

Abstract
Rhesus macaques (Macaca mulatta) are the most widely used nonhuman primate in biomedical research, have the largest natural geographic distribution of any nonhuman primate, and have been the focus of much evolutionary and behavioral investigation. Consequently, rhesus macaques are one of the most thoroughly studied nonhuman primate species. However, little is known about genome-wide genetic variation in this species. A detailed understanding of extant genomic variation among rhesus macaques has implications for the use of this species as a model for studies of human health and disease, as well as for evolutionary population genomics. Whole-genome sequencing analysis of 133 rhesus macaques revealed more than 43.7 million single-nucleotide variants, including thousands predicted to alter protein sequences, transcript splicing, and transcription factor binding sites. Rhesus macaques exhibit 2.5-fold higher overall nucleotide diversity and slightly elevated putative functional variation compared with humans. This functional variation in macaques provides opportunities for analyses of coding and noncoding variation, and its cellular consequences. Despite modestly higher levels of nonsynonymous variation in the macaques, the estimated distribution of fitness effects and the ratio of nonsynonymous to synonymous variants suggest that purifying selection has had stronger effects in rhesus macaques than in humans. Demographic reconstructions indicate this species has experienced a consistently large but fluctuating population size. Overall, the results presented here provide new insights into the population genomics of nonhuman primates and expand genomic information directly relevant to primate models of human disease.
The amount and nature of the genetic variation within species is a fundamental aspect of biology with significant implications for a number of research questions (Leffler et al. 2012; Yang et al. 2013; Rogers and Gibbs 2014; Romiguier et al. 2014; Sankararaman et al. 2014; The 1000 Genomes Project Consortium 2015; Li et al. 2015). Among humans, single-nucleotide variants (SNVs) have been studied within personal genomes (Levy et al. 2007; Wheeler et al. 2008; Ahn et al. 2009; Lupski et al. 2010; Schuster et al. 2010) and across populations (The 1000 Genomes Project Consortium 2012; Gudbjartsson et al. 2015). Whole-genome sequencing (WGS) and whole-exome sequencing (WES) have identified millions of human SNVs (The 1000 Genomes Project Consortium 2015), as well as more complex structural variation and gene copy number differences (Sudmant et al. 2015). Knowledge regarding variants that influence disease risk, as well as normal variation in human anatomy and physiology, is accumulating at an accelerating pace. Genomic information is also becoming available for diverse nonhuman species (Hayes et al. 2013; Schoenebeck and Ostrander 2014; Lack et al. 2015).
Rhesus macaques (Macaca mulatta) are one of the most evolutionarily successful and intensively studied nonhuman primates. This species has the largest natural geographic range of any nonhuman primate, extending from India in the west across Asia to the Pacific coast of China, and south into Vietnam and Thailand. Across that range, it exhibits outstanding ecological flexibility and adaptability (Richard et al. 1989; Thierry 2011). Rhesus macaques are Old World monkeys (Family Cercopithecidae, Superfamily Cercopithecoidea) and thus are phylogenetically closely related to humans, sharing a common ancestor roughly 25 million years ago (Perelman et al. 2011). Only the apes (like humans, members of Superfamily Hominoidea) are more closely related to humans than are Old World monkeys. In part because of their adaptability and overall genetic and physiological similarity to humans, rhesus macaques are widely used as an animal model for biomedical studies related to human health and disease (Phillips et al. 2014). These macaques are the premier models for investigations related to several human infectious diseases, are critical to research in neurobiology and psychobiology, and play a central role in studies of reproductive endocrinology, metabolism, and other basic aspects of biology and medicine.
Identification of functionally significant genetic variation among rhesus macaques will increase their value as models for human physiology and disease. Prior studies have used specific genetic variants in rhesus macaques to model human genetic effects (Champoux et al. 2002; Barr et al. 2004; Loffredo et al. 2007; Vallender et al. 2008, 2010; Rogers et al. 2013). The discovery of novel functional variation in this species will lead directly to new genetic models of human disease, better characterization of existing models, and will also support rational genetic management of research colonies. This will advance rhesus macaque models beyond face validity to construct validity and improve translational relevance.
In addition to the biomedical implications, knowledge of intraspecies genetic variation in this widely distributed primate will inform analyses of the ecological, demographic, and population genetic factors that drive differences among taxa (Leffler et al. 2012; Corbett-Detig et al. 2015; Xue et al. 2015). The complex demographic and evolutionary history of modern humans (The 1000 Genomes Project Consortium 2012; Moreno-Estrada et al. 2014; Allentoft et al. 2015; Schroeder et al. 2015) and our recent hominin ancestors (Sankararaman et al. 2012; Antón et al. 2014) includes several periods of broad geographic distribution and diverse ecology. In contrast to the African great apes, rhesus macaques share these characteristics with ancestral humans; thus, patterns of genetic diversity and population structure may provide informative parallels to the population genomics of deep human history.
With the primary goal of advancing opportunities for biomedical research using rhesus macaques, we generated WGS for 132 unrelated rhesus obtained from multiple research facilities. In addition, we resequenced the individual sampled for the first rhesus whole-genome assembly (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007). Our results provide new insight into primate biology and open new avenues for research, both using rhesus macaques as models for the genetics of human disease and in studies of evolutionary and population genomics.
Results
Genome-wide single-nucleotide variation
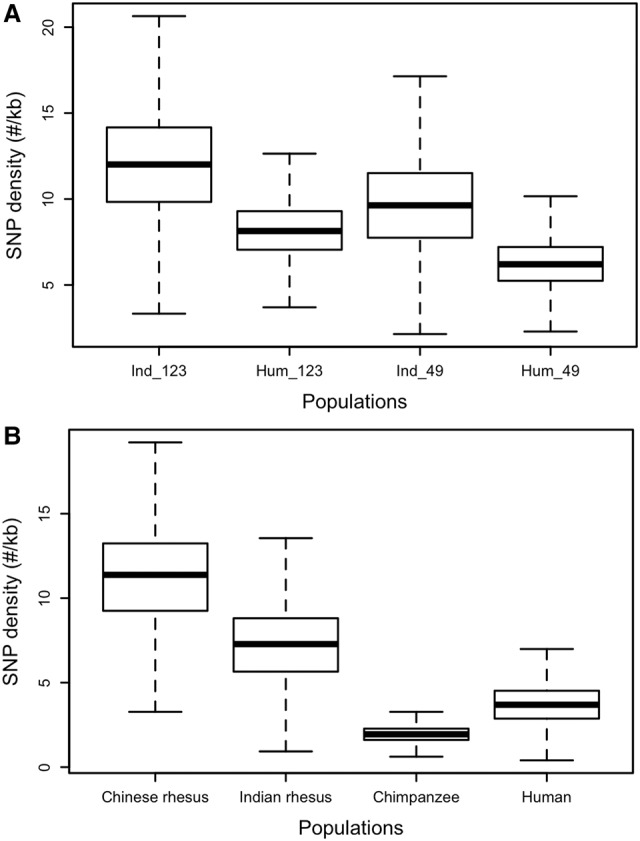
Whole-genome sequences for 133 rhesus macaques (Macaca mulatta) were generated using the Illumina HiSeq technology. The majority of study animals (n = 82) were sequenced to high coverage (mean 37.8×, range 23.2–60.7×) and the remainder to moderate coverage (mean 9.5×, range 7.0–11.7×), for an overall mean genome-wide coverage of 26.7×. The sample population includes the original Indian-origin female macaque used to generate the public reference genome (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007), 123 additional Indian-origin rhesus, and nine Chinese-origin animals. All animals were captive born in research colonies, with the exception of three wild-born Chinese rhesus macaques (Supplemental Table S1). SNVs were identified using both GATK (DePristo et al. 2011) and SNPTools (Wang et al. 2013). The intersection of the two variant call sets identified 43.7 million SNVs, 31.9 million among the 124 Indian-origin rhesus macaques (IRh) and 30.1 million variants in the nine Chinese-origin animals (CRh). The transition/transversion ratio across the SNV data set is 2.16. A subset of SNVs were validated by comparing these whole-genome results to SNV calls from whole-exome sequencing for a subset of individuals. A second subset of SNVs were reanalyzed using the Ion Torrent platform (Methods; Supplemental Material). Consistent with population differentiation observed in previous smaller studies (Smith and McDonough 2005; Hernandez et al. 2007), most SNVs (58%) are population specific, and many are rare (Minor Allele Frequency; MAF < 0.01). The proportion of rare alleles among the 124 IRh animals is 26.5%, whereas the sample size for CRh is too small to make this calculation meaningful.
Compared with humans, the average number of SNVs per sample in rhesus macaques is greater than twofold higher in IRh animals and greater than 2.5-fold higher in CRh individuals. The IRh show substantially higher SNV density than an equivalent sample of 123 humans from the 1000 Genomes Project (11.8/kb versus 7.9/kb), with a larger number of variants per sample (Table 1; Fig. 1A). This pattern holds when read depth coverage is equivalent at ∼9.5× per sample for both rhesus macaques and humans (9.7 SNV/kb versus 6.3 SNV/kb) (Fig. 1A). The average heterozygosity in both IRh (het = 0.0024) and CRh (het = 0.0027) is higher than most available estimates for nonhuman primates (e.g., chimpanzees, bonobos, and gorillas consistently less than 0.0019; Indian rhesus versus chimpanzee Mann-Whitney U test, P-value <2.2 × 10−16) while roughly equivalent to Sumatran orangutans (Prado-Martinez et al. 2013; data from Supplemental Fig. 6.1). The estimated heterozygosity per sample is also higher in rhesus than in human (IRh versus human Mann-Whitney U test, P-value <2.2 × 10−16), and the value for the CRh is larger than for IRh (Mann-Whitney U test, P-value <2.2 × 10−16) (Tables 1, 2; Fig. 1B). The CRh are expected to show a higher number of variants per animal than IRh because the reference genome used in this analysis was produced from an Indian-origin animal. Indeed the CRh exhibit an average of 4.3 million homozygous nonreference allele sites, whereas the IRh present only 3.0 million. The CRh also exhibit higher number of segregating sites and nucleotide diversity (π) than IRh (Table 2). We randomly sampled nine of the IRh animals and compared their SNV density (7.2/kb) to the nine CRh animals (11.1/kb), nine humans from the 1000 Genomes Project (3.7/kb), and nine western chimpanzees, Pan troglodytes verus (1.9/kb) (Auton et al. 2012) (IRh versus human: Mann-Whitney U test P < 2.2 × 10−16).
