

Abstract
Monoclonal antibodies can bind with high affinity and high selectivity to their targets. As a tool in therapeutics or diagnostics, however, their large size (∼150 kDa) reduces penetration into tissue and prevents passive cellular uptake. To overcome these and other problems, minimized protein scaffolds have been chosen or engineered, with care taken to not compromise binding affinity or specificity. An alternate approach is to begin with a minimal non‐antibody scaffold and select functional ligands from a de novo library. We will discuss the structure, production, applications, strengths, and weaknesses of several classes of antibody‐derived ligands, that is, antibodies, intrabodies, and nanobodies, and nonantibody‐derived ligands, that is, monobodies, affibodies, and macrocyclic peptides. In particular, this review is focussed on macrocyclic peptides produced by the Random non‐standard Peptides Integrated Discovery (RaPID) system that are small in size (typically ∼2 kDa), but are able to perform tasks typically handled by larger proteinaceous ligands.
Keywords: antibodies, cocrystallization ligands, macrocyclic peptides, RaPID system
1. Introduction
The development of a method to obtain monoclonal antibodies (mAbs) against a specific antigen in large quantity in mouse cells by Köhler and Milstein1 has led to a wide variety of successful applications with a profound impact on medicine. mAbs are used in a wide range of fields such as transplantation, autoimmune, and infectious diseases.2 In oncology, immune checkpoint therapy using antibodies that block CTLA‐4 (cytotoxic T lymphocyte antigen‐4) and programmed death ligand‐1 (PD‐1) pathways were successful in clinical trials.3 Other examples include Abciximab, an antagonist against the platelet membrane glycoprotein IIb/IIIa receptor,4 agonistic CD40 mAbs which have shown promising results,5 and ramucirumab, a recombinant human IgG1 neutralizing antibody for vascular endothelial growth factor receptor (VEGFR)−2.6 Antibodies have been developed against variety of diseases and pathogens for diagnostic use, for example, for the detection of Dengue virus7 or in a urine‐based multiplex assay against bladder cancer.8 Antibody‐inspired libraries have been engineered from smaller proteins with a well‐defined scaffold and randomized regions analogous in function to antibody's hypervariable loops (Figure 1A).
Figure 1.
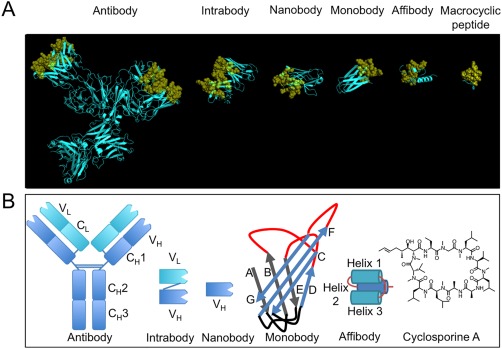
A comparison of ligands with different scaffolds. (A) From left to right, X‐ray crystal structures of: intact murine IgG1 monoclonal antibody for phenobarbital (PDB: 1IGY); single domain intrabody that binds to activated GTP‐bound RAS (PDB: 2UZI); humanized NbBcII10 nanobody that binds to BcII β‐lactamase (PDB: 3EAK); monobody ySMB‐9 that binds to human small ubiquitin‐like modifier 1 (PDB: 3RZW); affibody scaffold Z domain of Staphylococcal protein A (PDB: 1Q2N); macrocyclic peptide aCAP (PDB: 3WMG). Variable binding positions have been coloured yellow and are displayed in cartoon format and semi‐transparent spheres. Structural or non‐varied regions are coloured cyan and are displayed in cartoon format. X‐ray crystal structures were rendered in PyMOL v1.5.0.4. (B) Schematic structures of all ligands discussed, from left to right: monoclonal antibody, intrabody, nanobody, monobody, affibody and a chemical structure of a natural macrocyclic peptide, cyclosporin A
Mini‐proteins or peptides that are smaller than the aforementioned protein biologics, but larger than small molecules, are of great interest, mainly due to the potential for intracellular delivery. Unfortunately, peptide scaffolds have reduced intramolecular bonds and a well‐defined active structure is more difficult to maintain due to entropic penalties. One strategy used by nature and researchers is macrocyclization of the peptide scaffold using a non‐reducible bond to limit the conformational freedom of the peptide. In general, functionally active macrocyclic peptides show higher affinity for their targets and higher resistance to degradation by proteases when compared to their linear counterparts.9, 10 In addition, macrocyclization promotes the formation of intramolecular bonds, which reduces hydrogen bonding to water and increases membrane penetrating potential. Many non‐ribosomally synthesized peptides11 and ribosomally synthesized and post‐translationally modified peptides (RiPPs)12 possess a macrocyclic scaffold. In the search for therapeutic and diagnostic agents, researchers have noted the value of the macrocyclic scaffold and have integrated it into the development of peptide‐based ligands and ligand libraries.
Phage display is a powerful high throughput method for the production and in vitro screening of ligand libraries (Figure 2A).13 Because phage display does not depend on an animal's immune system, this method can be used to screen for ligands against highly conserved intracellular proteins,14 or even nonimmunogenic15 or toxic antigens.16 For the screening and production of smaller peptide‐based ligands, particularly macrocyclic peptides, other in vitro screening methods, such as mRNA display,17, 18, 19 are available. This review will briefly discuss the structure, production and application of certain biologic ligands and compare them to comparatively smaller macrocyclic peptide ligands, in particular in vitro selected macrocyclic peptides identified using the Random nonstandard Peptide Integrated Discovery (RaPID) system (Table 1).9, 20
Figure 2.
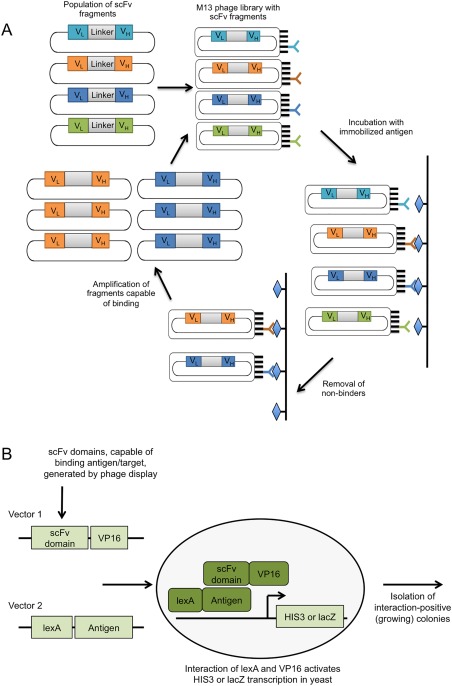
(A) Production of mAbs via antibody phage display. A library of scFvs is displayed on M13 phage particles. Phage particles are then incubated with immobilised antigen, followed by removal of phage particles expressing non‐binding scFvs. Remaining phages are eluted and amplified in E. coli for further screening or continued selection. (B) Production of intrabodies via Intracellular Antibody Capture Technology Phage display is used to screen a library of scFvs, generating a library enriched for antigen‐specific scFvs. These are then used as prey in the yeast antibody–antigen interaction assay and challenged intracellular with antigen bait. Interaction of lexA (bound to the antigen) and VP16 (bound to the scFv domain) activates reporter gene transcription (i.e., HIS3, LacZ), and indicates successful intracellular expression and binding of the scFvs
Table 1.
