele resulted in increased cell renewal capacity of HSPCs and reduced expression of transcription factors and other genes associated with lineage commitment. Hematopoietic progenitors of Scm3 heterozygous animals displayed increased accessibility in regions harboring binding sites for yet another transcription factor, STAT5.
Taken all together, these studies suggest that decreased cohesin levels may promote transformation of HSPCs through delaying or skewing differentiation and instead enforcing stem-cell programs. They appear to do so through modulation of chromatin accessibility of TFs involved in stem-cell maintenance. An alternative possibility is that the observed changes are a consequence rather that the cause of the differentiation block. Cohesin has been proposed to regulate the expression of cell identity genes together with CTCF (Dowen et al. 2014), or with tissue-specific TFs (Schmidt et al. 2010) through the control of chromosome structure. To extend the studies described here, it would be informative to actually compare cohesin binding to chromatin in cells carrying or not the cohesin mutants, or partially deficient in cohesin subunits. This comparison could be performed in bulk by using chromatin fractionation, at genome wide scale resolution by using ChIP-seq and at specific sites by ChIP-qPCR. It would also be of interest to look for changes in chromatin architecture near the promoters of genes required to promote or prevent terminal myeloid differentiation, including the above-mentioned TFs. For instance, cohesin-mediated contacts between cis-regulatory elements modulate tissue-specific RUNX1 expression in zebrafish embryos, and probably also in human hematopoietic cells (Horsfield et al. 2007; Marsman et al. 2014).
Importantly, SMC3 haploinsufficiency by itself did not result in AML, but it enhanced tumorigenesis when combined with A FLT3-internal tandem duplication (ITD) mutation often found in AML (Viny et al. 2015). Aged cohesin knockdown mice in the study by Mullenders et al. (2015) developed phenotypes resembling myeloid neoplasias, but did not develop frank AML. These observations are consistent with the idea that cohesin mutations cooperate with additional mutations to promote myeloid malignancies.
Cohesin Mutations in Bladder Cancer
Urothelial bladder cancer (UBC) is a heterogeneous disease. Tumors are classified according to the stage of invasion (Tis-T4), and graded based on their cellular characteristics. At diagnosis, around 60% of bladder cancers are non-muscle-invasive (NMIBC) papillary tumors of low grade. Stage T1 tumors, which have penetrated the epithelial basement membrane but have not invaded the muscle, are mostly of high grade. Also aggressive are the muscle-invasive bladder cancers (MIBCs). Sequencing of 99 low-grade tumors revealed mutations in STAG2 (16%), NIPBL (4%), SMC1A (3%), and SMC3 (2%), as well as in the gene-encoding separase, ESPL1 (6%) (Guo et al. 2013). Individuals with STAG2 mutations had worse prognosis and increased number of copy number variations (CNVs), indicative of increased genomic instability. Soon afterward, a discovery exome sequencing screen (n = 17), followed by a prevalence screen (n = 60), identified mutations in STAG2 (16%) and some other cohesin subunits in UBC (Balbas-Martinez et al. 2013). STAG2 was mutated mainly in tumors of low stage or grade, commonly genomically stable, and unlike the previous study, its loss was associated with improved outcome. Moreover, chromosome number changes were not associated with STAG2 deficiency. Another analysis of aggressive MIBCs identified STAG2 mutations in 14 out of 131 tumors (11%), often co-occurring with mutations in epigenetic regulators (Cancer Genome Atlas Research Network 2014). Tumors with mutations in other cohesin subunits (9%) and cohesin regulators (12%) were also identified (Fig. 4C).
Solomon et al. (2013) reported higher mutation frequencies after sequencing STAG2 in 111 tumors of different stages/grades. Around 36% and 27% of STAG2 mutations were found in pTa and pT1 NMIBCs, respectively, and 16% in MIBCs. In low-grade NMIBCs, loss of STAG2 expression was significantly associated with increased disease-free survival, whereas the opposite was observed in MIBCs. Chromosomal copy number aberrations were found in many tumor samples, but even in the presence of wild-type STAG2. Another study sequencing STAG2 in 307 bladder tumors confirmed higher mutation frequencies in NMIBC noninvasive tumors (33%) or superficially invasive tumors (21%), and lower in MIBCs (13%) (Taylor et al. 2013). No significant association was found with disease recurrence in either NMIBC or MIBCs. Whole chromosome copy number alterations measured by aCGH showed an inverse relationship to STAG2 mutation. No association between STAG2 mutation and outcome was found in another report analyzing 109 high grade UBCs (16% STAG2 mutation rate) (Kim et al. 2014).
Taken all together, we can conclude that in UBC: (1) STAG2 mutation frequencies are higher than for genes encoding other cohesin subunits, and also higher than in other cancers; (2) most mutations in STAG2 are truncating, whereas other cohesin genes harbor missense mutations, similar to what was observed in myeloid syndromes; (3) cohesin mutations appear more frequently in lower grade/stage UBCs; and (4) there is no clear correlation with prognosis or aneuploidy.
Functional Studies in Bladder Cancer Cell Lines
Very limited functional experiments in bladder cells have been reported to date. SA2 kd in UBC cell lines with normal SA2 expression did not consistently alter chromosome number in one study (Balbas-Martinez et al. 2013) but it did in another (Solomon et al. 2013). Similarly, reintroduction of STAG2 cDNA in UBC cell lines with truncating STAG2 mutations led to a significant decrease in colony formation in one study (Balbas-Martinez et al. 2013) but did not affect proliferation in vitro or in xenografts in the other (Solomon et al. 2013). To interpret these conflicting observations, it would be important to quantify the functional cohesin complexes remaining in the cell under each experimental condition. Given the role of cohesin-SA2 in centromeric cohesion, one would expect chromosome segregation defects when STAG2 expression is lost (Canudas and Smith 2009). Indeed, SA2 kd led to chromosome missegregation in some human cell lines (Barber et al. 2008; Solomon et al. 2011; Kleyman et al. 2014). In contrast, and similar to UBCs, premature sister chromatid separation was not observed after SA2 kd in mouse hematopoietic progenitor cells, and was detected in only a small percentage of cells after efficient kd of Smc1α, Smc3, or Rad21 (Mullenders et al. 2015). Arm cohesion mediated by cohesin-SA1 may compensate for the loss of centromeric cohesion in the absence of cohesin-SA2, and the relative levels of both complexes may differ among cell types. In addition, a low amount of cohesin may be sufficient to maintain cohesion in mitosis. In yeast, cohesin levels must be reduced below 13% to result in detectable cohesion and segregation defects (Heidinger-Pauli et al. 2010a).