Table 1.
Observed levels of autosomal nucleotide variation in rhesus macaques and humans
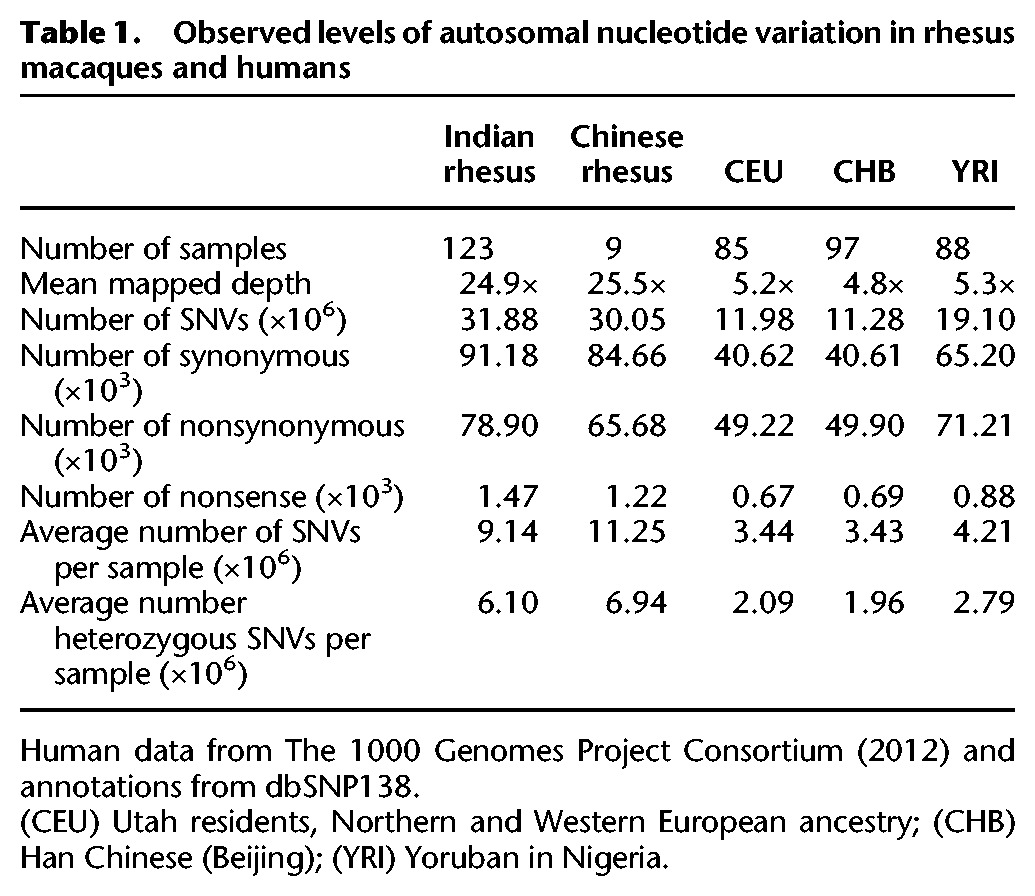
Figure 1.
(A) Density of autosomal SNVs observed in rhesus (Indian rhesus only, 123 samples) and human (1000 Genomes Project data, phase 1, 123 randomly selected samples). Ind_123 indicates results for 123 rhesus samples; Hum_123 for 123 human samples; Ind_49 indicates 49 low-coverage (average 9.47×) Indian rhesus samples; Hum_49 for 49 human samples with the average coverage 9.44×. (B) Density of autosomal SNVs observed in Chinese rhesus (sample size n = 9), Indian rhesus (nine samples randomly sampled from 123 Indian rhesus samples), chimpanzee (nine samples randomly sampled from 10 samples; downloaded from http://panmap.uchicago.edu), and human (1000 Genomes Project data, phase 1, nine samples randomly sampled from 1092 samples).
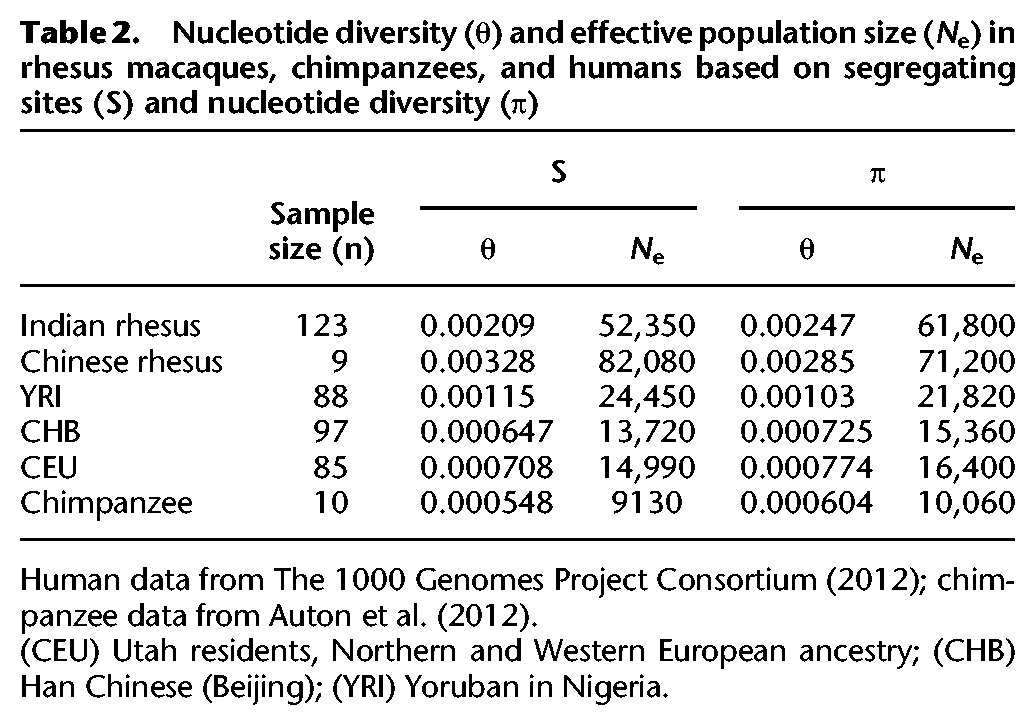
Table 2.
Nucleotide diversity (θ) and effective population size (Ne) in rhesus macaques, chimpanzees, and humans based on segregating sites (S) and nucleotide diversity (π)
Rhesus SNVs fall disproportionately in CpG dinucleotide sites. Just 2.06% of bases in the rhesus genome are contained in CpG dinucleotides, but 16.7% of rhesus SNVs fall in such positions. This is consistent with the higher mutation rate observed at these sites due to deamination of methylated cytosines. Given the utility of rhesus macaque models of human genetic disease, we are interested in identifying polymorphic sites that are shared between humans and rhesus. Of 36.9 million sites that are polymorphic in our sample of rhesus macaques and that survive reciprocal liftOver to the human genome (hg19) and back to the rhesus, 1.8 million sites are polymorphic in both rhesus and humans, and share both alleles in common. Furthermore, 42.1% of those shared polymorphisms are at CpG sites in the rhesus genome. Given the higher CpG mutation rate and the 6.5% sequence divergence between the human and rhesus genomes (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007), this high proportion of shared polymorphisms falling in CpG dinucleotides seems more consistent with independent parallel mutations in the two species than with retention of shared ancestral polymorphism over approximately 25 million years since their last common ancestor (for a discussion of shared ancestral polymorphisms between humans and chimpanzees, see Leffler et al. 2013). Regardless of their origin, these rhesus polymorphisms that are shared with humans will be extraordinarily useful as new macaque models of human genetic effects, facilitating the testing of specific genotype–phenotype relationships in a well-characterized primate model system.
Recombination map
We used LDhat software (McVean et al. 2004) to calculate a recombination map for the rhesus genome. Figure 2A shows that recombination is reduced across all chromosomes in the rhesus genome relative to the human, and the distribution of local rates for orthologous segments is shifted significantly to lower values (P = 3.74 × 10−9, Mann-Whitney U test) (Fig. 2B). Across the entire genome, the recombination rate for 100-kb windows within IRh autosomes is 0.433 ± 0.333 cM/Mb (mean ± SD), which is significantly lower (P < 1 × 10−20, Mann-Whitney U test) than recombination across human autosomes in Hapmap genetic map (1.322 ± 1.399 cM/Mb estimated for 100-kb windows) (The International HapMap Consortium 2007). A detailed comparison of one short segment (5 Mb; human Chromosome 6: coordinates 70–75 million base pairs) (Fig. 2C) demonstrates this lower recombination rate in rhesus compared to humans. A previously constructed low resolution recombination map for a rhesus macaque pedigree using crossovers among 241 microsatellite loci (Rogers et al. 2006) also suggested reduced recombination per megabase in rhesus relative to human.
Figure 2.

(A) Genome-wide comparison of recombination rates in syntenic regions scaled and averaged over 1-Mb segments in the Indian rhesus genome (blue and cyan, sample n = 49) and human population-average Hapmap genetic map (red and orange). (B) Distribution of 4Ner estimated directly for 534 autosomal syntenic regions that are orthologous in humans and rhesus. (C) Recombination rates scaled by 100 kb in a given syntenic region (human genome coordinate Chr 6: 70,000,000) for humans (orange line) and rhesus (blue line).