Approximate sizes and a comparison of the advantages and disadvantages of the ligands discussed in this review
| Molecule | Approximate Size (kDa) | Advantages | Disadvantages |
|---|---|---|---|
| Agonist or antagonist activity | Limited penetration | ||
| Activates immune system | Limited distribution | ||
| Antibody | 150 | Conjugation to molecules (therapeutics, imaging) | Production dependent on animal immunization |
| Blocks protein–protein interactions | Costly and laborious production | ||
| High avidity due to bivalency | |||
| Intrabody | 28 | Intracellular action | Selection not amenable for transcriptional activators or repressors |
| Toxic effects of some targets, when expressed in yeast | |||
| Binds epitopes unreachable by antibodies | High uptake in liver and kidneys | ||
| Nanobody | 25 | High tissue penetration | |
| Stability in adverse environments | |||
| Single domain facilitates cloning | |||
| Monobody | 10 | Stability (thermal/reducing conditions) | High renal clearance |
| Can be constructed as multi‐domain (modularity) | |||
| High tissue penetration | High renal clearance | ||
| Can be constructed as multi‐domain (modularity) | |||
| Affibody | 8 | Can block protein‐to‐protein interactions | |
| High solubility | |||
| High stability | |||
| Can be chemically synthetized and altered | |||
| mRNA display libraries reach 1014 unique sequences | Limited delivery across cell membrane | ||
| Chemically modifiable | High renal clearance | ||
| Protease resistant | |||
| Rapid screening | |||
| Use of npAAs | |||
| Macrocyclic | 2 | High tissue penetration | |
| peptide | Ability to bind flat surfaces and pockets | ||
| Produced in animal‐ and cell‐free systems | |||
| Semi‐rigid structure | |||
| Ability to bind non‐druggable targets |
2. Antibody‐Based Ligand Scaffold
2.1. Antibodies
Conventional mAbs1 consist of constant regions (F c) and variable regions (F v) linked by a flexible hinge (Figure 1B). Each F v region consists of a heavy variable domain (V H) attached to another heavy F c and light variable domain (V L) attached to a light F c. The two F v regions are identical and contain the same antigen‐binding regions. Anti‐antibody response was greatly reduced by constructing the human F c‐murine F v chimera. Smaller variants of mAbs are fragment antigen binding (Fab) (∼50 kDa)21 and single‐chain variable fragment (scFv) (∼25 kDa). Fabs consist of one V L and V H domain linked to their respective light and heavy constant domains, while scFvs consists of one V L and V H domain fused by a flexible linker.22
Their high target affinity and specificity makes mAbs valuable therapeutic and diagnostic agents.23 Antibodies can function either as agonists24 or antagonists,25 modulate the immune system via their F c portion,26 or deliver drugs conjugated to the antibodies to specific sites.27, 28 In structural biology, antibodies also have been used as cocrystallization ligands. X‐ray crystal structures of membrane proteins have proven to be difficult to obtain, as their crystallization faces several challenges. The abundant hydrophobic surface of membrane proteins, particularly multi‐pass transmembrane proteins, requires the use of detergents to keep the protein in solution. Unfortunately, the partial micelle surrounding the protein could obscure hydrophilic protein surfaces needed for specific crystal contacts. Even with the use of partial micelles, membrane proteins are often unstable and addition of lipids is required.29 A cocrystallization ligand can be used to stabilise the membrane protein and extend hydrophilic surfaces beyond the micelle and provide the protein surfaces needed for crystal contact.30 The conformational flexibility of transmembrane proteins is another major issue in crystallography. In a study addressing these difficulties, mAbs have been shown to function as cocrystallization ligands and stabilise membrane proteins for X‐ray crystallography, as was demonstrated with the crystallization of the human β2 adrenergic receptor, a G protein‐coupled receptor.31 mAbs are also used extensively in biological imaging.32, 33, 34 However, due to their relatively large size and dimensions,35 passive membrane penetration and distribution in tissue is often limited.36 The long half‐lives of several days to weeks may cause high background levels in imaging due the time needed before any nonspecific binding molecules are cleared from circulation. This long half‐life is caused by the protection the F c region provides against transport to the lysosome compartment and degradation.22, 37 In therapeutics, however, this protection and extended half‐life is advantageous as it reduces the dose and number of administrations required for therapeutic effect.38 This trade‐off is seen in most ligands.
2.2. Intrabodies
One class of antibodies most often used is the intracellular antibody (intrabody), a class of scFv.39 Intrabodies consist of just the variable regions of the heavy and light chains, linked together by a flexible polypeptide linker or a disulfide bond to form a complex of ∼28 kDa (Figure 1B). They retain essential antigen binding regions,40 and can be introduced and expressed inside cells with recombinant adenovirus as gene delivery system.41, 42, 43 Although use of full‐length mAb44, 45 within a cell is possible, scFv are more commonly used.
Various strategies exists for the selection of functional intrabodies.46 Phage display is often used for the in vitro selection and identification of functional intrabodies. Unfortunately, the reducing environment within the cell cytoplasm or nucleus prevents the formation of intrachain disulfide bonds that are vital for the structural integrity of most intrabodies, leading to insolubility, instability or incorrect folding of the in vitro selected protein ligand. Although techniques used for the production of functional ligands that are dependent on disulfide bonds are available, such as producing the disulfide bonds in the periplasmic space or altering the reducing environment of the cytoplasm,47, 48, 49 functional scFv ligands that are stable even in the reducing environment of the target cell cytoplasm are desired. Therefore, a subsequent selection procedure, intracellular antibody capture technology (IACT) was developed by Visintin et al. (Figure 2B).50 Libraries originating from selections using phage‐display are used as the input library for the yeast two‐hybrid system (Y2H) to select for intracellular antibodies based on their ability to bind a target inside cells, which could be indicative of the ligand's stability inside the cell.50 Limitations of the Y2H technique are that interactions involving transcriptional activators or repressors cannot be performed, while some proteins have toxic effects when targeting the yeast nucleus.51, 52
Intrabodies are used to determine the function of intracellular proteins by interfering with their function.46, 53 An intrabody against E6 oncoprotein of human papillomavirus 16 (HPV16) was shown to induce impaired growth of HPV16‐positive tumour cells.54 Other intrabodies have been selected against the precursor of nerve growth factor (proNGF) signalling,55 or against protein tyrosine kinase Syk to elucidate the exact mechanisms of FcεRI signal transduction.56 The potential of intrabodies as therapeutics was shown in mice, where intrabodies against a‐disintegrin‐and‐metalloproteinase‐with‐thrombospondin‐like‐sequences‐5 (ADAMTS5) (also termed aggrecanase‐2) delayed cartilage breakdown due to osteoarthritis.57 A successful clinical (Phase I) study using intrabodies for gene therapy‐mediated delivery targeting receptor tyrosine kinase ERBB‐2‐overexpressing ovarian cancer cells found limited side effects.43 Clinical benefits were also limited, as gene therapy requires gene transfer into a large proportion of tumour cells. Inefficiency of the gene delivery system could be a possible explanation for this outcome.43 While progress has been made in the development of viral and nonviral transduction methods, more studies on the safety profile and efficacy of such methods are required.43, 58, 59
2.3. Nanobodies
Nanobodies (Nbs) are Camelidae antibodies discovered by Hamers‐Casterman et al.60 Besides conventional antibodies, members of the Camelidae family also contain Nbs, antibodies consisting of only the variable heavy chain (VHH) domain (12–15 kDa)61 (Figure 1B). Humanization of the Nbs and their similarity to the human type 3 VH domain results in a low immunogenic potential. Because of their simple design, Nbs are easy to clone and express in high yields, making them amenable to phage display.62