Genes involved in chromatin regulation are more frequently mutated in urothelial carcinoma than in any other common cancer studied so far. Cohesin belongs to this category. These mutations likely modulate the activity levels of various TFs and pathways implicated in cancer (Cancer Genome Atlas Research Network 2014). Mutations in STAG2 may be more frequent because the gene is located in the X chromosome and because in the absence of cohesin-SA2, cohesin-SA1 may be sufficient to perform essential cohesin functions. It is also possible that transcriptional dysregulation of key genes involved in tumorigenesis depends on cohesin-SA2, not on cohesin-SA1. So far the functional specificities of cohesin-SA1 and cohesin-SA2 in terms of chromatin regulation are poorly understood. In MEFs, genome-wide distribution of both complexes is similar and overlaps with the distribution of CTCF. Upon ablation of STAG1, however, cohesin could be detected at additional sites that showed less overlap with promoters and CTCF (Remeseiro et al. 2012b). It was then proposed that cohesin-SA1 could be more important than cohesin-SA2 for transcriptional regulation (Cuadrado et al. 2012). Consistent with this possibility, the transcriptomes of paired human gliobastoma cell lines with and without STAG2 expression did not differ significantly (Solomon et al. 2011). However, in one of the studies described in the previous sections, bone marrow cells treated with shRNAs against SA2 did display significant alterations in gene expression. Moreover, these alterations were similar to those observed on kd of Smc1α, suggesting that cohesin-mediated transcriptional regulation in HPSCs relies specifically on cohesin-SA2 (Mullenders et al. 2015). The reasons underlying this specificity are unknown. Even the relative abundance of cohesin-SA1 versus cohesin-SA2 in different cell types could be different. It will be of great interest to compare gene expression profiles before and after SA2 kd in bladder cell lines, as well as other cell types, to better understand how the cohesin variants contribute to cell proliferation and gene expression in a tissue-specific manner.
Cohesin Mutations in Other Cancers
A look at TCGA database reveals the presence of cohesin mutations in many additional cancers (Fig. 5). Bladder cancer is the one in which alteration of the cohesin network components is more common, followed by melanoma, colorectal, and lung cancers. Other than bladder cancer, STAG2 mutations are most frequent in Ewing sarcoma (EWS). This a pediatric tumor of the bone and soft tissues characterized genetically by the presence of translocations involving ETS family transcription factors such as EWS-FLI (Brohl et al. 2014; Crompton et al. 2014; Tirode et al. 2014; Agelopoulos et al. 2015). In one study looking for secondary genetic lesions, somatic mutations were detected in STAG2 (17%), CDKN2A (12%), and TP53 (7%). Although mutations in STAG2 and CDKN2A were mutually exclusive, STAG2 and TP53 mutations co-occurred particularly in aggressive tumors (Tirode et al. 2014). Mutation rates ranged from 8% to 21% in the other studies. In some cases, loss of expression was detected without mutation, suggesting another mechanism of STAG2 inactivation (Crompton et al. 2014). EWS is among the most genetically stable cancers. No association with aneuploidy was detected although tumors without STAG2 showed an increased number of somatic CNVs. However, it was not clear if this was because of the association with TP53 mutations (Crompton et al. 2014; Tirode et al. 2014).
Figure 5.

Mutations in cohesin complex and related genes in cancer. Bar graph showing mutations in the indicated cancer types for cohesin subunits and cohesin-related genes (grouped as in Fig. 4). (Data obtained from the cBioPortal for Cancer Genomics [Cerami et al. 2012; Gao et al. 2013] and from studies mentioned in Fig. 4.)
One intriguing observation is the high frequency of mutations in meiosis-specific cohesin genes (labeled in yellow in Fig. 5). Whether these mutations have actual consequences (e.g., the genes are expressed in the tumor cells and act as dominant negative mutant proteins), or are just passenger or silent mutations, remains to be addressed. Similarly, it is also unclear whether meiotic versions of cohesin subunits become expressed in tumors with mutations in their somatic counterparts to compensate for their loss. For instance, truncating mutations in the X-linked SMC1A gene have been described in tumor samples from male patients. Maybe Smc1β is expressed in these cells and forms functional complexes with Rad21 and SA1/2 (Mannini et al. 2015).
In addition to mutations, CNVs encompassing cohesin genes have also been observed in several cancer types. Of all cohesin genes, RAD21 is the most frequently amplified (e.g., 20% in invasive breast cancer) (Ciriello et al. 2015). This could be because of its proximity to the MYC locus, also present in 8q24. Abnormal levels of cohesin may also contribute to tumorigenesis. Finally, whole-genome sequencing data from more than 200 samples of colorectal cancer (CRC) patients together with ChIP-seq analyses in a CRC cell line revealed a high incidence of mutations in cohesin/CTCF binding sites in the noncoding genome (Katainen et al. 2015). A fraction of these mutations are predicted to affect CTCF binding affinity to cis-regulatory elements and could therefore contribute to tumorigenesis through aberrant expression of their target genes. Another epigenetic mechanism recently described in gliomas involves disruption of boundary elements through hypermethylation of CTCF/cohesin binding sites leading to oncongene activation (Flavahan et al. 2016). Thus, there are multiple ways in which cohesin dysfunction may contribute to tumorigenesis.