Demographic analysis of two rhesus macaque populations
The estimated effective population sizes, based on the number of segregating sites (S) and observed genetic diversity (π) are approximately 52,000 and 62,000, respectively, for IRh and 82,000 and 71,000 for CRh (Table 2). For all demographic analyses, we use an estimated macaque mutation rate of 1.0 × 10−8 per site per generation, and generation time of 11 yr (for rationale behind these values, see Methods). We inferred the history of demographic change for IRh and CRh separately using the stairway plot method (Liu and Fu 2015) and the Pairwise Sequential Markovian Coalescent (PSMC) method (Fig. 3; Li and Durbin 2011). Those two methods were chosen as they complement each other: Stairway plots provide more accurate reconstructions of recent demographic history (<100,000 yr ago), whereas the PSMC method performs better for more ancient history, older than ∼100,000 yr (Liu and Fu 2015). Whole-genome site frequency spectra (SFS) for 75 high-coverage (>25×) IRh and six high-coverage (>20×) CRh were used for stairway plot estimation. Whole-genome sequence data from three high-coverage IRh and three high-coverage CRh were used for PSMC estimation.
Figure 3.
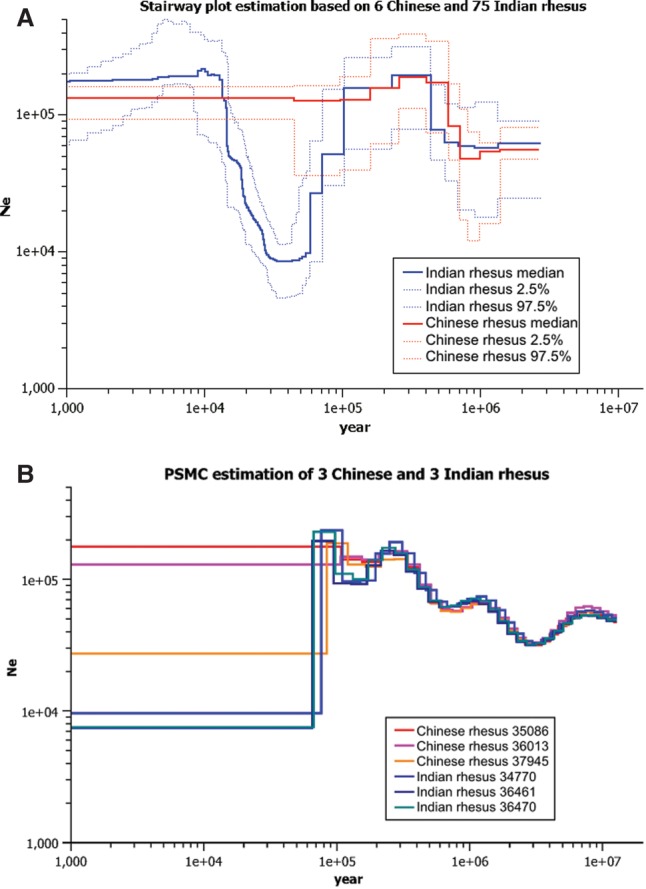
Demographic histories inferred by the stairway plot and PSMC. (A) Stairway plot estimation and 95% CI based on six high-coverage (>20×) Chinese rhesus and 75 high-coverage (>20×) Indian rhesus. (B) PSMC estimations with pattern “6+29*2” for three high-coverage Chinese rhesus (35086, 36013, and 37945) and three high-coverage Indian rhesus (34770, 36461, and 36470): (Ne) effective population size.
For Chinese rhesus, the stairway plot and PSMC (using pattern parameter of p “6+29*2”) inferred similar demographic histories (Fig. 3), i.e., a period of population growth starting ∼600,000–700,000 yr ago that reached its peak ∼400,000 yr ago. The models suggest that the population size remained more or less constant after that. The PSMC estimation for one CRh (37945) suggested a population size decrease ∼100,000 yr ago. However, given that PSMC is less accurate when reconstructing recent history, and that the estimations from the other two CRh do not support this population size decrease, we suspect this decrease may be an artifact of PSMC estimation. Alternatively, the three CRh individuals analyzed may have their origins in different regions within China that experienced different population histories. PSMC estimation using an alternative pattern parameter (p “4+25*2+4+6”), also suggested a similar demographic history (Supplemental Fig. S1). In the following discussion, we refer to PSMC analyses with pattern parameter of “6+29*2” as PSMC1 and those with pattern parameter “4+25*2+4+6” as PSMC2. We tested the fit of each model (stairway plot, PSMC1, and PSMC2) by comparing SFS predicted by each model's inferred demographic results against the true observed SFS (Supplemental Fig. S2) for Chinese rhesus. PSMC1 fit slightly better than the other two models (Supplemental Fig. S2; Supplemental Table S2). Furthermore, we note that there is no definitive consensus regarding the most appropriate mutation rate to be used for these estimations of demographic history (Ségurel et al. 2014; Rahbari et al. 2016). If a higher rate of mutation is used, the inferred dates would all be adjusted forward in time.
For Indian rhesus, both the stairway plot and PSMC (PSMC1 and PSMC2) again suggest population growth starting ∼600,000–700,000 yr ago and reaching a peak ∼400,000 yr ago. Both methods also suggest a population bottleneck ∼60–70,000 yr ago. However, there are also discrepancies between results. The most obvious discrepancy is that the stairway model infers a population size recovery beginning ∼30,000 yr ago, reaching full recovery ∼15,000 yr ago, while this recovery is not inferred by PSMC. Comparing the fit of these models to the data using the observed SFS for 52 low-coverage (<12×) IRh suggests that the stairway results fit the observed SFS significantly better than the PSMC models (Supplemental Fig. S2; Supplemental Table S2). The PSMC results significantly underestimate the frequency of rare SNPs. Further simulation studies suggest that PSMC may miss the recent population size recovery when individual genomes were used, whereas the stairway plot can infer such recovery using SFS from multiple individuals (Supplemental Fig. S3). Overall, the results support a recent population growth as inferred by stairway plot, which is missed by PSMC. Another discrepancy between results is that PSMC1 infers a population growth ∼100,000 yr ago, but this growth was not inferred by stairway plot or PSMC2. Finally, PSMC1 and PSMC2 infer an ancient population bottleneck ∼3,000,000 yr ago, but that time period is beyond the range of the stairway plot method.
Conservation and negative selection across the rhesus genome
Purifying selection is a potent force shaping the amount and nature of genetic variation within species. Both theory and prior evidence predict that genomic regions that are significantly conserved across mammalian species should exhibit lower levels of intraspecies variation than the rest of the genome (Lindblad-Toh et al. 2011; Corbett-Detig et al. 2015). We used reciprocal liftOver methods to identify regions of the rhesus genome that are orthologous to the 4.2% of the human genome that is significantly conserved across 29 mammals, based on the SiPhy approach (Lindblad-Toh et al. 2011), and we then compared rhesus macaque SNV density in the conserved and nonconserved segments. Using the full data set from IRh and CRh animals, 1,075,143 SNVs fall within the 112.1 Mb of conserved sequence regions. This density of 9.6 SNVs/kb contrasts significantly with the density in the remainder of the genome (17.8 SNVs/kb for combined IRh/CRh data, P < 0.0001).The CpG content in the conserved regions is 14.03 CpGs/kb compared to 7.93 outside of the conserved regions, and there are 1.60 SNVs in CpGs per 1000 bp of conserved region sequence compared to 2.38 SNVs in CpGs per 1000 bp outside of conserved regions. This indicates that although there is a higher density of CpG sites in conserved regions than in the rest of the genome, fewer of those CpG sites exhibit polymorphism in our study sample. This may be related to conservation of promoter function, affecting CpG islands in promoter regions (Lindblad-Toh et al. 2011). The distribution of MAF for rhesus SNVs in the conserved regions is also shifted to lower allele frequencies than for other regions (Supplemental Fig. S4). We are particularly interested in rhesus SNVs that fall in regions predicted to have the greatest functional effects in humans. Within the 0.4% of the genome that shows the strongest purifying selection within humans (Khurana et al. 2013), we again found significantly reduced variation (12.1 SNVs/kb versus 17.5 SNVs/kb; P < 0.0001).
Ratio of nonsynonymous to synonymous variants
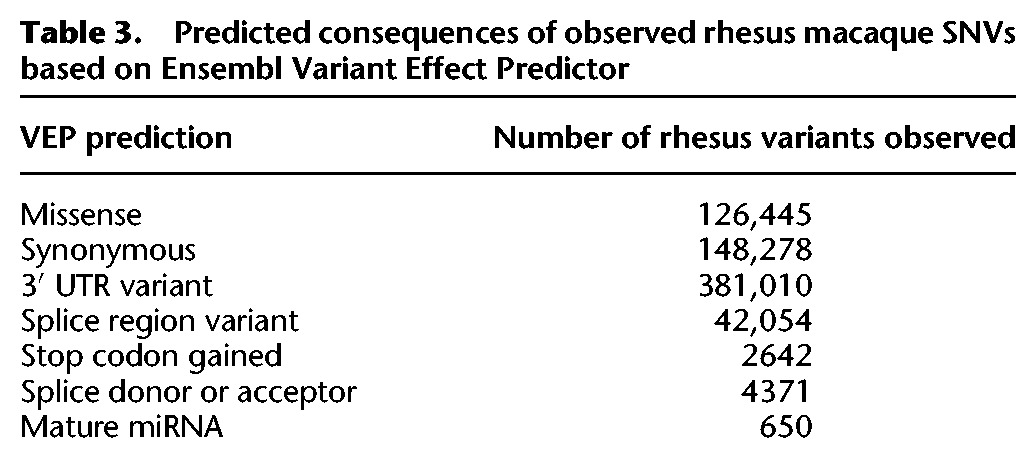
Purifying selection against slightly deleterious variants is expected to be more powerful in rhesus than in modern humans due to the larger Ne in rhesus. This predicts a reduction in the proportion of functionally deleterious mutations in the macaques relative to humans. We used liftOver and Ensembl Variant Effect Predictor (VEP) software to infer the functional consequences of the observed rhesus macaque SNVs. Counting only rhesus SNVs unambiguously mapped to human genome coordinates, and using the human VEP predicted consequences rather than the rhesus annotations, we identified substantial numbers of putative functional variants (Table 3). The ratio of nonsynonymous to synonymous variants is lower in rhesus than in a subset of 133 humans from the 1000 Genomes Project (Supplemental Fig. S5). The relative density of nonsynonymous SNVs (rdnsv) (Freudenberg et al. 2012) for humans = 0.46 and for rhesus = 0.32 (t-test P = 1.09 × 10−261). The average number of synonymous variants (homozygous alternative allele plus heterozygous) per individual in these two matched samples is substantially higher in rhesus than in human (11,113.9 versus 7369.5; t-test P = 2.81 × 10−115). The number of nonsynonymous variants per individual is also higher in rhesus, but more similar between species than the counts of synonymous variants (7810.1 in rhesus versus 6551.7 in humans; t-test P = 7.66 × 10−47) (see also Yuan et al. 2012). Consequently, the ratio of nonsynonymous-to-synonymous variants is lower in rhesus, consistent with population genetic theory. This is true despite the total pool of rhesus nonsynonymous variants (across a population sample or per individual) being higher than in humans (Table 1).