The first in‐human application of radiolabeled Nbs recently entered phase II studies. Human epidermal growth factor receptor (HER) 2‐Nbs were used as probes in positron emission tomography (PET)/computer tomography (CT) imaging of breast carcinoma patients.63 The high binding capacity of Nbs results in high sensitivity64 combined with high specificity.65 The relatively short half‐life, tumour penetration ability and fast extravasation lead to low background noise.66 Nbs accumulate in the kidneys, leading to a higher signal and radiation dose in the kidneys.67 Although the short serum half‐life profiles are favourable for use in nuclear imaging, most therapeutic applications require slower drug clearance. Fusion to serum albumin proved a successful strategy to prolong the half‐life of anti‐epidermal growth factor receptor (EGFR) Nbs.68 Nbs have also been used as crystallization chaperones to lock proteins in a particular conformation and stabilise flexible regions. Nbs were successfully used as cocrystallization ligands of the T2 mechanosensitive channel of small conductance (T2 MscS channel) from Thermoplasma volcanium.69
3. Artificial Ligands Based on Non‐Antibody Protein Scaffolds
3.1. Monobodies
Monobodies (also known as Trinectins70 or Adnectins71 are synthetic binding proteins based on the tenth human fibronectin type III domain (FN3) developed by the Koide group72 (Figure 1B). FN3 consists of ∼93 residues and has a size of ∼10 kDa.73, 74 FN3 is stable, even at high temperatures and can be produced in reducing conditions due to its lack of disulfide bridges or free cysteines.75 Three surface loops close to its N‐terminus function as antigen‐recognition loops (BC, DE, FG) and are structurally analogous to the complementarity‐determining regions of antibodies. Monobody libraries are produced by diversification of one to all of the antigen‐recognition loops. Incorporating a stretch of random nucleotides (the so‐called NNK codon, where N is A, C, G, or T, and K is T or G) in the antigen‐recognition loop regions of the monobody gene is a simple and relatively inexpensive method to create such diversity. Random libraries have successfully been generated via phage display,76 mRNA display,77 yeast surface display.78 Monobodies are currently in development for therapeutic use and have come as far as clinical trials, such as in the case of an inhibitor of VEGFR2.79
Human ephrin receptor tyrosine kinase A2 (EphA2) is a marker protein for various tumours and monobodies against EphA2 were developed by yeast surface display and are able to target tissue expressing EphA2, but showed no cytotoxicity.80 VEGFR‐2 is involved in VEGF‐driven tumours such as glioblastoma and showed promising results in mice when targeted with monobodies.81 In a phase II clinical study, this monobody targeting the extracellular domain of VEGFR‐2 was further tested, but failed to elicit a response.82
3.2. Affibodies
Affibodies were derived from the B‐domain of the immunoglobulin‐binding region of staphylococcal protein A by Nord et al.83 They are 58 amino acid long peptides without cysteine residues and quickly fold into a small three‐helix bundle structure (Figure 1B).84 Key positions of the B‐domain were mutated to enhance chemical stability and the B‐domain was renamed the Z‐domain.85 The highly thermostable and soluble final ligand has a molecular weight of ∼6 kDa.86 The amino acids of 13 solvent‐accessible surface positions on the helices that make up the Fc‐binding surface of the Z‐domain,87 helix one and two, can be randomized to create libraries.88, 89
Affibodies are small enough to be chemically synthesized and augmented with fluorescent probes90 and are often used in imaging due to their high in vivo stability, quick tumour targeting, high kidney uptake and lower uptake in normal tissue.91, 92 EGFR‐targeting radiolabeled affibodies were shown to be promising PET probes for detecting EGFR‐expression in mice.93 As therapeutics, they can be used against various targets, as was demonstrated with an affibody targeting HER2 bound to Pseudomonas exotoxin A.94 Another study investigated the potential of affibodies in radionuclide therapy and found higher accumulation of radionuclides in tumours than kidneys, making this a promising use of affibodies.95
4. Ligands Based on a Macrocyclic Peptide Scaffold
4.1. Macrocyclic peptide scaffold
Naturally occurring macrocyclic peptides are known to have several functions, such as immunosuppressant like cyclosporin A (Figure 1B).96 Macrocyclization via a non‐reducible covalent bond biases the peptide toward its active configuration.97, 98 A notable feature of cyclosporin A is the incorporation of several non‐proteinaceous amino acids (npAAs) such as L‐2‐aminobutyric acid, D‐Ala and (4R)‐4‐[(E)‐2‐butenyl]‐4‐methyl‐L‐threonine as well as seven N‐methylated peptide bonds.99 These npAAs can prevent protease degradation as these may limit recognition by proteases.100 Recognition by proteases is particularly difficult in the presence of chemical modifications, such as N‐methylation of the polyamide backbone. Another benefit is that the reduced number of hydrogen bond donors improves diffusion across the cellular membrane and enhances oral bioavailability101 and is most strongly demonstrated by cyclosporin A.102
Most small peptides suffer from fast renal clearance, as their small size makes them susceptible to rapid excretion by glomerular filtration. Increasing their size to the molecular weight threshold for glomerular filtration (50–70 kDa) increases in vivo half‐life. A commonly used method for increasing half‐life has been conjugation to large polymers such as polyethylene glycol (PEG),103 or plasma proteins with long serum half‐life such as albumin.104 An alternative approach is conjugation to albumin‐binding molecules such as fatty acids.105 While conjugation to proteins increases size and alters pharmacokinetics, the steric hindrance of macromolecules may be detrimental to not only target binding but also oral bioavailability and membrane penetration of small peptides, prompting the conjugation of smaller chemical groups that promote association with in vivo circulating proteins.106
Nature also employs bridging disulfide bonds in macrocyclic peptides to further promote stabilization of the active 3D structure. Disulfide‐rich head‐to‐tail macrocycles are extremely stable peptide scaffolds and are being engineered into therapeutic agents through the grafting of active peptides into the variable regions of the cyclic scaffolds for enhanced stability.107 In addition to the enhanced stability provided by the disulfide‐rich macrocyclic peptide scaffold, the cyclotide MCoTI‐II also displays cell‐penetrating ability, which opens the opportunity for targeting intracellular proteins.108 Furthermore, the cell‐penetrating ability of MCoTI‐II can be further enhanced through the grafting of a cell penetrating peptide, CTP512, into loop 1.109 Sunflower trypsin inhibitor (SFTI‐1) has been shown to be an excellent platform for the development of novel protease inhibitors,110, 111, 112, 113 and recently, θ‐defensin has been added to the macrocyclic peptide scaffolds used for grafting.114, 115, 116
The minimal macrocyclic scaffold is one that uses no predetermined template and is simply the variable loop with the end residues coupled. Although several new technologies for the production and selection of de novo macrocyclic peptide ligands exist,117, 118 due to space limitation we will focus on macrocyclic peptides produced the Random non‐standard Peptides Integrated Discovery (RaPID) system developed by the Suga group (Figure 3).20 The RaPID system combines the Flexible In vitro Translation (FIT) system119 for the production of macrocyclic peptide libraries with mRNA display17 for the selection of the functionally active ligands. Where phage display libraries are often in the order of billions, mRNA display libraries may contain trillions of variants, increasing the chance of identifying high‐affinity binders.120 Although the macrocyclization method commonly used for the RaPID system98 cannot be used to produce potentially stabilizing disulfide bonds114 that bridge residues of the macrocyclic scaffold,121 allowing the macrocyclic peptide scaffold to retain some flexibility may promote passive membrane penetration.122 The RaPID system was successfully used to discover inhibitors of E6 associated protein (E6AP),9 sirtuin 2 (SiRT2)123 and protein kinase AKT2.124 The former two studies demonstrate the integration of npAAs not only for the formation of the macrocyclic scaffold but also increasing bioavailability potential and mechanistic inhibition, respectively. Inspired by the versatility of antibodies, the Suga group is finding new applications for in vitro selected macrocyclic peptides beyond enzyme inhibition.