CONCLUSIONS AND PERSPECTIVES
Mutations in genes encoding cohesin subunits and regulators have been now identified in many cancer genomes, but their relevance for tumor initiation and progression is unknown. It is also unclear how they affect the functionality of the complex, particularly in the case of missense mutations. Because most mutations are heterozygous, they may either reduce the amount of fully functional cohesin complexes in the cell or even have a dominant negative effect. The diverse tasks accomplished by cohesin require different amounts of the complex and may rely on a particular variant. In the majority of tumors or cancer cell lines analyzed, there is no clear correlation between the presence of cohesin mutations and aneuploidy. Thus, cohesion is unlikely to be the function impaired in these tumor cells. Moreover, the recent functional studies performed in hematopoietic cells point to changes in chromatin accessibility and transcription as the most striking consequences of cohesin dysfunction. Future studies will tell if the same is true in other cell types. Identification of vulnerabilities in cancer cells harboring cohesin mutations will be an important step toward targeted therapies. A study in yeast and Caenorhabditis elegans identified a strong synthetic lethality between mutations in cohesin genes and genes involved in replication fork progression and stability, including Poly (ADP-ribose) polymerase or PARP (McLellan et al. 2012). Consistent with this observation, SMC1 down-regulation sensitized triple-negative breast cancer cells to PARP inhibition (Yadav et al. 2013). STAG2-mutated glioblastoma cell lines also displayed increased sensitivity to PARP inhibitors, especially when used in combination with DNA-damaging agents (Bailey et al. 2014). A better understanding of how cohesin works and how it contributes to proliferation, cell-identity determination, and homeostasis will hopefully guide improvements in diagnosis and treatment of cancer and other diseases related with cohesin dysfunction.
ACKNOWLEDGMENTS
The graphs in Figures 4 and 5 contain data obtained by TCGA Research Network (cancergenome.nih.gov) that have not been published yet. We therefore thank all researchers within this network as well as those involved in making these data available through the cBioPortal for Cancer Genomics (www.cbioportal.org). Our own research on cohesin is funded by the Spanish Ministry of Economy (MINECO) and The European Regional Development Fund (FEDER) (Grant BFU2013-48481-R to A.L. and Fellowship BES-2014-069166 to M.D.K.).
Footnotes
Editors: Scott A. Armstrong, Steven Henikoff, and Christopher R. Vakoc
Additional Perspectives on Chromatin Deregulation in Cancer available at www.perspectivesinmedicine.org
REFERENCES
- Agelopoulos K, Richter GH, Schmidt E, Dirksen U, von Heyking K, Moser B, Klein HU, Kontny U, Dugas M, Poos K, et al. 2015. Deep sequencing in conjunction with expression and functional analyses reveals activation of FGFR1 in Ewing sarcoma. Clin Cancer Res 21: 4935–4946. [DOI] [PubMed] [Google Scholar]
- Arumugam P, Gruber S, Tanaka K, Haering CH, Mechtler K, Nasmyth K. 2003. ATP hydrolysis is required for cohesin’s association with chromosomes. Curr Biol 13: 1941–1953. [DOI] [PubMed] [Google Scholar]
- Bailey ML, O’Neil NJ, van Pel DM, Solomon DA, Waldman T, Hieter P. 2014. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Mol Cancer Ther 13: 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbas-Martinez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Marquez M, Vazquez M, Lapi E, Castro-Giner F, Beltran S, Bayes M, et al. 2013. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat Genet 45: 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S, et al. 2008. Chromatid cohesin defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci 105: 3443–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchene NA, Diaz-Martinez LA, Furniss K, Hsu WS, Tsai HJ, Chamberlain C, Esponda P, Gimenez-Abian JF, Clarke DJ. 2010. Rad21 is required for centrosome integrity in human cells independently of its role in chromosome cohesion. Cell Cycle 9: 1774–1780. [DOI] [PubMed] [Google Scholar]
- Beckouet F, Hu B, Roig MB, Sutani T, Komata M, Uluocak P, Katis VL, Shirahige K, Nasmyth K. 2010. An Smc3 acetylation cycle is essential for establishment of sister chromatid cohesion. Mol Cell 39: 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard P, Schmidt CK, Vaur S, Dheur S, Drogat J, Genier S, Ekwall K, Uhlmann F, Javerzat JP. 2008. Cell-cycle regulation of cohesin stability along fission yeast chromosomes. EMBO J 27: 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges V, Lehane C, Lopez-Serra L, Flynn H, Skehel M, Rolef Ben-Shahar T, Uhlmann F. 2010. Hos1 deacetylates Smc3 to close the cohesin acetylation cycle. Mol Cell 39: 677–688. [DOI] [PubMed] [Google Scholar]
- Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, Patidar R, Hurd L, Chen L, Shern JF, et al. 2014. The genomic landscape of the Ewing sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 10: e1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buheitel J, Stemmann O. 2013. Prophase pathway-dependent removal of cohesin from human chromosomes requires opening of the Smc3-Scc1 gate. EMBO J 32: 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camdere G, Guacci V, Stricklin J, Koshland D. 2015. The ATPases of cohesin interface with regulators to modulate cohesin-mediated DNA tethering. eLife 4: e11315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. 2013. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. 2014. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507: 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canudas S, Smith S. 2009. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J Cell Biol 187: 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canudas S, Houghtaling BR, Kim JY, Dynek JN, Chang WG, Smith S. 2007. Protein requirements for sister telomere association in human cells. EMBO J 26: 4867–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron P, Aymard F, Iacovoni JS, Briois S, Canitrot Y, Bugler B, Massip L, Losada A, Legube G. 2012. Cohesin protects genes against γH2AX induced by DNA double-strand breaks. PLoS Genet 8: e1002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero M, Ruiz-Torres M, Rodriguez-Corsino M, Barthelemy I, Losada A. 2013. Pds5B is required for cohesion establishment and Aurora B accumulation at centromeres. EMBO J 32: 2938–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. 