Table 3.
Predicted consequences of observed rhesus macaque SNVs based on Ensembl Variant Effect Predictor
To independently test these conclusions, we used DFE-alpha (Eyre-Walker and Keightley 2009) to investigate the distribution of fitness effects (DFE) within Indian-origin rhesus across 10,944 rhesus–human orthologous genes. We compared IRh nonsynonymous (NS) sites potentially under selection to more neutrally evolving sites (synonymous sites, introns, and regions flanking known genes) (Supplemental Table S3). Akaike information criterion (AIC) values calculated for DFE-alpha maximum likelihood scores indicate that a three-epoch model of initial population decline followed by expansion provides the best fit to the data, and this is consistent with our stairway plot results for IRh animals. Among IRh, the three-epoch model using synonymous sites as the neutrally evolving data set suggests 19.5% of NS mutations behave as effectively neutral (Nes 0–1), whereas just over 75% of NS mutations are so strongly selected against that they almost never become fixed (Nes > 10). Equivalent analyses in humans (Eyre-Walker and Keightley 2009) suggest that 29%–38% of NS mutations behave as effectively neutral (Nes 0–1), and only 44%–64% of amino acid changing mutations are strongly selected against (Nes > 10). Prior comparisons of DFE between humans and chimpanzees (Hvilsom et al. 2012) found that, as we see in rhesus macaques, purifying selection is also stronger in chimpanzees than in humans (see also Boyko et al. 2008). Although not definitive, this analysis suggests that purifying selection has acted more strongly in both rhesus macaques and chimpanzees than it has in humans. However, the effect of purifying selection is weaker in all these primates than in Drosophila (Eyre-Walker and Keightley 2009). On the other hand, we estimate that alpha (α, the proportion of new NS mutations fixed as a result of positive selection) is 19.6% in the macaques. In both humans (Eyre-Walker and Keightley 2009; Veeramah et al. 2014) and chimpanzees (Hvilsom et al. 2012) α is estimated to be lower than our estimate for rhesus, although those previous studies did find significantly higher α levels on the X Chromosome compared with autosomes. We also performed additional analyses of the effects of selection on the rhesus genome (Residual Variance Intolerance Scores), and those results are presented in the Supplemental Material.
Functional annotation of rhesus SNVs
Each of the putatively functional rhesus variants is potentially useful for modeling genetic effects on human phenotypes, including risk for disease. As specific examples, we investigated rhesus macaque variants in 166 genes (Supplemental Table S4) known to cause human eye diseases, such as retinal degeneration or congenital blindness. Among those 166 genes, we identified 157,595 total rhesus variants, including 157 alleles predicted to adversely affect gene function. This includes 18 loss-of-function and other variants in MYO7A and ABCA4 that affect codons known to cause disease when altered in humans (Molday et al. 2009; Millán et al. 2011). These codon-specific mutations are particularly valuable in rhesus macaques because this species models human retinal disease more closely than rodent models (Lillo et al. 2003; Coleman et al. 2004; Francis et al. 2008; Colella et al. 2013). Across a variety of physiological systems, human genetic mechanisms can be modeled more effectively in primates than in other species (Barr et al. 2004; Loffredo et al. 2007; Vallender et al. 2010; Rogers et al. 2013; Phillips et al. 2014); thus, functional variants in macaque genes orthologous to human disease genes (eye diseases or others) will provide significant and unique opportunities to model genetic mechanisms or test therapies for those disorders.
Of particular interest are variants in rhesus macaques that affect nucleotide sites already shown to influence disease risk in humans. We used the WGSA human genome annotation pipeline (Liu et al. 2015) to annotate the rhesus SNVs that were reciprocally lifted over to the human genome. Among those rhesus SNVs, 164 variants were found that match human variants annotated as “disease causing” in HGMD or pathogenic in ClinVar (Supplemental Table S5). Those 164 rhesus variants affect genes that cause specific human diseases including leukemia, ALS, atrial fibrillation, ADHD, autism, breast cancer, cardiomyopathy, Charcot–Marie–Tooth 1B, cystic fibrosis, diabetes, hypercholesterolemia, polycystic kidney disease, and others (Supplemental Table S5).
One of the major challenges in human genetics is the prediction and validation of functional effects for noncoding sequence variants. Noncoding functional variation in macaques can be used for experimental investigation of its cellular and broader phenotypic consequences, taking advantage of the outstanding similarity of genetic pathways (Seok et al. 2013; Bakken et al. 2015, 2016). We used reciprocal liftOver to identify segments of the rhesus macaque genome that are orthologous to human DNA segments annotated by the ENCODE Project as transcription factor binding sites (TFBS) (Spivakov et al. 2012; The ENCODE Project Consortium 2012). We found 111,290 rhesus SNVs that affect the predicted TFBS. Among these, 24,449 are predicted by JASPAR (Mathelier et al. 2014) to alter TF binding, and 3554 affect sites that are also polymorphic in humans. Among those sites, 2192 are exact allelic matches for known human polymorphisms within TFBS, creating opportunities for targeted in vivo study of specific effects on gene expression.
Discussion
Rhesus macaques (M. mulatta) are critical to progress in many aspects of biomedical research and have also been central to fundamental analyses of primate behavior and evolution. The current census of rhesus in NIH-funded research colonies is approximately 20,000, with the vast majority derived from Indian-origin founders, although a smaller number of Chinese-origin rhesus are available. The analyses presented here demonstrate that an extensive array of genetic variation is segregating among rhesus macaques, and much of that variation is predicted to have functional consequences (e.g., stop codons, splice site variants, damaging nonsynonymous variants, and others that affect TFBS). The higher nucleotide diversity in rhesus macaques compared with humans is consistent with prior studies examining smaller data sets of mtDNA (Kanthaswamy and Smith 2004), microsatellites (Satkoski et al. 2008), or smaller sets of SNVs (Ferguson et al. 2007; Hernandez et al. 2007; Fawcett et al. 2011; Yuan et al. 2012). Our new data show for the first time that a random sample of 133 rhesus macaques exhibit hundreds of variants that match annotated, known human disease variants and thousands more that are predicted to be damaging within known human disease genes.
Although rhesus macaques have been valuable models for human disease research for many years (Phillips et al. 2014), the putative functional variants identified in this study can greatly increase their value as models for human genetics. Direct in vivo experimental analyses of the cellular and physiological consequences of both protein-coding and noncoding variation in primate models that closely mimic human biology are now feasible, including analyses that test specific hypotheses concerning genotype–phenotype relationships that develop out of human association studies. In addition, the macaque genetic variation can be used to investigate the developmental effects of putative functional variation early in embryogenesis or fetal development, as well as the consequences of host genetic variation on the progression of controlled infection with known pathogens, studies that would be unethical or impractical in humans and are often impossible with nonprimate models. Rhesus monkeys that carry specific functionally significant (disease-causing) alleles could also be used to test the efficacy of novel pharmaceutical therapies targeted to ameliorate dysfunction in specific genes or genetic pathways.
One unanticipated finding of this study is the evidence that recombination rates per megabase of autosomal sequence are significantly lower in rhesus macaques relative to humans. Prior studies of pedigree-based recombination had suggested the potential for reduced recombination in rhesus (Rogers et al. 2006), but given the number of potential confounding factors, the support from those microsatellite data for reduced recombination was not strong. The present results based on extensive whole-genome SNV data indicate that there is a significant difference in recombination rates between these two species, and recommends further investigation of recombination in rhesus and other closely related species such as other macaques, baboons, and other Old World monkeys. Previous investigators observed that although specific recombination hotspots are not conserved, the overall rate and pattern of recombination is quite similar in chimpanzees and humans (Auton et al. 2012). Our results argue for additional comparative analyses of PRDM9 function (Schwartz et al. 2014) and other aspects of the recombination machinery among Old World monkeys and other nonhuman primates that may reveal interesting and informative differences among primate genomes.
The large effective population size inferred for this species provides an interesting contrast with modern humans and most of the nonhuman primates studied to date (Prado-Martinez et al. 2013). In contrast to smaller mammals such as rodents or bats, average heterozygosity, linkage disequilibrium, degree of regional population differentiation, and historical dynamics of fluctuations in population size may differ in relatively large-bodied, longer-lived species such as primates. Most genome-scale studies of nonhuman primate population genetics have focused either on African great apes and hylobatids, which generally exhibit restricted geographic ranges and episodes of low effective population size (Prado-Martinez et al. 2013; Carbone et al. 2014; Gordon et al. 2016), or other endangered primates (Perry et al. 2012) that may not reflect the population genetics of widely distributed, ecologically flexible species. Because several human ancestors (Homo erectus, archaic Homo sapiens) had extensive geographic, ecological and temporal distributions (Stringer 2012; Antón et al. 2014), it is plausible that the population genomics and population structure of those species shares more in common with extant rhesus macaques than with extant gibbons, gorillas, chimpanzees or bonobos (Prado-Martinez et al. 2013; Xue et al. 2015; Gordon et al. 2016). The population genetics of extant humans is dominated by the dramatic recent population expansion, which can complicate efforts to estimate parameters such as the distribution of fitness effects (Eyre-Walker and Keightley 2009; Gazave et al. 2013). Inferences regarding the population genomics of archaic hominins can be placed into a broader comparative perspective by also considering this type of information for rhesus macaques and other widely distributed, ecologically successful nonhuman primates with relatively long lifespan.