Figure 3.
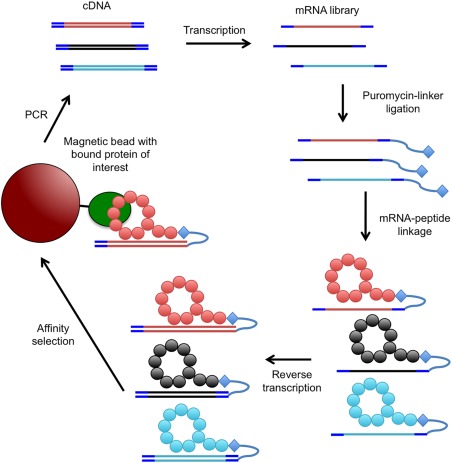
Production of macrocyclic peptides via the RaPID system. The FIT system and an mRNA library is used to express a macrocyclic peptide library. Peptides are covalently ligated to their respective mRNAs via a DNA oligonucleotide that is complementary to the 3'‐end of the mRNA templates and has a 3'‐PEG‐linked puromycin. mRNA is reverse transcribed to create a double‐stranded mRNA‐cDNA hybrid. The peptide‐mRNA‐cDNA complexes are then used for selection against the protein of interest bound to magnetic beads. cDNA bound to the protein of interest is amplified by PCR for use in a subsequent round of selection or analysis by sequencing
4.2. Applications for macrocyclic peptides discovered using the rapid system
Tanaka et al. hypothesized that macrocyclic peptide could be used as cocrystallization ligands in the same manner as proteinaceous cocrystallization ligands.125, 126 Using the RaPID system, several macrocyclic peptides ligands that bind to Pyrococcus furiosus multidrug and toxic compound extrusion (PfMATE) transporter were identified.125, 126 Without the use of a cocrystallization ligand, PfMATE crystallization was not consistently reproducible, presumably due to the transporter's flexibility in solution. The in vitro selected MaD5 and MaD3S peptides have lasso‐like structures and bind and lock the transporter in its outward‐open conformation. The minicycle of the lasso‐shaped peptides fill the substrate‐binding cavity located in the N‐lobe with high shape complementarity. The MaL6 peptide, in contrast, does not interact with the N‐lobe cavity, although it does bind the central cleft mainly through hydrophobic interactions (Figure 4A). These were the first 3D structures of macrocyclic peptides identified using the RaPID system bound to their target protein, and they were found to bind to pockets similar to the manner of binding of a small molecule. At the time, it was not known if macrocyclic peptides produced by the RaPID system were limited to pocket binding or could bind to less contoured surfaces like those involved in protein–protein interactions.
Figure 4.
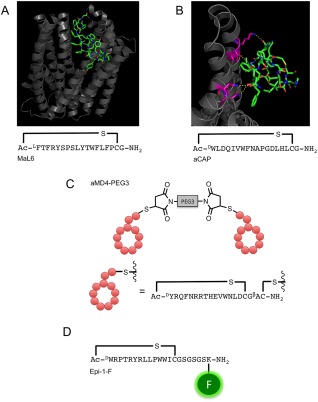
Examples of macrocyclic‐peptide ligands identified using the RaPID system. (A) Crystal structure of MaL6:PfMATE (PDB: 3WBN) and the sequence of MaL6. MaL6 is represented in stick format and PfMATE is represented in cartoon format. (B) Crystal structure of aCAP:CmABCB1 (PDB: 3WMG) and the sequence of aCAP. aCAP is represented in stick format and a single monomer unit of CmABCB1 is represented in cartoon format. CmABCB1 residues involved in specific interactions with aCAP are coloured magenta. Hydrogen bonds are shown in yellow dashes. (C) Schematic representation of a Met‐binding dimer‐macrocylic‐peptide, aMD4‐PEG3. Figure adapted from Ref. 10. (D) EpCAM‐binding fluorescent macrocyclic‐peptide Epi‐1‐F. X‐ray crystal structures were rendered in PyMOL v1.5.0.4
The concern over limited binding potential was addressed by a subsequent in vitro selection for macrocyclic peptides that bind to a homodimeric eukaryotic ABC transporter from Cyanidioschyzon merolae (CmABCB1).127 The ligand‐free structure of CmABCB1 was solved at a resolution of 2.75 Å. The in vitro selected anti‐CmABCB1 macrocyclic peptide, aCAP (Figures 1A and 4B), served as a cocrystallization ligand, improving the resolution to 2.4 Å. The authors suggest that the macrocyclic peptides (one aCAP molecule per transporter monomer) limit the movement of the transmembrane helices leading to the aforementioned improvement of resolution. Fortunately, the overall conformation of the transporter in the X‐ray crystal structures differed little in the presence or absence of aCAP. Despite its small size, aCAP was able to bind to the less contoured outer surface of the homo‐dimeric transporter in a protein–protein interaction‐like manner, providing crystallographic support for the use of macrocyclic peptides as potential protein–protein interaction inhibitors, a role small molecules are unable to fill.
The hepatocyte growth factor (HGF) receptor (also termed Met or cMet) is a class IV receptor tyrosine kinase (RTK) that interacts with HGF via its extracellular domain to form Met‐HGF dimers. Dimerization of two Met receptors promotes autophosphorylation of intracellular tyrosine residues, which in turn activates a range of intracellular signal transducers. Abnormal Met activation promotes oncogenesis and malignant transformation in various tissues. Met also plays a vital role in embryonic development and wound healing; its activation could have applications in regenerative medicine. Three anti‐Met macrocyclic peptides were identified using the RaPID system and were found to strongly bind to the Met ectodomain.10 Linear versions of these macrocyclic peptides showed much lower affinity, while scrambling the sequence resulted in a loss of binding activity. In contrast to human HGF, the peptides did not cross‐bind murine and canine ectodomains of Met. Although the peptides show high affinity for MET, they do not compete with human HGF binding nor inhibit signal activation by HGF, which suggests that they have different binding sites. To achieve dimerization of the Met receptor, the sulfhydryl groups of the two peptides’ C‐terminal cysteines were crosslinked using one of three bis‐maleimide cross‐linkers of different lengths (Carbon 6 (C6), PEG3, or PEG11) to produce macrocyclic peptide homodimers (Figure 4C). The different peptides required different cross‐linker lengths for optimal binding, suggesting that they bind to different regions. Despite differences in binding sites of HGF and the synthetic macrocyclic peptide homodimer, the cellular responses observed in various cell lines and normal human cells are identical to the response produced by human HGF, therefore these macrocyclic peptides are of great therapeutic interest, especially in regenerative medicine.