2012. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, Gligoris T, Upcher W, Kato Y, Shirahige K, Nasmyth K, Beckouet F. 2013. Pds5 promotes and protects cohesin acetylation. Proc Natl Acad Sci 110: 13020–13025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. 2015. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163: 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covo S, Westmoreland JW, Gordenin DA, Resnick MA. 2010. Cohesin is limiting for the suppression of DNA damage-induced recombination between homologous chromosomes. PLoS Genet 6: e1001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covo S, Puccia CM, Argueso JL, Gordenin DA, Resnick MA. 2014. The sister chromatid cohesion pathway suppresses multiple chromosome gain and chromosome amplification. Genetics 196: 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, Kiezun A, Carter SL, Shukla SA, Mehta SS, et al. 2014. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 4: 1326–1341. [DOI] [PubMed] [Google Scholar]
- Cuadrado A, Remeseiro S, Gomez-Lopez G, Pisano DG, Losada A. 2012. The specific contributions of cohesin-SA1 to cohesion and gene expression: Implications for cancer and development. Cell Cycle 11: 2233–2238. [DOI] [PubMed] [Google Scholar]
- Cuadrado A, Remeseiro S, Grana O, Pisano DG, Losada A. 2015. The contribution of cohesin-SA1 to gene expression and chromatin architecture in two murine tissues. Nucleic Acids Res 43: 3056–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum JR, Potapova TA, Sivakumar S, Daniel JJ, Flynn JN, Rankin S, Gorbsky GJ. 2011. Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Curr Biol 21: 1018–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, et al. 2012. HDAC8 mutations in Cornelia de Lange Syndrome affect the cohesin acetylation cycle. Nature 489: 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner SC, Verma-Gaur J, Wong TP, Bossen C, Iverson GM, Torkamani A, Vettermann C, Lin YC, Ju Z, Schulz D, et al. 2011. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc Natl Acad Sci 108: 9566–9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, Vos ES, Holwerda SJ, Valdes-Quezada C, Verstegen MJ, Teunissen H, Splinter E, Wijchers PJ, Krijger PH, de Laat W. 2015. CTCF binding polarity determines chromatin looping. Mol Cell 60: 676–684. [DOI] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. 2012. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481: 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, Mauch J, Kelkenberg-Schade S, Haldemann B, Fries T, Kronke J, Kuhn MW, Paschka P, et al. 2012. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood 120: e83–92. [DOI] [PubMed] [Google Scholar]
- Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schuijers J, Lee TI, Zhao K, et al. 2014. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159: 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier MR, Bekier ME II, Taylor WR. 2011. Regulation of sororin by Cdk1-mediated phosphorylation. J Cell Sci 124: 2976–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichinger CS, Kurze A, Oliveira RA, Nasmyth K. 2013. Disengaging the Smc3/kleisin interface releases cohesin from Drosophila chromosomes during interphase and mitosis. EMBO J 32: 656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng T, Guacci V, Koshland D. 2015. Interallelic complementation provides functional evidence for cohesin–cohesin interactions on DNA. Mol Biol Cell 26: 4224–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE. 2016. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529: 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi R, Gillespie PJ, Hirano T. 2006. Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr Biol 16: 2406–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelot C, Guirouilh-Barbat J, Le Guen T, Dardillac E, Chailleux C, Canitrot Y, Lopez BS. 2015. The cohesin complex prevents the end joining of distant DNA double-strand ends. Mol Cell 61: 15–26. [DOI] [PubMed] [Google Scholar]
- Gerlich D, Koch B, Dupeux F, Peters JM, Ellenberg J. 2006. Live-cell imaging reveals a stable cohesin-chromatin interaction after but not before DNA replication. Curr Biol 16: 1571–1578. [DOI] [PubMed] [Google Scholar]
- Gillespie PJ, Hirano T. 2004. Scc2 couples replication licensing to sister chromatid cohesion in Xenopus egg extracts. Curr Biol 14: 1598–1603. [DOI] [PubMed] [Google Scholar]
- Gligoris TG, Scheinost JC, Burmann F, Petela N, Chan KL, Uluocak P, Beckouet F, Gruber S, Nasmyth K, Lowe J. 2014. Closing the cohesin ring: Structure and function of its Smc3-kleisin interface. Science 346: 963–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn EF, Megee PC, Yu HG, Mistrot C, Unal E, Koshland DE, DeRisi JL, Gerton JL. 2004. Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoS Biol 2: E259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber S, Haering CH, Nasmyth K. 2003. Chromosomal cohesin forms a ring. Cell 112: 765–777. [DOI] [PubMed] [Google Scholar]
- Gruber S, Arumugam P, Katou Y, Kuglitsch D, Helmhart W, Shirahige K, Nasmyth K. 2006. Evidence that loading of cohesin onto chromosomes involves opening of its SMC hinge. Cell 127: 523–537. [DOI] [PubMed] [Google Scholar]
- Guacci V, Hogan E, Koshland D. 1997. Centromere position in budding yeast: Evidence for anaphase A. Mol Biol Cell 8: 957–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillou E, Ibarra A, Coulon V, Casado-Vela J, Rico D, Casal I, Schwob E, Losada A, Mendez J. 2010. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev 24: 2812–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, Dean M, Huang Y, Jia W, Zhou Q, et al. 2013. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet 45: 1459–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Mokalled MH, Montgomery RL, Olson EN. 2009. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev 23: 1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, Merkenschlager M. 2009. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature 460: 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haering CH, Gruber S. 2016. SnapShot: SMC protein complexes part I. Cell 164: 326–326.e321. [DOI] [PubMed] [Google Scholar]
- Haering CH, Lowe J, Hochwagen A, Nasmyth K. 2002. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol Cell 9: 773–788. [DOI] [PubMed] [Google Scholar]