The rhesus macaques in the major NIH-funded research colonies, approximately 20,000 in number, consist primarily of animals with ancestry in India. The inferred divergence of Indian from Chinese rhesus macaques ∼150,000 to 200,000 yr ago is consistent with reconstructions of macaque evolutionary history based on a smaller nuclear DNA sequence data set (Hernandez et al. 2007), mtDNA analyses (Smith and McDonough 2005; Hasan et al. 2014), and climate, sea level changes, and paleontology (Abegg and Thierry 2002). Fossils of ancient macaques, along with other forest dwelling mammals, are known from throughout the Zhoukoudian faunal assemblage in northeast China, dating from >500,000 yr ago to less than 200,000 (Li et al. 2014). The divergence of rhesus macaques from the closely related cynomolgus macaque (Macaca fascicularis) likely occurred in east or southeast Asia approximately 900,000 yr ago (Osada et al. 2008). The geographic range of the ancestral mulatta/fascicularis lineage is not entirely clear but was likely influenced during the Pleistocene by significant fluctuations in climate, temperature, rainfall, and sea level that would expand and contract the habitat for these monkeys (Morley 2012). The dispersal of modern rhesus macaques into eastern China and westward across Bangladesh into India established the broad geographic range and ecological diversity that now characterizes Macaca mulatta. Our results suggest that the dispersal of rhesus macaques from their origin in east or southeast Asia westward (Abegg and Thierry 2002) probably occurred between 150,000 and 200,000 yr ago (Smith and McDonough 2005; Hernandez et al. 2007; Hasan et al. 2014). Furthermore, the bottleneck inferred for the Indian-origin animals by the stairway analyses may be the result of this westward dispersal. Integration of genome-based reconstructions of demographic history with paleontological and climatic information can lead to a more complete understanding of the history and population dynamics of this important primate species. That population history has produced the pattern of intra-species genetic diversity that we now observe. This array of extant genetic variation provides substantial opportunity for further analyses of gene and genome function that can contribute to various aspects of biomedical and primatological research.
Methods
Description of DNA samples analyzed
DNA or blood samples were obtained from eight US primate research colonies, as listed in Supplemental Table S1. We also analyzed three wild-caught Chinese rhesus macaques from the Yellow Mountains region of Anhui Province using DNA samples provided by Dr. G. Fan (UCLA) and Drs. Liu and Sun (Anhui University, Hefei, China). One study animal we sequenced from the Southwest NPRC is the same animal used for the initial rheMac2 (NCBI Mmul_051212) whole-genome assembly (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007). The 133 animals used as the final data set show no evidence of close relatedness (defined as closer than second cousins) nor evidence of admixture between regional populations. All animals were broadly healthy, with no obvious congenital abnormalities at the time of sampling or reported subsequently.
Initial sample collection and sequencing
To begin this analysis, we sequenced the genomes of 152 rhesus macaques from the eight US primate research colonies. Among those animals, there were 144 presumed Indian-origin animals and eight presumed Chinese-origin animals. Whole-genome sequencing was performed using the Illumina HiSeq 2000 platform, generating 100-bp paired-end reads. Depth of sequence read coverage in Illumina paired-end reads for each study subject ranges from 7.0× to 60.7×, with a mean of 26.7× (Supplemental Fig. S6; Supplemental Table S6). Single-nucleotide variants (SNVs) were identified by mapping the quality-filtered sequence reads to the rheMac2 rhesus macaque whole-genome assembly (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007) using BWA (Li and Durbin 2009). SNVs were then called using SNPTools (Wang et al. 2013). This process identified slightly more than 53.7 million SNVs.
Evaluation of initial data set using PRIMUS
We next used the PRIMUS suite of analytical tools to evaluate and perform quality controls on this initial data (Staples et al. 2013, 2014). The algorithms in PRIMUS that estimate allele sharing were used to infer genealogical relatedness among all pairs. This identified several sample pairs with apparent relatedness, defined here as having estimated kinship closer than second cousins. We next investigated possible admixture (individuals with evidence of ancestry from both Chinese-origin and Indian-origin ancestors). Using the tools within PRIMUS to generate principal components analyses and plots, we found evidence of Indian-Chinese admixture in some animals and misidentification of ancestry in others (Supplemental Fig. S7). As a result of the relatedness testing and admixture analyses, 22 individuals were removed from the data set of 152. At this stage, we also added three wild-caught Chinese rhesus obtained from Drs. Fan, Liu, and Sun. Thus, the final sample size was set at 133 macaques.
Final SNV calling
Initial variant calling on the full set of 155 rhesus (152 initial study animals plus three additional Chinese animals) was performed using SNPTools (Wang et al. 2013), with the sequence reads mapped to the rheMac2 reference genome (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007) with BWA (Li and Durbin 2009). For final SNV calling, we again used the read mapping from BWA and SNV calls generated by SNPTools. The default cutoff value for variance ratio score (VRS) in SNPTools is set at 1.5. In this study, we used a VRS cutoff of 1.6 in order to balance the quality of SNP sites and the number of SNPs discarded (see Supplemental Fig. S8). Using this threshold, the overall transition/transversion ratio (Ti/Tv) is 2.092, which is slightly low compared to the human 1000 Genomes Project data set (=2.14 for 1092 samples) (The 1000 Genomes Project Consortium 2012). However, when the number of samples is adjusted to be the same (n = 133, by randomly sampling 133 human samples from the total 1092 samples), the Ti/Tv for our rhesus data set with the VRS cutoff set at 1.6 is actually higher than the Ti/Tv for the human 1000 Genomes data set (Supplemental Fig. S9).
Using SNPTools, we discovered 45,656,297 biallelic SNVs on the autosomes of the 133 rhesus, VRS >1.6 (Wang et al. 2013). To further improve the quality of sites reported, we then used GATK (DePristo et al. 2011) to perform the SNV calling again, independently. GATK identified 46,991,381 SNVs. The Ti/Tv ratio for the GATK variant calls is also higher than what is observed for the 1000 Genomes Project human data set with the same number of samples. The intersection of these two SNV call sets defines 43,767,770 variants (Supplemental Fig. S10). The Ti/Tv ratio for the intersection data set is slightly higher than that for SNPTools (Supplemental Fig. S9). Next, the intersection set of SNV calls was used as the input for genotype likelihood estimation and imputation using SNPTools. Following likelihood estimation and imputation, we removed SNVs with homozygous alternative alleles in the reference animal 17573 (n = 16,078 or 0.037%), and removed another 44,174 SNPs that had alternative allele frequency = 0 in the VCF file. This produced a final data set and SNV call list of 43,707,518 SNVs across the rhesus autosomes.
Independent validation of SNV calls
To assess the reliability of the final SNV call set, we resequenced eight randomly selected animals using whole-exome capture. The human Vcrome2.1 exome capture array was used to enrich sequencing libraries for exonic sequences, and resulting libraries were sequenced using the Illumina HiSeq 2000 platform. Macaque sequence reads were mapped to the rhesus reference genome using BWA and SNVs called using GATK. After using standard exome analysis procedures and quality filters, we compared the relevant SNV results (a total of 131,708 variable sites) across the eight individuals. Concordance of SNV identification between WGS and WES data was 95.03%. This observed discordance of ∼5% represents the sum of false positive calls in the WGS data and false negative calls in the WES data. We also performed another validation of a much smaller subset of several hundred SNVs using the Ion Torrent PGM platform (Thermo Fisher Scientific, Inc.) to resequence custom designed amplicons covering rhesus macaque SNVs with a range of minor allele frequencies (for further details, see Supplemental Material).
Selection of human samples for comparison to rhesus macaques
To compare patterns of base pair variation in humans and rhesus macaques, we randomly selected 123 human samples from the 1000 Genomes Phase 1 data set (The 1000 Genomes Project Consortium 2012) to compare with 123 samples from Indian-origin rhesus (excluding the original Indian-origin animal used to generate the reference genome assembly). In addition, we selected nine human samples from the same 1000 Genomes data to compare with our nine samples from Chinese rhesus. The background ethnicity information for these randomly selected human samples is shown in Supplemental Table S7.
Rhesus recombination map
We set up the workflow for estimating recombination rates using LDHat software (Auton and McVean 2007; Auton et al. 2012) and the recommended procedures. To check our workflow, we downloaded the chimpanzee genotype data (sample size n = 10) that was used to create the initial chimpanzee genetic recombination map (Auton et al. 2012), and then used our workflow to estimate the recombination rate across chimpanzee autosomes. The recombination rates we obtained are very close to the published results (Supplemental Fig. S11; Supplemental Table S8), indicating that our workflow functions appropriately. We next estimated the recombination rates across rhesus macaque autosomes and compared them with human autosomes. Sample size and sequence coverage may affect recombination rate estimates, so we only estimated recombination rates for IRh. In human 1000 Genomes data, the read coverage is relatively low, so we used 49 IRh samples with low-to-moderate sequence coverage (mean 9.5×) to estimate the rhesus recombination rates and selected 49 moderate-coverage samples each from CEU and YRI populations within the 1000 Genomes data to estimate recombination rates for these two human populations (Supplemental Fig. S12). The average coverage for 49 CEU samples is 6.18, and average coverage for 49 YRI samples is 6.3.