Epithelial cell adhesion molecule (EpCAM) is a transmembrane glycoprotein overexpressed on various carcinoma cells, especially cancer stem cells, and serves as a valuable biomarker and drug target. A macrocyclic peptide against the extracellular domain of EpCAM (ex‐EpCAM) was selected and developed into a fluorescently labelled probe by attaching fluorescein to the ε‐amino group of the C‐terminal lysine121 (Figure 4D). The probe could clearly bind to the membranes of live cells in a manner similar to that of conventional anti‐EpCAM antibodies. In contrast to antibody probes, the macrocyclic peptide probes were able to stain not only surface cells but also cells deeper within the tissue, indicating the great potential of macrocyclic peptide for targeting cells deep in tissues or tumours for specific binding and/or delivery of imagining agents or cytotoxic drugs.
5. Conclusions and Future Perspectives
Antibodies are still one of the most used and well‐investigated classes of high‐affinity molecules, with an enormous variety of applications in research, imaging and therapeutics. Various new ligands have been discovered, many of which are of much smaller size compared to mAbs. One particularly interesting class of molecules is the macrocyclic peptides. The macrocyclic scaffold increases the peptide's resistance to degradation by proteases and improves their binding affinity and specificity over their linear counterparts. As the average size of macrocyclic peptides falls between small molecules and biologics, they could potentially combine the beneficial characteristics of both classes.
The RaPID system allows for the incorporation of npAAs not only for the production of the macrocyclic scaffold but also increasing the chemical diversity of the library with N‐methyl amino acids9 and mechanistic warheads.123 Also exciting is their utility as co‐crystallization ligands.125, 126, 127 The methods of binding observed in the X‐ray cocrystal structures also have important implications in drug discovery. From the cocrystal structures, it has been observed that in vitro selected macrocyclic peptides can bind to accessible protein pockets125, 126 or to flat protein surfaces.127 The study of MET signal‐activating macrocyclic peptide dimers demonstrates a completely new mechanism of action for macrocyclic peptides in medicine. One may imagine that bi‐specific or multimeric macrocyclic peptide constructs will be studied in the near future. Finally, the EpCAM‐binding macrocyclic peptides were shown to penetrate deeper into tissue than antibody probes, suggesting that macrocyclic peptides may be more effective than antibodies at the delivery of therapeutic drugs to deeply buried cells in tissues or tumours. Macrocyclic peptides have proved to be as versatile as antibodies, and even surpass antibodies in some categories, that is, chemical diversity, deep pocket binding, tissue penetration. With recent developments that have decreased the time required for selection of macrocyclic peptides,118 macrocyclic peptide research will only continue to accelerate.
Funding information Contract grant sponsor: Ministry of Education, Culture, Sports, Science, and Technology
References
- 1. Köhler G., Milstein C., Nature 1975, 256, 495. [DOI] [PubMed] [Google Scholar]
- 2. Hansel T. T., Kropshofer H., Singer T., Mitchell J. A., George A. J. T., Nat Rev Drug Discov 2010, 9, 325. [DOI] [PubMed] [Google Scholar]
- 3. Yang Y., J Clin Invest 2015, 125, 3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drewe E., Powell R. J., J Clin Pathol 2002, 55, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vonderheide R. H., Glennie M. J., Clin Cancer Res 2013, 19, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoshino T., Yamazaki K., Gotoh M., Nasroulah F., Gao L., Yoshizuka N., Ohtsu A., Anticancer Res 2015, 35, 4003. [PubMed] [Google Scholar]
- 7. Hermann L. L., Thaisomboonsuk B., Poolpanichupatam Y., Jarman R. G., Kalayanarooj S., Nisalak A., Yoon I. K., Fernandez S., PLoS Negl Trop Dis 2014, 8, e3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shimizu Y., Furuya H., Bryant Greenwood P., Chan O., Dai Y., Thornquist M. D., Goodison S., Rosser C. J., J Transl Med 2016, 14, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamagishi Y., Shoji I., Miyagawa S., Kawakami T., Katoh T., Goto Y., Suga H., Chem Biol 2011, 18, 1562. [DOI] [PubMed] [Google Scholar]
- 10. Ito K., Sakai K., Suzuki Y., Ozawa N., Hatta T., Natsume T., Matsumoto K., Suga H., Nat Commun 2015, 6, 6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finking R., Marahiel M. A., Annu Rev Microbiol 2004, 58, 453. [DOI] [PubMed] [Google Scholar]
- 12. Arnison P. G., Bibb M. J., Bierbaum G., Bowers A. A., Bugni T. S., Bulaj G., Camarero J. A., Campopiano D. J., Challis G. L., Clardy J., Cotter P. D., Craik D. J., Dawson M., Dittmann E., Donadio S., Dorrestein P. C., Entian K. D., Fischbach M. A., Garavelli J. S., Göransson U., Gruber C. W., Haft D. H., Hemscheidt T. K., Hertweck C., Hill C., Horswill A. R., Jaspars M., Kelly W. L., Klinman J. P., Kuipers O. P., Link A. J., Liu W., Marahiel M. A., Mitchell D. A., Moll G. N., Moore B. S., Müller R., Nair S. K., Nes I. F., Norris G. E., Olivera B. M., Onaka H., Patchett M. L., Piel J., Reaney M. J. T., Rebuffat S., Ross R. P., Sahl H. G., Schmidt E. W., Selsted M. E., Severinov K., Shen B., Sivonen K., Smith L., Stein T., Süssmuth R. D., Tagg J. R., Tang G. L., Truman A. W., Vederas J. C., Walsh C. T., Walton J. D., Wenzel S. C., Willey J. M., van der Donk W. A., Nat Prod Rep 2013, 30, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith G. P., Science 1985, 228, 1315. [DOI] [PubMed] [Google Scholar]
- 14. Nissim A., Hoogenboom H. R., Tomlinson I. M., Flynn G., Midgley C., Lane D., Winter G., Embo J 1994, 13, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Winter G., Griffiths A. D., Hawkins R. E., Hoogenboom H. R., Annu Rev Immunol 1994, 12, 433. [DOI] [PubMed] [Google Scholar]