- Haering CH, Schoffnegger D, Nishino T, Helmhart W, Nasmyth K, Lowe J. 2004. Structure and stability of cohesin’s Smc1-kleisin interaction. Mol Cell 15: 951–964. [DOI] [PubMed] [Google Scholar]
- Haering CH, Farcas AM, Arumugam P, Metson J, Nasmyth K. 2008. The cohesin ring concatenates sister DNA molecules. Nature 454: 297–301. [DOI] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. 2014. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Zheng G, Qu Q, Liu H, Ouyang Z, Chen Z, Tomchick DR, Yu H. 2014. Structure of cohesin subcomplex pinpoints direct shugoshin–Wapl antagonism in centromeric cohesion. Nat Struct Mol Biol 21: 864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf S, Waizenegger IC, Peters JM. 2001. Cohesin cleavage by separase required for anaphase and cytokinesis in human cells. Science 293: 1320–1323. [DOI] [PubMed] [Google Scholar]
- Heidinger-Pauli JM, Mert O, Davenport C, Guacci V, Koshland D. 2010a. Systematic reduction of cohesin differentially affects chromosome segregation, condensation, and DNA repair. Curr Biol 20: 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidinger-Pauli JM, Onn I, Koshland D. 2010b. Genetic evidence that the acetylation of the Smc3p subunit of cohesin modulates its ATP-bound state to promote cohesion establishment in Saccharomyces cerevisiae. Genetics 185: 1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsfield JA, Anagnostou SH, Hu JK, Cho KH, Geisler R, Lieschke G, Crosier KE, Crosier PS. 2007. Cohesin-dependent regulation of Runx genes. Development 134: 2639–2649. [DOI] [PubMed] [Google Scholar]
- Horsfield JA, Print CG, Monnich M. 2012. Diverse developmental disorders from the one ring: Distinct molecular pathways underlie the cohesinopathies. Front Genet 3: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F, Zou H. 2005. Two human orthologues of Eco1/Ctf7 acetyltransferases are both required for proper sister-chromatid cohesion. Mol Biol Cell 16: 3908–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Itoh T, Mishra A, Katoh Y, Chan KL, Upcher W, Godlee C, Roig MB, Shirahige K, Nasmyth K. 2011. ATP hydrolysis is required for relocating cohesin from sites occupied by its Scc2/4 loading complex. Curr Biol 21: 12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huis in ‘t Veld PJ, Herzog F, Ladurner R, Davidson IF, Piric S, Kreidl E, Bhaskara V, Aebersold R, Peters JM. 2014. Characterization of a DNA exit gate in the human cohesin ring. Science 346: 968–972. [DOI] [PubMed] [Google Scholar]
- Ing-Simmons E, Seitan VC, Faure AJ, Flicek P, Carroll T, Dekker J, Fisher AG, Lenhard B, Merkenschlager M. 2015. Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res 25: 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi K, Nakato R, Zhang Z, Edmondson AC, Noon S, Dulik MC, Rajagopalan R, Venditti CP, Gripp K, Samanich J, et al. 2015. Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat Genet 47: 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. 2012. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med 4: 149ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. 2010. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467: 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katainen R, Dave K, Pitkanen E, Palin K, Kivioja T, Valimaki N, Gylfe AE, Ristolainen H, Hanninen UA, Cajuso T, et al. 2015. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat Genet 47: 818–821. [DOI] [PubMed] [Google Scholar]
- Kawauchi S, Calof AL, Santos R, Lopez-Burks ME, Young CM, Hoang MP, Chua A, Lao T, Lechner MS, Daniel JA, et al. 2009. Multiple organ system defects and transcriptional dysregulation in the Nipbl+/− mouse, a model of Cornelia de Lange Syndrome. PLoS Genet 5: e1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, et al. 2014. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia 28: 1586–1595. [DOI] [PubMed] [Google Scholar]
- Kim PH, Cha EK, Sfakianos JP, Iyer G, Zabor EC, Scott SN, Ostrovnaya I, Ramirez R, Sun A, Shah R, et al. 2014. Genomic predictors of survival in patients with high-grade urothelial carcinoma of the bladder. Eur Urol 67: 198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleyman M, Kabeche L, Compton DA. 2014. STAG2 promotes error correction in mitosis by regulating kinetochore-microtubule attachments. J Cell Sci 127: 4225–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, et al. 2013. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 45: 1232–1237. [DOI] [PubMed] [Google Scholar]
- Kong X, Ball AR Jr, Pham HX, Zeng W, Chen HY, Schmiesing JA, Kim JS, Berns M, Yokomori K. 2014. Distinct functions of human cohesin-SA1 and cohesin-SA2 in double-strand break repair. Mol Cell Biol 34: 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueng S, Hegemann B, Peters BH, Lipp JJ, Schleiffer A, Mechtler K, Peters JM. 2006. Wapl controls the dynamic association of cohesin with chromatin. Cell 127: 955–967. [DOI] [PubMed] [Google Scholar]
- Kumada K, Yao R, Kawaguchi T, Karasawa M, Hoshikawa Y, Ichikawa K, Sugitani Y, Imoto I, Inazawa J, Sugawara M, et al. 2006. The selective continued linkage of centromeres from mitosis to interphase in the absence of mammalian separase. J Cell Biol 172: 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladurner R, Bhaskara V, Huis in ‘t Veld PJ, Davidson IF, Kreidl E, Petzold G, Peters JM. 2014. Cohesin’s ATPase activity couples cohesin loading onto DNA with Smc3 acetylation. Curr Biol 24: 2228–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. 2014. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson MD, Vandin F, Wu HT, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al. 2015. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet 47: 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengronne A, Katou Y, Mori S, Yokobayashi S, Kelly GP, Itoh T, Watanabe Y, Shirahige K, Uhlmann F. 2004. Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature 430: 573–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. 2010. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 37: 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Jia L, Yu H. 2013a. Phospho-H2A and cohesin specify distinct tension-regulated Sgo1 pools at kinetochores and inner centromeres. Curr Biol 23: 1927–1933. [DOI] [PubMed] [Google Scholar]