The LDHat program interval was used to estimate the recombination rates. For human, θ = 0.001 and ρ = 100; for rhesus, θ = 0.025 and ρ = 100. The autosomal haplotype data was divided into windows, each including 4000 SNVs, with an overlap of 200 SNVs between adjacent windows. We performed 60 million iterations of interval, with a block penalty of 5 and the first burn-in interations (20 million) discarded. Finally, recombination rates were combined across adjacent (overlapping) windows using previously described methods (Auton and McVean 2007; Auton et al. 2012). Chromosomal regions exhibiting unusual patterns of linkage disequilibrium were removed from the data set using the filters described previously (Auton et al. 2012). If the value of 4Ner estimated between two adjacent SNVs is larger than 100 in humans or rhesus, or if a gap of >50 kb occurs within a window, we set the recombination rate to zero in the region of the surrounding 100 SNVs (±50 SNVs upstream and downstream). The total amount of sequence removed due to all filtering steps was 0.14%. Once the data were filtered as described, the genetic map was assembled. To obtain the statistics of recombination rates on IRh and human autosomes, we split autosomes into 100-kb windows and calculated the genetic distance for each window. We discarded windows in which the proportion of “N”s (unclear nucleotide in the reference genome) was >0.1 or the proportion of SNPs zeroed >0.1. Finally, we calculated the average genetic distance in the remaining 100-kb windows in units of cM/Mb for IRh and human. Supplemental Figures S13 and S14 present plots of the values of 4Ner and LD across 534 orthologous regions of the rhesus and human genomes.
Estimating effective population size and demographic history for rhesus macaques
For all demographic estimations, we have assumed a mutation rate of 1 × 10−8 per site per generation and a generation time of 11 yr. The most appropriate mutation rate to use for this type of analysis remains somewhat controversial for humans, in which a variety of methods have been used to determine the “best” estimate (Ségurel et al. 2014). For rhesus macaques, there is far less empirical evidence. We assume a mutation rate of 1.0 × 10−8 per site per generation for macaques, because a review of the data for humans suggests a rate of 1.0–1.5 × 10−8 per site per generation (Ségurel et al. 2014). Assuming the generation time for rhesus macaques is 11 yr and humans is 25 yr, the per year mutation rates are then 0.9 × 10−9 for macaques and 0.4–0.6 × 10−9 for humans, an appropriate ratio given the demonstrated slowdown in humans and other hominoids. Generation time is set at 11 yr based on the field data that indicate rhesus macaques begin reproduction ∼6 yr of age and can breed until their late teens, resulting in age at median birth of ∼11 yr.
A total of 75 high-coverage (>25×, average 38.1×) Indian rhesus and six high-coverage (>20×, average 34.4×) Chinese rhesus were used to infer demographic history using the stairway plot 2.0b software, the June 2016 v2 beta release (Liu and Fu 2015). Two hundred SFS subsamples with each containing a random selection of two-thirds of the sites with called SNV genotypes were produced. For each subsample, a stairway plot was estimated using a random selection of 120 or six “break points” for the IRh and CRh subsamples, respectively. We used the median of the 200 estimations as the final estimation and the 2.5% and 97.5% estimations as the 95% pseudo-CI of the final estimation. We also used three high-coverage IRh (34770, 36461, and 36470) and three high-coverage CRh (35086, 36013, and 37945) for PSMC estimation. The suggested data processing pipeline was used, starting from BAM files. We used default parameters, except for the option for “pattern.” The pattern “6+29*2” previously used for inferring cynomolgus macaque demographic history (Higashino et al. 2012) and the pattern “4+25*2+4+6” previously used for inferring great apes demographic histories (Prado-Martinez et al. 2013) were used in this study. Due to technical reasons, PSMC was not able to infer the demographic history for one IRh (36461).The final historical reconstructions from the stairway plot analyses based on 75 high-coverage (>25×, average 38.1×) IRh and six high-coverage (>20×, average 34.4×) CRh were used as “Stairway plot – Indian rhesus” and “Stairway plot – Chinese rhesus,” respectively. The PSMC model (p 6+29*2) – Indian rhesus was obtained by averaging the inferred demographic histories of three high-coverage IRh (34770, 36461, and 36470). The PSMC model (p 4+25*2+4+6) – Indian rhesus was obtained by averaging the inferred demographic histories of two high-coverage IRh (34770 and 36470). The PSMC model (p 6+29*2) – Chinese rhesus and the PSMC model (p 4+25*2+4+6) – Chinese rhesus were obtained by averaging the inferred demographic histories of three high-coverage CRh (35086, 36013, and 37945).
Comparison of demographic histories and testing for fit against SFS
Three demographic models, Stairway plot – Indian rhesus, PSMC (p 6+29*2) – Indian rhesus, and PSMC (p 4+25*2+4+6) – Indian rhesus were compared for the fit of the data to the observed SFS for 52 low-coverage (<12×, average 9.5×) Indian rhesus. Similarly, three demographic models, Stairway plot – Chinese rhesus, PSMC (p 6+29*2) – Chinese rhesus, and PSMC (p 4+25*2+4+6) – Chinese rhesus were compared for the fit of the data to the observed SFS for three low-coverage (<12×, average 9.8×) Chinese rhesus. We expect a slight downward bias in the frequencies of rare allele counts, especially singletons, due to low coverage. To avoid bias due to potentially incorrectly called ancestral allele, the observed SFSs were folded, i.e., minor allele counts were used for SFS.
For each model for IRh, 1-Gb length DNA sequences of 52 individuals were simulated using the coalescent-based algorithm SMC (McVean and Cardin 2005; Marjoram and Wall 2006) implemented in the scrm software package (Staab et al. 2015). Similarly, for each model for CRh, 1-Gb length DNA sequences of three individuals were simulated. The simulated folded SFS were compared to the observed SFS from the low-coverage samples (Supplemental Fig. S2). Composite likelihood of the observed SFS was approximated as
where n is the sample size (individuals); ηi is the count of observed sites with a minor allele count of i; pi is the frequency of ηi in the simulated samples; and .
Simulation study assuming a population size recovery
To investigate our ability to infer recent population size growth/recovery, we simulated 200 DNA sequence samples using the scrm software package (Staab et al. 2015). The stairway plot estimation based on 75 high-coverage IRh is assumed to be the true demographic model. The ratio of recombination rate and mutation rate is assumed to be 0.4. For each simulation, 75 individuals were simulated each with 500-Mb DNA sequences. Then stairway plot and PSMC with pattern parameter “6+29*2” and “4+25*2+4+6” were used to infer demographic histories based on the simulated DNA sequences. Only the first individual (of the 75 individuals) was used for PSMC estimations, whereas all 75 individuals were used for the stairway plot estimation.
Estimation of long-term Ne
To estimate the long-term Ne for rhesus macaques, we first estimated θ, where θ = 4 Ne μ and μ is the mutation rate per base pair per generation. We calculated S (the number of segregating sites) and π (genetic diversity) for 100-kb windows on rhesus autosomes to estimate θ (Watterson 1975; Tajima 1983; Fu and Li 1993; Hartl and Clark 2007). We then estimated Ne by dividing θ by 4μ.
SNV density in conserved regions of the genome
A search for conserved genomic regions across low-coverage assemblies of 29 mammalian genomes (Lindblad-Toh et al. 2011) used two different criteria to identify regions of evolutionary conservation. We used the results of the Lindblad-Toh et al. (2011) SiPhy-ω analysis, which identified 4.2% of the human genome as significantly conserved across these other mammals. We used liftOver tools to perform reciprocal liftOver and identify the regions of the rhesus macaque genome that are homologous to this 4.2% of the human genome. We then counted the total number of SNVs and calculated their density per kilobite within this 112.1 Mb. We also calculated the average density of SNVs across the rhesus genome outside this designated 112.1 Mb of conserved sequences. To produce a second look at the same issue, we performed parallel analyses using the 0.4% of the human genome identified by Khurana et al. (2013) as “sensitive” regions. Again, we compared the density per kb of SNVs within and out of the Khurana et al. (2013) “sensitive” segments.
rdnsv analysis of ratio of synonymous to nonsynonymous SNVs
To compare relative densities of nonsynonymous and synonymous variants across distinct data sets, Freudenberg et al. (2012) developed the rdnsv statistic. This parameter is based on the ratio of synonymous and nonsynonymous variants in a set of SNVs, adjusting for the expected proportion under assumptions of neutrality. To compare rdnsv in rhesus macaques versus humans, we identified one-to-one gene homologs in the two species, based on the Ensembl database. We next identified 133 humans from the 1000 Genomes project with whole-genome sequence coverage most closely approximating the rhesus coverage. We used VEP to identify synonymous and nonsynonymous variants (variants scored as nonsynonymous in any transcript were considered nonsynonymous). Population-level rdnsv was calculated using the equations in Freudenberg et al. (2012). Individual sample-level rdnsv was calculated for each individual (rhesus and human).
Analysis of fitness effects
To estimate fitness effects of the rhesus macaque SNVs, we calculated DFE-alpha (Eyre-Walker and Keightley 2009) using the folded site frequency spectrum for a nonsynonymous, and therefore potentially selected data set, comparing that separately with downstream (50 kbp), intron, synonymous, and upstream (50 kbp) neutral data sets drawn from 10,944 rhesus–human orthologous genes. DFE-alpha was estimated using three demographic models: one epoch (constant population), two epoch (one population size change) and three epoch (two population changes) models. The calculated alpha values for each model were used to estimate the proportion of deleterious mutations with effects in four different ranges of fitness effects, on a scale of Nes.