- 16. Stoyanova V., Aleksandrov R., Lukarska M., Duhalov D., Atanasov V., Petrova S., Toxicon Off J Int Soc Toxinol 2012, 60, 802. [DOI] [PubMed] [Google Scholar]
- 17. Nemoto N., Miyamoto‐Sato E., Husimi Y., Yanagawa H., FEBS Lett 1997, 414, 405. [DOI] [PubMed] [Google Scholar]
- 18. Ellington A. D., Szostak J. W., Nature 1990, 346, 818. [DOI] [PubMed] [Google Scholar]
- 19. Roberts R. W., Szostak J. W., Proc Natl Acad Sci USA 1997, 94, 12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hipolito C. J., Suga H., Curr Opin Chem Biol 2012, 16, 196. [DOI] [PubMed] [Google Scholar]
- 21. Miller K. R., Koide A., Leung B., Fitzsimmons J., Yoder B., Yuan H., Jay M., Sidhu S. S., Koide S., Collins E. J., PLoS One 2012, 7, e43746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oliveira S., Heukers R., Sornkom J., Kok R. J., van Bergen En Henegouwen P. M. P., J Control Release 2013, 172, 607. [DOI] [PubMed] [Google Scholar]
- 23. Alley S. C., Okeley N. M., Senter P. D., Curr Opin Chem Biol 2010, 14, 529. [DOI] [PubMed] [Google Scholar]
- 24. Khalil M., Vonderheide R. H., Update Cancer Ther 2007, 2, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu Y., Zhong Z., Huber J., Bassi R., Finnerty B., Corcoran E., Li H., Navarro E., Balderes P., Jimenez X., Koo H., Mangalampalli V. R. M., Ludwig D. L., Tonra J. R., Hicklin D. J., Clin Cancer Res 2006, 12, 6573. [DOI] [PubMed] [Google Scholar]
- 26. Scott A. M., Wolchok J. D., Old L. J., Nat Rev Cancer 2012, 12, 278. [DOI] [PubMed] [Google Scholar]
- 27. Hamblett K. J., Kozlosky C. J., Siu S., Chang W. S., Liu H., Foltz I. N., Trueblood E. S., Meininger D., Arora T., Twomey B., Vonderfecht S. L., Chen Q., Hill J. S., Fanslow W. C., Mol Cancer Ther 2015, 14, 1614. [DOI] [PubMed] [Google Scholar]
- 28. Li J. Y., Perry S. R., Muniz‐Medina V., Wang X., Wetzel L. K., Rebelatto M. C., Hinrichs M. J. M., Bezabeh B. Z., Fleming R. L., Dimasi N., Feng H., Toader D., Yuan A. Q., Xu L., Lin J., Gao C., Wu H., Dixit R., Osbourn J. K., Coats S. R., Cancer Cell 2016, 29, 117. [DOI] [PubMed] [Google Scholar]
- 29. Carpenter E. P., Beis K., Cameron A. D., Iwata S., Curr Opin Struct Biol 2008, 18, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hino T., Iwata S., Murata T., Curr Opin Struct Biol 2013, 23, 563. [DOI] [PubMed] [Google Scholar]
- 31. Day P. W., Rasmussen S. G. F., Parnot C., Fung J. J., Masood A., Kobilka T. S., Yao X. J., Choi H. J., Weis W. I., Rohrer D. K., Kobilka B. K., Nat Methods 2007, 4, 927. [DOI] [PubMed] [Google Scholar]
- 32. Oosting S. F., Brouwers A. H., Es S. C. V., Nagengast W. B., Munnink T. H. O., Hooge M. N. L., Hollema H., Jong J. R. D., Jong I. J. D., Haas S. D., Scherer S. J., Sluiter W. J., Dierckx R. A., Bongaerts A. H. H., Gietema J. A., Vries E. G. E. D., J Nucl Med 2015, 56, 63. 25476536 [Google Scholar]
- 33. Rolle A. M., Hasenberg M., Thornton C. R., Solouk‐Saran D., Männ L., Weski J., Maurer A., Fischer E., Spycher P. R., Schibli R., Boschetti F., Stegemann‐Koniszewski S., Bruder D., Severin G. W., Autenrieth S. E., Krappmann S., Davies G., Pichler B. J., Gunzer M., Wiehr S., Proc Natl Acad Sci USA 2016, 113, E1026–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fan L., Tian Y., Yin R., Lou D., Zhang X., Wang M., Ma M., Luo S., Li S., Gu N., Zhang Y., Nanoscale 2016, 8, 8553–8558. [DOI] [PubMed] [Google Scholar]
- 35. Silverton E. W., Navia M. A., Davies D. R., Proc Natl Acad Sci USA 1977, 74, 5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hughes B., Nat Rev Drug Discov 2010, 9, 665. [DOI] [PubMed] [Google Scholar]
- 37. Wu A. M., Methods San Diego Calif 2014, 65, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kontermann R. E., Curr Opin Biotechnol 2011, 22, 868. [DOI] [PubMed] [Google Scholar]
- 39. Biocca S., Neuberger M. S., Cattaneo A., Embo J 1990, 9, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jendreyko N., Popkov M., Beerli R. R., Chung J., McGavern D. B., Rader C., Barbas C. F., J Biol Chem 2003, 278, 47812. [DOI] [PubMed] [Google Scholar]
- 41. Bird R. E., Hardman K. D., Jacobson J. W., Johnson S., Kaufman B. M., Lee S. M., Lee T., Pope S. H., Riordan G. S., Whitlow M., Science 1988, 242, 423. [DOI] [PubMed] [Google Scholar]
- 42. Wheeler Y. Y., Chen S. Y., Sane D. C., Mol Ther 2003, 8, 355. [DOI] [PubMed] [Google Scholar]
- 43. Alvarez R. D., Barnes M. N., Gomez‐Navarro J., Wang M., Strong T. V., Arafat W., Arani R. B., Johnson M. R., Roberts B. L., Siegal G. P., Curiel D. T., Clin Cancer Res 2000, 6, 3081. [PubMed] [Google Scholar]
- 44. Fang J., Qian J. J., Yi S., Harding T. C., Tu G. H., VanRoey M., Jooss K., Nat Biotechnol 2005, 23, 584. [DOI] [PubMed] [Google Scholar]
- 45. Jiang M., Shi W., Zhang Q., Wang X., Guo M., Cui Z., Su C., Yang Q., Li Y., Sham J., Liu X., Wu M., Qian Q., Clin Cancer Res 2006, 12, 6179. [DOI] [PubMed] [Google Scholar]
- 46. Marschall A. L., Dübel S., Böldicke T., MAbs 2015, 7, 1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Glockshuber R., Malia M., Pfitzinger I., Plückthun A., Biochemistry (Mosc.) 1990, 29, 1362. [DOI] [PubMed] [Google Scholar]
- 48. Bessette P. H., Åslund F., Beckwith J., Georgiou G., Proc Natl Acad Sci USA 1999, 96, 13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jurado P., de Lorenzo V., Fernández L. A., J Mol Biol 2006, 357, 49. [DOI] [PubMed] [Google Scholar]
- 50. Visintin M., Quondam M., Cattaneo A., Methods San Diego Calif 2004, 34, 200. [DOI] [PubMed] [Google Scholar]
- 51. Visintin M., Meli G. A., Cannistraci I., Cattaneo A., J Immunol Methods 2004, 290, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Allen J. B., Walberg M. W., Edwards M. C., Elledge S. J., Trends Biochem Sci 1995, 20, 511. [DOI] [PubMed] [Google Scholar]