- Liu H, Rankin S, Yu H. 2013b. Phosphorylation-enabled binding of SGO1-PP2A to cohesin protects sororin and centromeric cohesion during mitosis. Nat Cell Biol 15: 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, Sougnez C, Knoechel B, Gould J, Saksena G, et al. 2014. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 25: 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A, Hirano M, Hirano T. 1998. Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev 12: 1986–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A, Yokochi T, Kobayashi R, Hirano T. 2000. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J Cell Biol 150: 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A, Hirano M, Hirano T. 2002. Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev 16: 3004–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A, Yokochi T, Hirano T. 2005. Functional contribution of Pds5 to cohesin-mediated cohesion in human cells and Xenopus egg extracts. J Cell Sci 118: 2133–2141. [DOI] [PubMed] [Google Scholar]
- Mannini L, Cucco F, Quarantotti V, Amato C, Tinti M, Tana L, Frattini A, Delia D, Krantz ID, Jessberger R, et al. 2015. SMC1B is present in mammalian somatic cells and interacts with mitotic cohesin proteins. Sci Rep 5: 18472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman J, O’Neill AC, Kao BR, Rhodes JM, Meier M, Antony J, Monnich M, Horsfield JA. 2014. Cohesin and CTCF differentially regulate spatiotemporal runx1 expression during zebrafish development. Biochim Biophys Acta 1839: 50–61. [DOI] [PubMed] [Google Scholar]
- Mazumdar C, Shen Y, Xavy S, Zhao F, Reinisch A, Li R, Corces MR, Flynn RA, Buenrostro JD, Chan SM, et al. 2015. Leukemia-associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell 17: 675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness BE, Hirota T, Kudo NR, Peters JM, Nasmyth K. 2005. Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells. PLoS Biol 3: e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan JL, O’Neil NJ, Barrett I, Ferree E, van Pel DM, Ushey K, Sipahimalani P, Bryan J, Rose AM, Hieter P. 2012. Synthetic lethality of cohesins with PARPs and replication fork mediators. PLoS Genet 8: e1002574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei J, Huang X, Zhang P. 2001. Securin is not required for cellular viability, but is required for normal growth of mouse embryonic fibroblasts. Curr Biol 11: 1197–120. [DOI] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K. 1997. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell 91: 35–45. [DOI] [PubMed] [Google Scholar]
- Minamino M, Ishibashi M, Nakato R, Akiyama K, Tanaka H, Kato Y, Negishi L, Hirota T, Sutani T, Bando M, et al. 2015. Esco1 acetylates cohesin via a mechanism different from that of Esco2. Curr Biol 25: 1694–1706. [DOI] [PubMed] [Google Scholar]
- Mishiro T, Ishihara K, Hino S, Tsutsumi S, Aburatani H, Shirahige K, Kinoshita Y, Nakao M. 2009. Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J 28: 1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misulovin Z, Schwartz YB, Li XY, Kahn TG, Gause M, Macarthur S, Fay JC, Eisen MB, Pirrotta V, Biggin MD, et al. 2008. Association of cohesin and Nipped-B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma 117: 89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi T, Fudenberg G, Mehta S, Belton JM, Taneja N, Folco HD, FitzGerald P, Dekker J, Mirny L, Barrowman J, et al. 2014. Cohesin-dependent globules and heterochromatin shape 3D genome architecture in S. pombe. Nature 516: 432–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr L, Buheitel J, Schockel L, Karalus D, Mayer B, Stemmann O. 2015. An Alternatively spliced bifunctional localization signal reprograms human shugoshin 1 to protect centrosomal instead of centromeric cohesin. Cell Rep 12: 2156–2168. [DOI] [PubMed] [Google Scholar]
- Mullenders J, Aranda-Orgilles B, Lhoumaud P, Keller M, Pae J, Wang K, Kayembe C, Rocha PP, Raviram R, Gong Y, et al. 2015. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med 212: 1833–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, Uhlmann F. 2014. Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature 505: 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, Uhlmann F. 2015. DNA entry into and exit out of the cohesin ring by an interlocking gate mechanism. Cell 163: 1628–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K. 2011. Cohesin: A catenase with separate entry and exit gates? Nat Cell Biol 13: 1170–1177. [DOI] [PubMed] [Google Scholar]
- Nasmyth K, Haering CH. 2009. Cohesin: Its roles and mechanisms. Annu Rev Genet 43: 525–558. [DOI] [PubMed] [Google Scholar]
- Nativio R, Wendt KS, Ito Y, Huddleston JE, Uribe-Lewis S, Woodfine K, Krueger C, Reik W, Peters JM, Murrell A. 2009. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet 5: e1000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols MH, Corces VG. 2015. A CTCF code for 3D genome architecture. Cell 162: 703–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama T, Ladurner R, Schmitz J, Kreidl E, Schleiffer A, Bhaskara V, Bando M, Shirahige K, Hyman AA, Mechtler K, et al. 2010. Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell 143: 737–749. [DOI] [PubMed] [Google Scholar]
- Nishiyama T, Sykora MM, Huis in ‘t Veld PJ, Mechtler K, Peters JM. 2013. Aurora B and Cdk1 mediate Wapl activation and release of acetylated cohesin from chromosomes by phosphorylating Sororin. Proc Natl Acad Sci 110: 13404–13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo-Hafalla MT, Uhlmann F. 2011. Cohesin loading and sliding. J Cell Sci 124: 685–691. [DOI] [PubMed] [Google Scholar]
- Orgil O, Matityahu A, Eng T, Guacci V, Koshland D, Onn I. 2015. A conserved domain in the scc3 subunit of cohesin mediates the interaction with both mcd1 and the cohesin loader complex. PLoS Genet 11: e1005036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Z, Zheng G, Tomchick DR, Luo X, Yu H. 2016. Structural basis and IP requirement for Pds5-dependent cohesin dynamics. Mol Cell 62: 248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, et al. 2008. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132: 422–433. [DOI] [PubMed] [Google Scholar]