Functional annotation of SNVs
A total of 36,886,925 of the 43,707,518 rhesus SNVs analyzed in this study were successfully mapped to the human reference sequence version hg19 using liftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver). Among them, 28,237,561 SNVs’ reference alleles and 7,083,882 SNVs’ alternative alleles match the corresponding human reference alleles. Those SNVs were then treated as human SNVs and annotated using the WGSA pipeline version 0.5 (Liu et al. 2015). All annotation resources available for version 0.5 were used for the annotation, including five functional prediction scores, eight conservation scores, allele frequencies from four large-scale resequencing studies, and variants in four disease-related databases, among others. Further annotation results are available upon request. The annotation results were further filtered using HGMD version 2015.1 and ClinVar (downloaded 3/15/2015). A total of 164 SNVs were annotated as disease-causing (tagged as DM or DM?) in HGMD or pathogenic (clnsig equals 5) in ClinVar. Based on the functional predictions of SnpEff (Cingolani et al. 2012), dbscSNV (Jian et al. 2014), MetaLR (Dong et al. 2015), GERP++ (Davydov et al. 2010), CADD (Kircher et al. 2014), and fathmm-MKL (Shihab et al. 2015), a list of 179,424 SNVs were nominated as potentially functional and are available on request.
Data access
The sequencing data from this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA251548. NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) accession numbers are listed in Supplemental Table S6. The rhesus SNV calls have been submitted to dbSNP (https://www.ncbi.nlm.nih.gov/snp) under accession numbers ss2031476753–ss2075184272. These calls, along with the SNV sites lifted over to the human genome, are also available for visualization through the UCSC track hub accessible from https://www.hgsc.bcm.edu/non-human-primates/rhesus-monkey-genome-project.
Supplementary Material
Acknowledgments
This work was supported by the Office of Extramural Research, National Institutes of Health (NIH) grants R24-OD11173 to J.R.; R01-EY026045 to J.R. and R.C.; and U54-HG003273 to R.A.G. We thank three anonymous reviewers for constructive criticism of an earlier draft.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.204255.116.
Freely available online through the Genome Research Open Access option.
References
- The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abegg C, Thierry B. 2002. Macaque evolution and dispersal in insular south-east Asia. Biol J Linnean Soc 75: 555–576. [Google Scholar]
- Ahn SM, Kim TH, Lee S, Kim D, Ghang H, Kim DS, Kim BC, Kim SY, Kim WY, Kim C, et al. 2009. The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res 19: 1622–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allentoft ME, Sikora M, Sjögren KG, Rasmussen S, Rasmussen M, Stenderup J, Damgaard PB, Schroeder H, Ahlström T, Vinner L, et al. 2015. Population genomics of Bronze Age Eurasia. Nature 522: 167–172. [DOI] [PubMed] [Google Scholar]
- Antón SC, Potts R, Aiello LC. 2014. Human evolution. Evolution of early Homo: an integrated biological perspective. Science 345: 1236828. [DOI] [PubMed] [Google Scholar]
- Auton A, McVean G. 2007. Recombination rate estimation in the presence of hotspots. Genome Res 17: 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Fledel-Alon A, Pfeifer S, Venn O, Ségurel L, Street T, Leffler EM, Bowden R, Aneas I, Broxholme J, et al. 2012. A fine-scale chimpanzee genetic map from population sequencing. Science 336: 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken TE, Miller JA, Luo R, Bernard A, Bennett JL, Lee CK, Bertagnolli D, Parikshak NN, Smith KA, Sunkin SM, et al. 2015. Spatiotemporal dynamics of the postnatal developing primate brain transcriptome. Hum Mol Genet 24: 4327–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken TE, Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, Dalley RA, Royall JJ, Lemon T, et al. 2016. A comprehensive transcriptional map of primate brain development. Nature 535: 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr CS, Newman TK, Lindell S, Shannon C, Champoux M, Lesch KP, Suomi SJ, Goldman D, Higley JD. 2004. Interaction between serotonin transporter gene variation and rearing condition in alcohol preference and consumption in female primates. Arch Gen Psychiatry 61: 1146–1152. [DOI] [PubMed] [Google Scholar]
- Boyko AR, Williamson SH, Indap AR, Degenhardt JD, Hernandez RD, Lohmueller KE, Adams MD, Schmidt S, Sninsky JJ, Sunyaev SR, et al. 2008. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet 4: e1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone L, Harris RA, Gnerre S, Veeramah KR, Lorente-Galdos B, Huddleston J, Meyer TJ, Herrero J, Roos C, Aken B, et al. 2014. Gibbon genome and the fast karyotype evolution of small apes. Nature 513: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champoux M, Bennett A, Shannon C, Higley JD, Lesch KP, Suomi SJ. 2002. Serotonin transporter gene polymorphism, differential early rearing, and behavior in rhesus monkey neonates. Mol Psychiatry 7: 1058–1063. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella P, Sommella A, Marrocco E, Di Vicino U, Polishchuk E, Garcia Garrido M, Seeliger MW, Polishchuk R, Auricchio A. 2013. Myosin7a deficiency results in reduced retinal activity which is improved by gene therapy. PLoS One 8: e72027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JE, Zhang Y, Brown GA, Semple-Rowland SL. 2004. Cone cell survival and downregulation of GCAP1 protein in the retinas of GC1 knockout mice. Invest Ophthalmol Vis Sci 45: 3397–3403. [DOI] [PubMed] [Google Scholar]
- Corbett-Detig RB, Hartl DL, Sackton TB. 2015. Natural selection constrains neutral diversity across a wide range of species. PLoS Biol 13: e1002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. 2010. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6: e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X. 2015. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 24: 2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD. 2009. Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol Biol Evol 26: 2097–2108. [DOI] [PubMed] [Google Scholar]
- Fawcett GL, Raveendran M, Deiros DR, Chen D, Yu F, Harris RA, Ren Y, Muzny DM, Reid JG, Wheeler DA, et al. 2011. Characterization of single-nucleotide variation in Indian-origin rhesus macaques (Macaca mulatta). BMC Genomics 12: 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson B, Street SL, Wright H, Pearson C, Jia Y, Thompson SL, Allibone P, Dubay CJ, Spindel E, Norgren RB Jr. 2007. Single nucleotide polymorphisms (SNPs) distinguish Indian-origin and Chinese-origin rhesus macaques (Macaca mulatta). BMC Genomics 8: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PJ, Appukuttan B, Simmons E, Landauer N, Stoddard J, Hamon S, Ott J, Ferguson B, Klein M, Stout JT, et al. 2008. Rhesus monkeys and humans share common susceptibility genes for age-related macular disease. Hum Mol Genet 17: 2673–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenberg J, Gregersen PK, Freudenberg-Hua Y. 2012. A simple method for analyzing exome sequencing data shows distinct levels of nonsynonymous variation for human immune and nervous system genes. PLoS One 7: e38087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YX, Li WH. 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazave E, Chang D, Clark AG, Keinan A. 2013. Population growth inflates the per-individual number of deleterious mutations and reduces their mean effect. Genetics 195: 969–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Huddleston J, Chaisson MJ, Hill CM, Kronenberg ZN, Munson KM, Malig M, Raja A, Fiddes I, Hillier LW, et al. 2016. Long-read sequence assembly of the gorilla genome. Science 352: aae0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, Besenbacher S, Magnusson G, Halldorsson BV, Hjartarson E, et al. 2015. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet 47: 435–444. [DOI] [PubMed] [Google Scholar]
- Hartl DH, Clark AG. 2007. Principles of population genetics. Sinauer Associates, Inc, Sunderland, MA. [Google Scholar]
- Hasan MK, Feeroz MM, Jones-Engel L, Engel GA, Kanthaswamy S, Smith DG. 2014. Diversity and molecular phylogeny of mitochondrial DNA of rhesus macaques (Macaca mulatta) in Bangladesh. Am J Primatol 76: 1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes BJ, Lewin HA, Goddard ME. 2013. The future of livestock breeding: genomic selection for efficiency, reduced emissions intensity, and adaptation. Trends Genet 29: 206–214. [DOI] [PubMed] [Google Scholar]
- Hernandez RD, Hubisz MJ, Wheeler DA, Smith DG, Ferguson B, Rogers J, Nazareth L, Indap A, Bourquin T, McPherson J, et al. 2007. Demographic histories and patterns of linkage disequilibrium in Chinese and Indian rhesus macaques. Science 316: 240–243. [DOI] [PubMed] [Google Scholar]
- Higashino A, Sakate R, Kameoka Y, Takahashi I, Hirata M, Tanuma R, Masui T, Yasutomi Y, Osada N. 2012. Whole-genome sequencing and analysis of the Malaysian cynomolgus macaque (Macaca fascicularis) genome. Genome Biol 13: R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvilsom C, Qian Y, Bataillon T, Li Y, Mailund T, Sallé B, Carlsen F, Li R, Zheng H, Jiang T, et al. 2012. Extensive X-linked adaptive evolution in central chimpanzees. Proc Natl Acad Sci 109: 2054–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian X, Boerwinkle E, Liu X. 2014. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res 42: 13534–13544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthaswamy S, Smith DG. 2004. Effects of geographic origin on captive Macaca mulatta mitochondrial DNA variation. Comp Med 54: 193–201. [PubMed] [Google Scholar]
- Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, et al. 2013. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science 342: 1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack JB, Cardeno CM, Crepeau MW, Taylor W, Corbett-Detig RB, Stevens KA, Langley CH, Pool JE. 2015. The Drosophila genome nexus: a population genomic resource of 623 Drosophila melanogaster genomes, including 197 from a single ancestral range population. Genetics 199: 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler EM, Bullaughey K, Matute DR, Meyer WK, Ségurel L, Venkat A, Andolfatto P, Przeworski M. 2012. Revisiting an old riddle: What determines genetic diversity levels within species? PLoS Biol 10: e1001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler EM, Gao Z, Pfeifer S, Ségurel L, Auton A, Venn O, Bowden R, Bontrop R, Wall JD, Sella G, et al. 2013. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 339: 1578–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2011. Inference of human population history from individual whole-genome sequences. Nature 475: 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang Y, Wu X, Ao H, Li L, An Z. 2014. Mammalian evolution in Asia linked to climate changes. In Late Cenozoic climate change in Asia (ed. An Z), pp. 435–490. Springer Science+Business Media, Dordrecht, Netherlands. [Google Scholar]