- 53. Marschall A. L. J., Single F. N., Schlarmann K., Bosio A., Strebe N., van den Heuvel J., Frenzel A., Dübel S., MAbs 2014, 6, 1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Amici C., Visintin M., Verachi F., Paolini F., Percario Z. D., Bonito P., Mandarino A., Affabris E., Venuti A., Accardi L., Oncotarget 2016, 7, 15539–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Paoletti F., Malerba F., Konarev P. V., Visintin M., Scardigli R., Fasulo L., Lamba D., Svergun D. I., Cattaneo A., Arch Biochem Biophys 2012, 522, 26. [DOI] [PubMed] [Google Scholar]
- 56. Dauvillier S., Mérida P., Visintin M., Cattaneo A., Bonnerot C., Dariavach P., J Immunol 2002, 169, 2274. [DOI] [PubMed] [Google Scholar]
- 57. Chiusaroli R., Visentini M., Galimberti C., Casseler C., Mennuni L., Covaceuszach S., Lanza M., Ugolini G., Caselli G., Rovati L. C., Visintin M., Osteoarthr Cartil OARS Osteoarthr Res Soc 2013, 21, 1807. [DOI] [PubMed] [Google Scholar]
- 58. Lundstrom K., Technol Cancer Res Treat 2004, 3, 467. [DOI] [PubMed] [Google Scholar]
- 59. Yang N., Int J Pharm Investig 2012, 2, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hamers‐Casterman C., Atarhouch T., Muyldermans S., Robinson G., Hamers C., Songa E. B., Bendahman N., Hamers R., Nature 1993, 363, 446. [DOI] [PubMed] [Google Scholar]
- 61. Cortez‐Retamozo V., Lauwereys M., Hassanzadeh Gh G., Gobert M., Conrath K., Muyldermans S., De Baetselier P., Revets H., Int J Cancer J Int Cancer 2002, 98, 456. [DOI] [PubMed] [Google Scholar]
- 62. De Meyer T., Muyldermans S., Depicker A., Trends Biotechnol 2014, 32, 263. [DOI] [PubMed] [Google Scholar]
- 63. Keyaerts M., Xavier C., Heemskerk J., Devoogdt N., Everaert H., Ackaert C., Vanhoeij M., Duhoux F. P., Gevaert T., Simon P., Schallier D., Fontaine C., Vaneycken I., Vanhove C., Greve J. D., Lamote J., Caveliers V., Lahoutte T., J Nucl Med 2016, 57, 27. [DOI] [PubMed] [Google Scholar]
- 64. Hassanzadeh‐Ghassabeh G., Devoogdt N., De Pauw P., Vincke C., Muyldermans S., Nanomed 2013, 8, 1013. [DOI] [PubMed] [Google Scholar]
- 65. Abbady A. Q., Al‐Daoude A., Al‐Mariri A., Zarkawi M., Muyldermans S., Vet Immunol Immunopathol 2012, 146, 254. [DOI] [PubMed] [Google Scholar]
- 66. Huang L., Gainkam L. O. T., Caveliers V., Vanhove C., Keyaerts M., De Baetselier P., Bossuyt A., Revets H., Lahoutte T., Mol Imaging Biol MIB Off Publ Acad Mol Imaging 2008, 10, 167. [DOI] [PubMed] [Google Scholar]
- 67. Vaneycken I., Devoogdt N., Gassen N. V., Vincke C., Xavier C., Wernery U., Muyldermans S., Lahoutte T., Caveliers V., FASEB J 2011, 25, 2433. [DOI] [PubMed] [Google Scholar]
- 68. Tijink B. M., Laeremans T., Budde M., Walsum M. S., Dreier T., Haard H. J. D., Leemans C. R., Dongen G. A. M. S. V., Mol Cancer Ther 2008, 7, 2288. [DOI] [PubMed] [Google Scholar]
- 69. Löw C., Yau Y. H., Pardon E., Jegerschöld C., Wåhlin L., Quistgaard E. M., Moberg P., Geifman‐Shochat S., Steyaert J., Nordlund P., PLoS One 2013, 8, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xu L., Aha P., Gu K., Kuimelis R. G., Kurz M., Lam T., Lim A. C., Liu H., Lohse P. A., Sun L., Weng S., Wagner R. W., Lipovsek D., Chem Biol 2002, 9, 933. [DOI] [PubMed] [Google Scholar]
- 71. Bloom L., Calabro V., Drug Discov Today 2009, 14, 949. [DOI] [PubMed] [Google Scholar]
- 72. Koide S., Koide A., Lipovšek D., Methods Enzymol 2012, 503, 135–156. [DOI] [PubMed] [Google Scholar]
- 73. Koide A., Bailey C. W., Huang X., Koide S., J Mol Biol 1998, 284, 1141. [DOI] [PubMed] [Google Scholar]
- 74. Stockbridge R. B., Koide A., Miller C., Koide S., Nat Commun 2014, 5, 5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lipovšek D., Protein Eng Des Sel 2011, 24, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wojcik J., Hantschel O., Grebien F., Kaupe I., Bennett K. L., Barkinge J., Jones R. B., Koide A., Superti‐Furga G., Koide S., Nat Struct Mol Biol 2010, 17, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Getmanova E. V., Chen Y., Bloom L., Gokemeijer J., Shamah S., Warikoo V., Wang J., Ling V., Sun L., Chem Biol 2006, 13, 549. [DOI] [PubMed] [Google Scholar]
- 78. Hackel B. J., Kapila A., Wittrup K. D., J Mol Biol 2008, 381, 1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tolcher A. W., Sweeney C. J., Papadopoulos K., Patnaik A., Chiorean E. G., Mita A. C., Sankhala K., Furfine E., Gokemeijer J., Iacono L., Eaton C., Silver B. A., Mita M., Clin Cancer Res Off J Am Assoc Cancer Res 2011, 17, 363. [DOI] [PubMed] [Google Scholar]
- 80. Park S. H., Park S., Kim D. Y., Pyo A., Kimura R. H., Sathirachinda A., Choy H. E., Min J. J., Gambhir S. S., Hong Y., PLoS One 2015, 10, e0132976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Waters J. D., Sanchez C., Sahin A., Futalan D., Gonda D. D., Scheer J. K., Akers J., Palanichamy K., Waterman P., Chakravarti A., Weissleder R., Morse B., Marsh N., Furfine E., Chen C. C., Carvajal I., Carter B. S., J. Neurooncol 2012, 110, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schiff D., Kesari S., Groot J. D., Mikkelsen T., Drappatz J., Coyle T., Fichtel L., Silver B., Walters I., Reardon D., Invest New Drugs 2014, 33, 247. [DOI] [PubMed] [Google Scholar]
- 83. Uhlén M., Guss B., Nilsson B., Gatenbeck S., Philipson L., Lindberg M., J Biol Chem 1984, 259, 1695. [PubMed] [Google Scholar]
- 84. Myers J. K., Oas T. G., Nat Struct Biol 2001, 8, 552. [DOI] [PubMed] [Google Scholar]
- 85. Nilsson B., Moks T., Jansson B., Abrahmsén L., Elmblad A., Holmgren E., Henrichson C., Jones T. A., Uhlén M., Protein Eng 1987, 1, 107. [DOI] [PubMed] [Google Scholar]
- 86. Orlova A., Magnusson M., Eriksson T. L. J., Nilsson M., Larsson B., Höidén‐Guthenberg I., Widström C., Carlsson J., Tolmachev V., Ståhl S., Nilsson F. Y., Cancer Res 2006, 66, 4339. [DOI] [PubMed] [Google Scholar]
- 87. Löfblom J., Feldwisch J., Tolmachev V., Carlsson J., Ståhl S., Frejd F. Y., FEBS Lett 2010, 584, 2670. [DOI] [PubMed] [Google Scholar]
- 88. Nord K., Gunneriusson E., Ringdahl J., Ståhl S., Uhlén M., Nygren P. A., Nat Biotechnol 1997, 15, 772. [DOI] [PubMed] [Google Scholar]