- Pellagatti A, Fernandez-Mercado M, Di Genua C, Larrayoz MJ, Killick S, Dolatshad H, Burns A, Calasanz MJ, Schuh A, Boultwood J. 2014. Whole-exome sequencing in del(5q) myelodysplastic syndromes in transformation to acute myeloid leukemia. Leukemia 28: 1148–1151. [DOI] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, et al. 2013. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153: 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Jones MJ, Jallepalli PV. 2015. Cohesin recruits the Esco1 acetyltransferase genome wide to repress transcription and promote cohesion in somatic cells. Proc Natl Acad Sci 112: 11270–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remeseiro S, Cuadrado A, Carretero M, Martínez P, Drosopoulos WC, Cañamero M, Schildkraut CL, Blasco MA, Losada A. 2012a. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J 31: 2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remeseiro S, Cuadrado A, Gómez-López G, Pisano DG, Losada A. 2012b. A unique role of cohesin-SA1 in gene regulation and development. EMBO J 31: 2090–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remeseiro S, Cuadrado A, Kawauchi S, Calof AL, Lander AD, Losada A. 2013. Reduction of Nipbl impairs cohesin loading locally and affects transcription but not cohesion-dependent functions in a mouse model of Cornelia de Lange Syndrome. Biochim Biophys Acta 1832: 2097–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolef Ben-Shahar T, Heeger S, Lehane C, East P, Flynn H, Skehel M, Uhlmann F. 2008. Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science 321: 563–566. [DOI] [PubMed] [Google Scholar]
- Rubio ED, Reiss DJ, Welcsh PL, Disteche CM, Filippova GN, Baliga NS, Aebersold R, Ranish JA, Krumm A. 2008. CTCF physically links cohesin to chromatin. Proc Natl Acad Sci 105: 8309–8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuno T, Tada K, Watanabe Y. 2009. Kinetochore geometry defined by cohesion within the centromere. Nature 458: 852–858. [DOI] [PubMed] [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. 2015. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci 112: E6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CA, Kwak H, Koenig A, Misulovin Z, Gohara DW, Watson A, Zhou Y, Lis JT, Dorsett D. 2013. Genome-wide control of RNA polymerase II activity by cohesin. PLoS Genet 9: e1003382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Schwalie PC, Ross-Innes CS, Hurtado A, Brown GD, Carroll JS, Flicek P, Odom DT. 2010. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res 20: 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz J, Watrin E, Lenart P, Mechtler K, Peters JM. 2007. Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr Biol 17: 630–636. [DOI] [PubMed] [Google Scholar]
- Schockel L, Mockel M, Mayer B, Boos D, Stemmann O. 2011. Cleavage of cohesin rings coordinates the separation of centrioles and chromatids. Nat Cell Biol 13: 966–972. [DOI] [PubMed] [Google Scholar]
- Seitan VC, Hao B, Tachibana-Konwalski K, Lavagnolli T, Mira-Bontenbal H, Brown KE, Teng G, Carroll T, Terry A, Horan K, et al. 2011. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature 476: 467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitan VC, Faure AJ, Zhan Y, McCord RP, Lajoie BR, Ing-Simmons E, Lenhard B, Giorgetti L, Heard E, Fisher AG, et al. 2013. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res 23: 2066–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. 2009. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138: 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo N, Kumada K, Hirota T. 2012. Separase sensor reveals dual roles for separase coordinating cohesin cleavage and cdk1 inhibition. Dev Cell 23: 112–123. [DOI] [PubMed] [Google Scholar]
- Sjogren C, Nasmyth K. 2001. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr Biol 11: 991–995. [DOI] [PubMed] [Google Scholar]
- Smith E, Shilatifard A. 2013. Transcriptional elongation checkpoint control in development and disease. Genes Dev 27: 1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TG, Laval S, Chen F, Rock MJ, Strachan T, Peters H. 2014. Neural crest cell-specific inactivation of Nipbl or Mau2 during mouse development results in a late onset of craniofacial defects. Genesis 52: 687–694. [DOI] [PubMed] [Google Scholar]
- Sofueva S, Yaffe E, Chan WC, Georgopoulou D, Vietri Rudan M, Mira-Bontenbal H, Pollard SM, Schroth GP, Tanay A, Hadjur S. 2013. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J 32: 3119–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, Toretsky JA, Rosenberg SA, Shukla N, Ladanyi M, et al. 2011. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333: 1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DA, Kim JS, Bondaruk J, Shariat SF, Wang ZF, Elkahloun AG, Ozawa T, Gerard J, Zhuang D, Zhang S, et al. 2013. Frequent truncating mutations of STAG2 in bladder cancer. Nat Genet 45: 1428–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, Matsusaka T, Morrison C, Vagnarelli P, Hoshi O, Ushiki T, Nojima K, Fukagawa T, Waizenegger IC, Peters JM, et al. 2001. Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells. Dev Cell 1: 759–770. [DOI] [PubMed] [Google Scholar]
- Sumara I, Vorlaufer E, Gieffers C, Peters BH, Peters JM. 2000. Characterization of vertebrate cohesin complexes and their regulation in prophase. J Cell Biol 151: 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumara I, Vorlaufer E, Stukenberg PT, Kelm O, Redemann N, Nigg EA, Peters JM. 2002. The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol Cell 9: 515–525. [DOI] [PubMed] [Google Scholar]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. 2015. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163: 1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CF, Platt FM, Hurst CD, Thygesen HH, Knowles MA. 2013. Frequent inactivating mutations of STAG2 in bladder cancer are associated with low tumour grade and stage and inversely related to chromosomal copy number changes. Hum Mol Genet 23: 1964–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, Wutz G, Huet S, Jaritz M, Wuensche A, Schirghuber E, Davidson IF, Tang W, Cisneros DA, Bhaskara V, et al. 2013. Wapl is an essential regulator of chromatin structure and chromosome segregation. Nature 501: 564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thol F, Bollin R, Gehlhaar M, Walter C, Dugas M, Suchanek KJ, Kirchner A, Huang L, Chaturvedi A, Wichmann M, et al. 2014. Mutations in the cohesin complex in acute myeloid leukemia: Clinical and prognostic implications. Blood 123: 914–920. [DOI] [PubMed] [Google Scholar]