- Li AH, Morrison AC, Kovar C, Cupples LA, Brody JA, Polfus LM, Yu B, Metcalf G, Muzny D, Veeraraghavan N, et al. 2015. Analysis of loss-of-function variants and 20 risk factor phenotypes in 8,554 individuals identifies loci influencing chronic disease. Nat Genet 47: 640–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillo C, Kitamoto J, Liu X, Quint E, Steel KP, Williams DS. 2003. Mouse models for Usher syndrome 1B. Adv Exp Med Biol 533: 143–150. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. 2011. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478: 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Fu YX. 2015. Exploring population size changes using SNP frequency spectra. Nat Genet 47: 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, White S, Peng B, Johnson AD, Brody JA, Li AH, Huang Z, Carroll A, Wei P, Gibbs R, et al. 2015. WGSA: an annotation pipeline for human genome sequencing studies. J Med Genet 53: 111–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffredo JT, Maxwell J, Qi Y, Glidden CE, Borchardt GJ, Soma T, Bean AT, Beal DR, Wilson NA, Rehrauer WM, et al. 2007. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J Virol 81: 8827–8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, et al. 2010. Whole-genome sequencing in a patient with Charcot–Marie–Tooth neuropathy. N Engl J Med 362: 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjoram P, Wall JD. 2006. Fast “coalescent” simulation. BMC Genet 7: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen CY, Chou A, Ienasescu H, et al. 2014. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 42(Database issue): D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GA, Cardin NJ. 2005. Approximating the coalescent with recombination. Philos Trans R Soc Lond B Biol Sci 360: 1387–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P. 2004. The fine-scale structure of recombination rate variation in the human genome. Science 304: 581–584. [DOI] [PubMed] [Google Scholar]
- Millán JM, Aller E, Jaijo T, Blanco-Kelly F, Gimenez-Pardo A, Ayuso C. 2011. An update on the genetics of Usher syndrome. J Ophthalmol 2011: 417217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday RS, Zhong M, Quazi F. 2009. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochim Biophys Acta 1791: 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Estrada A, Gignoux CR, Fernandez-López JC, Zakharia F, Sikora M, Contreras AV, Acuña-Alonzo V, Sandoval K, Eng C, Romero-Hidalgo S, et al. 2014. Human genetics. The genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science 344: 1280–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley RJ. 2012. A review of the Cenozoic palaeoclimate history of Southeast Asia. In Biotic evolution and environmental change in Southeast Asia (ed. Gower DJ, Johnson K), pp. 79–114. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Osada N, Hashimoto K, Kameoka Y, Hirata M, Tanuma R, Uno Y, Inoue I, Hida M, Suzuki Y, Sugano S, et al. 2008. Large-scale analysis of Macaca fascicularis transcripts and inference of genetic divergence between M. fascicularis and M. mulatta. BMC Genomics 9: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuánez HN, Horvath JE, Moreira MA, Kessing B, Pontius J, Roelke M, Rumpler Y, et al. 2011. A molecular phylogeny of living primates. PLoS Genet 7: e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Melsted P, Marioni JC, Wang Y, Bainer R, Pickrell JK, Michelini K, Zehr S, Yoder AD, Stephens M, et al. 2012. Comparative RNA sequencing reveals substantial genetic variation in endangered primates. Genome Res 22: 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KA, Bales KL, Capitanio JP, Conley A, Czoty PW, ’t Hart BA, Hopkins WD, Hu SL, Miller LA, Nader MA, et al. 2014. Why primate models matter. Am J Primatol 76: 801–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, Veeramah KR, Woerner AE, O'Connor TD, Santpere G, et al. 2013. Great ape genetic diversity and population history. Nature 499: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, Dominiczak A, Morris A, Porteous D, Smith B, et al. 2016. Timing, rates and spectra of human germline mutation. Nat Genet 48: 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhesus Macaque Genome Sequencing and Analysis Consortium, Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, Mardis ER, Remington KA, Strausberg RL, Venter JC, et al. 2007. Evolutionary and biomedical insights from the rhesus macaque genome. Science 316: 222–234. [DOI] [PubMed] [Google Scholar]
- Richard AF, Goldstein SJ, Dewar RE. 1989. Weed macaques: the evolutionary implications of macaque feeding ecology. Int J Primatol 10: 569–594. [Google Scholar]
- Rogers J, Gibbs RA. 2014. Comparative primate genomics: emerging patterns of genome content and dynamics. Nat Rev Genet 15: 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Garcia R, Shelledy W, Kaplan J, Arya A, Johnson Z, Bergstrom M, Novakowski L, Nair P, Vinson A, et al. 2006. An initial genetic linkage map of the rhesus macaque (Macaca mulatta) genome using human microsatellite loci. Genomics 87: 30–38. [DOI] [PubMed] [Google Scholar]
- Rogers J, Raveendran M, Fawcett GL, Fox AS, Shelton SE, Oler JA, Cheverud J, Muzny DM, Gibbs RA, Davidson RJ, et al. 2013. CRHR1 genotypes, neural circuits and the diathesis for anxiety and depression. Mol Psychiatry 18: 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romiguier J, Gayral P, Ballenghien M, Bernard A, Cahais V, Chenuil A, Chiari Y, Dernat R, Duret L, Faivre N, et al. 2014. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature 515: 261–263. [DOI] [PubMed] [Google Scholar]
- Sankararaman S, Patterson N, Li H, Pääbo S, Reich D. 2012. The date of interbreeding between Neandertals and modern humans. PLoS Genet 8: e1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, Patterson N, Reich D. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507: 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satkoski J, George D, Smith DG, Kanthaswamy S. 2008. Genetic characterization of wild and captive rhesus macaques in China. J Med Primatol 37: 67–80. [DOI] [PubMed] [Google Scholar]
- Schoenebeck JJ, Ostrander EA. 2014. Insights into morphology and disease from the dog genome project. Annu Rev Cell Dev Biol 30: 535–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder H, Ávila-Arcos MC, Malaspinas AS, Poznik GD, Sandoval-Velasco M, Carpenter ML, Moreno-Mayar JV, Sikora M, Johnson PL, Allentoft ME, et al. 2015. Genome-wide ancestry of 17th-century enslaved Africans from the Caribbean. Proc Natl Acad Sci 112: 3669–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SC, Miller W, Ratan A, Tomsho LP, Giardine B, Kasson LR, Harris RS, Petersen DC, Zhao F, Qi J, et al. 2010. Complete Khoisan and Bantu genomes from southern Africa. Nature 463: 943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JJ, Roach DJ, Thomas JH, Shendure J. 2014. Primate evolution of the recombination regulator PRDM9. Nat Commun 5: 4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ségurel L, Wyman MJ, Przeworski M. 2014. Determinants of mutation rate variation in the human germline. Annu Rev Genomics Hum Genet 15: 47–70. [DOI] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci 110: 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day IN, Gaunt TR, Campbell C. 2015. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31: 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DG, McDonough J. 2005. Mitochondrial DNA variation in Chinese and Indian rhesus macaques (Macaca mulatta). Am J Primatol 65: 1–25. [DOI] [PubMed] [Google Scholar]
- Spivakov M, Akhtar J, Kheradpour P, Beal K, Girardot C, Koscielny G, Herrero J, Kellis M, Furlong EE, Birney E. 2012. Analysis of variation at transcription factor binding sites in Drosophila and humans. Genome Biol 13: R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staab PR, Zhu S, Metzler D, Lunter G. 2015. scrm: efficiently simulating long sequences using the approximated coalescent with recombination. Bioinformatics 31: 1680–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples J, Nickerson DA, Below JE. 2013. Utilizing graph theory to select the largest set of unrelated individuals for genetic analysis. Genetic Epidemiol 37: 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples J, Qiao D, Cho MH, Silverman EK, University of Washington Center for Mendelian G, Nickerson DA, Below JE. 2014. PRIMUS: rapid reconstruction of pedigrees from genome-wide estimates of identity by descent. Am J Hum Genet 95: 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer CB. 2012. The status of Homo heidelbergensis (Schoetensack 1908). Evol Anthropol 21: 101–107. [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry B. 2011. The macaques: a double-layered social organization. In Primates in perspective, 2nd ed. (ed Campbell CJ, et al. ), pp. 229–241. Oxford University Press, Oxford, UK. [Google Scholar]
- Vallender EJ, Priddy CM, Hakim S, Yang H, Chen GL, Miller GM. 2008. Functional variation in the 3′ untranslated region of the serotonin transporter in human and rhesus macaque. Genes Brain Behav 7: 690–697. [DOI] [PubMed] [Google Scholar]
- Vallender EJ, Ruedi-Bettschen D, Miller GM, Platt DM. 2010. A pharmacogenetic model of naltrexone-induced attenuation of alcohol consumption in rhesus monkeys. Drug Alcohol Depend 109: 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, Gutenkunst RN, Woerner AE, Watkins JC, Hammer MF. 2014. Evidence for increased levels of positive and negative selection on the X chromosome versus autosomes in humans. Mol Biol Evol 31: 2267–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lu J, Yu J, Gibbs RA, Yu F. 2013. An integrative variant analysis pipeline for accurate genotype/haplotype inference in population NGS data. Genome Res 23: 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. 1975. On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. 2008. The complete genome of an individual by massively parallel DNA sequencing. Nature 452: 872–876. [DOI] [PubMed] [Google Scholar]
- Xue Y, Prado-Martinez J, Sudmant PH, Narasimhan V, Ayub Q, Szpak M, Frandsen P, Chen Y, Yngvadottir B, Cooper DN, et al. 2015. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 348: 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. 2013. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369: 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Zhou Z, Lindell SG, Higley JD, Ferguson B, Thompson RC, Lopez JF, Suomi SJ, Baghal B, Baker M, et al. 2012. The rhesus macaque is three times as diverse but more closely equivalent in damaging coding variation as compared to the human. BMC Genet 13: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.