- 89. Nord K., Nilsson J., Nilsson B., Uhlén M., Nygren P. A., Protein Eng 1995, 8, 601. [DOI] [PubMed] [Google Scholar]
- 90. Engfeldt T., Renberg B., Brumer H., Nygren P. A., Karlström A. E., Chembiochem Eur J Chem Biol 2005, 6, 1043. [DOI] [PubMed] [Google Scholar]
- 91. Miao Z., Levi J., Cheng Z., Amino Acids 2011, 41, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sörensen J., Sandberg D., Sandström M., Wennborg A., Feldwisch J., Tolmachev V., Åström G., Lubberink M., Garske‐Román U., Carlsson J., Lindman H., J Nucl Med 2014, 55, 730. [DOI] [PubMed] [Google Scholar]
- 93. Su X., Cheng K., Jeon J., Shen B., Venturin G. T., Hu X., Rao J., Chin F. T., Wu H., Cheng Z., Mol Pharm 2014, 11, 3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zielinski R., Lyakhov I., Jacobs A., Chertov O., Kramer‐Marek G., Francella N., Stephen A., Fisher R., Blumenthal R., Capala J., J Immunother Hagerstown MD 1997 2009, 32, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Honarvar H., Westerlund K., Altai M., Sandström M., Orlova A., Tolmachev V., Karlström A. E., Theranostics 2016, 6, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Petcher T. J., Weber H. P., Rüegger A., Helv Chim Acta 1976, 59, 1480. [DOI] [PubMed] [Google Scholar]
- 97. Sako Y., Goto Y., Murakami H., Suga H., ACS Chem Biol 2008, 3, 241. [DOI] [PubMed] [Google Scholar]
- 98. Goto Y., Ohta A., Sako Y., Yamagishi Y., Murakami H., Suga H., ACS Chem Biol 2008, 3, 120. [DOI] [PubMed] [Google Scholar]
- 99. Dittmann J., Wenger R. M., Kleinkauf H., Lawen A., J Biol Chem 1994, 269, 2841. [PubMed] [Google Scholar]
- 100. Madala P. K., Tyndall J. D. A., Nall T., Fairlie D. P., Chem Rev 2010, 110, PR1. [DOI] [PubMed] [Google Scholar]
- 101. Nielsen D. S., Hoang H. N., Lohman R. J., Hill T. A., Lucke A. J., Craik D. J., Edmonds D. J., Griffith D. A., Rotter C. J., Ruggeri R. B., Price D. A., Liras S., Fairlie D. P., Angew Chem Int Ed Engl 2014, 53, 12059. [DOI] [PubMed] [Google Scholar]
- 102. Giordanetto F., Kihlberg J., J Med Chem 2014, 57, 278. [DOI] [PubMed] [Google Scholar]
- 103. Veronese F. M., Pasut G., Drug Discov Today 2005, 10, 1451. [DOI] [PubMed] [Google Scholar]
- 104. Baggio L. L., Huang Q., Cao X., Drucker D. J., Gastroenterology 2008, 134, 1137. [DOI] [PubMed] [Google Scholar]
- 105. Havelund S., Plum A., Ribel U., Jonassen I., Vølund A., Markussen J., Kurtzhals P., Pharm Res 2004, 21, 1498. [DOI] [PubMed] [Google Scholar]
- 106. Penchala S. C., Miller M. R., Pal A., Dong J., Madadi N. R., Xie J., Joo H., Tsai J., Batoon P., Samoshin V., Franz A., Cox T., Miles J., Chan W. K., Park M. S., Alhamadsheh M. M., Nat Chem Biol 2015, 11, 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Northfield S. E., Wang C. K., Schroeder C. I., Durek T., Kan M. W., Swedberg J. E., Craik D. J., Eur J Med Chem 2014, 77, 248. [DOI] [PubMed] [Google Scholar]
- 108. Ji Y., Majumder S., Millard M., Borra R., Bi T., Elnagar A. Y., Neamati N., Shekhtman A., Camarero J. A., J Am Chem Soc 2013, 135, 11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Huang Y. H., Chaousis S., Cheneval O., Craik D. J., Henriques S. T., Front Pharmacol 2015, 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Swedberg J. E., Nigon L. V., Reid J. C., de Veer S. J., Walpole C. M., Stephens C. R., Walsh T. P., Takayama T. K., Hooper J. D., Clements J. A., Buckle A. M., Harris J. M., Chem Biol 2009, 16, 633. [DOI] [PubMed] [Google Scholar]
- 111. Swedberg J. E., de Veer S. J., Sit K. C., Reboul C. F., Buckle A. M., Harris J. M., PLoS One 2011, 6, e19302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. de Veer S. J., Swedberg J. E., Parker E. A., Harris J. M., Biol Chem 2012, 393, 331. [DOI] [PubMed] [Google Scholar]
- 113. de Veer S. J., Swedberg J. E., Akcan M., Rosengren K. J., Brattsand M., Craik D. J., Harris J. M., Biochem J 2015, 469, 243. [DOI] [PubMed] [Google Scholar]
- 114. Conibear A. C., Craik D. J., Angew Chem Int Ed 2014, 53, 10612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Conibear A. C., Bochen A., Rosengren K. J., Stupar P., Wang C., Kessler H., Craik D., J Chembiochem Eur J Chem Biol 2014, 15, 451. [DOI] [PubMed] [Google Scholar]
- 116. Conibear A. C., Rosengren K. J., Daly N. L., Henriques S. T., Craik D. J., J Biol Chem 2013, 288, 10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Heinis C., Winter G., Curr Opin Chem Biol 2015, 26, 89. [DOI] [PubMed] [Google Scholar]
- 118. Ishizawa T., Kawakami T., Reid P. C., Murakami H., J Am Chem Soc 2013, 135, 5433. [DOI] [PubMed] [Google Scholar]
- 119. Murakami H., Ohta A., Ashigai H., Suga H., Nat Methods 2006, 3, 357. [DOI] [PubMed] [Google Scholar]
- 120. Rogers J. M., Suga H., Org Biomol Chem 2015, 13, 9353. [DOI] [PubMed] [Google Scholar]
- 121. Iwasaki K., Goto Y., Katoh T., Yamashita T., Kaneko S., Suga H., J Mol Evol 2015, 81, 210. [DOI] [PubMed] [Google Scholar]
- 122. Hewitt W. M., Leung S. S. F., Pye C. R., Ponkey A. R., Bednarek M., Jacobson M. P., Lokey R. S., J Am Chem Soc 2015, 137, 715. [DOI] [PubMed] [Google Scholar]
- 123. Morimoto J., Hayashi Y., Suga H., Angew Chem Int Ed 2012, 51, 3423. [DOI] [PubMed] [Google Scholar]
- 124. Hayashi Y., Morimoto J., Suga H., ACS Chem Biol 2012, 7, 607. [DOI] [PubMed] [Google Scholar]
- 125. Tanaka Y., Hipolito C. J., Maturana A. D., Ito K., Kuroda T., Higuchi T., Katoh T., Kato H. E., Hattori M., Kumazaki K., Tsukazaki T., Ishitani R., Suga H., Nureki O., Nature 2013, 496, 247. [DOI] [PubMed] [Google Scholar]
- 126. Hipolito C. J., Tanaka Y., Katoh T., Nureki O., Suga H., Molecules 2013, 18, 10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kodan A., Yamaguchi T., Nakatsu T., Sakiyama K., Hipolito C. J., Fujioka A., Hirokane R., Ikeguchi K., Watanabe B., Hiratake J., Kimura Y., Suga H., Ueda K., Kato H., Proc Natl Acad Sci USA 2014, 111, 4049. [DOI] [PMC free article] [PubMed] [Google Scholar]