- Thota S, Viny AD, Makishima H, Spitzer B, Radivoyevitch T, Przychodzen B, Sekeres MA, Levine RL, Maciejewski JP. 2014. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 124: 1790–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, Zhang Z, Lapouble E, Grossetete-Lalami S, Rusch M, et al. 2014. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4: 1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tittel-Elmer M, Lengronne A, Davidson MB, Bacal J, Francois P, Hohl M, Petrini JH, Pasero P, Cobb JA. 2012. Cohesin association to replication sites depends on Rad50 and promotes fork restart. Mol Cell 48: 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda Y, Yanagida M. 2006. Coordinated requirements of human topo II and cohesin for metaphase centromere alignment under Mad2-dependent spindle checkpoint surveillance. Mol Biol Cell 17: 2287–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F, Wernic D, Poupart MA, Koonin EV, Nasmyth K. 2000. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell 103: 375–386. [DOI] [PubMed] [Google Scholar]
- Unal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, Koshland DE. 2008. A molecular determinant for the establishment of sister chromatid cohesion. Science 321: 566–569. [DOI] [PubMed] [Google Scholar]
- Vass S, Cotterill S, Valdeolmillos AM, Barbero JL, Lin E, Warren WD, Heck MM. 2003. Depletion of Drad21/Scc1 in Drosophila cells leads to instability of the cohesin complex and disruption of mitotic progression. Curr Biol 13: 208–218. [DOI] [PubMed] [Google Scholar]
- Vaur S, Feytout A, Vazquez S, Javerzat JP. 2012. Pds5 promotes cohesin acetylation and stable cohesin-chromosome interaction. EMBO Rep 13: 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viny AD, Ott CJ, Spitzer B, Rivas M, Meydan C, Papalexi E, Yelin D, Shank K, Reyes J, Chiu A, et al. 2015. Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J Exp Med 212: 1819–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waizenegger IC, Hauf S, Meinke A, Peters JM. 2000. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 103: 399–410. [DOI] [PubMed] [Google Scholar]
- Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, Larson DE, McLellan MD, Dooling D, Abbott R, et al. 2012. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 366: 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, Miller CA, Niu B, McLellan MD, Dees ND, et al. 2013. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 27: 1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yang Y, Duan Q, Jiang N, Huang Y, Darzynkiewicz Z, Dai W. 2008. sSgo1, a major splice variant of Sgo1, functions in centriole cohesion where it is regulated by Plk1. Dev Cell 14: 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrin E, Schleiffer A, Tanaka K, Eisenhaber F, Nasmyth K, Peters JM. 2006. Human Scc4 is required for cohesin binding to chromatin, sister-chromatid cohesion, and mitotic progression. Curr Biol 16: 863–874. [DOI] [PubMed] [Google Scholar]
- Weitzer S, Lehane C, Uhlmann F. 2003. A model for ATP hydrolysis-dependent binding of cohesin to DNA. Curr Biol 13: 1930–1940. [DOI] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. 2012. The origin and evolution of mutations in acute myeloid leukemia. Cell 150: 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, et al. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451: 796–801. [DOI] [PubMed] [Google Scholar]
- Whelan G, Kreidl E, Wutz G, Egner A, Peters JM, Eichele G. 2012. Cohesin acetyltransferase Esco2 is a cell viability factor and is required for cohesion in pericentric heterochromatin. EMBO J 31: 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JK, Gerdin AK, Karp NA, Ryder E, Buljan M, Bussell JN, Salisbury J, Clare S, Ingham NJ, Podrini C, et al. 2013. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell 154: 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, et al. 2010. Combinatorial transcriptional control in blood stem/progenitor cells: Genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 7: 532–544. [DOI] [PubMed] [Google Scholar]
- Wirth KG, Wutz G, Kudo NR, Desdouets C, Zetterberg A, Taghybeeglu S, Seznec J, Ducos GM, Ricci R, Firnberg N, et al. 2006. Separase: A universal trigger for sister chromatid disjunction but not chromosome cycle progression. J Cell Biol 172: 847–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Kong X, Ji Z, Zeng W, Potts PR, Yokomori K, Yu H. 2012. Scc1 sumoylation by Mms21 promotes sister chromatid recombination through counteracting Wapl. Genes Dev 26: 1473–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Wallace J, Felsenfeld G. 2011. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin-dependent insulation activity. Mol Cell Biol 31: 2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Balakrishnan K, Malaterre J, Beasley M, Yan Y, Essers J, Appeldoorn E, Tomaszewski JM, Vazquez M, Verschoor S, et al. 2010. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS ONE 5: e12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S, Sehrawat A, Eroglu Z, Somlo G, Hickey R, Liu X, Awasthi YC, Awasthi S. 2013. Role of SMC1 in overcoming drug resistance in triple negative breast cancer. PLoS ONE 8: e64338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada HY, Yao Y, Wang X, Zhang Y, Huang Y, Dai W, Rao CV. 2012. Haploinsufficiency of SGO1 results in deregulated centrosome dynamics, enhanced chromosomal instability and colon tumorigenesis. Cell Cycle 11: 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Toki T, Okuno Y, Kanezaki R, Shiraishi Y, Sato-Otsubo A, Sanada M, Park MJ, Terui K, Suzuki H, et al. 2013. The landscape of somatic mutations in Down syndrome–related myeloid disorders. Nat Genet 45: 1293–1299. [DOI] [PubMed] [Google Scholar]
- Zhang B, Jain S, Song H, Fu M, Heuckeroth RO, Erlich JM, Jay PY, Milbrandt J. 2007. Mice lacking sister chromatid cohesion protein PDS5B exhibit developmental abnormalities reminiscent of Cornelia de Lange syndrome. Development 134: 3191–3201. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi X, Li Y, Kim BJ, Jia J, Huang Z, Yang T, Fu X, Jung SY, Wang Y, et al. 2008. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell 31: 143–151. [DOI] [PubMed] [Google Scholar]
- Zhang B, Chang J, Fu M, Huang J, Kashyap R, Salavaggione E, Jain S, Kulkarni S, Deardorff MA, Uzielli ML, et al. 2009. Dosage effects of cohesin regulatory factor PDS5 on mammalian development: Implications for cohesinopathies. PLoS ONE 4: e5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin J, Dixon JR, van der Reijden MI, Ye Z, Kolovos P, Brouwer RW, van de Corput MP, van de Werken HJ, Knoch TA, van Ijcken WF, et al. 2014a. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci 111: 996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin J, Franke V, van Ijcken WF, van der Sloot A, Krantz ID, van der Reijden MI, Nakato R, Lenhard B, Wendt KS. 2014b. A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet 10: e1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]